

Abstract
Alkenyl sulfides have gained increasing prominence in medicinal chemistry and materials. Hydrothiolation of alkynes for the diverse synthesis of alkenyl sulfides is an appealing method. Herein, we report a cobalt-catalyzed Markovnikov hydromethylthiolation of alkynes to afford branched alkenyl methylsulfanes with good yields and high regioselectivity. This method also enables the diverse synthesis of branched alkenyl sulfides. The reaction shows good functional group tolerance and could be scaled up. The mechanistic studies including control experiments, deuterium-labeling experiments, and Hammett plot indicated alkynes insertion followed by electrophilic thiolation pathway.
Similar content being viewed by others

Introduction
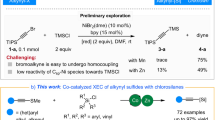
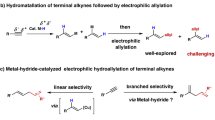
Alkenyl sulfides have important applications in antibiotic drugs, analytical detection, and functional materials (Fig. 1A), driving continuous developments in synthetic methods for their preparation1,2,3,4,5. The use of readily available and cost-effective alkynes as starting materials for hydrothiolation represents an appealing method (Fig. 1B)6,7,8,9,10. It should be noted that the synthetic methods for branched configuration products are still limited. Representative examples include transition metal-catalysis, such as Pd11,12,13,14,15, Rh16,17,18,19,20, Th21,22, Zr23, Ni24,25,26,27, and photocatalysis28, using thiols or thiophenols as raw materials. Although the methylthio group is recognized for its utility in modifying drug properties by enhancing solubility and altering metabolic pathways4,29,30, the hydromethylthiolation of alkynes with methanethiol is rarely explored31,32,33, due to the high toxicity, low boiling point (279 K at 1 atm), repugnant odor, and strong coordination with transition metal catalysts (as an example, the bond energy of Pd–SMe (92–99 kcal/mol) is much higher than that of CAr–SMe (82–85 kcal/mol), making the reduction elimination process difficult)34. Furthermore, hydrothiolation reactions are significantly influenced by the groups of alkynes and sulfur sources14, and there is still no protocol applicable to four types of hydrothiolation (reactions between alkyl, aryl alkynes and alkyl, aryl sulfur sources), so the development of more versatile protocols for the diversity hydrothiolation of alkynes remains desirable.

A Alkenyl-thio moiety in drug candidates and materials. B Catalytic hydrothiolation of terminal alkynes. C This work: Markovnikov hydrothiolation of terminal alkynes.
While significant progress has been made in developing masked sulfurizing reagents for the green synthesis of sulfur-containing compounds31,35,36,37,38,39, to the best of our knowledge, metal-catalyzed regioselective hydrothiolation of alkynes with masked sulfurization reagents has not been reported. The intrinsic challenge of regioselective hydrothiolation involves addressing the issues of chemoselectivity (the masked sulfurizing reagents could be reduced to deliver side products, such as thiols and disulfides), regioselectivity (branched versus linear alkenyl sulfides), and potential metal poisoning40,41. Here, we report a cobalt-catalyzed Markovnikov hydromethylthiolation of alkynes with masked sulfurizing reagents, which also enables diverse reactions between alkyl, aryl alkynes and alkyl, aryl sulfur sources (Fig. 1C).
Results
Reaction optimization
The investigation was initiated by examining the hydromethylthiolation of 1-ethynyl-4-methoxybenzene (1a) with sulfur electrophile (2a). After screening various masked sulfurizing reagents, it was observed that using L1 as the ligand, OMTS (1,1,1,3,5,7,7,7-octamethyltetrasiloxane) as the silane, and lithium methoxide as the base, 3a could be obtained with excellent yield (90%) and excellent regioselectivity (b/l = 97/3) (Table 1, entry 1). However, with phthalimide, methanethio, or methanesulfonyl masked sulfurizing reagents, the reactions afforded 3a in 5–15% yields (entries 2–4), demonstrating the substantial influence of the masks. Employing L2 and L3 as ligands although the yields were reduced, products are predominantly afforded in the branched configuration; however, the bisphosphine ligand L4 did not facilitate this reaction (entries 5–7). Other bases such as sodium methoxide, lithium tert-butoxide or CsF significantly reduced the yield of 3a (entries 11–13). Finally, the pre-catalyst complex was used instead of the in situ catalyst to control the reaction, and the similar yield and selectivity of 3a were obtained (entry 14).
