
Abstract
Metallic iron (Fe0) has shown outstanding performances for water decontamination and its efficiency has been improved by the presence of sand (Fe0/sand) and manganese oxide (Fe0/MnOx). In this study, a ternary Fe0/MnOx/sand system is characterized for its discoloration efficiency of methylene blue (MB) in quiescent batch studies for 7, 18, 25 and 47 days. The objective was to understand the fundamental mechanisms of water treatment in Fe0/H2O systems using MB as an operational tracer of reactivity. The premise was that, in the short term, both MnO2 and sand delay MB discoloration by avoiding the availability of free iron corrosion products (FeCPs). Results clearly demonstrate no monotonous increase in MB discoloration with increasing contact time. As a rule, the extent of MB discoloration is influenced by the diffusive transport of MB from the solution to the aggregates at the bottom of the vessels (test-tubes). The presence of MnOx and sand enabled the long-term generation of iron hydroxides for MB discoloration by adsorption and co-precipitation. Results clearly reveal the complexity of the Fe0/MnOx/sand system, while establishing that both MnOx and sand improve the efficiency of Fe0/H2O systems in the long-term. This study establishes the mechanisms of the promotion of water decontamination by amending Fe0-based systems with reactive MnOx.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
Since the 1950s the world is conventionally divided into two groups with regards to the access to safe drinking water: (i) developed countries striving for selecting the best available technology for water treatment, and (ii) developing countries striving for making some appropriate technologies available for their mostly low-income and rural populations1,2,3. In the meantime, cities have grown, but drinking water systems in peri-urban areas are comparable to that of rural environments without piped water4,5,6. Appropriate technologies are essentially considered as interim solutions wherever a centralized water supply is not yet available1. Factors relevant for selecting an appropriate solution for safe drinking water supply include1,7,8: (i) Simplicity in operation (no special skilled personnel), (ii) robustness (no frequent break downs), (iii) affordability (low installation and operation costs), (iv) ability to function without electricity, and (v) use of local skills and readily available resources. Research during the past two decades has rediscovered filtration systems based on metallic iron (Fe0) as an affordable, applicable, and efficient water treatment technology for decentralized water supply (e.g. households and small communities)3,7,9,10,11,12,13. Such Fe0 filters are only sustainable upon admixing Fe0 with other aggregates like granular activated carbon, biochar, gravel, magnetite (Fe3O4), manganese oxides (MnOx), pyrite (FeS2), and sand10,14,15.
There are two fundamental challenges in designing Fe0 filtration systems: (i) “reactivity loss” and (ii) permeability loss16,17,18,19,20. Permeability loss is not addressed herein. For the presentation, it suffices to recall that this issue has been partly resolved in earlier studies demonstrating that only hybrid Fe0 filters are sustainable14,21. The remaining task is a temporal issue regarding the reaction kinetics of Fe0 corrosion, which is material-specific and has received limited attention22,23,24. Reactivity loss is the expression of the inherent time-dependent decrease of the Fe0 corrosion rate as well-documented in the corrosion literature and referred to as ‘passivation’25,26,27,28. However, “reactivity loss” has been introduced in the post-1990 literature to characterize the limited electron transfer from the metal body to some dissolved contaminants16,19. Given that under natural conditions Fe0 is corroded only by protons from water dissociation (Eq. 1)29, Miyajima and Noubactep30 argued that reactivity loss is a mirage. In fact, “reactivity loss” has also occurred in Fe0-based permeable reactive barriers successfully working for up to two decades31,32,33,34. On the other hand, Roh et al.35 reported on Fe0 specimens from World War I still corroding in soils. Clearly, it can be argued that the old motto “rust never rests” is valid for Fe0 filters where corrosion additionally occurs under immersed conditions. The question is, how to ensure that Fe0 oxidation with changing corrosion rates still secures clean water in the long-term?
During the past decade, substantial experiences have been accumulated on increasing the efficiency of Fe0/H2O systems by admixing Fe0 with other materials (Table 1)15,36. However, these efforts were mostly misled by the misconception that Fe0 is a reducing agent36. Fortunately, available data can be re-interpreted based on the chemistry of the system. It suffices to consider that reduction is not a relevant contaminant removal mechanism, and that contaminant reduction is never mediated by electrons from the metal body11. For example, MnO2 is not reduced by Fe0 (Eq. 2), but rather by Fe2+ (Eq. 3a) (Fig. 1). Equation 3b depicts that MnO2 reductive dissolution by Fe2+ induces acidification of the system (releases protons). O2 and other dissolved species are equally reduced by Fe2+ and other reductive species present in the Fe0/H2O system (e.g., H2, Fe3O4, green rust)37,38,39,40. Thus, it is established that contaminants are reduced by an indirect mechanism (Fig. 1), and that this process continues even after virtual surface passivation (which is thus not a “loss of their reactivity”). Successful efforts to overcome Fe0 passivation include the addition of gravel41,42, magnetite15,43, MnOx44,45, pyrite36,46, and sand47,48.

Scheme showing the pathways of contaminant reductive transformations in Fe0/MnO2/H2O systems. Only water has access to the metal surface. Fe2+ and H2 are stand-alone reducing agents. MnO2 and other relevant dissolved species (e.g. RX) are reduced by Fe2+ and H2. Upon the oxidation of Fe2+, various solid iron hydroxides/oxides (e.g. Fe(OH)3) precipitate and act as contaminant scavengers. RX stands for an halogenated hydrocarbon. The key information is that MnO2 is not reduced by Fe0.
The presence of inert sand improves the efficiency of even batch Fe0/H2O systems for water treatment49. However, the studies testing other reactive materials have not considered inert systems (e.g., sand) as operational references. Only Ndé-Tchoupé et al.50 did such a comparison. However, the objective was to test pozzolan as an alternative filling material to sand for Fe0 filters. In other words, while testing magnetite (Fe3O4) as admixing agent for the reductive transformation of contaminants43, a reference Fe0/sand should have been considered in parallel experiments. The inclusion of an operational reference enables a better understanding of the specific action of the reactive additive (here Fe3O4). Following the science of aqueous iron corrosion under environmental conditions25,28, this study premises that iron passivation is delayed by avoiding the precipitation of iron corrosion products in the vicinity of the metal. Thus, Fe2+ and Fe3+ ions are consumed instead of coating sand in Fe0/sand systems, and Fe2+ ions are additionally consumed in the reductive dissolution of MnOx (Eq. 2) in the Fe0/MnOx/sand systems (Fig. 1). Note that all other aggregates including Fe3O4 and granular activated carbon are in-situ coated by FeCPs like sand and the postulated effects are not realizable in the long-term (Table 1).
