
Abstract
Simultaneous in situ immobilisation of uranium (U) and radium (226Ra) by injectible amounts of grey cast iron (gcFe), nano-scale iron (naFe) and a gcFe/MnO2 mixture (1:1) was studied in batch and column tests. Both 0.5 g/L naFe and gcFe are effective in 226Ra and U removal from mine water, whereas MnO2 addition clearly increased the efficiency of gcFe for 226Ra and U immobilisation. In a column test with 0.6 wt% gcFe/MnO2 mixture (1:1), neither 226Ra nor U was detected in the effluent after replacement of 45 pore volumes. A sequential extraction under flow condition revealed 226Ra to be mostly occluded in manganese oxides. Uranium was mostly sorbed onto poorly crystalline iron hydroxides, but a significant part was found to be occluded in manganese oxides also. The results of this study suggest that MnO2 promotes iron hydroxide formation under slightly reducing environmental conditions resulting in an increased pollutant retention capacity.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The intensive uranium ore mining which took place until the mid-1970s has devised many abandoned mine processing sites in Eastern Germany. Hydraulic connections to aquifers, which are present at many sites, resulted in numerous, smaller and decentralised groundwater run-offs contaminated with heavy metals, arsenic and radio-nuclides including uranium (U) and 226Radium (226Ra). Their concentrations frequently exceed groundwater standards for remedial actions at inactive U processing sites (Schneider et al. 2001). For Germany, the 226Ra exemption limit is regulated at 0.7 Bq/L (StrSchV 2001), the uranium groundwater standard for remedial actions is recommended at 0.3 mg/L (SSK 1992).
In order to prevent an ongoing contamination of ground- and surface water, relevant abandoned mine processing sites need restoration. In Germany, classical pump-and-treat technologies and conventional water treatment methods are common remediation practice. This is a cost-intensive approach which leaves in addition concentrated residues for disposal (Neitzel et al. 2000). Therefore, the development of long-term active and inexpensive in situ treatment methods has become a research topic of major importance and interest.
The use of zero valent iron (ZVI) for in situ containment of chlorinated solvents, arsenic and some metals within permeable reactive barriers (PRB) is already almost standard approach in the United States (US Department of Energy 1996). In contrast to conventional water treatment methods, whereas different treatment steps for several contaminants are feasible, a simultaneous contaminant removal is aimed for in situ treatment methods. Furthermore, the application of PRB requires excavation and is still limited to shallow aquifers (Simpkin 2003), whereby this method is often ineffective for smaller, decentralised groundwater run-offs.
A potential alternative to PRB are reactive zones (RZ). They are created by installing a line of overlapping injection points perpendicular to the groundwater flow direction. For this purpose, metallic iron to be injected is necessarily in relatively small quantities (≤1 wt% of aquifer material) (Simpkin 2003) and very small size (usually <<100 μm in diameter, depending on the aquifer’s pore channel size). Because of small quantities of metallic iron, reducing conditions and contaminant removal capacity of RZ are not as high as PRB, whose iron content is usually greater than 20 wt% of aquifer material. However, due to the use of injection lances, the RZ technology is more flexible and is only limited by achievable depth of the drilling equipment used. Therefore, this innovative technology seems to be more economic to sites with smaller, decentralised run-offs and was focused in this study.
Uranium immobilisation can take place by sorption onto metal (hydr)oxides (Bargar et al. 2000; Hsi and Langmuir 1985) and iron sulphides (Moyes et al. 2000), coprecipitation with carbonate minerals (Reeder et al. 2000; Abdelouas et al. 1998) and partly by reductive precipitation catalysed by iron sulphide surfaces (Livens et al. 2004; Wersin et al. 1994). Radium immobilisation occurs predominantly by coprecipitation with sulphate minerals (e.g. Ba, RaSO4) (Jenk et al. 2004), but also by sorption onto metal (hydr)oxides (IAEA 1990). In contrast to uranium, radium immobilisation is ineffective under strongly reducing environmental conditions due to the release of metal (hydr)oxides and sulphate minerals (Knappik et al. 1996; Fedorak et al. 1986). Consequently, a simultaneous immobilisation of radium and uranium, which is aimed for in situ treatment methods can only be stimulated by metal (hydr)oxide supply.