Substrate scope
After optimizing the reaction conditions, a series of alkynes as substrates were investigated (Fig. 2). Due to the instability of branched alkenyl sulfides in air, all separation steps were carried out in a nitrogen atmosphere whenever possible. The electron-donating and electron-withdrawing groups on the phenyl ring were tolerated to afford 3b-3k in 50–83% yields with 90/10– > 99/1 b/l. Particularly, Csp2–Br (3d), Csp2–I (3e) and phenylamino (3k) were well compatible, providing more chances for further molecule cross-linkage. Meta-substituted and ortho-substituted alkynes 1l–1o could also participate in the reaction to afford 3l–3o in 49–85% yields with 92/8–97/3 b/l. The alkynes containing a polycyclic ring or heterocycle, such as piperonyl (1p), 2-naphthyl (1q), and 3-thienyl (1r), could be converted to the corresponding products 3p–3r in 54–77% yields with 95/5–97/3 b/l. Notably, aliphatic alkynes were also applicable to the reaction. Ethynylcyclohexane (1 s) could participate to deliver the hydromethylthiolation product 3 s in 51% yield with 95/5 b/l. The linear aliphatic alkynes (1t–1v) could be reacted to afford 3t–3v in 72–82% yields with 88/12–98/2 b/l. Additionally, alkynes that incorporate bioactive molecules, such as sesamol (1w), naproxen (1x), ibuprofen (1y), and the fragment of empagliflozin (1z), could be employed to deliver the corresponding products 3w-3z in 46–71% yields with 88/12–96/4 b/l, further demonstrating the utilities of the approach.

a1 (0.50 mmol), 2 (0.25 mmol), CoBr2 (5 mol%), L1 (6 mol%), OMTS (1.2 eq.), LiOMe (2.5 eq.), THF (2.0 mL), 40 °C for 12 h, yields were determined by 1H NMR using 1,1,2,2-tetrachloroethane or 1,3,5-trimethoxybenzene as an internal standard, isolated yields are shown in parentheses. bL1•CoBr (5 mol%) instead of CoBr2 and L1. cAt 45 °C. dAt 35 °C.
Inspired by the effectiveness of thiol-ene and thiol-yne reactions in connecting two molecular structures through the robust creation of carbon-sulfur bonds42,43,44,45,46,47, the diverse synthesis based on this protocol has been conducted (Fig. 3). The sulfur source available for this protocol can be easily obtained from alcohols, halides, and disulfides. Aryl alkynes could undergo hydrothiolation with aliphatic sulfurizing reagents such as deuterated methyl (2ab), ethyl (2ac), cyclopropylmethyl (2ad), butyl (2ae), isopropyl (2af), and trifluoropropyl (2ag), obtaining the corresponding products (3ab–3af) in 28–84% yields with 90/10–97/3 b/l. Sulfurizing reagents bearing substituted benzyl groups (2ah, 2ai), could also be transformed into the desired products (3ah, 3ai). To our delight, the reaction between 1aj and 2aj resulted in 3aj in 86% yield and 92/8 b/l, proving the suitability of aryl alkynes with aryl sulfurizing reagents. The reaction between alkyl alkynes and alkyl or aryl sulfurizing reagents is also suitable to obtain the corresponding products (3ak–3am) in 35–81% yields with 86/14–93/7 b/l. Masked sulfurizing reagents derived from 1-adamantaneethanol (2an), geraniol (2ao), and naproxen (2ap), successfully reacted with alkynes to obtain 3an–3ap in 51–80% yields with 96/4– > 99/1 b/l, confirming the universality of diverse synthesis.

