

Abstract
Two-dimensional (2D) materials and their heterostructures show a promising path for next-generation electronics1,2,3. Nevertheless, 2D-based electronics have not been commercialized, owing mainly to three critical challenges: i) precise kinetic control of layer-by-layer 2D material growth, ii) maintaining a single domain during the growth, and iii) wafer-scale controllability of layer numbers and crystallinity. Here we introduce a deterministic, confined-growth technique that can tackle these three issues simultaneously, thus obtaining wafer-scale single-domain 2D monolayer arrays and their heterostructures on arbitrary substrates. We geometrically confine the growth of the first set of nuclei by defining a selective growth area via patterning SiO2 masks on two-inch substrates. Owing to substantial reduction of the growth duration at the micrometre-scale SiO2 trenches, we obtain wafer-scale single-domain monolayer WSe2 arrays on the arbitrary substrates by filling the trenches via short growth of the first set of nuclei, before the second set of nuclei is introduced, thus without requiring epitaxial seeding. Further growth of transition metal dichalcogenides with the same principle yields the formation of single-domain MoS2/WSe2 heterostructures. Our achievement will lay a strong foundation for 2D materials to fit into industrial settings.
Similar content being viewed by others

Main
2D transition metal dichalcogenides (TMDs) and their heterostructures are promising platforms for next-generation electronics, spintronics and valleytronics, as well as optoelectronics applications1,2,3,4,5,6,7,8,9,10,11,12,13. However, so far, the integration of such semiconducting 2D heterostructures onto industrial platforms has been challenging owing to limited scalability. The most common method to construct 2D heterostructures is through mechanical exfoliation and stacking of 2D flakes, which is a trial-and-error-based operation, and thus suffers from severely limited size of the structures and long assembly times. So far, substantial progress has been made to address the scalability issue by developing an ‘epitaxial’ growth method to obtain single-crystalline monolayer TMDs on single-crystalline hexagonal substrates such as sapphire14. However, there still exist major challenges in growing large-scale 2D heterostructures, owing to the lack of layer-by-layer growth strategies of single-domain TMDs. The current state-of-the-art growth method requires an undesirable transfer process of 2D materials to infuse them into Si technology. Although a ‘non-epitaxial’ method for producing single-domain TMD arrays has been reported recently using laser irradiation on the nucleation spots15, this method will inevitably accompany undesirable damages in TMD. Moreover, lateral heterostructures will preferably form when a vertical growth of the second heterolayer is attempted.
Here we report the two major features of this study, while addressing possible concerns on the feasibility of layer-by-layer growth of 2D materials on arbitrary substrates for commercialization. First, we demonstrate single-domain heterojunction TMDs at the wafer-scale by layer-by-layer growth. Second, we report our non-epitaxial strategy to grow single-domain TMDs on amorphous oxides, thus enabling direct growth of single-crystalline 2D materials on an Si wafer coated with an oxide layer. To achieve these, we have attempted to tackle the fundamental kinetic issues in TMD growth, where the first set of nuclei must merge with each other before the next set of nuclei to form a continuous monolayer of TMDs. We confine the growth of the first set of nuclei by defining a selective growth area of less than a few micrometres via patterning amorphous (a)-SiO2 masks on c-plane Al2O3 (c-Al2O3) or a-HfO2-deposited Si substrates. We confirm higher binding energy of TMDs on those substrates compared to that on a-SiO2 via density function theory (DFT) calculations, and thus the nucleation of TMDs is concentrated on the substrate surface instead of on a-SiO2. The reduced size of the a-SiO2 trenches substantially reduces the duration of the growth, yielding a fully merged first set of nuclei within the incubation time of introducing the second set of nuclei. Thus, the grown monolayer TMD layers are all single-domain across the wafer. The confined geometry allows precise control of the number of layers such that the next TMD monolayers can be subsequently grown to fill up the trenches. Field-effect transistors (FETs) fabricated on the arrays of single-domain WSe2 over the entire 2-inch wafer exhibit excellent performance, close to the level of mechanically exfoliated WSe2 flakes; for example, effective mobility up to 72.8 cm2 V–1 s–1 for monolayer (ML)-WSe2 and 103.5 cm2 V–1 s–1 for bilayer (BL)-WSe2. Moreover, we successfully demonstrated a layer-by-layer confined growth of MoS2/WSe2 heterostructures at the wafer scale and measured their valley lifetime on the arrays of single-domain MoS2/WSe2 heterostructures, which exhibit comparable values from those obtained from single-domain flakes12. We believe that our confined-growth technique can bring all the useful physics of 2D materials to the level of commercialization by allowing the construction of single-domain layer-by-layer homojunctions or heterojunctions on ‘non-epitaxial’ surfaces at the wafer scale.
