

Abstract
Small-ring cage hydrocarbons are popular bioisosteres (molecular replacements) for commonly found para-substituted benzene rings in drug design1. The utility of these cage structures derives from their superior pharmacokinetic properties compared with their parent aromatics, including improved solubility and reduced susceptibility to metabolism2,3. A prime example is the bicyclo[1.1.1]pentane motif, which is mainly synthesized by ring-opening of the interbridgehead bond of the strained hydrocarbon [1.1.1]propellane with radicals or anions4. By contrast, scaffolds mimicking meta-substituted arenes are lacking because of the challenge of synthesizing saturated isosteres that accurately reproduce substituent vectors5. Here we show that bicyclo[3.1.1]heptanes (BCHeps), which are hydrocarbons for which the bridgehead substituents map precisely onto the geometry of meta-substituted benzenes, can be conveniently accessed from [3.1.1]propellane. We found that [3.1.1]propellane can be synthesized on a multigram scale, and readily undergoes a range of radical-based transformations to generate medicinally relevant carbon- and heteroatom-substituted BCHeps, including pharmaceutical analogues. Comparison of the absorption, distribution, metabolism and excretion (ADME) properties of these analogues reveals enhanced metabolic stability relative to their parent arene-containing drugs, validating the potential of this meta-arene analogue as an sp3-rich motif in drug design. Collectively, our results show that BCHeps can be prepared on useful scales using a variety of methods, offering a new surrogate for meta-substituted benzene rings for implementation in drug discovery programmes.
Similar content being viewed by others
Main
Strategies for the structural modification of lead molecules that improve physicochemical and pharmacokinetic properties such as metabolic stability are increasingly sought in drug development6. One example is the replacement of aromatic rings with non-classical bioisosteres such as small-ring cage hydrocarbons1,3,5,7. Such structures display a higher fraction of saturated carbon atoms compared with their parent arenes (Fsp3, corresponding to greater three-dimensionality), which is a property that is linked to greater clinical success rates8. Among these motifs, the replacement of planar para-substituted arenes with bicyclo[1.1.1]pentanes (BCPs, Fig. 1a), which have similar dimensions and identical substituent vectors to the parent aromatic, has emerged as a popular strategy2,9,10. For instance, substitution of the fluorinated arene in the Alzheimer’s treatment avagacestat with a BCP resulted in an analogue that maintained the bioactivity of the parent compound, but displayed an improved pharmacokinetic profile2. More generally, cage hydrocarbons expand the vector space around a molecular core, offering new opportunities in drug design.

a, BCPs derived from [1.1.1]propellane (2) are bioisosteres for para-substituted benzene rings. b, Previous mimics of meta-substituted arenes, which do not accurately reproduce the geometry of the aromatic. c, BCHeps derived from [3.1.1]propellane (1) exactly mimic the geometry of meta-substituted arenes. d, A multigram scale synthesis of [3.1.1]propellane.
Meta-substituted arenes are also commonplace in pharmaceuticals and agrochemicals11. However, in stark contrast to the numerous sp3-rich bioisosteres for ortho- and para-substituted arenes2,9,10,12, a geometrically accurate bioisostere for meta-arenes is yet to be discovered. Recent reports on the use of (hetero)bicyclo[2.1.1]hexanes13,14,15,16,17 and bridge-substituted BCPs18,19,20 have contributed to this arena (Fig. 1b). However, those motifs fail to recreate the bond vectors displayed in the meta-substituted aromatic, and a precise and accessible mimic remains absent from the arsenal of the medicinal chemist.
Here we report a solution to this challenge in the form of the saturated carbocycle bicyclo[3.1.1]heptane (BCHep, Fig. 1c), the bridgehead substituent vectors of which precisely replicate those of the parent meta-arene (approximately 119° and 120°, respectively). Although BCHeps have been prepared by ring expansion of BCPs21 and by cyclization of cyclohexane dicarboxylates22, these approaches can be limited in substituent scope or involve lengthy synthetic sequences. We show that BCHeps can instead be conveniently and directly accessed from [3.1.1]propellane (1), a homologue of [1.1.1]propellane (2) that is widely used as the near-ubiquitous source of BCPs4. We found 1 to be a versatile precursor that undergoes a variety of radical-based transformations to access a wide range of functionalized BCHeps, including drug analogues. Profiling of the ADME properties of these analogues reveals that, like BCPs, BCHeps substantially improve physicochemical properties compared to their arene parents. As such, we anticipate that this new scaffold should offer a readily accessible bioisostere for broad implementation in drug discovery programmes.