a1 (0.50 mmol), 2 (0.25 mmol), CoBr2 (5 mol%), L1 (6 mol%), OMTS (1.2 eq.), LiOMe (2.5 eq.), THF (2.0 mL), 40 °C for 12 h, yields were determined by 1H NMR using 1,1,2,2-tetrachloroethane or 1,3,5-trimethoxybenzene as an internal standard, isolated yields are shown in parentheses. bL1•CoBr (5 mol%) was used instead of CoBr2 and L1. cAt 45 °C. dAt room temperature. e59 h. fPh2SiH2 instead of OMTS.
Gram-scale reaction and synthetic applications
The reaction could be smoothly conducted on a 10 mmol scale, yielding 1.34 grams of the methyl(1-phenylvinyl)sulfane 3aa with an impressive 89% yield (Fig. 4a), facilitating subsequent derivatization with ease. The Ts- group utilzied in the reaction can be efficiently recovered in the form of lithium 4-methylbenzenesulfinate (5) and regenerated into the starting material (2a) through a two-step conversion. This capability showcases the remarkable recyclability and sustainability of the employed protocol. To showcase the utility of the branched alkenyl sulfides, further transformations of 3aa were investigated (Fig. 4b). The product 3aa could undergo nickel-catalyzed Kumada coupling with methylmagnesium bromide to obtain 1,1-dialkene (6) in 80% yield18,48. With copper catalysis, 3aa could proceed hydroboronation to deliver compound 7 in 95% yield49,50. The double bond of 3aa could be reduced with p-toluenesulfonyl hydrazide to obtain α-methylthioethylbenzene (8) in 90% yield51. The [2 + 1] cycloaddition reaction of difluorocarbene to 3aa could give difluorocyclopropane (9) in 87% yield52. Additionally, given that selenium belongs to the same chalcogen family as sulfur and has a similar electronegativity, Markovnikov hydroselenation of alkynes was likewise found to be a viable reaction pathway (Fig. 4c)53.

a Gram-scale reaction and recycling of byproduct. b Synthetic applications of the product. c Hydroselenation of alkynes. d Aerobic oxidation of the product in air.
Unlike linear alkenyl sulfides, branched alkenyl sulfides are highly sensitive to air (Fig. 4d). Exemplified by 3aa, it could readily undergo oxidation in the presence of air, transforming into 2-(methylthio)-1-phenylethan-1-one (10). This reactivity explains why 3aa can be easily stained with (2,4-dinitrophenyl)hydrazine on thin-layer chromatography plates. To our knowledge, this phenomenon has never been reported. However, upon scrutinizing the supporting information from prior works on the synthesis of branched alkenyl sulfides, traces of oxidized products can be observed in the 1H NMR spectra. A possible mechanism involving radical addition has been proposed (see SI). The sensitivity of branched alkenyl sulfides to oxygen provides insights into the potential applications in ROS probes54. It should be emphasized that branched alkenyl sulfides are quite stable in a nitrogen-filled glove box, with no oxidation products observed after six months of storage.