Iron hydroxides could be formed in situ by aerobic corrosion of metallic iron which is often used for PRB and RZ technologies. However, this pathway produces voluminous iron hydroxides, which causes an early surface passivation of metallic iron and a decreasing permeability of RZs, respectively (Mackenzie et al. 1999). As a result of this, they were bypassed by the flowing groundwater and no more chemical reactions with contaminants are possible (Noubactep 2003). Therefore, this pathway should not be favoured for in situ treatment methods.
A more suitable pathway for in situ iron hydroxide formation under anaerobic environmental conditions was described by Noubactep (2001) and Postma and Appelo (2000). Ferrous iron, formed by the anaerobic corrosion of metallic iron, could be oxidised effectively by manganese dioxide reduction. As ferrous iron is oxidised on the surface of manganese dioxide, the formation of precipitates on the surface of metallic iron was avoided or delayed. Furthermore, the produced iron hydroxides are less voluminous, whereas the clogging risk of RZs decreases. A mixture of metallic iron and manganese dioxide encourages not only an effective iron corrosion but also pollutant immobilisation under reducing environmental conditions. Additionally, manganese (hydr)oxides are known as good sorbents for radium (Valentine 1992; Mott et al. 1993).
Because of these advantages, a mixture of injectible grey cast iron (gcFe) and MnO2 should be investigated comparatively to sole gcFe and nano-scale iron (naFe) for simultaneous U and 226Ra immobilisation from a mine water.
Materials and methods
Solid materials
Zero valent iron materials, which were often used in PRB technologies (US-EPA 1998), tend to an early surface passivation caused by formation of precipitates on the surface of ZVI material due to their high purity degree (Friedrich and Knappik 2001). Because of this, a particulate, impure gcFe was selected for batch and column tests. Impurities like carbides counter an early passivation of the iron surface and therefore encouraging iron corrosion. The gcFe was delivered by the Maier Metallpulver GmbH, Rheinfelden. The particles were gas classified <30 μm before their use.
Nano-scale iron was selected as second iron material for batch and column tests. Injectible naFe is a promising new technology for groundwater remediation. A markedly improved reactivity due to greater surface area (33.5 m2/g compared to about 0.9 m2/g for commercial iron powder) was described for in situ reductive dehalogenation by Vance (2003). The colloid size of naFe was denoted with 0.1 μm by the producer (Toda Kogyo Europe GmbH).
For MnO2 used in this study (Sigma-Aldrich No. 1324) a particle size <63 μm was denoted. According to Eqs. 3 and 4, 1 mol MnO2 (M=87 g/mol) is required to complete the oxidation of 2 mol metallic iron (M=56 g/mol) to FeOOH. Therewith, 0.78 g MnO2 is necessary to complete the anaerobic oxidation of 1 g metallic iron, corresponding to a gcFe:MnO2 ratio of 56:44. Because of this, a 1:1 mixture of gcFe and MnO2 was used for batch and column tests.
Contaminated water
Two flooding waters from the Königstein mine (Saxony) were used for batch and column experiments by courtesy of the Wismut GmbH. For the batch test series, the acid flooding water k-7080 was used, which contained about 42 Bq 226Ra/L and 2 mg U/L. The column was supplied with the nearly neutral flooding water k-7300, which contained about 24 Bq 226Ra/L and 5 mg U/L. Table 1 contains an excerpt from the composition of the flooding waters k-7080 and k-7300.
Geochemical modelling
To find out dominant redox processes in batch and column tests, a coupled python script/phreeqC-equilibrium modelling was used. Simulations were carried out for both flooding waters, the addition of 1 mmol Fe/L and 1 mmol Mn/L, atmospheric pressure as well as test-relevant pH and redox ranges. The calculated saturation indices of Mn and Fe mineral phases were exported in excel files. Subsequently, stability fields of supersaturated mineral phases could be depicted together with measured E h/pH values in E h/pH diagrams.
