

Abstract
Here, we reported a cobalt-hydride-catalyzed Markovnikov-type hydroallylation of terminal alkynes with allylic electrophile to access valuable and branched skipped dienes (1,4-dienes) with good regioselectivity. This operationally simple protocol exhibits excellent functional group tolerance and exceptional substrate scope. The reactions could be carried out in gram-scale with TON (turn over number) up to 1160, and the products could be easily derivatized. The preliminary mechanism of electrophilic allylation of α-selective cobalt alkenyl intermediate was proposed based on deuterium labeling experiment and kinetic studies.
Similar content being viewed by others

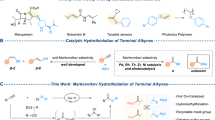
Introduction
Skipped dienes derivatives are important motifs in biologically active natural products and medicines1,2,3. Traditionally, there are several methods for the preparation of these useful compounds, such as allylation of alkenyl metallic reagents4,5,6,7, allyl metalation of alkynes8,9,10,11,12, and Alder ene reaction of alkynes13,14. However, these methods are mainly restricted to using stoichiometric amounts of metallic reagents or limited substrate scope. Compared to currently available methodologies for non-catalytic regioselective allylation of alkynes (Fig. 1a)8,9,10,11,12,15,16, metal-catalyzed hydroallylation of alkynes provides another step-economical approach to access skipped dienes. In 1998, Trost and co-workers reported ruthenium-catalyzed addition of alkenes with terminal alkynes to access skipped dienes with excellent functional group tolerance under mild conditions via ruthenacyclopentene intermediate17,18. This methodology was developed to assemble complex building blocks rapidly from simple alkenes and alkynes17,18,19,20,21. However, the aromatic terminal alkynes were not explored. In 2007, Hilt and co-workers reported cobalt-catalyzed addition of alkenes with internal alkynes to deliver 1,4-dienes with high chemo- and regio-selectivity22. However, the terminal alkynes were not suitable due to the preference to form polymerization products23,24.