Mechanistic investigation
To elucidate the reaction mechanism, a range of comparative experiments were undertaken. The addition of 2.0 equivalents of radical scavengers to the reaction system did not obstruct the course of the reaction (Fig. 5a). Introduction of deuterated phenylacetylene or deuterated diphenylsilane into the reaction elucidated the emergence of two identical deuterated ratio at the β-position of the product (Fig. 5b). To excluded the possibility of the possibility of the process that β-H elimination followed by branched alkenyl methylsulfanes inserting into Co–H pathway, 3aa was added to the reaction with deuterated diphenylsilane, and no significant deuterium substitution was observed in the recovered 3aa. To ruled out the possible H-D exchange between terminal alkynes and silanes, 1,2-diphenylacetylene was used as a raw material for the reaction, resulting in 11 in equivalent yield with E/Z = 1/4 (Fig. 5c). The observations suggest the conceivable occurrence of a Crabtree-Ojima isomerization process. When L1•CoOMe was used as catalyst, the targeted product was obtained in 84% yield. When lithium methoxide was removed, only trace of product was obtained (Fig. 5d). This means that in this system, despite being classified as a cobalt oxide species, the coordination between the toluenesulfonate anion and the central metal was relatively robust, making it impossible to regenerate cobalt hydride species from silane. The reaction exhibited an induction period before reaching maximum yield approximately after eight hours (see SI). An in-depth analysis of the influence of different substituents on phenylacetylene on the reaction kinetics facilitatedthe construction of a Hammett plot (Supplementary Data 1), which unveiled a positive slope. The observation implies that the turnover-limiting step is dominated by negative charge accumulation, providing valuable insights into the reaction mechanism (Fig. 5e).

a Radical trapping experiments. b Deuterium-labeling experiments. c Hydromethylthiolation of the internal alkyne. d Control experiments. e Hammett plot.
Based on these mechanistic experiments and previous literatures55,56,57,58,59,60,61,62,63,64, a possible reaction mechanism is proposed (Fig. 6). The initial steps involve cobalt bromide species undergoing ligand exchange with lithium methoxide to obtain cobalt hydride species II upon interaction with silane. Subsequent alkyne coordination is followed by cobalt hydride insertion, resulting in the formation of an alkenyl cobalt intermediate IV. This species further undergoes reaction with the masked sulfurizing reagent, proceeding through a five-membered ring pathway. Ultimately, the branched alkenyl sulfide and cobalt p-toluenesulfonate V are obtained65. Then V undergoes ligand exchange with lithium methoxide, initiating the subsequent catalytic cycle.

Proposed reaction pathway starts from Co–OMe, then alkyne inserts into Co–H, followed by the reaction with the masked sulfurizing reagent.
Discussion
In summary, a cobalt-catalyzed hydromethylthiolation reaction of alkynes have been developed, which also enables the diverse synthesis of branched alkenyl sulfides and exhibits remarkable regioselectivity and compatibility within a wide range of substrates, including alkenes, esters, amines, and aryl halides. This protocol provides an efficient route obtaining a series of alkenyl sulfides from readily available starting materials. Preliminary investigations into the reaction mechanism provide evidence for alkyne insertion into cobalt hydrides, followed by reaction with the masked sulfurizing reagents. The studies on aerobic oxidation of alkenyl sulfides revealed blind spots in literatures. Further studies on the synthesis and applications of alkenyl sulfides are undergoing.
Methods
General procedure for the synthesis of branched alkenyl sulfides
In a nitrogen-filled glove box, a 10 mL vial equipped with a stir bar was charged with CoBr2 (0.0125 mmol, 5 mol%), L1 (0.015 mmol, 6 mol%), LiOMe (0.625 mmol, 2.5 eq.), and THF (2.0 mL). The mixture was stirred for about 20 min to afford a golden solution. Then OMTS (1,1,1,3,5,7,7,7-octamethyltetrasiloxane, 1.2 eq., ρ = 0.863 g/mL), alkyne (0.50 mmol, 2.0 eq.), and TsSR2 (0.25 mmol, 1.0 eq.) were added sequentially (dropwise if liquid). The vial was sealed, removed from the glove box, and stirred at 40 °C for 12 h. The reaction mixture was quenched by PE (20 mL), filtered through a short pad of silica, and eluted with ether or PE/EA (5/1). The combined filtrate was concentrated under reduced pressure at 40 °C and flushed with nitrogen gas, then purified by flash column chromatography to give the corresponding product (Caution: the flask must be flushed with nitrogen gas after concentration steps due to the instability of the product under the air).