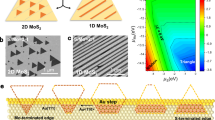
Figure 1a shows the conventional process of TMD growth. Initially, the first set of nuclei are formed and their orientations are random, because they are not typically aligned with the substrate. Whereas the first set of nuclei are laterally grown to meet each other, the second set of nuclei are formed on top of the first set of nuclei. Without additional control of the nucleation, this process is repeated, resulting in the growth of irregular TMD layer thicknesses upon merging of the first set of nuclei. When they merge with each other, a continuous polycrystalline layer is formed, owing to the random orientation of the initial nuclei, which will eventually degrade the intrinsic properties of the TMDs. To solve this issue, in Fig. 1b we present our strategy to digitally control the thickness and crystallinity of the TMD growth. First, we coated the c-Al2O3 or a-HfO2-deposited Si wafers with a-SiO2, followed by forming a micrometre-sized trench array on a-SiO2. Then, we selectively grew WSe2 inside the a-SiO2 trench. The size of the trench is small enough that only a single-domain nucleation event is allowed, which later grows and fills up the entire trench. The resulting WSe2 films are single-domain monolayer. This process is repeated to obtain single-domain MoS2/WSe2 heterostructures by performing confined growth of a second MoS2 layer (Fig. 1c). A single-domain homobilayer has been also achieved by additional growth of WSe2. Their growth selectivity is shown in Fig. 1d, where the binding energies of the precursors WO3 and Se, as well as the product WSe2 clusters on c-Al2O3, a-HfO2 and a-SiO2, are calculated from DFT. Compared to the surface of a-SiO2, WO3 cluster (W3O9, black box), Se cluster (Se2, red box), and WSe2 cluster (W3Se6, blue box) have stronger binding interactions with c-Al2O3 and a-HfO2. This indicates that the clusters preferentially bind to substrates instead of a-SiO2, leading to selective WSe2 growth (see Supplementary Information for details). We also have experimentally confirmed their selectivity by simultaneously growing WSe2 on c-Al2O3, a-HfO2 and a-SiO2 substrates under the same chemical vapor deposition (CVD) growth conditions, as shown in atomic force microscopy (AFM) images (Extended Data Fig. 1).

a, Schematic of the general process by which TMDs are grown. Stage I shows the same chance of nucleation across the entire area. b, Schematic of the selective single-domain synthesis strategy to address the limitations of conventional TMD growth. c, Single-domain MoS2/WSe2 heterostructures by confined growth of a second MoS2 layer. d, The binding energies of W3O9 (black box), Se2 (red box) and W3Se6 (blue box) clusters on c-Al2O3, a-HfO2 and a-SiO2 substrates.
To determine the appropriate size of the a-SiO2 trenches—sufficiently small to allow single-domain WSe2 formation at the confined area—we measured the lateral growth rate of WSe2 and the nucleation incubation time of the second set of nuclei after the first nucleation layer (see Extended Data Fig. 2). The measured lateral growth rate of WSe2 and the incubation time of the second nucleation were approximately 0.4 μm min−1 and 5 min, respectively. As shown in Fig. 2a, growing WSe2 on the 10-μm trench patterns for longer than the incubation time led to multiple domains in the trench. Whereas a single WSe2 triangle occupied approximately 70% of the trench area (indicated as yellow), the rest of the area contained multiple domains (green for two domains, and red for three domains). Further increasing the growth time resulted in the formation of multidomain polycrystalline WSe2 (Fig. 2b). Substantial reduction of photoluminescence intensity was observed for the multidomain area compared to the single-domain area (see Supplementary Fig. 1). Our statistics show that among multiple trenches with a size of 10 μm, approximately 25% contain more than two domains (see Extended Data Fig. 3). On the other hand, when the size of the trench was restricted to 2 μm, only a single domain was formed in every inspected area (484 trenches) regardless of the position of the initial nucleation event, either at the centre leading to homogeneous nucleation or at the edge of trenches for heterogeneous nucleation (see Extended Data Fig. 4). Further growth leads to merging of the nuclei, filling the entire trench as shown in Fig. 2c,d. Because the final arrays of WSe2 filled in the confined trenches originated from a single-domain nucleation event, all WSe2 arrays are single-crystalline across the wafer. We also confirmed that approximately 97% of the trench area was filled by WSe2 and complete filling of the trench was accomplished during the incubation time of the second-layer nuclei formation on top of the first WSe2 layer. Raman mapping at the E12g peak position and photoluminescence mapping at 1.65 eV confirmed that the WSe2 grown in all trenches was indeed monolayer (Fig. 2e,f). The average of the full-width at half-maximum (FWHM) of the photoluminescence spectrum of our single-domain WSe2 in the trenches was measured to be approximately 55 meV at room temperature (300 K), which is similar to that of high-quality single-domain WSe2 flakes that are mechanically isolated from bulk (see Supplementary Fig. 2)16. Our thorough characterization reveals that single-domain WSe2 arrays exhibit excellent crystallinity and composition (see AFM, high-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) and X-ray photoelectron spectroscopy (XPS) data shown in Extended Data Fig. 5).

a–d, Single-domain WSe2 selectively synthesized in 10-μm (a,b) and 2-μm (c,d) sapphire pockets fabricated with a-SiO2 trenches. Growth times are indicated at the top right. e–h, Raman and photoluminescence mapping images of confined ML-WSe2 (e,f) and BL-WSe2 (g,h) in 2-μm sapphire pockets. i,j, Cross-sectional high-resolution transmission electron microscopy (HRTEM) images of confined ML-WSe2 and BL-WSe2 in a sapphire pocket. a.u., arbitrary units. Scale bars, 10 μm (a,b), 2 μm (c–h), 2 nm (i,j).