Higher [n.1.1]propellanes such as 1 have until now been of predominantly theoretical interest23,24; these elusive molecules have probably been overlooked in synthetic and medicinal chemistry because of the challenge of their synthesis and reported instability25,26. We devised a strategy to synthesize 1 on a multigram scale (Fig. 1d), which began with Kulinkovich cyclopropanation of commercially available γ-chloroester 3 (ref. 27). Mesylation of the resulting alcohol 4, followed by TiCl4-mediated cyclopropyl–allyl chlorinative rearrangement and dibromocyclopropanation, afforded cyclopropane 5 in 58% yield over four steps on a scale larger than 30 mmol with only one chromatographic purification. Reaction of 5 with two equivalents of phenyllithium generated 1 in yields of 43–61% after distillation; the resulting solution of 1 (0.25–0.50 M in dibutyl ether) can be stored at −20 °C under an inert atmosphere for several months with negligible decomposition.
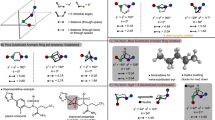
Aside from solvolysis25, previous reports on the chemistry of 1 detail only one productive reaction—the addition of thiophenol to generate BCHep phenyl thioether26. As a prelude to exploring the wider reactivity of 1, we first compared the calculated reaction barriers for the addition of a prototypical radical (CH3•) and nucleophile (NH2−) with those for 2 (ref. 28) (Fig. 2a). These calculations predict that 1 should be similarly susceptible to the addition of radicals to the interbridgehead C–C bond (difference between free energies of activation ΔΔG‡ = −1.1 kcal mol−1 between 1 and 2), but that the reaction of 1 with anions is less favourable (ΔΔG‡ = +5.4 kcal mol−1). This in part may relate to the greater increase of charge density inside the propellane cage in anionic additions, which can be better accommodated by 2 owing to the presence of a third three-membered ring that enables enhanced charge delocalization28,29. Our calculations therefore suggest that 1 should be amenable to the same array of radical functionalization chemistry established in the [1.1.1]propellane/BCP arena.

a, Reactivity profile of 1 calculated at the SMD(THF)-DLPNO-CCSD(T)/ma-def2-QZVPP//SMD(THF)-B2PLYP-D3BJ/def2-TZVP (ma-def2-TZVP on N) level of theory. THF, tetrahydrofuran. b, Carbon/halogen-substituted BCHeps prepared from organohalides using Ir(ppy)3 (2.5 mol%), blue light-emitting diode (LED) irradiation (a), Et3B (10 mol%) as initiator (b) or without an initiator (c). c, Nitrogen-substituted BCHeps prepared using dual photoredox/Cu-catalysed coupling of iodonium dicarboxylates and N-heteroarenes (d), pyrazole/I2 (e) or α-iodoaziridine, Ir(ppy)3 (2.5 mol%), blue LEDs (f). d, Chalcogen-substituted BCHeps prepared by direct reaction with the chalcogen–X precursor (g). e, Cysteine-selective conjugation studies using the (l,l,d) δ-(α-aminoadipolyl)-Cys-Val (ACV) tripeptide in aqueous phosphate buffer (50 mM, pH 8.0). Reactions run on a 0.1–0.2 mmol scale unless shown otherwise. See the Supplementary Information for details.
This theoretical analysis correlated well with experimental findings. We first explored atom transfer radical addition (ATRA) reactions, which are a powerful method to access disubstituted BCPs from 1. Both Et3B-initiated30 and Ir(ppy)3-catalysed31 addition of a variety of C–I bonds to 1 proceeded efficiently to afford diverse BCHep scaffolds (Fig. 2b). The photoredox-catalysed variant (Ir(ppy)3) proved more general and higher yielding, producing iodo-BCHeps from α-iodocarbonyls (6a–6d), benzyl iodides (6e–6f), alkyl iodides (6g–6k), α-amino acids (6l) and heteroaryl iodides (6m). In contrast to ATRAs with [1.1.1]propellane30, Et3B initiation was suitable mainly for electrophilic radicals such as α-iodocarbonyls (6a, 6b) and azetidines (6h). Notably, the addition of iodotrifluoromethane to 1 proceeded in the absence of an external initiator to afford 6n, which could be a valuable building block for the synthesis of bioisosteres of meta-CF3-substituted arenes. Addition to alkyl bromides such as bromomalonate (6o, 57%) and bromotrichloromethane (6p, 68%) also proved feasible, the latter proceeding without an initiator. The chemistry could further be applied to the late-stage bicycloheptylation of various drug analogues, producing BCHep derivatives of corticosterone (6q), nicotinic acid (6r), brequinar (6s) and indomethacin (6t), which were obtained from the corresponding alkyl iodides. Notably, in contrast to equivalent ATRA reactions with [1.1.1]propellane, no ‘staffane’ by-products arising from [3.1.1]propellane oligomerization were observed.