Data availability
The authors declare that the data Supplementary the findings of this study are available within the paper and its Supplementary Information file. The experimental procedures and characterization of all new compounds are provided in the Supplementary Information. Data supporting the findings of this manuscript are also available from the authors upon request.
References
Tewari, N. et al. An improved procedure for preparation of carbapenem antibiotic: Meropenem. Org. Process Res. Dev. 11, 773–775 (2007).
Greger, H., Zechner, G., Hofer, O., Hadacek, F. & Wurz, G. Sulphur-containing amides from Glycosmis species with different antifungal activity. Phytochemistry 34, 175–179 (1993).
Nakabayashi, K., Abiko, Y. & Mori, H. RAFT polymerization of S-vinyl sulfide derivatives and synthesis of block copolymers having two distinct optoelectronic functionalities. Macromolecules 46, 5998–6012 (2013).
Wang, N., Saidhareddy, P. & Jiang, X. Construction of sulfur-containing moieties in the total synthesis of natural products. Nat. Prod. Rep. 37, 246–275 (2020).
Velasco, N., Virumbrales, C., Sanz, R., Suarez-Pantiga, S. & Fernandez-Rodriguez, M. A. General synthesis of alkenyl sulfides by palladium-catalyzed thioetherification of alkenyl halides and tosylates. Org. Lett. 20, 2848–2852 (2018).
Beletskaya, I. P. & Ananikov, V. P. Transition-metal-catalyzed C−S, C−Se, and C−Te bond formation via cross-coupling and atom-economic addition reactions. Chem. Rev. 111, 1596–1636 (2011).
Wang, Z. et al. Research progress in C—S bond formation reaction of olefins with organic sulfur reagents under photocatalyst-free and non-electrochemical conditions. Chin. J. Org. Chem. 41, 171–184 (2021).
Beletskaya, I. P. & Ananikov, V. P. Transition-Metal-Catalyzed C-S, C-Se, and C-Te bond formations via cross-coupling and atom-economic addition reactions. achievements and challenges. Chem. Rev. 122, 16110–16293 (2022).
Castarlenas, R., Di Giuseppe, A., Perez-Torrente, J. J. & Oro, L. A. The emergence of transition-metal-mediated hydrothiolation of unsaturated carbon-carbon bonds: a mechanistic outlook. Angew. Chem. Int. Ed. 52, 211–222 (2013).
Chen, J., Wei, W.-T., Li, Z. & Lu, Z. Metal-catalyzed Markovnikov-type selective hydrofunctionalization of terminal alkynes. Chem. Soc. Rev. 53, 7566–7589 (2024).
Ogawa, A. Activation and reactivity of Group 16 inter-element linkage—transition-metal-catalyzed reactions of thiols and selenols. J. Organomet. Chem. 611, 463–474 (2000).
Ogawa, A., Ikeda, T., Kimura, K. & Hirao, T. Highly regio- and stereocontrolled synthesis of vinyl sulfides via transition-metal-catalyzed hydrothiolation of alkynes with thiols. J. Am. Chem. Soc. 121, 5108–5114 (1999).
Kuniyasu, H. et al. The first example of transition-metal-catalyzed addition of aromatic thiols to acetylenes. J. Am. Chem. Soc. 114, 5902–5903 (1992).
Degtyareva, E. S. et al. Pd-NHC catalytic system for the efficient atom-economic synthesis of vinyl sulfides from tertiary, secondary, or primary thiols. ACS Catal. 5, 7208–7213 (2015).
Ananikov, V. P. et al. New approach for size- and shape-controlled preparation of Pd nanoparticles with organic ligands. Synthesis and Application in Catalysis. J. Am. Chem. Soc. 129, 7252–7253 (2007).
Di Giuseppe, A. et al. Ligand-controlled regioselectivity in the hydrothiolation of alkynes by rhodium N-heterocyclic carbene catalysts. J. Am. Chem. Soc. 134, 8171–8183 (2012).