Batch experiments
Contaminant immobilisation was studied in batch tests for 14 days at 12°C under N2 atmosphere. Each test contained 1.1 L flooding water k-7080 and 550 mg gcFe, naFe, MnO2 or a 1:1 mixture gcFe/MnO2. The aqueous phase was analysed for pH, redox, O2, major and trace elements as well as 226Ra at the beginning and at the end of the experiments.
Column study
The most effective reactive material from the batch tests (1:1 mixture of gcFe/MnO2) was investigated under flow conditions using a glass column of 10 cm diameter and 20 cm length. The column was packed with 9 g gcFe and 9 g MnO2 in 3 kg sand. Therewith, the percentage of gcFe/MnO2 mixture (0.6 wt%) remained below 1 wt% quoted for RZs by Simkin (2003). The column was charged with mine water k-7300 for 120 days at 12°C. The influent vessel was connected to a nitrogen bag using a special transfer sealing to prevent oxygen access. The mine water was delivered with a flow rate of 0.2 L/day (resulting in the replacement of 0.4 pore volumes per day) using a peristaltic pump (Ismatec Reglo). Every third replaced pore volume was analysed for pH, redox, oxygen, major and trace elements as well as 226Ra.
At the end of the test, a sequential sediment extraction (Zeien 1995) was carried out under flow conditions in order to identify the binding forms of both U and 226Ra. For this purpose, the column was successively supplied with groundwater k-6773E (Table 1) from the Königstein mine and extractants summarised in Table 2. About three replaced pore volumes were collected in the effluent in each extraction phase and analysed for major and trace elements and 226Ra.
Analytical methods
Redox potential and pH were measured using electrode methods and a Multi 340i-instrument (WTW). Uranium, major and trace elements were measured by ICP-OES (CirosCCD, Co. Spectro, Kleve, Germany). The 226Ra was analysed via γ-spectrometry (n-type HPGe-detectors, Co. eurisys, München, Germany/EG&G ORTEC) by the VKTA Rossendorf e.V.
Results and discussion
Batch tests
In order to identify the redox processes in the batch tests, E h/pH diagrams were constructed by the aid of a coupled python script/phreeqC-equilibrium modelling for flooding water k-7080 and supersaturated Mn and Fe phases (Figs. 1, 2). Thereafter, the measured pH and redox (E h) values could be depicted in these diagrams. An insignificant pH and E h change was measured in the MnO2 test. This fact indicated that no significant MnO2 reduction took place. In contrast, distinct environmental changes were observed in the other batch test indicating an intensive iron corrosion. The redox potential decreased at 602 mV (from 735 to 133 mV) in the grey cast iron test at 699 mV (from 735 to 36 mV) in the nano-scale iron experiment and at 796 mV (from 735 to −61 mV) in the test with the gcFe/MnO2 mixture. Corresponding with decreasing redox potentials, an increase of pH was measured. The pH increased each from 3.3 to 6.7 in the gcFe and naFe tests and to 6.3 in the gcFe/MnO2 test. Considering the calculated field of supersaturated Mn mineral phases (Fig. 1), MnO2 was reduced to Mn2+ in both tests with MnO2 addition. Regarding Fig. 2, ferrous iron from the anaerobic iron corrosion (Eq. 1) was obviously oxidised to Fe3(OH)8 by the reduction of water (Eq. 2a) or oxygen (Eq. 2b) in the naFe and gcFe batch tests:

pH/E h change and supersaturated Mn mineral phases in the batch test with MnO2 addition

pH/E h change and supersaturated Fe mineral phases in the batch tests with Fe0 addition
In the test with the gcFe/MnO2 mixture (1:1), ferrous iron was obviously oxidised to FeOOH due to the reduction of MnO2 to Mn2+:
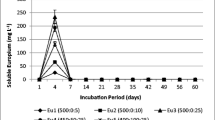
The percentage U and 226Ra removal from the mine water is shown in Fig. 3. No uranium was removed in the test with 0.5 g/L manganese oxide. This could be attributed to the low pH, at which no manganese (hydr)oxide formation was possible. However, a pro rata Ra removal (13.5 Bq 226Ra/L) by sorption onto MnO2 could be evidenced with this test. In the test with 0.5 g/L gcFe, 20.8 Bq 226Ra/L and about 2 mg U/L were removed from the mine water, probably by sorption onto iron (hydr)oxides like hematite (Fe2O3, evidenced by XRD analyses on the original gcFe) or newly formed Fe3(OH)8. The addition of 0.5 g/L naFe resulted in the immobilisation of 17.4 Bq 226Ra/L as well as a complete U removal. Considering the slightly reducing environmental conditions at the end of these batch tests (Figs. 1, 2), U and Ra removal may caused by sorption onto newly formed Fe3(OH)8. The mixture of 0.25 g/L gcFe and 0.25 g/L MnO2 dioxide provides the most sorption places for radium, 22.8 Bq/L and about 2 mg U/L were removed from the flooding water. Ra and U were probably occluded in hematite and newly formed FeOOH, but also in manganese dioxide.