Bridgehead amine substituents are highly attractive as potential meta-substituted aniline bioisosteres. We found that the three-component metallaphotoredox catalysed coupling of iodonium dicarboxylates, [1.1.1]propellane and N-heteroarenes described by the Macmillan group32 translated smoothly to [3.1.1]propellane (Fig. 2c), producing azole- and sulfonamide-substituted BCHeps 7a–7g in good yields, including pharmaceutical derivatives (gemfibrozil, 7g). The synthesis of N-substituted iodo-BCHeps was achieved using other methods, such as pyrazole BCHep 7h by reaction of 1 with pyrazole/I2 (ref. 33), and allyl sulfonamide BCHep 7i from radical fragmentation of an iodomethyl aziridine34. As well as C- and N-centred radicals, other heteroatoms proved excellent substrates for reactions with 1 (Fig. 2d): thioether 8a and selenoether 8b were formed in quantitative yields at room temperature, sulfonothioate addition (8c, 8d) proceeded efficiently under heating35,36 and reaction with a disulfide could be achieved under ultraviolet irradiation (8e)37. The successful bicycloheptylation of protected cysteine (8f) in diethyl ether highlights the potential for applications in peptide modification38; unexpectedly, reaction of a similar cysteine residue in a tripeptide ((l,l,d) δ-(α-aminoadipolyl)-Cys-Val) in aqueous buffer afforded the rearranged adducts 9 and 10 (Fig. 2e), which may arise from a cationic reaction pathway. Although the reason for this reactivity difference is unknown, it is clear that selective S-alkylation of cysteine is possible under physiologically relevant conditions.
Iodinated BCHeps offer opportunities for C–I functionalization towards medicinally relevant difunctionalized scaffolds. Investigation of iron-catalysed Kumada cross-coupling39 revealed efficient reaction of iodo-BCHeps with both aryl and heteroaryl Grignard reagents to afford (hetero)aryl BCHeps in excellent yields (11a–11f, Fig. 3a). BCHep functionalization was also possible by lithiation of the iodide; reaction of the resulting bridgehead carbanion with CO2 or i-PrOBpin gave carboxylic acid 11g and hydroxy-BCHep 11h (after in situ oxidation), respectively, the latter of which corresponds to a meta-phenol bioisostere.

a, Functionalization of iodo-BHeps through iron-catalysed Kumada coupling (a) or lithiation/electrophilic quenching (b). b, Comparison of angles between substituent vectors from single-crystal X-ray structures (X-ray), and computed structures for BCHeps and the parent arenes (BCHepcalc and m-Arcalc, CPCM(THF)-B2PLYP-D3BJ/def2-TZVP level of theory). See the Supplementary Information for details.
X-ray structural determination of several crystalline BCHeps enabled us to study the geometry of the scaffold in more detail (Fig. 3b). Two substituent vector angles were considered: the exit vector angle α (around 120° for m-arenes), and the out-of-plane vector angle ϕ (the dihedral angle along the BCHep interbridgehead axis, around 0° for m-arenes). Comparison of the BCHep solid state structures with computed structures of the BCHep and the equivalent meta-arene showed excellent agreement for both angles (Δα = 0–7°, Δϕ = 3–11°), validating our hypothesis that the replacement of meta-substituted arenes with a BCHep conserves the critical substituent geometry.
Kumada cross-coupling was deployed to synthesize two BCHep drug analogues (Fig. 4a). The BCHep analogue of the anticancer agent sonidegib was accessed from coupling product 11e by pivaloate ester hydrolysis, oxidation of the resulting primary alcohol to the carboxylic acid 13 and amide formation with aminopyridine 14 (53% yield over three steps, 19% from 1). BCHep-URB597, the parent meta-arene of which was developed as a fatty acid amide hydrolase inhibitor, was synthesized from 6j by a similar cross-coupling–hydrolysis–oxidation sequence, followed by amide formation, debenzylation and carbamoylation with cyclohexyl isocyanate (16% over five steps). Computational conformer sampling once again revealed a similar global topology between BCHep-sonidegib and the parent drug for the vector angles α and ϕ (Fig. 4b). Here an additional parameter was considered: the rotational orientation of the planes between the two substituent groups as defined by the dihedral angle ψ (Δψ = 13°). The BCHep displayed a shallow potential energy profile for rotation around the BCHep–substituent C–C bond (free energy barrier ΔG = 1.5 kcal mol−1), reflecting a low conformational preference of the substituents adjacent to the quaternary carbons of the BCHep, whereas for the parent arene more defined minima exist (ΔG = 12 kcal mol−1, see the Supplementary Information for details). This may suggest that BCHeps offer significant flexibility in substituent conformation, which could be a valuable property for drug design by facilitating a more adaptable association with protein targets.