On the basis of the obtained single-domain ML-WSe2 arrays, we attempted to consecutively grow another layer of WSe2 to obtain uniform homobilayers, which are electrically superior to monolayer. We have observed that each trench allowed only single-domain nucleation events and that further increasing the growth time resulted in the formation of single-domain BL-WSe2 by fully filling up the trenches (Extended Data Fig. 6a,b). This was additionally verified by the shift of the Raman spectra from A1g peak (approximately 259.6 cm−1) for a confined monolayer to the B12g peak (approximately 308.5 cm−1) for the confined bilayer (Extended Data Fig. 6c). In addition, the photoluminescence spectra peak shift from 1.65 to 1.6 eV confirms its transition from direct-gap to indirect-gap (Extended Data Fig. 6d). Raman mapping at the B12g peak and photoluminescence mapping at 1.6 eV verified our achievement in obtaining uniform bilayer 2D materials across the entire wafer (Fig. 2g,h). We additionally performed atomic-scale characterization on the ML- and BL-WSe2. High-resolution transmission electron microscopy (HRTEM) showed 0.8-nm-thick ML-WSe2 and 1.6-nm-thick BL-WSe2 as shown in Fig. 2i,j. In particular, from STEM images we observed that BL-WSe2 was grown without visible strain effects that can potentially arise at the edge of the a-SiO2 trench compared to the centre, confirming the uniformity of the BL-WSe2 (Extended Data Fig. 7). In addition, plan-view HAADF-STEM analysis revealed that BL-WSe2 is epitaxially aligned with AA′ stacking (Supplementary Fig. 3).
To verify the electrical quality of our single-domain TMDs prepared by the confined-growth method, we have fabricated arrays of FETs on the BL-WSe2 grown on a 2-inch wafer; Fig. 3a,b shows representative Ids–Vgs characteristics measured from one of the BL-WSe2 FET arrays. The FET exhibits an on/off current ratio of greater than 108, a subthreshold swing of 240.5 mV dec−1, a maximum on-current (Ion) density of up to 155.8 μA μm–1 and a field-effect mobility (μeff = gmL/WCgVds, Cg = 11.6 nF cm–2, where gm, L, W, and Cg are the transconductance, channel length, width, and gate capacitance, respectively) of up to 103.5 cm2 V–1 s–1, at Vds = −1 V. Furthermore, a saturation current of up to 465 μA μm–1 was observed via output characteristics (Fig. 3c). We benchmarked the results of WSe2 FETs reported so far (Fig. 4d)15,17,18,19,20,21,22,23,24,25. We highlight that the electrical properties of FETs fabricated with confined ML-/BL-WSe2 are comparable to the best properties reported for single-crystalline WSe2-based FETs (see Supplementary Fig. 4 and Supplementary Table 1 for details), and similar to (or better than) those of as-exfoliated flake-based ML-/BL-WSe2 FETs (Fig. 3e and Supplementary Fig. 5). We next performed statistical analysis on the FET arrays with respect to Ion per width and μeff (Fig. 3f). The FETs exhibited a Gaussian distribution in both Ion per width and μeff; the average and variation values are 89.9 μA μm–1 and 17.3% for Ion density and 79.1 cm2 V–1 s–1 and 24.1% for μeff. Detailed statistical investigation with respect to Ion density, μeff, subthreshold swing, on/off current ratio and threshold voltage (Vth) on 213 FETs fabricated with confined BL-WSe2 are provided in Extended Data Figs. 8,9, with an estimated yield of 93.9%. In addition, we also characterized FETs fabricated with confined ML-WSe2 (see Supplementary Fig. 6), achieving electrical performances comparable to FETs fabricated with confined BL-WSe2.

a, Photograph of an integrated confined BL-WSe2 FET arrays on an a-SiO2/Si wafer with size 5.1 cm × 5.1 cm. Inset shows micrograph of an individual FET array, where 20 FETs are integrated. Scale bar, 10 μm. b, Transfer characteristics of confined BL-WSe2 FET at Vds = –1 V, where LCH (channel length) is 0.7 μm. Obtained results show maximum on-current density of up to 155.8 μA μm–1 and a field-effect mobility of up to 103.5 cm2 V–1 s–1. c, Output characteristics of confined BL-WSe2 FET. d, Benchmark of WSe2 FETs, highlighting the performance of this work (red stars) with respect to Ion and μeff. Criteria of selection for other works: CVD-grown single-crystalline (SC) 1–3 ML-WSe2 (filled blue squares)15,17,18,19,20,21,22, CVD-grown polycrystalline (PC) 1–3 ML-WSe2 films (cross-filled orange squares)23,24, as-exfoliated WSe2 flakes (violet pentagons)25. Vds and LCH are –1 V and nearby 1 μm, respectively. e, Comparison of results with respect to Ion and μeff between FETs fabricated with as-exfoliated ML-/BL-WSe2 flakes and confined ML-/BL-WSe2 films. f, Statistical distribution with respect to Ion and μeff achieved from confined BL-WSe2 FET arrays.