Fe0 is used to efficiently remove various contaminants such as turbidity, pathogens, and dissolved species from aqueous solutions51,52,53,54,55,56,57,58,59,60. Chemical pollutants in the aqueous phase can be ions, molecules, and colloids. For reducible dissolved species, there is a trend to consider Fe0-based materials (E0 = − 0.44 V) as (strong) reducing agent51,56,60, and contaminant reductive transformation as an electrochemical process58. It is evident that colloids, pathogens, and suspended solids (turbidity) are not removed by any chemical reaction. Rather, they are removed via coagulation and co-precipitation. The previous text has already demonstrated that no electrochemical contaminant reduction is possible in a Fe0/H2O system58. Yet, published results using the Fe0/MnO2 mixtures are mainly premised on the wrong assumption that contaminant removal occurs via reduction by electrons from Fe0 (Fig. 1) (Table 1)6,54,57. Accordingly, there is still a need to further clarify the role of MnO2 in sustaining the efficiency of Fe0/H2O systems for water treatment59. In particular, there is need to elucidate how insoluble Fe(OH)3 contribute to the co-precipitation of pollutants from the aqueous phase.
The objective of this study is to investigate the impact of MnOx addition on the efficiency of Fe0/H2O systems for MB discoloration as a function of the experimental duration (contact time). The specific objective is to confirm the suitability of ‘MB discoloration’ as powerful tool for the characterization of decontaminantion processes in Fe0/H2O systems while using MnOx and sand to control the availability of ‘free’ FeCPs. The extent of MB discoloration is investigated in five different systems: (i) Fe0 alone, (ii) sand alone, (iii) Fe0/sand, (iv) Fe0/MnOx, and (v) Fe0/MnOx/sand for 7, 18, 25 and 47 days. A comparison of the results from the five systems provides critical information on the contaminant removal mechanisms and the role of MnOx.
Materials and methods
The theory of iron and manganese cycle in a Fe0/MnOx/sand system
Initially (t0 = 0), when Fe0, MnOx and sand are put into the solution, there is no dissolved iron and no dissolved manganese in the system (Table 2). At t > t0, Fe0 is dissolved by protons (water) to generate H2 and Fe2+ (Eq. 1). Fe2+ induces the reductive dissolution of MnOx (Eq. 3)61,62,63,64,65. At t > t0, the Fe0/MnOx/sand system hosts dynamic processes which might continue after Fe0 depletion. In fact, the mixture of Fe and MnOx minerals is a very complex reactive system that has been investigated for more that a century66,67,68. The uniqueness of the Fe0/MnOx/sand system is that Fe minerals are generated in-situ and are comparatively more reactive than aged minerals like goethite or hematite. Because the pH of the system is larger than 5.0, in the absence of ligands, Fe and Mn hydroxides have very low solubility and precipitate not far away from their points of nucleation65,69. The dynamics within the Fe0/MnOx/sand entail a series of interchanges of iron and manganese from older to younger forms as follows: (i) dissolution of Fe0 and MnOx, (ii) migration of Fe2+, Fe3+ and Mn2+ from the areas of their generation to areas where precipitation will occur, and (iii) precipitation in one or more forms of iron and manganese hydroxide.
In the Fe0/MnOx/sand system, iron and manganese chemically precipitate at the surface of MnOx, sand or in the bulk solution. Due to the good adsorptive affinities of Fe2+ and Fe3+ for sand surface, it is assumed that the deposition of Fe hydroxides at its surface (coating) will compete with Fe2+ consumption by the reductive dissolution of MnOx until sand coating is completed. Thereafter, the “free” precipitation of iron and manganese occurs and the final products are deposits of more or less pure iron and manganese ores66,71. In other words, the investigated Fe0/MnOx/sand system is a ternary system only at the start of the experiment. It then turns to a mixture of Fe0, iron oxide-coated sand, iron oxide-coated MnOx, Fe/Mn shales, etc. Even after Fe0 depletion, the Fe/Mn mineral mixture will still be a reactive one, with a great potential for water treatment by both abiotic and biotic processes67,68,72.
Experimental details
This experimental section is adapted from Cao et al.70 using the same experimental design and two more MnO2 minerals.
Solutions
The used methylene blue (MB—Basic Blue 9 from Merck) was of analytical grade. The working solution was 10.0 mg L–1 prepared by diluting a 1000 mg L–1 stock solution. The stock solution was prepared by dissolving accurately weighted MB in tap water. The use of tap water rather than deionised water was motivated by the fact that tap water is closer to natural water in its chemical composition. The MB molecular formula is C16H18N3SCl corresponding to a molecular weight of 319.85 g. MB was chosen in this study because of its well-known strong adsorption onto solids70.
Solid materials
Metallic iron (Fe0)
The used Fe0 material was purchased from iPutech (Rheinfelden, Germany). The material is available as filings with a particle size between 0.3 and 2.0 mm. Its elemental composition as specified by the supplier was: C: 3.52%; Si: 2.12%; Mn: 0.93%; Cr: 0.66% while the balance was Fe. The material was used without any further pre-treatment. Fe0 was proven as a powerful discoloration agent for MB given that discoloration agents in the form of FeCPs are progressively generated in-situ70.
Manganese dioxide (MnO2)
The tested natural MnO2-bearing minerals was Manganit from Ilfeld/Harz, Thüringen (Germany). The mineral was crushed and fractionated by sieving. The fraction 0.5–1.0 mm was used without any further pre-treatment. No chemical, mineralogical nor structural characterizations were performed. MnO2 is a reactive mineral73,74,75 and is used to delay the availability of ‘free’ iron corrosion products (FeCPs) in the system. This results in a delay of quantitative MB discoloration30.
Sand
The used sand was a commercial material for aviculture (“Papagaiensand” from RUT—Lehrte/Germany). The sand was used as received without any further pre-treatment. The particle size was between 2.0 and 4.0 mm. Sand was used as an adsorbent because of its worldwide availability and its use as admixing agent in Fe0 barriers50,76. The adsorption capacity of sand for MB has been systematically documented as early as in 1955 by Mitchell et al.77.