a, Synthesis of BCH analogues of sonidegib and URB597. EDC, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide; HOBt, 1-hydroxybenzotriazole; NMM, N-methylmorpholine; TMEDA, N,N,N',N'-tetramethylethylenediamine. b, Computational investigation of the topology of BCHep-sonidegib. DFT, density functional theory. c, Physicochemical and metabolic profile of BCHep-sonidegib and BCHep-URB597 along with their parent compounds. See the Supplementary Information for details. Cli, intrinsic clearance; Papp, apparent permeability; T1/2, half-life.
Synthesis of these drug analogues raises the question of how the physicochemical and pharmacological properties of the BCHep compare with the parent arene (Fig. 4c). The clogP, topological polar surface area and solubility of each drug–analogue pair are remarkably similar, demonstrating that BCHeps can be readily deployed in drug design as true meta-arene bioisosteres. In keeping with their well-established BCP cousins, BCHeps showed reduced clearance rates in mouse and human liver microsomes compared to their arene equivalents, and membrane permeability (Caco-2) was improved. The BCHep analogues were also tested for CYP inhibition and also generally showed an improvement compared to their corresponding arenes (Fig. 4c). URB597 inhibits CYP1A2 and CYP2C9 with half-maximal inhibitory concentration (IC50) values below 10 μM, but BCHep-URB597 is seven- and threefold weaker against these two polymorphic enzymes. Collectively, these data underline the potential power of the BCHep scaffold as a beneficial motif for improving the pharmacokinetic and physicochemical properties of drug candidates.
Methods
Synthesis of [3.1.1]propellane
1-(3-Chloropropyl)cyclopropan-1-ol, S1
A solution of ethyl 4-chlorobutanoate (5.60 ml, 40.0 mmol, 1.0 equiv.), and Ti(Oi-Pr)4 (1.20 ml, 4.0 mmol, 0.10 equiv.) in anhydrous diethyl ether (60 ml) was cooled to 0 °C and a solution of EtMgBr (33.3 ml, 3.0 M in Et2O, 100 mmol, 2.5 equiv.) was added dropwise over 90 min. The mixture was stirred for a further 30 min at 0 °C, and then the mixture was slowly quenched by dropwise addition of 10% aqueous H2SO4 (50 ml). The organic layer was washed sequentially with H2O (70 ml), NaHCO3 (sat., aq., 70 ml) and brine (70 ml), and then dried (Na2SO4), filtered and concentrated in vacuo to produce S1 (4.73 g, 35.1 mmol, 88%) as a colourless oil, which was used without further purification.
1-(2-Chloroethyl)cyclopropyl methanesulfonate, 4
A solution of S1 (4.73 g, 35.1 mmol, 1.00 equiv.) and triethylamine (7.05 ml, 50.7 mmol, 1.44 equiv.) in anhydrous CH2Cl2 (60 ml) was cooled to 0 °C and methanesulfonyl chloride (3.08 ml, 40.0 mmol, 1.14 equiv.) was added dropwise over 30 min. The mixture was stirred for a further 30 min at 0 °C, and then quenched with water (30 ml). The layers were separated, and the organic layer was washed sequentially with H2O (60 ml), 10% H2SO4 (aq., 50 ml), NaHCO3 (sat., aq., 50 ml) and brine (50 ml), and then dried (Na2SO4), filtered and concentrated in vacuo to produce 4 (7.20 g, 33.9 mmol, 96%) as a pale-yellow oil, which was used without further purification.
5-Chloro-2-(chloromethyl)pent-1-ene, S2
TiCl4 (5.72 ml, 52.4 mmol, 1.55 equiv.) was slowly added over 20 min to a solution of 4 (7.20 g, 33.8 mmol, 1.0 equiv.) in anhydrous CH2Cl2 (60 ml) at room temperature. The mixture was stirred at room temperature for 3 h, and then slowly quenched with H2O (60 ml) at 0 °C with vigorous stirring. The layers were separated, and the organic layer was washed sequentially with H2O (2 × 70 ml), NaHCO3 (sat., aq., 70 ml) and brine (70 ml), and then dried (MgSO4), filtered and concentrated in vacuo (300 mbar, 30 °C) to produce S2 (4.46 g, 29.1 mmol, 86%) as a clear pale-yellow liquid. Note that the product is volatile under reduced pressure (isolated with residual solvent) and was taken forward without further purification. The state yield makes allowance for residual solvent.