a Hydroallylation of terminal alkynes with stoichiometric metallic reagents. b Hydromatallation of terminal alkynes followed by electrophilic allylation. c Metal-hydride-catalyzed electrophilic hydroallylation of terminal alkynes. d Cobalt-catalyzed branched selective hydroallylation of terminal alkynes.
Metal-hydride catalyzed selective electrophilic hydroallylation of terminal alkyne could be considered as an alternative and step-economic method for the synthesis of skipped dienes (Fig. 1b)25,26,27,28,29,30,31. However, selective electrophilic hydroallylation of terminal alkyne with allylic electrophile via metal hydride strategy is still challenging: (1) Due to the higher reactivity and instability of in-situ generated alkenyl-metal intermediate, its compatibility with other reagents, such as ligand exchange with metal hydride, carbometallation of alkynes, would affect the chemoselectivity of the reaction. (2) Compared to the direct coupling of unactivated alkyl electrophiles with metal-alkenyl intermediate32,33,34, activated allylic electrophiles were prone to process substitution reaction with other nucleophiles, such as in-situ generated metal-hydride intermediate. (3) Due to the weak electronic and steric effects of terminal alkynes, the metal hydride species would like to undergo the anti-Markovnikov type insertion rather than Markovnikov type insertion25,26,27,28,29,30,31,35,36,37,38. In 2017, Lalic and co-workers reported an elegant copper-hydride-catalyzed anti-Markovnikov type hydroallylation of terminal alkynes for the synthesis of skipped dienes with moderate to excellent regioselectivity (Fig. 1c)35. Subsequently, Xiong and Zhang reported one example of anti-Markovnikov hydroallylation of terminal alkynes36. Recently, Lu and Y. Fu developed a cobalt catalyzed regio- and enantioselective hydroalkylation of fluoroalkenes37. To the best of our knowledge, highly branched selective electrophilic hydroallylation of terminal alkyne with allylic electrophile via metal hydride strategy has not been reported. With our continuous interests on base-metal-catalyzed selective hydrofunctionalization reactions (hydrogenation, hydroboration, and hydrosilylation) of unsaturated bond39,40,41,42,43,44,45,46, we set out to explore base-metal-catalyzed selective hydroallylation of terminal alkyne.
In this work, we report cobalt-hydride-catalyzed branched selective hydroallylation of terminal alkynes with allylic bromides as electrophiles to access branched terminal skipped dienes with good regioselectivity and excellent functional group tolerance (Fig. 1d).
Results
Reaction optimization
We performed the study by using 4-ethynylanisole 1a as a model substrate, 2-methylallylbromide 2a as an allylic electrophile, and PMHS as a hydride source (Table 1). The Co(OAc)2 was used as a catalyst, N-(2-(4,5-dihydrooxazol-2-yl)phenyl)quinoline-2-carboxamide (L1) and LiOtBu were used as the ligand and base, respectively. The reaction was performed in a solution of tetrahydrofuran (THF) at 50 °C for 24 h to afford electrophilic hydroallylation product in 42% yield with 83/17 b/l (entry 1). A significant increase in the regioselectivity was observed when a larger gem-dimethyl group was used on the oxazoline moiety, which gave rise to skipped diene in 70% yield with >95/5 rr (ratio of regioselectivity; entries 1-3). However, when the size of the substituent was further increased, the regioselectivity slightly decreased (entries 4-6) which implied that the steric hindrance of the substituents on the oxazoline moiety might affect the yield and regioselectivity. Changing the steric effect on pyridine moiety, the selectivity of the reaction decreased slightly (entries 7-8). Using various hydrosilane, such as PhSiH3, (EtO)3SiH, and Ph2MeSiH led to poor yield and selectivity (entries 9-11). Using Ni(OAc)2 instead of Co(OAc)2, a poor yield of hydroallylation was observed (entry 12). Additionally, Cu(OAc)2 could not promote this transformation. However, the Sonogashira coupling reaction of allyl bromides with terminal alkynes could be promoted under mild conditions (entry 13) (We should thank one of the reviewers for the suggestion of using nickel or copper catalyst to performing the control experiments.). Using CoBr2 instead of Co(OAc)2, this transformation could process smoothly (entry 14). The model reaction could be completed in 20 min (entry 15). The (L3-H)•CoBr complex reported in our previous studies46 could also be used as an efficient catalyst (entry 16). The standard conditions were identified as 1.0 mmol of terminal alkyne, 0.50 mmol of allylic electrophile, 5 mol% of (L3-H)•CoBr, 0.75 mmol of LiOtBu, and 0.75 mmol of PMHS in a solution of THF (1 mL) at 50 °C.
Substrate scope
Compared to other olefins, 2,4-disubstituted skipped dienes are difficult to obtain through classic Wittig reaction, due to the keto-enol tautomerism of 1,3-diketone. Thus, with the optimized conditions in hand, we mainly examined the substrate scope of 2,4-disubstituted skipped dienes (Fig. 2). However, due to the presence of Lewis acid metal catalysts and corresponding bases, the inevitable side reaction of terminal alkynes self-polymerization and dehalogenation of allylic bromides would reduce the yield of allylation products. Respectively, 3b and 3c could be obtained on gram scale. The volatile dienes (3d, 3af) could also be obtained via distillation on gram-scale. Various allylic bromide (2d-2i) could be tolerated in this transformation to deliver skipped diene with moderate yield and excellent regioselectivity. Z−1,2-disubstituted bromide (2j) was also investigated. Interestingly, SN2′-type skipped alkene (3j) was obtained as a major product in this transformation. The significant regioselectivitity differences between 3 f and 3j might owe to the different E/Z stereo-configurations and steric hindrance of allyl bromides between 2 f and 2j. Besides allylic bromides, different allylic electrophilic reagents such as allyl iodide and allyl phosphate, could also be tolerated in this transformation with slightly decreased yield and regioselectivity (3b, 3d). Benzyl electrophiles, which exhibit similar properties with allylic reagent, could also be transformed smoothly on gram-scale with excellent regioselectivity47,48. The 1,4-bis(bromomethyl)benzene (2 l) could also react with terminal alkynes smoothly to obtained the 1,1-disubstituted alkenes with excellent regioselectivity. For broader synthetic interests, a variety of functional groups on phenyl rings were investigated. Methyl, trifluoromethyl, fluorine, bromine, chlorine, protected alcohol, and ester could be well tolerated to afford the skipped dienes (3m-3v) in moderate yields with good to excellent regioselectivity (55-76% yield, 92/8 - >95/5 b/l). The alkynes containing heterocycles, such as pyridine 1w and thiophene 1x, could also be tolerated to deliver branched terminal skipped dienes in 48-50% yield. Conjugated enyne (1 y), silyl alkyne (1z), and cyclopropyl acetylene (1aa) could undergo this hydroallylation reaction smoothly. Simple terminal alkynes (1ab-1ae) were also amenable to this transformation to deliver the corresponding product in 55-67% yields with 91/9 to >95/5 rr. Additionally, terminal alkynes contained in bioactive molecules were investigated. Naproxen, menthol, and geraniol derivative (1ag-1ai) could be employed to deliver corresponding products in 45-69% yield.