MB discoloration
Quiescent batch experiments (non-shaken) were conducted in assay tubes for experimental durations of 7, 18, 25 and 47 d. The batches consisted of 0.0 or 1.0 g of sand, 0.0 or 0.1 g to Fe0, 0.0 or 0.05 g of MnO2 and mixtures thereof in 22.0 mL of a 10.0 mg L–1 MB solution. The investigated systems were: (i) Fe0 alone, (ii) sand alone, (iii) MnO2 alone, (iv) Fe0/sand, (v) Fe0/MnO2 and (vi) Fe0/sand/MnO2. The efficiency of individual systems at discolouring MB was characterized at laboratory temperature (about 22 °C). Initial pH was about 8.2. After equilibration, up to 3.0 mL of the supernatant solutions were carefully retrieved (no filtration) for MB measurements (no dilution). Each experiment was performed in triplicates, and averaged values are presented. Table 3 summarizes the aggregate content of the 6 Fe0/MnO2/sand systems investigated herein. The operational reference (blank experiment) is also added. Note that the pure Fe0 system (Fe0 alone) is regarded as a ‘Fe0/MnO2/sand system’ without MnO2 nor sand.

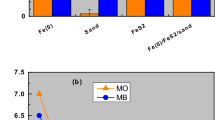
Comparison of the efficiency of tested materials for methylene blue (MB) discoloration for 7 (a) and 47 days (b). Experimental conditions: [Fe0] = 0 to 45 g L–1; [sand] = 45 g L–1; and [MnO2] = 2.3 g L–1.
Analytical methods
Iron and MB aqueous concentrations were determined by a Cary 50 UV–Vis spectrophotometer (Varian). The working wavelengths for MB and iron were 664.5 and 510.0 nm, respectively. Cuvettes with 1.0 cm light path were used. The spectrophotometer was calibrated for Fe and MB concentrations ≤ 10.0 mg L–1. The pH value was measured by combined glass electrodes (WTW Co., Germany).
Expression of MB discoloration results (E value)
In order to characterize the magnitude of the tested systems for MB discoloration, the discoloration efficiency (E) was calculated (Eq. 4). After the determination of the residual MB concentration (C), the corresponding percent MB discoloration (E value) was calculated as:
where, C0 is the initial aqueous MB concentration (ideally 10.0 mg L–1), while C gives the MB concentration after the experiment. The operational initial concentration (C0) for each case was acquired from a triplicate control experiment without additive material (so-called blank). This procedure was to account for experimental errors during dilution of the stock solution, MB adsorption onto the walls of the reaction vessels, and all other possible side reactions during the experiments.
Results and discussion
Evidence for the complexity of the Fe0/MnO2/sand systems
Figure 2 compares the extent of MB discoloration in the six investigated systems for 7 and 47 days. Figure 2a clearly shows that, after 7 d, only MnO2 had not significantly discolored MB (4%) while Fe0 alone depicts the best discoloration efficiency (62%). The E values for the other systems varied between 31 and 40%. The increasing order of efficiency was: MnO2 < sand < Fe0/MnO2 < Fe0/MnO2/sand < Fe0/sand < Fe0. These results can be regarded as counter-intuitive since binary (Fe0/MnO2, Fe0/sand) and ternary (Fe0/MnO2/sand) performed less than Fe0 alone. In conventional shaken or stirred batch experiments, involved processes are accelerated to the extent that achieved results are the intuitive ones observed after 47 days (Fig. 2b)78.
Figure 2b compares the extent of MB discoloration in the six systems after 47 days. Compared to the results after 7 days, the extent of MB discoloration has increased to more than 50% in all systems, except MnO2 alone. Based on the absolute E values, the increasing order of efficiency was: MnO2 (4%) < sand (51%) < Fe0/sand (66%) < Fe0/MnO2 = Fe0/MnO2/sand = Fe0 (72%). It is interesting to note that Fe0/sand performed less than Fe0 alone and the two MnO2-bearing systems. This observation alone confirms that MnO2-amendment enhances the efficiency of Fe0/H2O systems by “reinforcing” corrosion (Eq. 3), but only in the long-term. Thus, the complexity of the ternary system as well as the need to understand its operation model is apparent. This is achieved herein by investigating the systems for 7, 18, 25 and 47 days. This corresponds to following the fate of aqueous MB (discoloration) as the contact time increases from 7 to 47 days59,70,71. In particular the variation of the pH value in the systems will be discussed in detail.
Effect of the contact time on the Fe0/MnO2/sand system
Figure 3a compares the extent of MB discoloration in Fe0/MnO2/sand systems for the four tested contact times (7, 18, 25 and 47 d) and Fig. 3b depicts the corresponding changes in pH values. It is seen that the lowest extent of MB discoloration corresponds to 18 d contact time. This means that after 7 days the system performed better than after 18 d. The observation can be regarded as counter-intuitive, while the monotonous increase of the pH value (Fig. 3b) is intuitive. The investigated systems were 0 ≤ [Fe0] (g L–1) ≤ 45, with [MnO2] = 2.3 g L–1 and [sand] = 45 g L–1. This means that [Fe0] = 0.0 g L–1 corresponds to a MnO2/sand system or simplified to the sand system as MnO2 has no adsorptive affinities for MB (Fig. 2). In other words, the counter-intuitive observation corresponds to the effect of MnO2 on the Fe0/H2O system.

Changes in Fe0/sand/MnO2 systems as impacted by the addition of various Fe0 loading for 7, 18, 25 and 47 days: (a) Methylene blue discoloration, and (b) pH value. Experimental conditions: [Fe0] = 2.3 to 45 g L–1; [sand] = 22.5 g L–1; and [MnO2] = 2.3 g L–1. The lines are not fitting functions, they simply connect points to facilitate visualization.
A key feature from Fig. 3a is that there is an intuitive monotonous increase of the E value with increasing Fe0 loading for all four tested contact times. This suggests that if the experiments were performed by different investigators, the given interpretations would have been conclusive and even convincing. The tested experimental conditions were selected based on past works30,79 to achieved such results. In addition, most of the observations made by researchers in Fe0/H2O systems are just static snap-shots (mostly inaccurately measured) of processes occurring over an enormous range of time scales80. Following this premise, it was necessary to further vary the experimental conditions to maximize the chance to make more relevant observation81. One really intriguing observation is that the MB previously removed (t < 7 days) was released back to the solution at day 18 such that MB discoloration was lower even in the sand system (Fig. 3a).