1,1-Dibromo-2-(chloromethyl)-2-(3-chloropropyl)cyclopropane, 5
A 50% NaOH solution (22 ml) was added dropwise over 20 min to a vigorously stirred (1,000 r.p.m.) solution of S2 (4.46 g, 29.1 mmol, 1.00 equiv.), CHBr3 (20.4 ml, 233 mmol, 8.00 equiv.), dibenzo-18-crown-6 (524 mg, 1.47 mmol, 0.05 equiv.) and pinacol (137 mg, 1.15 mmol, 0.04 equiv.) in CH2Cl2 (37 ml) at 50 °C. The resulting mixture was stirred for 5 h at 50 °C, and then cooled to room temperature and diluted with n-pentane (100 ml) and distilled water (100 ml). The resulting suspension was filtered through a pad of celite and washed with additional n-pentane (100 ml). Additional distilled water (100 ml) was added to the filtrate. The layers were separated and the organic layer was washed with brine (150 ml), and then dried (Na2SO4), filtered and concentrated in vacuo. The crude product was purified by column chromatography (column diameter 4 cm, 40 g SiO2, gradient 100% pentane to 95:5 pentane:EtOAc) to produce 5 (7.50 g, 23.1 mmol, 79%) as a colourless oil.
[3.1.1]Propellane, 1
Phenyllithium (31.8 ml, 60.4 mmol, 2.01 equiv., 1.9 M in n-Bu2O) was slowly added to a cooled (−78 °C) solution of 5 (9.74 g, 30.0 mmol, 1.0 equiv.) in anhydrous Et2O (160 ml) . The resulting mixture was stirred at −78 °C for 15 min then warmed to room temperature and stirred for 7 h. The mixture was then distilled using a rotary evaporator (25 °C water bath temperature) equipped with a dry-ice cold finger condenser, with the receiving flask immersed in a dry-ice–acetone bath. The Et2O fraction was removed by slowly decreasing the applied pressure to 150 mbar. This fraction was then discarded. The remaining solution was distilled by slowly reducing the applied pressure to less than 10 mbar to produce a solution of [3.1.1]propellane 1 in n-Bu2O, which was stored under an inert atmosphere at −20 °C. The yield was determined by 1H nuclear magnetic resonance spectroscopy using 1,2-dichloroethane as an internal standard (see below). The concentration of the [3.1.1]propellane solution ranged between 0.25 M and 0.50 M, with yields of 43–61%. Note that the resulting propellane stock solution contains bromobenzene, which does not influence the reactions presented herein.
General procedures for reactions of [3.1.1]propellane
Photoredox-catalysed ATRA
fac-Ir(ppy)3 (2.5 mol%), alkyl or aryl halide (1.0 equiv.), t-BuCN (0.1 M) and [3.1.1]propellane (1.1 to 2.0 equiv. of a solution in n-Bu2O) was added to a flame-dried, screw-capped vial equipped with a stirrer bar. The vial was placed under nitrogen, and the solution was degassed via a modified freeze–pump–thaw cycle (the vacuum was only applied while the reaction mixture was frozen owing to the volatility of [3.1.1]propellane). The stirred mixture was irradiated with blue LEDs (Kessil PR160 456 nm) with fan cooling for the indicated time. The reaction mixture was concentrated and the residue was purified by column chromatography. See ref. 31 and Fig. 2b (condition a).
ATRA with BEt3
Under air, a solution of alkyl iodide (1.0 equiv.) in [3.1.1]propellane (1.1 to 1.5 equiv. of a solution in n-Bu2O) was cooled to 0 °C and Et3B (10 mol%, 1.0 M in hexane) was added via syringe (with the needle tip in the solution). The mixture was stirred until the reaction reached completion as monitored by thin-layer chromatography. The reaction mixture was then concentrated and the residue purified by column chromatography. See ref. 30 and Fig. 2b (condition b)
Ir/Cu-catalysed additions to [3.1.1]propellane
fac-Ir(ppy)3 (2.0 mol%), amine starting material (1.0 equiv.), iodomesitylene biscarboxylic acid (2.0 equiv.), Cu(acac)2 or Cu(TMHD)2 (0.60 equiv), 2-tert-butyl-1,1,3,3-tetramethylguanidine (BTMG) (3.0 equiv.) and anhydrous 1,4-dioxane (0.03 M) were added to a flame dried, screw-capped vial equipped with a stirrer bar. The solution was sparged with Ar for 10 min, and then [3.1.1]propellane (1.5 equiv. of a solution in n-Bu2O) was added and the vial capped and sealed with parafilm. The mixture was stirred and irradiated with blue LEDs (Kessil PR160 456 nm) with fan cooling for 16 h. The reaction mixture was diluted with EtOAc and washed with 30% aqueous ammonia solution. The phases were separated, and the aqueous phase was extracted with EtOAc (3×). The combined organics were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by column chromatography. See ref. 32 and Fig. 2c (condition d).