a, Photograph and schematic image of a-HfO2 deposited by atomic layer deposition (ALD) system on an Si wafer in a reduced trench size (1 μm). b,c, Single-domain MoS2 selectively synthesized in 1-μm a-HfO2 pockets fabricated with a-SiO2 trenches. Scale bars, 5 μm. d, Transfer characteristics of FETs fabricated with confined ML-/BL-MoS2 (blue and red, respectively) on an a-HfO2 substrate. Inset denotes optical microscopic (OM) image of confined MoS2 FET arrays fabricated on a-HfO2. e, Statistical distribution with respect to Ion per width and μeff achieved from confined ML-/BL-MoS2 FET arrays. f, Cross-sectional HAADF-STEM image of the heterointerface, MoS2 (upper layer) and WSe2 (lower layer), overlain with the energy dispersive X-ray (EDX) spectra for Mo Kα (purple), S Kα (yellow), W Lα (grey) and Se Kα (green). g, Time-resolved circular dichroism (CD) response in ML-WSe2 at 300 K (black) and heterobilayer (MoS2/WSe2) at 300 K and 77 K (red and blue, respectively), where the red and blue solid lines are the cumulative fits. Scale bars, 5 μm (a–d), 2 nm (f).
We have further developed our confined-growth strategy to fulfil the requirement of industrialization in the logic and memory sectors, which requires single-crystalline TMD formation on amorphous surfaces on Si wafers and single-crystalline heterostructures at the wafer scale. First, we have applied our learning in confined TMD growths on single-crystalline sapphire wafers to achieve single-crystalline TMDs on a-HfO2 deposited on an Si wafer with a reduced trench size (Fig. 4a). As shown in Fig. 4b,c, all single-domain nucleation events occur fully within a 1-μm trench, and grows successfully, filling up the trenches and resulting in single-crystalline ML-MoS2 on an amorphous surface at the wafer scale. Also, additional confined growths of MoS2 on a-HfO2 allows layer-by-layer growth of bilayer and trilayer MoS2 as shown in STEM images (see Supplementary Fig. 7). We observe excellent electrical characteristics of the confined TMDs on a-HfO2 by fabricating the FETs on as-grown ML-/BL-MoS2 on a-HfO2/Si wafers without any transfer process. Fig. 4d shows Ids–Vds measured from fabricated ML-/BL-MoS2 FETs. The FETs exhibit a maximum Ion density of up to 86.7 μA μm–1 (ML-MoS2) and 129.3 μA μm–1 (BL-MoS2), a μeff of up to 62.2 cm2 V–1 s–1 and 88.61 cm2 V–1 s–1, wherein \({C}_{{{\rm{g}}}_{{{\rm{HfO}}}_{2}}}=600\;{\rm{nF}}\;{{\rm{cm}}}^{-2}\) and Vds = 1 V. In addition, we next performed statistical analysis on the 42 MoS2 FETs (21 ML-MoS2 FETs and 21 BL-MoS2 FETs) with respect to the Ion per width and μeff, wherein the average values of Ion and μeff are 71.4 μA μm−1 (for Ion per width of ML-MoS2 FETs), 99.3 μA μm−1 (for Ion per width of BL-MoS2 FETs), 51.2 cm2 V−1 s−1 (for μeff of ML-MoS2 FETs), and 71.2 cm2 V−1 s−1 (for μeff of BL-MoS2 FETs), respectively. Here, we estimated 23.8% and 25.2% of variation for Ion per width values of ML-/BL-MoS2 FETs, and 20.3% and 24.9% of variation for μeff values of ML-/BL-MoS2 FETs, respectively, with a 90.5% device yield (Fig. 4e).
Second, we have attempted consecutive growths of single-domain heterobilayer TMD semiconductors. To prove the concept, we performed the growth of ML-MoS2 on the single-domain ML-WSe2 arrays. We confirmed that single-MoS2-domain nucleation events can also occur within the confined WSe2-filled trench, resulting in a full area coverage of MoS2, forming single-domain MoS2/WSe2 heterobilayers. Raman mapping and photoluminescence spectra confirmed uniform formation of MoS2/WSe2 heterobilayers (see Extended Data Fig. 10 for crystalline quality). In addition, a cross-sectional HAADF-STEM image showed a sharp van der Waals heterointerface between the confined ML-MoS2 and ML-WSe2 without any alloying (Fig. 4f). A uniform heterointerface without secondary nucleation was also observed in the HAADF-STEM image at low magnification (see Supplementary Fig. 8). These data encouraged us to further characterize valleytronic performance of our MoS2/WSe2 heterobilayer arrays. We investigated valley-polarized carrier dynamics of heterobilayer (MoS2/WSe2) arrays via ultrafast circular dichroism based on time-resolved pump–probe spectroscopy (Fig. 4g). We measured valley lifetime from our single-domain MoS2/WSe2 arrays (approximately hundreds of picoseconds at 300 K and a few nanoseconds at 77 K).
In conclusion, we have demonstrated the synthesis of single-domain arrays of 2D TMDs at the wafer scale using a confined-growth technique. This growth technique enables layer-by-layer synthesis with critical Gibbs free energy difference, realizing wafer-scale single-domain homobilayers and heterobilayers on arbitrary substrates. In addition, our confined TMD layers show excellent electrical performance, comparable to that of flake TMDs. Therefore, our confined-growth technique can not only overcome the difficulty in controlling the kinetics of 2D materials at the wafer scale, which has been a major obstacle for 2D TMDs, but also show great potential for creating various single-crystalline van der Waals integration at a large scale, providing a route for building a 2D-material-based electronics platform.