This MB desorption is rationalized by the pH decrease accompanying MnO2 reductive precipitation as given in Eq. (3b). By decreasing the pH value, further adsorption onto sand is inhibited and the previously adsorbed MB is desorbed and released into solution (Fig. 3a). On the other hand, the process of Fe0 dissolution by MnO2 implies intensified interactions at the bottom of the assay tubes which slowed down the diffusion of MB from the bulk solution. Iron corrosion determined the extent of MB discoloration and the intuitive increase of MB discoloration with increasing Fe0 loading is observed in all systems only after a pseudo-steady state is established in the systems or the capacity of MnO2 is exhausted. Alyoussef59,70,71 tested a parallel system with 4.5 g L–1 of MnO2 and observed a larger decrease of MB discoloration for 18 days. Similar observations were made by Noubactep et al.82 in their experiments for uranium removal.
Figure 3b shows that for [Fe0] > 7.5 g L–1, the pH value monotonously increases with increasing Fe0 loading. For [Fe0] < 7.5 g L–1, there were some fluctuations justified by the co-occurrence of Fe0 corrosion (consuming protons—Eq. 1) and MnO2 reductive dissolution (producing protons—Eq. 3b) to fix the pH of the systems. Again, once the oxidation capacity of MnO2 is exhausted, iron corrosion controls the pH of the system.
The comparative evaluation of the time-dependent changes of E and pH values has clarified the operating mode of MnO2 in enhancing the efficiency of the Fe0/H2O system without any solid phase characterization. This discussion has equally not considered the redox reactivity of MnO2 for Fe0 (and MB). Only the availability of “free” FeCPs was considered in the investigated single, binary and ternary aggregate systems. Achieved results corroborate the usefulness of varying several operational parameters to better understand complex dynamic systems81,83,84.
Significance of the findings
Operating mode of remediation Fe0/H2O systems
This study has confirmed that Fe0 in engineered filtration systems is oxidized by protons to ferrous ion (Fe2+) (Eq. 1). Fe2+ ions are partly transferred to the surface of available aggregates (e.g. MnO2 and sand) and is oxidized further to ferric ion (Fe3+) and deposited on the aggregates as hydroxides (in-situ coating) (Fig. 1). Iron oxide-coated sand is a good adsorbent for several contaminants including chromium39,85, pathogens86,87 and phosphates88,89. Fe0 oxidation also contributes to produce anoxic conditions which are favorable for the abiotic reductive transformation of several dissolved species including chlorinated compounds38,90. Unlike sand and other inert aggregates, MnO2 is reactive and uses Fe2+ for its reductive dissolution (Eq. 3). Because the reaction occurs at the surface of MnO2 (Fig. 1), Fe0 passivation is delayed until the oxidative capacity of MnO2 is exhausted. Results presented herein have demonstrated these mechanisms excellently, while benefiting from the tracer nature of methylene blue (MB method)30,79. In fact, mechanistic discussions are often complicated by the need to consider the redox reactivity of both Fe0 and MnO2 with the contaminant of concern72. In other words, one major output of this research is that the popular hypothesis to rationalize reductive transformations in Fe0/H2O systems is faulty91. The hypothesis that Fe0 is an electron donor for dissolved has been seriously challenged during the past 15 years, however, the questioned view is still prevailing11,36, 59.
The stoichiometry of electrochemical reactions (similar to Eq. 2) has been routinely used to design Fe0 remediation systems92,93. The evidence that twice more Fe0 is needed to exchange the same number of electrons when reduction is induced by Fe2+ implies that the service life of Fe0-based systems has been wrongly estimated57. The statement is valid regardless of the approach used to estimate the efficiency of the system. However, the main problem has been the failure to properly consider the expansive nature of iron corrosion, which makes only hybrid systems viable in the long term14.
The importance of hybrid Fe0/H2O systems
The long history of Fe0 filtration systems teaches that only hybrid systems are sustainable. The Bischof filters, applied both for household and large-scale uses, contained a reactive zone made up of 25% sponge iron (vol/vol) mixed with gravel41,94. The Multi-Soil-Layering of Wakatsuki et al.89 contained only 15% Fe0 (w/w) (iron fillings) mixed with 15% Fe0 (w/w) pelletized jute and balanced with zeolite (60%). The phosphate filters of Erickson et al.95 contained only up to 5% steel wool balanced with sand. All these systems operated for more that 1 year without clogging. In the framework of subsurface permeable reactive barriers, O'Hannesin and Gillham31 tested a reactive wall containing 22% Fe0 balanced with gravel and reported on good hydraulic properties in the long term. Other systems with 100% Fe0 have failed because of loss of porosity coupled with the early development of preferential flow paths in the Fe0 permeable reactive barrier96. However, the availability of preferential flow paths was globally attributed to mineral precipitation (e.g. calcium carbonate, iron oxides, sulfide minerals). The key point is that iron oxides resulting from corrosion products are more abundant and universally present, and their generation should be reduced by “diluting” Fe0 with non-expansive aggregates like gravel or sand.
All systems containing a pure Fe0 layer (100%) were reported to be efficient but not sustainable97,98,99. The most prominent example is probably the use of iron filings for selenium removal from agricultural drainage water by the Harza Process98,100. In 1985, Harza Engineering Co. tested a pilot-scale process using iron filings in flow-through beds. The testing was discontinued because the beds quickly cemented with precipitates100. The study concluded that the advantage of Fe0 filtration is to decrease Se concentration to very low concentrations. The mechanism of Se removal was further investigated and it was established that Se is not reduced by an electrochemical mechanism98. Furthermore, Fe0 filters were suggested as a polishing step following microbial treatments100. Despite this evidence, it is disappointing to observe that available works on Se removal in Fe0/based systems have not built on existing knowledge as Se is still reported to be reduced by electrons from the metal body60,101,102,103. Following the state-of-the-art knowledge on the sustainability of Fe0 filtration, hybrid Fe0 systems should have been tested as stand-alone technology for Se removal. In essence, such work was independently conducted by Huang and his colleagues15,43,104,105 who developed and demonstrated the efficiency of a hybrid Fe0/Fe3O4 for the removal of several micro-pollutants, including MoVI, NO3– and SeVI, and recently for the mitigation of pathogens (bacteria) from dairy manure. The fact that a hybrid system, initially developed for chemically reducible micro-pollutants is performing well for pathogens corroborate the idea that it suffices to sustain iron corrosion to achieve water treatment37,52,53,54,106. As discussed in the “Introduction”, Huang et al.43 have not convincingly demonstrated the specificity of their hybrid system (Fe0/Fe3O4). This is particularly the case in a context where Fe0/sand systems are already essentially more sustainable than pure Fe0 (100%)14,49. The present work also confirms previous results that any additive to Fe0 basically delay the availability of corrosion products under typical field conditions. The observed enhanced performance results from sustained iron corrosion in the whole system. The question then arises, what makes MnO2 a specific admixing aggregate for Fe0 filters?