Addition of thiols
[3.1.1]Propellane (1.0 equiv., of a solution in n-Bu2O) was added dropwise to a solution of thiol (1.1 equiv.) in anhydrous diethyl ether. The mixture was stirred for 1 h, and then diluted with diethyl ether and washed with 1 M aqueous NaOH solution (3×). The organic layer was dried over Na2SO4, filtered and concentrated in vacuo. The obtained crude product was either purified by column chromatography or trituration. See Fig. 2d (condition g).
Disulfide addition
Disulfide (3.0 equiv.) and [3.1.1]propellane (1.0 equiv. of a solution in n-Bu2O) was added to a flame dried, screw-capped vial. The mixture was irritated with a LED lamp (HepatoChem Evoluchem HCK1012-01-011 365 nm) with fan cooling for 20 h. The solvents were removed in vacuo and the resulting residue was purified by column chromatography. See ref. 37 and Fig. 2d (condition g).
Addition of sulfonylthionates
[3.1.1]Propellane solution (1.5 equiv.) was added to a solution of the specific thiosulfonate (1.0 equiv.) in anhydrous MeCN. The flask was sealed and heated to 60 °C for 16 h, and then cooled to room temperature and concentrated in vacuo. The crude product was purified by column chromatography. See ref. 35 and Fig. 2d (condition g).
Iron-catalysed Kumada coupling
BCHep iodide (1.0 equiv.) and Fe(acac)3 (20 mol%) was added to a flame-dried vial. The vial was then evacuated and refilled with N2 (g) three times. To this was added anhydrous THF (0.2 ml) and N,N,N',N'-tetramethylethylenediamine (TMEDA) (40 mol%), and the resulting mixture was stirred for 5 min. The Grignard reagent (1.6 equiv.) was then added via a syringe pump over approximately 1 h at room temperature. The reaction was stirred for a further 1 h, and then quenched by addition of aqueous NH4Cl (2 ml, saturated). The layers were separated, and the aqueous layer was extracted with Et2O (3 × 1 ml). The combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. The crude product was purified by column chromatography. See ref. 39 and Fig. 3a.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
References
Subbaiah, M. A. M. & Meanwell, N. A. Bioisosteres of the phenyl ring: recent strategic applications in lead optimization and drug design. J. Med. Chem. 64, 14046–14128 (2021).
Stepan, A. F. et al. Application of the bicyclo[1.1.1]pentane motif as a nonclassical phenyl ring bioisostere in the design of a potent and orally active γ-secretase inhibitor. J. Med. Chem. 55, 3414–3424 (2012).
Auberson, Y. P. et al. Improving nonspecific binding and solubility: bicycloalkyl groups and cubanes as para-phenyl bioisosteres. Chem. Med. Chem. 12, 590–598 (2017).
Ma, X. & Pham, Nhat L. Selected topics in the syntheses of bicyclo[1.1.1]pentane (BCP) analogues. Asian J. Org. Chem. 9, 8–22 (2020).
Mykhailiuk, P. K. Saturated bioisosteres of benzene: where to go next? Org. Biomol. Chem. 17, 2839–2849 (2019).
Meanwell, N. A. Improving drug design: an update on recent applications of efficiency metrics, strategies for replacing problematic elements, and compounds in nontraditional drug space. Chem. Res. Toxicol. 29, 564–616 (2016).
Chalmers, B. A. et al. Validating Eaton’s hypothesis: cubane as a benzene bioisostere. Angew. Chem. Int. Edn 55, 3580–3585 (2016).
Lovering, F., Bikker, J. & Humblet, C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 52, 6752–6756 (2009).
Pu, Q. et al. Discovery of potent and orally available bicyclo[1.1.1]pentane-derived indoleamine-2,3-dioxygenase 1 (IDO1) inhibitors. ACS Med. Chem. Lett. 11, 1548–1554 (2020).
Measom, N. D. et al. Investigation of a bicyclo[1.1.1]pentane as a phenyl replacement within an LpPLA2 inhibitor. ACS Med. Chem. Lett. 8, 43–48 (2017).