U and 226Ra immobilisation in 14-days batch tests under N2 atmosphere with 0.5 g/L reactive materials
Column test
Until the eighth replaced pore volume, a pH increase of about 1.2 pH units as well as a redox (E h) decrease from 480 to 280 mV indicated the corrosion of grey cast iron. This initial environmental reduction was accompanied by an increase of manganese concentrations in the column effluent (Figs. 4, 5).

Change of pH/E h values and Mn concentrations in the column test with flooding water k-7300

226Ra and U concentration changes in the column test with flooding water k-7300
To find out the redox processes within the column, E h/pH diagrams were constructed for flooding water k-7300 and supersaturated Mn and Fe mineral phases, respectively (Figs. 6, 7). The measured pH and E h values at the column effluent were depicted by bubbles; the reaction progress was marked by an arrow.

E h/pH diagram for flooding water k-7300 and supersaturated Mn mineral phases. The bubble diameter represents the relative Mn 2+(aq) concentration

E h/pH diagram for flooding water k-7300 and supersaturated Fe mineral phases.
So it can be shown, that measured pH and E h values moved from the field of supersaturated rhodochrosite (MnCO3) to aqueous Mn2+ at the initial stage of the experiment. Following to the eighth pore volume, pH and E h values in the column effluent reached influent levels. Additionally, only slightly increased manganese concentrations (about 1 mg/L) were analysed. Regarding the E h/pH diagram (Fig. 6), MnO2 was obviously reduced to \( {\text{Mn}}^{{{\text{2+ }}}}_{{{\text{(aq)}}}} \) at these environmental conditions. During the whole test period, no ferrous iron was detected in the column effluent. Regarding Fig. 7, ferrihydrite (Fe(OH)3) was supersaturated. Therewith, the dominant redox processes in the column experiment can be written as follows:
Only two hydroxyl ions from Eq. 3 were consumed in Eq. 5, whereby a pH increase should be resulted from the sum of these reactions. However, no significant pH increase was measured at the column effluent after the eighth replaced pore volume. So it must be assumed, that some other H+ producing reactions occurred on the flow path of the column, for example:
The concentration changes of radium and uranium were given in Fig. 5. During 45 replaced pore volumes, neither radium nor uranium was detected in the effluent. According to the flooding water supply, a sequential extraction was carried out under flow conditions in order to evaluate the binding forms of uranium and radium. Figure 8 shows relevant results.

Binding forms of immobilised U und 226Ra in the column test with flooding water k-7300
Neither radium nor uranium could be dissolved by groundwater from the Königstein mine. Radium was predominantly occluded in manganese oxides (5.8 Bq/100 g sand) and sorbed onto the cation exchanger (3.9 Bq/100 g sand.). A minor part was occluded in poorly crystalline iron hydroxides (2.6 Bq/100 g sand) and specifically adsorbed (1.2 Bq/100 g sand). Conversely, uranium was mostly occluded in poorly crystalline iron hydroxides (0.77 mg/100 g sand). However, a significant part (0.45 mg/100 g sand) was also occluded in manganese oxides.