Methods
DFT calculation for selective confined growth of TMDs
DFT calculations were performed using the Vienna ab initio Simulation Package (VASP) which uses projector augmented wave pseudopotentials26 and a plane-wave basis set27. Generalized gradient approximation of Perdew−Burke−Ernzerhof (PBE) functional was used to describe the electronic exchange-correlation interaction28. The valence electron configurations of W, Se, O, Al and Si are 6s25d4, 4s24p4, 2s22p4, 3s23p1 and 3s23p2, respectively. The energy cut-off for plane-wave expansion was set at 420 eV. Because large cells (lattice constant > 10 Å) were used for calculation, the Brillouin zone was sampled by using Γ-point only k-point grid. The surface binding interaction was investigated by placing the WO3, Se and WSe2 clusters on top of the a-HfO2, Al2O3 (0001) and a-SiO2 slab, respectively. a-HfO2, a-SiO2 and c-plane Al2O3 surfaces were passivated by H atoms to mimic the Ar/H2 ambient growth environment. The amorphous HfO2 and SiO2 atomic structure was obtained by subjecting their crystalline structure to a melt–quench process simulated by ab initio molecular dynamics29. Structures were optimized by relaxing top adsorbent atoms with substrate atoms fixed. The criterion for structure relaxation is that the force exerted on each atom should be less than 0.01 eV Å−1. Electronic minimization is reached when the system energy difference between two consecutive iterations is smaller than 10−5 eV. The surface binding energy for adsorbent A on substrate B is calculated as Eb = EA/B – EA – EB where EA/B, EA and EB are the energies of the adsorbing system A/B, isolated adsorbent A and substrate B, respectively.
Confined pattern fabrication
For confined growth of TMDs, we coated LOR 3A and photoresist (S1805) on a sapphire substrate and patterned it with an AS200 i-line stepper (AutoStep 200). A ~25-nm-thick a-SiO2 was deposited on a photoresist (PR)-patterned sapphire substrate with an electron-beam evaporator. Then, to fabricate sapphire pockets, the a-SiO2 pattern was lifted off with Remover PG (Kayaku Advanced Materials) and rinsed in acetone and isopropanol for 15 min each.
Synthesis of WSe2 and MoS2
Confined TMDs were synthesized in a quartz tube with 4-inch diameter. 300 mg of Se or S powders for (zone I), and 30 mg of WO3 or MoO3 powders for (zone II) were placed, and the distance between them were fixed at 33 cm. The sapphire substrate patterned with a-SiO2 was vertically loaded 6 cm behind the WO3 or MoO3 powders, and the front and back of the substrate were covered with quartz plates to minimize direct reaction. Before synthesizing confined ML-WSe2, the air in the quartz tube was removed with a vacuum pump. After closing the vacuum valve, a ratio of Ar (50 sccm)/H2 (50 sccm) was used as the carrier gas to fill the tube before opening the atmospheric valve. The ratio of Ar/H2 was maintained constantly. The growth temperatures of zones I and II were obtained by heating at ramp rates of 15 °C min−1 and 30 °C min−1, respectively, then held at 450 °C (zone I) and 890 °C (zone II) for 10 min before naturally cooling down to room temperature. For the confined BL-WSe2, a second WSe2 layer synthesis was performed with carrier gas having a ratio of Ar (35 sccm)/H2 (65 sccm). For the confined heterostructure (MoS2/WSe2), MoS2 synthesis was performed at 200 °C (zone I) and 750 °C (zone II) with ramp rates of 8 °C min−1 and 30 °C min−1, respectively. In particular, to improve the growth selectivity on the a-HfO2 substrate, we reduced the size of the a-SiO2 trenches to 1 μm and increased the overall flow rate of Ar/H2 from 100 sccm (Ar (50 sccm)/H2 (50 sccm)) to 200 sccm (Ar (100 sccm)/H2 (100 sccm)). All reactions were performed at atmospheric pressure, and all TMD powders were from Sigma-Aldrich with purity of more than 99.99%. We emphasize that it is important that various parameters must be kept consistent for growth reproducibility of confined TMDs (see Supplementary Information for details).
Characterization of confined TMDs
Raman and photoluminescence spectra were measured using a Renishaw InVia Reflex micro-spectrometer with 532-nm laser. The light was dispersed by a holographic grating with 2,400 grooves mm−1. For Raman and photoluminescence mapping images, samples were scanned on an x–y piezo stage with laser illumination. Scanning electron microscope (SEM) images were measured with an in-Lens detector using a high-resolution SEM (ZEISS Merlin). The working distance was 6 mm at an accelerating voltage of 2 kV and a probe current of 70 pA. Transmission electron microscopy (TEM) characterization was performed using a JEOL JEM-2100F with an accelerating voltage of 200 kV and STEM (Titan Themis Z G3 Cs-Corrected) with an accelerating voltage of 60 kV. Energy dispersive X-ray (EDX) line profiles were taken with the Velox software in STEM mode using the characteristic Mo Kα, S Kα, W Lα and Se Kα X-ray signals. XPS spectra are measured with a magnesium Kα source (MultiLab 2000, Thermo VG), and the peak energies were calibrated by the C 1s peak at 284.8 eV. AFM morphology analysis was performed using an XE 100 (Park Systems).