Nilova, A., Campeau, L.-C., Sherer, E. C. & Stuart, D. R. Analysis of benzenoid substitution patterns in small molecule active pharmaceutical ingredients. J. Med. Chem. 63, 13389–13396 (2020).
Denisenko, A., Garbuz, P., Shishkina, S. V., Voloshchuk, N. M. & Mykhailiuk, P. K. Saturated bioisosteres of ortho-substituted benzenes. Angew. Chem. Int. Edn 59, 20515–20521 (2020).
Levterov, V. V., Panasyuk, Y., Pivnytska, V. O. & Mykhailiuk, P. K. Water-soluble non-classical benzene mimetics. Angew. Chem. Int. Edn 59, 7161–7167 (2020).
Kleinmans, R. et al. Intermolecular [2π+2σ]-photocycloaddition enabled by triplet energy transfer. Nature 605, 477–482 (2022).
Agasti, S. et al. A catalytic alkene insertion approach to bicyclo[2.1.1]hexane bioisosteres. Preprint at https://doi.org/10.26434/chemrxiv-22022-v26493kv (2022).
Guo, R. et al. Strain-release [2π + 2σ] cycloadditions for the synthesis of bicyclo[2.1.1]hexanes Initiated by energy transfer. J. Am. Chem. Soc. 144, 7988–7994 (2022).
Dhake, K. et al. Beyond bioisosteres: divergent synthesis of azabicyclohexanes and cyclobutenyl amines from bicyclobutanes. Angew. Chem. Int. Edn 61, e202204719 (2022).
Zhao, J. X. et al. 1,2-Difunctionalized bicyclo[1.1.1]pentanes: long-sought-after mimetics for ortho/meta-substituted arenes. Proc. Natl Acad. Sci. USA 118, e2108881118 (2021).
Yang, Y. et al. An intramolecular coupling approach to alkyl bioisosteres for the synthesis of multisubstituted bicycloalkyl boronates. Nat. Chem. 13, 950–955 (2021).
Anderson, J. M., Measom, N. D., Murphy, J. A. & Poole, D. L. Bridge functionalisation of bicyclo[1.1.1]pentane derivatives. Angew. Chem. Int. Edn 60, 24754–24769 (2021).
Harmata, A. S., Spiller, T. E., Sowden, M. J. & Stephenson, C. R. J. Photochemical formal (4 + 2)-cycloaddition of imine-substituted bicyclo[1.1.1]pentanes and alkenes. J. Am. Chem. Soc. 143, 21223–21228 (2021).
Della, E. W. & Elsey, G. M. Bridgehead carbocations: solvolysis of a series of 5-substituted bicycle [3.1.1]heptyl bromides. Nucleophilic solvent assistance to ionization of 1-bromobicyclo[3.1.1]heptane. Aust. J. Chem. 48, 967–985 (1995).
Dilmac, A. M., Spuling, E., de Meijere, A. & Brase, S. Propellanes-from a chemical curiosity to “explosive” materials and natural products. Angew. Chem. Int. Edn 56, 5684–5718 (2017).
Wiberg, K. B. Small ring propellanes. Chem. Rev. 89, 975–983 (1989).
Gassman, P. G. & Proehl, G. S. [3.1.1]Propellane. J. Am. Chem. Soc. 102, 6862–6863 (1980).
Fuchs, J. S. G. Synthese von [n.l.l]propellanen (n = 2, 3, 4). Chem. Ber. 125, 2517–2522 (1992).
Kulinkovich, O. G., Kozyrkov, Y. Y., Bekish, A. V., Matiushenkov, E. A. & Lysenko, I. L. A convenient way for the conversion of carboxylic esters into 2-substituted allyl halides. Synthesis 2005, 1713–1717 (2005).
Sterling, A. J., Dürr, A. B., Smith, R. C., Anderson, E. A. & Duarte, F. Rationalizing the diverse reactivity of [1.1.1]propellane through σ–π-delocalization. Chem. Sci. 11, 4895–4903 (2020).
Sterling, A., Smith, R., Anderson, E. & Duarte, F. Beyond strain release: delocalisation-enabled organic reactivity. Preprint at https://doi.org/10.26434/chemrxiv-22021-n26430xm26439-v26432 (2022).
Caputo, D. F. J. et al. Synthesis and applications of highly functionalized 1-halo-3-substituted bicyclo[1.1.1]pentanes. Chem. Sci. 9, 5295–5300 (2018).
Nugent, J. et al. A general route to bicyclo[1.1.1]pentanes through photoredox catalysis. ACS Catal. 9, 9568–9574 (2019).
Zhang, X. et al. Copper-mediated synthesis of drug-like bicyclopentanes. Nature 580, 220–226 (2020).