Conclusion
The immobilisation of radium and uranium by injectible amounts of reactive materials presented the main focus of this study. Radium and uranium could only be removed simultaneously by sorption onto metal (hydr)oxides, which are still stable under easily reducing environmental conditions. However, the in situ formation of iron hydroxides due to the aerobic corrosion of metallic iron leads to an early passivation of iron surfaces as well as a permeability loss of RZs. Below the aspect of a long-term active immobilisation technology, this pathway should not be favoured for an in situ immobilisation technology.
Because of this, grey cast iron, nano-scale iron, manganese dioxide and a 1:1 mixture of grey cast iron and manganese dioxide were tested on their qualification for an anaerobic radium and uranium immobilisation in a batch test series. Surprisingly, the 1:1 mixture of grey cast iron and manganese dioxide could provide more sorption sites for radium than the highly reactive nano-scale iron. In contrast, the immobilisation of uranium was marginally better in the nano-scale iron test. The batch tests have shown that manganese dioxide could effectively encourage iron corrosion and iron hydroxide formation.
The high immobilisation potential for radium and uranium was confirmed under flow conditions using a column with 0.6 wt% gcFe/MnO2 mixture (1:1)/kg silica sand. Neither radium nor uranium was detected in the effluent during 45 replaced pore volumes. Ferrous iron, produced by the grey cast iron corrosion, was obviously oxidised to ferrihydrite due to the reduction of MnO2. Furthermore, manganese oxides could provide basic contribution to the in situ immobilisation of pollutants.
References
Abdelouas A, Lutze W, Nuttall HE (1998) Chemical reactions of uranium in groundwater at a mill tailings site. J Contam Hydrol 34:343–361
Bargar JR, Reitmeyer R, Davis JA (2000) Characterisation of U(VI)-carbonato ternary complexes on hematite: EXAFS and electrophoretic mobility measurements. Geochim Cosmochim Acta 64:2737–2749
Fedorak PM, Westlake DWS, Anders C, Kratochvil B, Motokosky N, Anderson WB, Huck PM (1986) Microbial release of 226Ra2+ from (Ba,Ra)SO4 sludges from uranium mine wastes. Appl Environ Microbiol 52/2:262–268
Friedrich HJ, Knappik R (2001) Entwicklung pH-redoxaktiver Sicherungssysteme zur Sanierung von Uran- und Arsenkontaminationen im Bereich von Grundwasserleitern. VKTA Rossendorf e.V.
Hsi CKD, Langmuir D (1985) Adsorption of uranyl onto ferric oxyhydroxides: application of the surface complexation site-binding model. Geochim Cosmochim Acta 49:1931–1941
IAEA (1990) International Atomic Energy Agency: Technical Reports Series No. 310: the environmental behaviour of radium, vols 1 and 2, Vienna, Austria
Jenk U, Paul M, Ziegenbalg G, Klinger Ch (2004) Alternative methods of mine water treatment—feasibility and technical limitations for a full-scale application at WISMUT’s Königstein mine site (Germany). In: Mine Water 2004 Proceedings of the symposium, vol 1, pp 245–252, ISBN 0-9543827-4-9
Knappik R, Mocker D, Friedrich HJ (1996) Migrationsverhalten von Radionukliden in Tailings unter besonderer Berücksichtigung des Oxidation spotentials in alten Tailingsablagerungen. VKTA Rossendorf Abschlussbericht
Livens RF, Jones MJ, Hynes AJ, Charnock JM, Mosselmans J, Hennig Ch, Steele H, Collison D, Vaughan DJ, Pattrick R, Reed WA, Moyes L (2004) X-ray absorption spectroscopy studies of reactions of technetium, uranium and neptunium with mackinawite. J Environ Radioact 74:211–219
Mackenzie PD, Horney DP, Sivavec TM (1999) Mineral precipitation and porosity losses in granular iron columns. J Hazard Mater 68:1–17
Mott HV, Singh S, Kondapally VR (1993) Factors affecting radium removal using mixed iron–manganese oxides. J AWWA 85(10):114–121