Device fabrication and electrical measurements
For device fabrication using confined ML-/BL-WSe2, a 600-nm-thick Au film was deposited on confined WSe2/sapphire by electron-beam evaporation. The Au/WSe2 stack was peeled off using a thermal release tape as a handling layer and transferred onto a 300-nm-thick SiO2/heavily p-doped silicon wafer. The thermal release tape was removed on a hot plate at 120 °C, followed by oxygen plasma treatment to remove tape residues from the Au film. Then, the Au film was etched with Au etchant and rinsed with deionized water (to compare the electrical characteristics, a few-layer WSe2 flake was also transferred in the same way). After the transfer of confined ML-/BL-WSe2 on a-SiO2 substrate, align marks for electron-beam lithography (EBL) process were patterned on an a-SiO2 substrate using an optical lithography process, followed by the deposition of 2.5-nm-thick Ti and 7.5-nm-thick Au using an electron-beam evaporator. Then, drain and source contact regions with a width of 2 μm were patterned using an EBL process. For EBL photoresists, polymethyl methacrylate (PMMA) A4 and PMMA A6 were spin-coated at 3,000 rpm and baked at 180 °C for 150 s. After developing the PMMA, 10-nm-thick Pt and 80-nm-thick Au layers were deposited using an electron-beam evaporator. Finally, the areas, except the source/drain contact metal regions, were removed by a lift-off process. For device fabrication using confined ML-/BL-MoS2 on a-HfO2, the same processes from patterning align marks for EBL process to developing the PMMA were performed. Then 10-nm-thick Ni and 80-nm-thick Au layers were deposited using an electron-beam evaporator, followed by a lift-off process. Here we emphasize that we directly fabricated confined ML-/BL-MoS2 FETs on a-HfO2-deposited Si wafers without a transfer process. The current–voltage characteristics were measured with an Agilent B2900A. All measurements were conducted at room temperature in air. In addition, 2-inch confined BL-WSe2 was transferred onto 300-nm-thick SiO2/Si substrate with a size of 5.1 × 5.1 cm2. Kelvin probe force microscopy (KPFM) confirmed highly uniform distribution of work functions (5.08 eV) on confined BL-WSe2 (Supplementary Information and Supplementary Fig. 9). Source and drain electrodes with a channel length (LCH) of 0.7 μm were then integrated using platinum; a hole barrier height of 0.31 eV was estimated via modified Richardson plotting (Supplementary Information and Supplementary Fig. 10)30.
Time-resolved pump–probe spectroscopy
To investigate valley-polarized carrier dynamics, we have measured ultrafast circular dichroism (CD) based on time-resolved pump–probe spectroscopy. A 100-kHz Yb-based regenerative amplifier system (Light Conversion PHAROS) provided a femtosecond laser pulse, and a sequential optical parametric amplifier (ORPHEUS) served wavelength-tunable pump and probe pulses resonant with A-exciton resonance of WSe2 with pulse duration of 50 fs and spectral bandwidth of 50 meV. Samples on cryostat have been illuminated by a pump excitation pulse using a 40× objective lens. Pump-induced changes in probe reflectance have been recorded as a function of time delay given by a mechanical translational stage and lock-in amplifier. The polarization profiles of pump and probe pulses are individually controlled by a pair consisting of a half-wave plate and a quarter-wave plate. We have measured the signal when the pump and probe pulses exhibit same helicity of circular polarization (co-polarized), and opposite helicity of the polarization (cross-polarized). Valley-dependent ultrafast CD responses shown in Fig. 4f,h were acquired by the differences between co-polarized and cross-polarized pump–probe responses. Experimental details can be found in the previous work31.
Data availability
The data that support the findings of this study are available from the corresponding author J.K. upon reasonable request.
References
Akinwande, D. et al. Graphene and two-dimensional materials for silicon technology. Nature 573, 507–518 (2019).
Fiori, G. et al. Electronics based on two-dimensional materials. Nat. Nanotechnol. 9, 768–779 (2014).
Chhowalla, M., Jena, D. & Zhang, H. Two-dimensional semiconductors for transistors. Nat. Rev. Mater. 1, 16052 (2016).
Xu, X. et al. Spin and pseudospins in layered transition metal dichalcogenides. Nat. Phys. 10, 343–350 (2014).
Sie, E. J. et al. Valley-selective optical Stark effect in monolayer WS2. Nat. Mater. 14, 290–294 (2015).
Kim, J. et al. Ultrafast generation of pseudo-magnetic field for valley excitons in WSe2 monolayers. Science 346, 1205–1208 (2014).
Mak, K. F. et al. The valley Hall effect in MoS2 transistors. Science 344, 1489–1492 (2014).
Schaibley, J. R. et al. Valleytronics in 2D materials. Nat. Rev. Mater. 1, 1–15 (2016).
Scuri, G. et al. Electrically tunable valley dynamics in twisted WSe2/WSe2 bilayers. Phys. Rev. Lett. 124, 217403 (2020).
Robert, C. et al. Exciton radiative lifetime in transition metal dichalcogenide monolayers. Phys. Rev. 93, 205423 (2016).
Rivera, P. et al. Interlayer valley excitons in heterobilayers of transition metal dichalcogenides. Nat. Nanotechnol. 13, 1004–1015 (2018).