The suitability of hybrid Fe0/MnO2 systems
The presentation until now has demonstrated that applying Fe0 for water treatment is promising as mixing Fe0 with other aggregates delays passivation or sustain treatment efficiency. Moreover, substantial experiences have been accumulated on the functionality of hybrid systems for water treatment ("The importance of hybrid Fe0/H2O systems"). The knowledge that Fe0 acts as generator of contaminant scavengers (and never as reducing agent) implies that adsorption and co-precipitation are the fundamental mechanisms of contaminant removal in Fe0/H2O systems. Hybrid systems tested as means to prevent iron passivation include amendment with granular activated carbon (GAC), magnetite (Fe3O4), manganese oxides (MnOx), pyrite (FeS2), and sand15. Among these aggregates, MnOx and FeS2 are the most chemically reactive36,71. Both aggregates induce a pH shift to more acidic values. However, because iron corrosion increases the pH, it is possible to find the optimal Fe0/FeS2 and/or Fe0/MnO2 ratio for case-specific water treatment. Therefore, long-term systematic testing with well-characterized materials is necessary.
Note that Fe0 is a generator of iron oxides, and adding Mn oxides (MnOx) to the system creates a very complex system, which is not new to geochemists, but which is yet to be investigated in the context of water treatment68,72. In fact, taken individually, the redox reactivity of these minerals plays important roles in the fate and transformation of many contaminants in natural environments59,61,62,63,64,70,71,72. Available works mostly investigate simple model systems with few contaminants68,72. To bridge the gap between simple model systems and complex environmental systems, a profound understanding of the redox reactivity of Mn- and Fe-oxides in complex model systems toward water decontamination is urgently needed. The effects of natural ligands (Cl–, HCO3–, PO43–, SO42–) and natural organic matter (NOM) on the redox reactivity of Fe0/MnO2 systems need to be investigated as well. Moreover, there is need to investigate the following: (i) fate of contaminants in Fe0 systems, and (ii) the safe disposal of spent Fe0 materials, including their use as filler material in novel construction materials, and the behavior of contaminants in such materials.
Concluding remarks
This study clearly delineates the important role of reactive MnOx minerals on the process of water treatment using Fe0-based systems. The presence of MnOx induces Fe2+ oxidation at the mineral surface, resulting in a significant delay of Fe0 passivation compared to that attained in Fe0 and Fe0/sand systems. Being a natural mineral or a soil resource, its incorporation in Fe0 filters reinforces the frugality of this already demonstrated affordable system. It is expected that adding MnOx to Fe0/H2O will create geochemical dynamics in the system which would sustain iron corrosion and maintain the efficiency of system for water decontamination for the long term. This would make Fe0 filters a sustainable solution for decentralized safe drinking water provision and enable the realization of universal access to safe drinking water and even on a self-reliant manner. To bridge the existing knowledge gaps, the need for further research entailing long-term testing of Fe0 systems was highlighted.
References
Schumacher, E. F. Small is Beautiful: Economics as If People Mattered (Harper & Row, 1973).
Howe, K. J., Hand, D. W., Crittenden, J. C., Trussell, R. R. & Tchobanoglous, G. Principles of Water Treatment (Wiley, 2012).
Liu, P., Gernjak, W. & Keller, J. Long-term performance of enhanced-zero valent iron for drinking water treatment: A lab-scale study. Chem. Eng. J. 315, 124–131 (2017).
Tepong-Tsindé, R., Ndé-Tchoupé, A. I., Noubactep, C., Nassi, A. & Ruppert, H. Characterizing a newly designed steel-wool-based household filter for safe drinking water provision: Hydraulic conductivity and efficiency for pathogen removal. Processes 7, 966 (2019).
Nya, E. L. Access to drinking water and sanitation in Nde Division. Cameroon. PhD Dissertation, University of Yaoundé I. (2020).
Nya, E. L. & Mougoué, B. Access to safe drinking water and sanitary risks in the town of Bangangté (West Region of Cameroon). Saudi J. Hum. Soc. Sci. https://doi.org/10.36348/sjhss.2020.v05i03.007 (2020).
Banerji, T. & Chaudhari, S. A cost-effective technology for arsenic removal: Case study of zerovalent iron-based IIT Bombay arsenic filter in West Bengal. In Water and Sanitation in the New Millennium (eds Nath, K. & Sharma, V.) (Springer, 2017).
Kearns, J. P., Bentley, M. J., Mokashi, P., Redmon, J. H. & Levine, K. Underrepresented groups in WaSH—The overlooked role of chemical toxicants in water and health. J. Water Sanit. Hyg. Dev. 9, 786–793 (2019).
Hussam, A. & Munir, A. K. M. A simple and effective arsenic filter based on composite iron matrix: Development and deployment studies for groundwater of Bangladesh. J. Environ. Sci. Health A 42, 1869–1878 (2007).
Antia, D. D. J. Water treatment and desalination using the eco-materials n-Fe0 (ZVI), n-Fe3O4, n-FexOyHz[mH2O], and n-Fex[Cation]nOyHz[Anion]m [rH2O]. In Handbook of Nanomaterials and Nanocomposites for Energy and Environmental Applications (eds Kharissova, O. V. et al.) (Springer Nature, 2020). https://doi.org/10.1007/978-3-030-11155-7_66-1.
Cao, V. et al. Tracing the scientific history of Fe0-based environmental remediation prior to the advent of permeable reactive barriers. Processes 8, 977 (2020).