Yang, J., Sabarre, A., Fraser, L. R., Patrick, B. O. & Love, J. A. Synthesis of 1,1-disubstituted alkyl vinyl sulfides via rhodium-catalyzed alkyne hydrothiolation: Scope and limitations. J. Org. Chem. 74, 182–187 (2009).
Sabarre, A. & Love, J. Synthesis of 1,1-disubstituted olefins via catalytic alkyne hydrothiolation/Kumada cross-coupling. Org. Lett. 10, 3941–3944 (2008).
Palacios, L. et al. Hydroxo–rhodium–N-heterocyclic carbene complexes as efficient catalyst precursors for alkyne hydrothiolation. ACS Catal. 3, 2910–2919 (2013).
Cao, C., Fraser, L. R. & Love, J. A. Rhodium-catalyzed alkyne hydrothiolation with aromatic and aliphatic thiols. J. Am. Chem. Soc. 127, 17614–17615 (2005).
Weiss, C. J., Wobser, S. D. & Marks, T. J. Organoactinide-mediated hydrothiolation of terminal alkynes with aliphatic, aromatic, and benzylic thiols. J. Am. Chem. Soc. 131, 2062–2063 (2009).
Weiss, C. J., Wobser, S. D. & Marks, T. J. Lanthanide- and actinide-mediated terminal alkyne hydrothiolation for the catalytic synthesis of markovnikov vinyl sulfides. Organometallics 29, 6308–6320 (2010).
Weiss, C. J. & Marks, T. J. Organozirconium complexes as catalysts for markovnikov-selective intermolecular hydrothiolation of terminal alkynes: Scope and mechanism. J. Am. Chem. Soc. 132, 10533–10546 (2010).
Zhang, Y., Xu, X. & Zhu, S. Nickel-catalysed selective migratory hydrothiolation of alkenes and alkynes with thiols. Nat. Commun. 10, 1752–1762 (2019).
Malyshev, D. A. et al. Homogeneous nickel catalysts for the selective transfer of a single arylthio group in the catalytic hydrothiolation of alkynes. Organometallics 25, 4462–4470 (2006).
Han, L., Zhang, C., Yazawa, H. & Shimada, S. Efficient and selective Nickel-catalyzed addition of H−P(O) and H−S bonds to alkynes. J. Am. Chem. Soc. 126, 5080–5081 (2004).
Ananikov, V. P., Orlov, N. V. & Beletskaya, I. P. Efficient and convenient synthesis of β-vinyl sulfides in nickel-catalyzed regioselective addition of thiols to terminal alkynes under solvent-free conditions. Organometallics 25, 1970–1977 (2006).
Burykina, J. V., Shlapakov, N. S., Gordeev, E. G., Konig, B. & Ananikov, V. P. Selectivity control in thiol-yne click reactions via visible light induced associative electron upconversion. Chem. Sci. 11, 10061–10070 (2020).
Beno, B. R., Yeung, K. S., Bartberger, M. D., Pennington, L. D. & Meanwell, N. A. A survey of the role of noncovalent sulfur interactions in drug design. J. Med. Chem. 58, 4383–4438 (2015).
Barreiro, E. J., Kümmerle, A. E. & Fraga, C. A. M. The methylation effect in medicinal chemistry. Chem. Rev. 111, 5215–5246 (2011).
Teders, M. et al. The energy-transfer-enabled biocompatible disulfide-ene reaction. Nat. Chem. 10, 981–988 (2018).
Kokin, K., Tsuboi, S., Motoyoshiya, J. & Hayashi, S. Selective synthesis of cis-α,β-unsaturated sulfoxides and sulfides by the horner-wittig reaction with bis(2,2,2-trifluoroethyl)phosphono sulfoxides and aromatic aldehydes. Synthesis 1996, 637–640 (1996).
Barre, V. & Uguen, D. Cyclopentanation with β-methylthio-allyl phenyl sulfone. Tetrahedron Lett. 28, 6045–6048 (1987).