Rivera, P. et al. Observation of long-lived interlayer excitons in monolayer MoSe2–WSe2 heterostructures. Nat. Commun. 6, 6242 (2015).
Jin, C. et al. Imaging of pure spin-valley diffusion current in WS2–WSe2 heterostructures. Science 360, 893–896 (2018).
Li, T. et al. Epitaxial growth of wafer-scale molybdenum disulfide semiconductor single crystals on sapphire. Nat. Nanotechnol. 16, 1201–1207 (2021).
Li, J. et al. General synthesis of two-dimensional van der Waals heterostructure arrays. Nature 579, 368–374 (2020).
Cadiz, F. et al. Excitonic linewidth approaching the homogeneous limit in MoS2-based van der Waals heterostructures. Phys. Rev. X 7, 021026 (2017).
Kozhakhmetov, A. et al. Scalable substitutional re-doping and its impact on the optical and electronic properties of tungsten diselenide. Adv. Mater. 32, 2005159 (2020).
Park, K. et al. Uniform, large-area self-limiting layer synthesis of tungsten diselenide. 2D Mater. 3, 014004 (2016).
Zhang, X. et al. Defect-controlled nucleation and orientation of WSe2 on hBN: a route to single-crystal epitaxial monolayers. ACS Nano 13, 3341–3352 (2019).
Lin, Y.-C. et al. Realizing large-scale, electronic-grade two-dimensional semiconductors. ACS Nano 12, 965–975 (2018).
Wang, X. et al. High-performance n-type transistors based on CVD-grown large-domain trilayer WSe2. APL Mater. 9, 071109 (2021).
Zhou, H. et al. Large area growth and electrical properties of p-type WSe2 atomic layers. Nano Lett. 15, 709–713 (2015).
Liu, B. et al. High-performance WSe2 field-effect transistors via controlled formation of in-plane heterojunctions. ACS Nano 10, 5153–5160 (2016).
Chen, M. et al. Gold-vapor-assisted chemical vapor deposition of aligned monolayer WSe2 with large domain size and fast growth rate. Nano Res. 13, 2625–2631 (2020).
Kim, W. et al. Field-dependent electrical and thermal transport in polycrystalline WSe2. Adv. Mater. Interfaces 5, 1701161 (2018).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953 (1994).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169 (1996).
Perdew, J. P., Kieron, B. & Matthias, E. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865 (1996).
Buchholz, D. B. et al. The structure and properties of amorphous indium oxide. Chem. Mater. 26, 5401–5411 (2014).
Lin, Y.-F. et al. Barrier inhomogeneities at vertically stacked graphene-based heterostructures. Nanoscale 6, 795–799 (2014).
Jin, G. et al. Heteroepitaxial van der Waals semiconductor superlattices. Nat. Nanotechnol. 16, 1092–1098 (2021).
Zhang, R. et al. Controlled layer thinning and p‐type doping of WSe2 by vapor XeF2. Adv. Funct. Mater. 27, 1702455 (2017).
Lin, Y. C. et al. Atomically thin resonant tunnel diodes built from synthetic van der Waals heterostructures. Nat. Commun. 6, 7311 (2015).
Kim, K. S. et al. Ultrasensitive MoS2 photodetector by serial nano-bridge multi-heterojunction. Nat. Commun. 10, 4701 (2019).
Acknowledgements
This material is based on work supported by DARPA (award no. 029584-00001)), NSF (grant no. CMMI-1825731), Intel, IARPA MicroE4AI program, MicroLink Devices, Inc., ROHM Co., and Samsung. Y.H. and K.C. were supported by nanomaterials development program through the National Research Foundation of Korea (NRF) (2022M3H4A1A04096496) funded by the Ministry of Science and ICT, Korea. S.C. and M.-H.J. acknowledge support from the Institute for Basic Science (IBS-R034-D1).
Author information
Authors and Affiliations
Contributions
K.S.K., S.-H.B. and J.K. conceived the idea and designed the experiments. K.S.K. performed confined TMD growth and coordinated the experiments and characterization. D.L. transferred confined TMDs for multiple characterizations. C.S.C. performed and analysed the TEM measurements. S.S., Ju-Hee Lee and J.-H.P. analysed the electrical characteristics. Y.H. and K.C. performed the DFT calculations. S.C. and M.-H.J. performed the ultrafast laser spectroscopy. K.S.K., D.L., C.S.C., H.K., J.S., S.L., J.S.K., K.H.K., J.M.S., Y.M., B.-I.P., Ju-Hee Lee, H.-S.P., H.S.K., G.Y.Y. and S.-H.B. performed material characterizations. K.S.K., J.-H.P., S.-H.B., and J.K. wrote the manuscript. All authors contributed to and commented on the analysis and discussion of the results.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Deji Akinwande, Kian Ping Lo and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Growth selectivity on both c-Al2O3, a-HfO2 and a-SiO2 substrates.
a–c, AFM images of WSe2 grown on c-Al2O3 (a), a-HfO2 (b), and a-SiO2 (c) under the same CVD process conditions. WSe2 only nucleates on the c-Al2O3 and a-HfO2 instead of a-SiO2 during 20 min of growth duration. This led to a successful selective confined growth of WSe2 on the exposed substrate surface of the micropatterned a-SiO2 trench arrays.