Yang, H. et al. Designing the next generation of Fe0-based filters for decentralized safe drinking water treatment. Processes 8, 745 (2020).
Huang, Z., Cao, V., Nya, E. L., Gwenzi, W. & Noubactep, C. Kanchan arsenic filters and the future of Fe0-based filtration systems for single household drinking water supply. Processes 9, 58 (2021).
Domga, R., Togue-Kamga, F., Noubactep, C. & Tchatchueng, J. B. Discussing porosity loss of Fe0 packed water filters at ground level. Chem. Eng. J. 263, 127–134 (2015).
Han, S., Huang, Y. & Liu, Z. Bacterial indicator reduction in dairy manure using hybridzero-valent iron (h-ZVI) system. Environ. Sci. Pollut. Res. 26, 10790–10799 (2019).
Henderson, A. D. & Demond, A. H. Long-term performance of zero-valent iron permeable reactive barriers: A critical review. Environ. Eng. Sci. 24, 401–423 (2007).
Bartzas, G. & Komnitsas, K. Solid phase studies and geochemical modelling of low-cost permeable reactive barriers. J. Hazard Mater. 183, 301–308 (2010).
Li, L. & Benson, C. H. Evaluation of five strategies to limit the impact of fouling in permeable reactive barriers. J. Hazard Mater. 181, 170–180 (2010).
Guan, X. et al. The limitations of applying zero-valent iron technology in contaminants sequestration and the corresponding countermeasures: The development in zero-valent iron technology in the last two decades (1994–2014). Water Res. 75, 224–248 (2015).
Gheju, M. & Balcu, I. Sequential abatement of FeII and CrVI water pollution by use of walnut shell-based adsorbents. Processes 9, 218 (2021).
Caré, S. et al. Modeling the permeability loss of metallic iron water filtration systems. Clean: Soil, Air, Water 41, 275–282 (2013).
Moraci, N., Lelo, D., Bilardi, S. & Calabrò, P. S. Modelling long-term hydraulic conductivity behaviour of zero valent iron column tests for permeable reactive barrier design. Can. Geotech. J. 53, 946–961 (2016).
Li, J. et al. Characterization methods of zerovalent iron for water treatment and remediation. Water Res. 148, 70–85 (2019).
Lufingo, M., Ndé-Tchoupé, A. I., Hu, R., Njau, K. N. & Noubactep, C. A novel and facile method to characterize the suitability of metallic iron for water treatment. Water 11, 2465 (2019).
Romanoff, M. Underground Corrosion. United States Department of Commerce, National Bureau of Standards. Circular 579 (1957).
Melchers, R. E. & Petersen, R. B. A reinterpretation of the Romanoff NBS data for corrosion of steels in soils. Corros. Eng. Sci. Technol. 53, 131–140 (2018).
Stefanonia, M., Zhanga, Z., Angsta, U. & Elsener, B. The kinetic competition between transport and oxidation of ferrous ions governs precipitation of corrosion products in carbonated concrete. RILEM Tech. Lett. 3, 8–16 (2018).
Stefanoni, M., Angst, U. M. & Elsener, B. Kinetics of electrochemical dissolution of metals in porous media. Nat. Mater. 18, 942–947 (2019).
Whitney, W. R. The corrosion of iron. J. Am. Chem. Soc. 25, 394–406 (1903).
Miyajima, K. & Noubactep, C. Characterizing the impact of sand addition on the efficiency of granular iron for water treatment. Chem. Eng. J. 262, 891–896 (2015).
O’Hannesin, S. F. & Gillham, R. W. Long-term performance of an in situ “iron wall” for remediation of VOCs. Ground Water 36, 164–170 (1998).
Phillips, D. H. et al. Ten year performance evaluation of a field-scale zero-valent iron permeable reactive barrier installed to remediate trichloroethene contaminated groundwater. Environ. Sci. Technol. 44, 3861–3869 (2010).
Wilkin, R. T. et al. Fifteen-year assessment of a permeable reactive barrier for treatment of chromate and trichloroethylene in groundwater. Sci. Tot. Environ. 468–469, 186–194 (2014).
Wilkin, R. T. et al. Geochemical and isotope study of trichloroethene degradation in a zero-valent iron permeable reactive barrier: A twenty-two-year performance evaluation. Environ. Sci. Technol. 53, 296–306 (2019).
Roh, Y., Lee, S. Y. & Elless, M. P. Characterization of corrosion products in the permeable reactive barriers. Environ. Geol. 40, 184–194 (2000).
Hu, R., Cui, X., Xiao, M., Gwenzi, W. & Noubactep, C. Characterizing the impact of pyrite addition on the efficiency of Fe0/H2O systems. Sci. Rep. 11, 2326 (2021).
Noubactep, C. Processes of contaminant removal in “Fe0–H2O” systems revisited. The importance of co-precipitation. Open Environ. Sci. 1, 9–13 (2007).
Jiao, Y. et al. Reductive dechlorination of carbon tetrachloride by zero-valent iron and related iron corrosion. Appl. Catal. B Environ. 91, 434–440 (2009).
Gheju, M. Hexavalent chromium reduction with zero-valent iron (ZVI) in aquatic systems. Water Air Soil Pollut. 222, 103–148 (2011).
Ghauch, A. Iron-based metallic systems: An excellent choice for sustainable water treatment. Freiberg Online Geosci. 32, 1–80 (2015).
Bischof, G. On the purification of water. Proc. R. Philos. Soc. Glasgow 8, 357–372 (1973).
Mackenzie, P. D., Horney, D. P. & Sivavec, T. M. Mineral precipitation and porosity losses in granular iron columns. J. Hazard Mater. 68, 1–17 (1999).
Huang, Y. H., Tang, C. & Zeng, H. Removing molybdate from water using a hybridized zero-valent iron/magnetite/Fe(II) treatment system. Chem. Eng. J. 200–202, 257–263 (2012).
Noubactep, C., Meinrath, G., Dietrich, P. & Merkel, B. Mitigating uranium in groundwater: Prospects and limitations. Environ. Sci. Technol. 37, 4304–4308 (2003).
Noubactep, C., Btatkeu-K, B. D. & Tchatchueng, J. B. Impact of MnO2 on the efficiency of metallic iron for the removal of dissolved metal. Chem. Eng. J. 178, 78–84 (2011).