Moyes LN, Parkman RH, Charnock JM, Vaughan DJ, Livens FR, Hughes CR, Braithwaite A (2000) Uranium uptake from aqueous solution by interaction with goethite, lepidocrocit, muscovite and mackinawite: an X-ray adsorption spectroscopy study. Environ Sci Technol 34:1062–1068
Neitzel PL, Schneider P, Hurst S (2000) Feldversuche zur in-situ Entfernung von Uran(NAT.) und Ra-226 aus Berge- und Flutungswässern. In: Pohl A, Häfner F, Schmidt J, Merkel B (eds) Wasserwirtschaftliche Sanierung von Bergbaukippen, Halden und Deponien. Freiberger Forschungshefte C 482 Geoingenieurwesen pp 196–206
Noubactep C (2001) In-situ immobilisation of uranium in groundwater. Wissenschaftliche Mitteilungen Institut für Geologie der TU Bergakademie Freiberg Band 16 pp 149–164, ISSN 1433–1284
Noubactep C (2003) Untersuchungen zur passiven in-situ-Immobilisierung von U(VI) aus Wasser. Dissertation, Wissenschaftliche Mitteilungen Institut für Geologie der TU Bergakademie Freiberg, Band 21, ISSN 1433–1284
Postma D, Appelo CAJ (2000) Reducing of Mn-oxides by ferrous iron in a flow system: column experiment and reactive transport modelling. Geochim Cosmochim Acta 64/7:1237–1247
Reeder RJ, Nugent M, Lamble GM, Tait CD, Morris DE (2000) Uranyl incorporation into calcite and aragonite: XAFS and luminescence studies. Environ Sci Technol 34:638–644
Schneider P, Neitzel PL, Osenbrück K, Noubactep Ch, Merkel B, Hurst S (2001) In-situ-treatment of radioactive mine water using reactive materials: results of laboratory and field experiments in uranium ore mines in Germany. Acta Hydrochim Hydrobiol 29:123–138
Simpkin (2003) Comparison of ZVI treatment zones and standard PRBs as groundwater containment barriers. In: AFCEE Technology Transfer Workshop, Texas, USA
SSK (1992) Strahlenschutzgrundsätze für die Verwahrung, Nutzung oder Freigabe von kontaminierten Materialien, Gebäuden, Flächen oder Halden aus dem Uranerzbergbau, Empfehlungen der Strahlenschutzkommission mit Erläuterungen, Veröffentlichung der Strahlenschutzkommission, B. 23, G. Fischer Verlag, Stuttgart
StrSchV (2001) Verordnung über den Schutz vor Schäden durch ionisierte Strahlung, Strahlenschutzverordnung 1989, Novellierung
US-EPA (1998) Permeable reactive barrier technologies for contaminant remediation. EPA 600-R-98–125
US Department of Energy (1996) Research and application of permeable reactive barriers. DOE-K0002000 (Subcontract No. DE-AC 13–96DJ87335)
Valentine RL (1992) Radium removal using preformed hydrous manganese oxides. American Water Works Association Research Foundation, Subject Area: Water Treatment and Operations
Vance DB (2003) Treatment of chlorinated hydrocarbon contaminated groundwater with injectible nano scale reactive particles. white paper, http://www.2the4.net
Wersin P, Hochella MFJR, Persson P, Redden G, Leckie JO, Harris DW (1994) Interaction between aqueous uranium(VI) and sulfide minerals: spectroscopic evidence for sorption and reduction. Geochim Cosmochim Acta 58:2829–2843
Zeien H (1995) Chemisch Extraktionen zur Bestimmung der Bindungsformen von Schwermetallen in Böden. Inaugural-Dissertation Institut für Bodenkunde Bonn, ISSN 0939–7809
Acknowledgments
Funding of this research project (FKZ 02WB0221) was provided by the BMBF (German federal ministry of education and research). We would like to thank the Wismut GmbH for flooding water supply, Dr. M. Köhler from VKTA Rossendorf for 226Ra analyses as well as M. Michel for ICP measurements.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Burghardt, D., Kassahun, A. Development of a reactive zone technology for simultaneous in situ immobilisation of radium and uranium. Environ Geol 49, 314–320 (2005). https://doi.org/10.1007/s00254-005-0093-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00254-005-0093-0