Zarate, C. et al. Development of scalable routes to 1-bicyclo[1.1.1]pentylpyrazoles. Org. Proc. Res. Dev. 25, 642–647 (2021).
Pickford, H. D. et al. Twofold radical-based synthesis of N,C-difunctionalized bicyclo[1.1.1]pentanes. J. Am. Chem. Soc. 143, 9729–9736 (2021).
Wu, Z., Xu, Y., Wu, X. & Zhu, C. Synthesis of selenoether and thioether functionalized bicyclo[1.1.1]pentanes. Tetrahedron 76, 131692 (2020).
Bär, R. M., Kirschner, S., Nieger, M. & Bräse, S. Alkyl and aryl thiol addition to [1.1.1]propellane: scope and limitations of a fast conjugation reaction. Chem. Eur. J. 24, 1373–1382 (2018).
Bär, R. M., Heinrich, G., Nieger, M., Fuhr, O. & Bräse, S. Insertion of [1.1.1]propellane into aromatic disulfides. Beilstein J. Org. Chem. 15, 1172–1180 (2019).
Tokunaga, K. et al. Bicyclobutane carboxylic amide as a cysteine-directed strained electrophile for selective targeting of proteins. J. Am. Chem. Soc. 142, 18522–18531 (2020).
Nugent, J. et al. Synthesis of all-carbon disubstituted bicyclo[1.1.1]pentanes by iron-catalyzed Kumada cross-coupling. Angew. Chem. Int. Edn 59, 11866–11870 (2020).
Acknowledgements
N.F. thanks Studienstiftung des Deutschen Volkes e.V. for a scholarship. J.N. thanks the Marie Skłodowska-Curie actions for an Individual Fellowship (GA No 786683). E.A.A. and A.J.S. thank the EPSRC for support (grant nos EP/S013172/1 and EP/T517811/1). B.R.S., A.J.S. and H.D.P. thank the EPSRC Centre for Doctoral Training in Synthesis for Biology and Medicine for studentships (EP/ L015838/1). T.Z.-T., T.G. and P.E.B. thank Alzheimer’s Research UK for support (grant no. ARUK-2021DDI-OX). P.R. thanks the Deutsche Akademie für Naturforscher Leopoldina for a fellowship.
Author information
Authors and Affiliations
Contributions
E.A.A., N.F., J.N. and A.J.S. conceived the project. Experimental work was carried out by N.F., J.N., B.R.S., P.R., T.Z.-T. and T.G. H.D.P. and A.L.T. collected the crystallographic data. N.F. and A.J.S. carried out the computational analysis. The project was supervised by E.A.A., F.D., P.E.B. and C.J.S. E.A.A., N.F., J.N. and F.D. wrote the initial manuscript which was reviewed and edited by E.A.A., N.F., J.N., R.C.S., P.E.B. and F.D.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks Tian Qin, Antonia Stepan and the other, anonymous, reviewer(s) for their contribution to the peer reivew of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Sections 1–18 containing materials and methods, procedures, crystallographic data, further computational analysis, peptide labelling studies, evaluation of ADME properties, unsuccessful reactions, coordinates and spectra data.
Supplementary Data (CIF)
Crystallographic data for 6s, 7f, 7h, 8d and 12. Deposited with the CCDC (CCDC 2175923–2175926 and CCDC 2176171).
Supplementary Data (Coordinates)
Folder containing all necessary .xyz files of computational analysis. Deposited in the Oxford Research Archive.
Supplementary Data (source data)
Physicochemical and pharmacokinetic property data. Deposited in the Oxford Research Archive.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Frank, N., Nugent, J., Shire, B.R. et al. Synthesis of meta-substituted arene bioisosteres from [3.1.1]propellane. Nature 611, 721–726 (2022). https://doi.org/10.1038/s41586-022-05290-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-022-05290-z
- Springer Nature Limited
This article is cited by
-
Stereoselective polar radical crossover for the functionalization of strained-ring systems
Communications Chemistry (2024)
-
Light-enabled scalable synthesis of bicyclo[1.1.1]pentane halides and their functionalizations
Nature Synthesis (2024)
-
Synthetic techniques for thermodynamically disfavoured substituted six-membered rings
Nature Reviews Chemistry (2024)
-
Enantioselective formal (3 + 3) cycloaddition of bicyclobutanes with nitrones enabled by asymmetric Lewis acid catalysis
Nature Communications (2024)
-
C−F bond activation enables synthesis of aryl difluoromethyl bicyclopentanes as benzophenone-type bioisosteres
Nature Communications (2024)