Lipczynska-Kochany, E., Harms, S., Milburn, R., Sprah, G. & Nadarajah, N. Degradation of carbon tetrachloride in the presence of iron and sulphur containing compounds. Chemosphere 29, 1477–1489 (1994).
Westerhoff, P. & James, J. Nitrate removal in zero-valent iron packed columns. Water Res. 37, 1818–1830 (2003).
Bi, E., Devlin, J. F. & Huang, B. Effects of mixing granular iron with sand on the kinetics of trichloroethylene reduction. Ground Water Monit. Remed. 29, 56–62 (2009).
Song, D. I., Kim, Y. H. & Shin, W. S. A simple mathematical analysis on the effect of sand in Cr(VI) reduction using zero valent iron. Korean J. Chem. Eng. 22, 67–69 (2005).
Ndé-Tchoupé, A. I., Makota, S., Nassi, A., Hu, R. & Noubactep, C. The suitability of pozzolan as admixing aggregate for Fe0-based filters. Water 10, 417 (2018).
Bigg, T. & Judd, S. J. Zero-valent iron for water treatment. Environ. Technol. 21, 661–670 (2000).
Furukawa, Y., Kim, J.-W., Watkins, J. & Wilkin, R. T. Formation of ferrihydrite and associated iron corrosion products in permeable reactive barriers of zero-valent iron. Environ. Sci. Technol. 36, 5469–5475 (2002).
Purenović, M., Perović, J., Bojić, A., Andelković, T. & Bojić, D. Cu and Cd removal from wastewater by a microalloyed aluminium composite. Environ. Chem. Lett. 2, 59–63 (2004).
Burghardt, D. & Kassahun, A. Development of a reactive zone technology for simultaneous in situ immobilisation of radium and uranium. Environ. Geol. 49, 314–320 (2005).
Yoon, I. H., Kim, K. W., Bang, S. & Kim, M. G. Reduction and adsorption mechanisms of selenate by zero-valent iron and related iron corrosion. Appl. Catal. B Environ. 104, 185–192 (2011).
Xu, J. et al. Reactivity, selectivity, and long-term performance of sulfidized nanoscale zerovalent iron with different properties. Environ. Sci. Technol. 53, 5936–5945 (2019).
Cao, V., Ndé-Tchoupé, A. I., Hu, R., Gwenzi, W. & Noubactep, C. Discussing the mechanism of contaminant removal in Fe0/H2O systems: The burden of a poor literature review. Chemosphere 280, 130614 (2021).
Zhang, L., Zhang, Y., Gao, X. & Xu, C. Insights on the effects of pH and Fe(II) regeneration during the chromate sequestration by sulfidated zero-valent iron. Chem. Eng. J. 378, 122115 (2019).
Alyoussef, G. Characterizing the impact of contact time in investigating processes in Fe0/H2O systems. Freiberg Online Geosci. 58, 1–80 (2021).
Wang, X., Zhang, X., Wang, Z., Xu, C. & Tratnyek, P. G. Advances in metal(loid) oxyanion removal by zerovalent iron: Kinetics, pathways, and mechanisms. Chemosphere https://doi.org/10.1016/j.chemosphere.2021.130766 (2021).
Appelo, C. A. J. & Postma, D. Variable dispersivity in a column experiment containing MnO2 and FeOOH-coated sand. J. Cont. Hydrol. 40, 95–106 (1999).
Appelo, C. A. J. & Postma, D. A consistent model for surface complexation on birnessite (MnO2) and its application to a column experiment. Geochim. Cosmochim. Acta 63, 3039–3048 (1999).
Post, J. E. Manganese oxide minerals: Crystal structures and economic and environmental significance. Proc. Natl. Acad. Sci. 96, 3447–3454 (1999).
Tebo, B. M. et al. Biogenic manganese oxides: Properties and mechanisms of formation. Annu. Rev. Earth Planet Sci. 32, 287–328 (2004).
Vodyanitskii, Y. N. Mineralogy and geochemistry of manganese: A review of publications. Eurasian Soil Sci. 42, 1170–1178 (2009).
Penrose, R. A. F. The chemical relation of iron and manganese in sedimentary rocks. J. Geol. 1, 356–370 (1893).
Fischel, M. H. H., Fischel, J. S., Lafferty, B. J. & Sparks, D. L. The influence of environmental conditions on kinetics of arsenite oxidation by manganese-oxides. Geochem. Trans. 16, 15 (2015).
Huang, J. & Zhang, H. Redox reactions of iron and manganese oxides in complex systems. Front. Environ. Sci. Eng. 14, 76 (2020).
Lewis, A. E. Review of metal sulphide precipitation. Hydrometallurgy 104, 222–234 (2010).
Cao, V., Alyoussef, G., Gatcha-Bandjun, N., Gwenzi, W. & Noubactep, C. The suitability of methylene blue discoloration (MB method) to investigate the Fe0/MnO2 system. Processes 9, 548 (2021).
Cao, V., Alyoussef, G., Gatcha-Bandjun, N., Gwenzi, W. & Noubactep, C. Characterizing the impact of MnO2 addition on the efficiency of Fe0/H2O systems. Sci. Rep. https://doi.org/10.1038/s41598-021-89318-w (2021).
Michel, M. M. et al. Mineral materials coated with and consisting of MnOx—Characteristics and application of filter media for groundwater treatment: A review. Materials 13, 2232 (2020).
Shindo, H. & Huang, P. M. Catalytic effects of manganese(IV), iron(III), aluminum, and silicon oxides on the formation of phenolic polymers. Soil Sci. Soc. Am. J. 48, 927–934 (1984).
Sparrow, L. A. & Uren, N. C. Manganese oxidation and reduction in soils: Effects of temperature, water potential, pH and their interactions. Soil Res. 52, 483–494 (2014).
Ye, Z., Giraudon, J. M., De Geyter, N., Morent, R. & Lamonier, J. F. The design of MnOx based catalyst in post-plasma catalysis configuration for toluene abatement. Catalysts 8, 91 (2018).
Varlikli, C. et al. Adsorption of dyes on Sahara desert sand. J. Hazard Mater. 170, 27–34 (2009).
Mitchell, G., Poole, P. & Segrove, H. D. Adsorption of methylene blue by high-silica sands. Nature 176, 1025–1026 (1955).