Extended Data Fig. 2 Lateral growth rate and secondary nucleation time for confined growth.
After an incubation time of 5 min, WSe2 was laterally grown up to 10 min, and secondary nucleation occurred after a confined monolayer was maintained for an additional 2 min. Scale bar, 2 μm.
Extended Data Fig. 3 Statistics of WSe2 single domains and multidomains grown in 10-μm-size trench patterns.
Approximately 25% were observed as multidomains (indicated as green and red) in the 10-μm-size trench patterns.
Extended Data Fig. 4 Statistics of WSe2 single domains grown in 2-μm-size trench patterns.
Each denoted top (red), bottom (blue), left (orange), and right (violet) region on the wafer includes 96 dies, whereas the denoted centre region (black) on the wafer includes 100 dies.
Extended Data Fig. 5 Characterization of confined single-domain WSe2.
a, AFM morphology of confined ML-WSe2. b, Root-mean-square (RMS) roughness in a cropped image of a confined ML-WSe2. c, Plan-view HAADF-STEM image of confined ML-WSe2. From plan-view STEM images, selenium vacancy was observed as the dominant point defect with a density of roughly less than 1% (~1.6 × 1013 cm−2). d,e, The atomic percentages of W 4f (d) and Se 3d (e) were 33.72% and 66.28%, respectively by XPS, confirming stoichiometric ratios, and the peak positions of W (4f5/2 and 4f7/2) and Se (3d3/2 and 3d5/2) were 34.8, 32.6, 55.7 and 55 eV, respectively. This result matches well with pure single-crystalline WSe232.
Extended Data Fig. 6 Layer-by-layer confined growth of TMDs.
a, SEM images of second set of nuclei formed on confined monolayer. b, The resulting growth of confined BL-WSe2.
Extended Data Fig. 7 Evaluation of the effect of trench edge on confined BL-WSe2.
a,b STEM images of BL-WSe2 grown at the edge (a) and centre (b) of SiO2 trench. The BL-WSe2 grown at the edge of the trench shows no difference from that at the centre, showing no obvious step bunching, distortions, or curling of BL-WSe2 due to possible strain that can be imposed by the edge.
Extended Data Fig. 8 Yield estimation and statistical analysis on FETs fabricated with confined BL-WSe2.
a, Schematic of wafer enabling to estimate a yield of 93.9% (200/213). Each denoted top region (red), bottom region (blue), left region (orange), and right region (violet) on the wafer includes 43 dies, and denoted centre region (black) on the wafer includes 41 dies. In each die, a centre-located FET was investigated for estimating yield. Coloured and ‘X’-marked dies denote good and defective dies, respectively. b–f, Statistical analysis on 200 FETs fabricated with confined BL-WSe2, where field-effect mobility (b), on current (c), subthreshold slope (d), current on/off ratio (e), and threshold voltage (f) were extracted and investigated. g, Summary table for performance labels.
Extended Data Fig. 9 Transfer characteristics of confined BL-WSe2 FETs.
Each graph shows transfer characteristics for over 40 FETs; top region (red), bottom region (blue), left region (orange), right region (violet), and centre region (black) of FET arrays fabricated on 2-inch wafer. Grey curves denote transfer characteristics of defective FETs.
Extended Data Fig. 10 Confined heterobilayer growth of TMDs (MoS2/WSe2) and their characterization.
a–c, Raman mapping of the E12g peak on the MoS2/WSe2 heterobilayer at each trench shows that ML-MoS2 is uniformly filled up (~94%) on top of ML-WSe2 at the wafer-scale (a). For the MoS2 layer of the heterobilayer, E12g (384.9 cm−1) and A1g (403.1 cm−1) peaks appeared in the Raman spectra (b). As shown in the representative photoluminescence spectra (c), two peaks associated with direct gaps of MoS2 and WSe2 were detected, confirming the ML-MoS2 formation. A slight shift of the peak from the original direct gap occurs due to a well-known interlayer exciton coupling33. d,e, The high crystalline quality of the MoS2 was further confirmed by ideal XPS spectra observed in narrow-scan data of Mo (3d3/2 and 3d5/2; d) and S (2p1/2 and 2p3/2; e), which showed no signs of defects or atomic mixing related Mo6+ peak at ~236 eV34.
Supplementary information
Supplementary Information
This file contains Supplementary Notes, Figures, Tables and References.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kim, K.S., Lee, D., Chang, C.S. et al. Non-epitaxial single-crystal 2D material growth by geometric confinement. Nature 614, 88–94 (2023). https://doi.org/10.1038/s41586-022-05524-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-05524-0
- Springer Nature Limited
This article is cited by
-
The future of two-dimensional semiconductors beyond Moore’s law
Nature Nanotechnology (2024)
-
Emerging probing perspective of two-dimensional materials physics: terahertz emission spectroscopy
Light: Science & Applications (2024)
-
200-mm-wafer-scale integration of polycrystalline molybdenum disulfide transistors
Nature Electronics (2024)
-
Remote epitaxy of single-crystal rhombohedral WS2 bilayers
Nature Communications (2024)
-
Flash-within-flash synthesis of gram-scale solid-state materials
Nature Chemistry (2024)