Wang, M., Qiao, Z., Zhao, J. & Jiang, X. Palladium-catalyzed thiomethylation via a three-component cross-coupling strategy. Org. Lett. 20, 6193–6197 (2018).
Qiao, Z., Ge, N. & Jiang, X. CO2-promoted oxidative cross-coupling reaction for C-S bond formation via masked strategy in an odourless way. Chem. Commun. 51, 10295–10298 (2015).
Xiao, X., Feng, M. & Jiang, X. New design of a disulfurating reagent: Facile and straightforward pathway to unsymmetrical disulfanes by copper-catalyzed oxidative cross-coupling. Angew. Chem. Int. Ed. 55, 14121–14125 (2016).
Qiao, Z. & Jiang, X. Recent developments in sulfur–carbon bond formation reaction involving thiosulfates. Org. Biomol. Chem. 15, 1942–1946 (2017).
Wei, Y., Gao, W., Chang, H. & Jiang, X. Recent advances in thiolation via sulfur electrophiles. Org. Chem. Front. 9, 6684–6707 (2022).
Zhang, C. et al. Cesium carbonate-promoted synthesis of aryl methyl sulfides using S-methylisothiourea sulfate under transition-metal-free conditions. Org. Biomol. Chem. 16, 6316–6321 (2018).
Martín, A. J., Mitchell, S., Mondelli, C., Jaydev, S. & Pérez-Ramírez, J. Unifying views on catalyst deactivation. Nat. Catal. 5, 854–866 (2022).
Aubart, M. A. & Bergman, R. G. Activation of organic disulfides by a paramagnetic heterobimetallic tantalum/cobalt complex and a comparison of their reactions with cobaltocene. Evidence for a dependence of mechanism on the electronic properties of the disulfide. J. Am. Chem. Soc. 118, 1793–1794 (1996).
Cao, T., Xu, T., Xu, R., Shu, X. & Liao, S. Decarboxylative thiolation of redox-active esters to free thiols and further diversification. Nat. Commun. 11, 5340 (2020).
Dénès, F., Pichowicz, M., Povie, G. & Renaud, P. Thiyl radicals in organic synthesis. Chem. Rev. 114, 2587–2693, (2014).
Boutureira, O. & Bernardes, G. J. L. Advances in chemical protein modification. Chem. Rev. 115, 2174–2195 (2015).
Boyd, D. A. Sulfur and its role in modern materials science. Angew. Chem. Int. Ed. 55, 15486–15502 (2016).
Kumar, R. et al. Thiol-ene “click” reaction triggered by neutral ionic liquid: the “ambiphilic” character of [hmim]Br in the regioselective nucleophilic hydrothiolation. Angew. Chem. Int. Ed. 54, 828–832 (2015).
Xiang, S., Ding, W., Wang, Y. & Tan, B. Catalytic atroposelective synthesis. Nat. Catal. 7, 483–498 (2024).
Huang, S., Wang, M. & Jiang, X. Ni-catalyzed C-S bond construction and cleavage. Chem. Soc. Rev. 51, 8351–8377 (2022).
Corberán, R., Mszar, N. W. & Hoveyda, A. H. NHC-Cu-catalyzed enantioselective hydroboration of acyclic and exocyclic 1,1-disubstituted aryl alkenes. Angew. Chem. Int. Ed. 50, 7079–7082 (2011).
Laitar, D. S., Müller, P. & Sadighi, J. P. Efficient homogeneous catalysis in the reduction of CO2 to CO. J. Am. Chem. Soc. 127, 17196–17197 (2005).
Cloarec, J.-M. & Charette, A. B. Highly efficient two-step synthesis of C-sp3-centered geminal diiodides. Org. Lett. 6, 4731–4734 (2004).