Kurth, A. M. Discoloration of methylene blue by elemental iron—Influence of the shaking intensity. Bachelor thesis, Universität Göttingen, 45 (2008).
Miyajima, K. Optimizing the design of metallic iron filters for water treatment. Freiberg Online Geosci. 32, 1–60 (2012).
Brenner, S. Sequences and consequences. Philos. Trans. R. Soc. B 365, 207–212 (2010).
Lavine, B. K., Auslander, G. & Ritter, J. Polarographic studies of zero valent iron as a reductant for remediation of nitroaromatics in the environment. Microchem. J. 70, 69–83 (2001).
Noubactep, C., Schöner, A. & Meinrath, G. Mechanism of uranium (VI) fixation by elemental iron. J. Hazard Mater. 132, 202–212 (2006).
Gatcha-Bandjun, N., Noubactep, C. & Loura-Mbenguela, B. Mitigation of contamination in effluents by metallic iron: The role of iron corrosion products. Environ. Technol. Innov. 8, 71–83 (2017).
Touomo-Wouafo, M. et al. Electrochemical monitoring of heavy metals removal from aqueous solutions by aged metallic iron. Competitive effects of cations Zn2+, Pb2+ and Cd2+. Monatsh. Chem. 151, 1511–1523 (2020).
Powell, M. R., Puls, W. R., Hightower, K. S. & Sebatini, A. D. coupled iron corrosion and chromate reduction: Mechanisms for subsurface remediation. Environ. Sci. Technol. 29, 1913–1922 (1995).
Bojic, A. et al. The inactivation of Escherichia coli by microalloyed aluminium based composite. Facta Universitatis 2, 115–124 (2001).
You, Y., Han, J., Chiu, P. C. & Jin, Y. Removal and inactivation of waterborne viruses using zerovalent iron. Environ. Sci. Technol. 39, 9263–9269 (2005).
James, B. R., Rabenhorst, M. C. & Frigon, G. A. Phosphorus sorption by peat and sand amended with iron oxides or steel wool. Water Environ. Res. 64, 699–705 (1992).
Wakatsuki, T., Esumi, H. & Omura, S. High performance and N, P removable on-site domestic wastewater treatment system by multi-soil-layering method. Water Sci. Technol. 27, 31–40 (1993).
Khudenko, B. M. Feasibility evaluation of a novel method for destruction of organics. Water Sci. Technol. 23, 1873–1881 (1991).
Matheson, L. J. & Tratnyek, P. G. Reductive dehalogenation of chlorinated methanes by iron metal. Environ. Sci. Technol. 28, 2045–2053 (1994).
Sarr, D. Zero-valent-iron permeable reactive barriers—How long will they last?. Remediation 11, 1–18 (2001).
Obiri-Nyarko, F., Grajales-Mesa, S. J. & Malina, G. An overview of permeable reactive barriers for in situ sustainable groundwater remediation. Chemosphere 111, 243–259 (2014).
Devonshire, E. The purification of water by means of metallic iron. J. Frankl. Inst. 129, 449–461 (1890).
Erickson, A. J., Gulliver, J. S. & Weiss, P. T. Enhanced sand filtration for storm water phosphorus removal. J. Environ. Eng. 133, 485–497 (2007).
Morrison, S. J., Mushovic, P. S. & Niesen, P. L. Early breakthrough of molybdenum and uranium in a permeable reactive barrier. Environ. Sci. Technol. 40, 2018–2024 (2006).
Murphy, A. P. Removal of selenate from water by chemical reduction. Ind. Eng. Chem. Res. 27, 181–191 (1988).
Anderson, M. A. Fundamental aspects of selenium removal by Harza process. Rep San Joaquin Valley Drainage Program, US Dep Interior, Sacramento (1989).
Hussam, A. Contending with a development disaster: Sono filters remove arsenic from well water in Bangladesh. Innovations 4, 89–102 (2009).
Frankenberger, W. T. Jr. et al. Advanced treatment technologies in the remediation of seleniferous drainage waters and sediments. Irrig. Drain. Syst. 18, 19–41 (2004).
Liang, L. et al. Efficient selenate removal by zero-valent iron in the presence of weak magnetic field. Sep. Purif. Technol. 156, 1064–1072 (2015).
Liang, L. et al. Kinetics of selenite reduction by zero-valent iron. Desalin. Water Treat. 53, 2540–2548 (2015).
Qin, H. et al. Unexpected effect of buffer solution on removal of selenite and selenate by zerovalent iron. Chem. Eng. J. 334, 296–304 (2018).
Huang, Y. H., Peddi, P. K., Zeng, H., Tang, C.-L. & Teng, X. Pilot-scale demonstration of the hybrid zero-valent iron process for treating flue-gas-desulfurization wastewater: Part I. Water Sci. Technol. 67, 16–23 (2013).
Huang, Y. H., Peddi, P. K., Zeng, H., Tang, C.-L. & Teng, X. Pilot-scale demonstration of the hybrid zero-valent iron process for treating flue-gas-desulfurization wastewater: Part II. Water Sci. Technol. 67, 239–246 (2013).
Noubactep, C. A critical review on the mechanism of contaminant removal in Fe0–H2O systems. Environ. Technol. 29, 909–920 (2008).
Acknowledgements
For providing the iron material investigated in this study the authors would like to express their gratitude to iPutec GmbH (Rheinfelden, Germany). The natural MnO2-mineral was provided by the Department of Geology of the Technical University Bergakademie Freiberg/Germany (Mineralsammlung). Mohammad Azizur Rahman (Technovative Solutions, Manchester Science Park, Manchester/UK) is thanked for technical support. The manuscript was improved by the insightful comments of anonymous reviewers from Scientific Reports. We acknowledge support by the German Research Foundation and the Open Access Publication Funds of the Göttingen University.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
G.A., V.C., N.G.-B. and C.N. conceived the presented idea and developed the theory. G.A. carried out the experiments. C.N. supervised this work. W.G. supervised the redaction of the first draft by V.C. and N.G.-B. All authors discussed the results and contributed to the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cao, V., Alyoussef, G., Gatcha-Bandjun, N. et al. The key role of contact time in elucidating the mechanisms of enhanced decontamination by Fe0/MnO2/sand systems. Sci Rep 11, 12069 (2021). https://doi.org/10.1038/s41598-021-91475-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-91475-x
- Springer Nature Limited