Wang, F. et al. Synthesis of gem-difluorinated cyclopropanes and cyclopropenes: Trifluoromethyltrimethylsilane as a difluorocarbene source. Angew. Chem. Int. Ed. 50, 7153–7157 (2011).
Slocumb, H. S., Nie, S., Dong, V. M. & Yang, X.-H. Enantioselective selenol-ene using Rh-hydride catalysis. J. Am. Chem. Soc. 144, 18246–18250 (2022).
Lou, Z., Li, P. & Han, K. Redox-responsive fluorescent probes with different design strategies. Acc. Chem. Res. 48, 1358–1368 (2015).
Chen, J., Shen, X. & Lu, Z. Cobalt-catalyzed markovnikov-type selective hydroboration of terminal alkynes. Angew. Chem. Int. Ed. 60, 690–694 (2021).
Chen, J., Ying, J. & Lu, Z. Cobalt-catalyzed branched selective hydroallylation of terminal alkynes. Nat. Commun. 13, 4518–4528 (2022).
Ojima, I., Clos, N., Donovan, R. J. & Ingallina, P. Hydrosilylation of 1-hexyne catalyzed by rhodium and cobalt-rhodium mixed-metal complexes. Mechanism of apparent trans addition. Organometallics 9, 3127–3133 (1990).
Zhang, R. et al. Thiyl radical trapped by cobalt catalysis: An approach to markovnikov thiol–ene reaction. Org. Lett. 26, 591–596 (2024).
Zhang, Y., Hu, L., Yuwen, L., Lu, G. & Zhang, Q. Nickel-catalysed enantioselective hydrosulfenation of alkynes. Nat. Catal. 6, 487–494 (2023).
Li, Y., Lu, X. & Fu, Y. Recent advances in cobalt-catalyzed regio- or stereoselective hydrofunctionalization of alkenes and alkynes. CCS Chem. 0, 1–27 (2024).
Ai, W., Zhong, R., Liu, X. & Liu, Q. Hydride transfer reactions catalyzed by cobalt complexes. Chem. Rev. 119, 2876–2953 (2019).
Chen, J., Guo, J. & Lu, Z. Recent advances in hydrometallation of alkenes and alkynes via the first row transition metal catalysis. Chin. J. Chem. 36, 1075–1109 (2018).
Liu, R. Y. & Buchwald, S. L. CuH-catalyzed olefin functionalization: From hydroamination to carbonyl addition. Acc. Chem. Res. 53, 1229–1243 (2020).
Uehling, M. R., Rucker, R. P. & Lalic, G. Catalytic anti-markovnikov hydrobromination of alkynes. J. Am. Chem. Soc. 136, 8799–8803 (2014).
Fang, Y., Rogge, T., Ackermann, L., Wang, S. & Ji, S. Nickel-catalyzed reductive thiolation and selenylation of unactivated alkyl bromides. Nat. Commun. 9, 2240–2249 (2018).
Acknowledgements
Financial supports were provided by the National Key R&D Program of China (2021YFF0701600 and 2021YFA1500200), the NSFC (22271249), the Fundamental Research Funds for the Central Universities (226-2022-00224 and 226-2024-00003), and Zhejiang Provincial Natural Science Foundation of China (LDQ24B020001). Our sincere thanks to Dr. Jieping Chen for providing the initial results, Mr. Chengong Zheng for his attempts to single crystal growth, and Mr. Lingtao Wang for polishing the manuscript.
Author information
Authors and Affiliations
Contributions
Z.L. proposed this project. J.Y. and Y.T. performed the experiments. Z.L. and J.Y. prepared the manuscript. J.Y. and Y.T. prepared the Supplementary Information.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Lan-Gui Xie, and the other, anonymous, reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ying, J., Tan, Y. & Lu, Z. Cobalt-catalyzed hydrothiolation of alkynes for the diverse synthesis of branched alkenyl sulfides. Nat Commun 15, 8057 (2024). https://doi.org/10.1038/s41467-024-52249-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-52249-x
- Springer Nature Limited