aCondition A: 1 (2 equiv.), 2 (0.50 mmol), (L3-H)•CoBr (5 mol%), PMHS (1.5 equiv.), and LiOtBu (1.5 equiv.) in THF (1 mL) under N2 at rt, isolated yield of 3. bCondition B: 1 (2 equiv.), 2 (0.50 mmol), Co(OAc)2 (5 mol%), L3 (6 mol%), PMHS (1.5 equiv.), and LiOtBu (1.5 equiv.) in THF (1 mL) under N2 at rt, isolated yield of 3. cLG = OP(O)(OPh)2 instead of LG = Br; LG means leaving group. d(L3-H)•CoBr (2 mol%) instead of (L3-H)•CoBr (5 mol%). eLG = I instead of LG = Br. fL7 instead of L3; Si(OSiHMe2)4 instead of PMHS. gL7 instead of L3. h(L3-H)•CoBr (2.5 mol%) instead of (L3-H)•CoBr (5 mol%). iSi(OSiHMe2)4 (2.0 equiv.) instead of PMHS; 0.2 mmol scale.
Catalytic efficiency and synthetic applications
In order to verify the catalytic efficiency of the catalyst, the reaction using 0.05 mol% of catalyst was carried out to afford skipped dienes in 58% yield which indicated that the TON was up to 1160 (Fig. 3a). The 1,4-diene could undergo double hydrosilylation under different conditions to deliver silyl heterocycle 549 or 1,5-disilyl compound 650 in 63% and 66% yield respectively (Fig. 3b). Double bonds on skipped diene 3d could be selectively converted via hydrosilylation reaction to deliver 4-vinyl silane compound 751. The 3d could aslo proceed alkylation-peroxidation with 1,3-dicarbonyl compounds and tert-butyl hydroperoxide to deliver functionalized carbonyl compound 8 (Fig. 3c)52. The reaction of 1,4-bis(bromomethyl)benzene with alkyne under standard conditions could deliver a disubstituted alkene which could be further converted to conjugated trisubstituted alkene 9 via cobalt-catalyzed alkene isomerization (Fig. 3d)53. The conjugated alkene displayed promising aggregation-induced emission (AIE) properties54,55,56.

a The catalytic efficiency of the catalyst. b Double hydrosilylation of skipped dienes. c Selective conversion of skipped diene’s double bonds. d Synthesis of conjugated alkene with AIE properties.
Mechanistic studies
To elucidate the C(sp2)-C(sp3) bond forming process in this transformation, control experiments were conducted. 3(E)-deuterated allylic bromide 10 was prepared to distinguish substitution at the 1- and 3-positions of the electrophile. This reaction was performed to give the deuterated skipped dienes 11 in 42% yield with 0.17 D at terminal carbon and 0.69 D at sp3 carbon (Fig. 4a). This result indicated that substitution occurred through the SN2′-like process accompanied with the partial SN2 process via attack of the postulated cobalt species at the 1,3-position of the allylic bromides. To elucidate the hydrometallation process, the hydroallylation of deuterium labeling phenylacetylene 12 was performed in 30 min to give the deuterated skipped alkene in 59% NMR yield with 0.40 D at C(sp2) 1(E)-position and 0.40 D at C(sp2) 1(Z)-position (Fig. 4b). Due to the presence of strong bases, deuterium atoms might loss during this process. This result combined with our previous mechanistic studies indicated that the E/Z ratio of deuterated product might owe to the relatively fast Crabtree-Ojima-type isomerization42,46. The reaction could undergo smoothly in the presence of 1,1-diphenylethylene or butylated hydroxytoluene (BHT), which might rule out the radical reaction pathway (Fig. 4c). The hydroallylation product (3 g, h, j, k) showed that the configuration of the allylic bromides would affect the SN2 and SN2′ selectivity of this transformation. Additionally, the π-allyl pathway could not be exclusively ruled out.

a The reaction with deuterium labeling allylic bromide. b The reaction with deuterium labeling terminal alkynes. c The hydroallylation reaction under additional radical scavenger.
Quantitative kinetic studies were also performed to determine the roles of alkyne, allylic bromide, hydrosilane, and (L3-H)•CoBr complex. Kinetic studies on alkyne showed that with a zero-order rate dependence on alkyne, however, as the concentrations of alkyne increased, the initial rates (kin) of the reaction decreased (Fig. 5a). This result demonstrated excessive alkyne might be a ligand to coordinate with cobalt catalyst leading to the reduction of the rate of reaction. Measurements of the initial rates (kin) of the reaction with different concentrations of allylic bromide and (L3-H)•CoBr complex showed a corresponding rise in the rates of the reactions. Plots of kin versus the concentrations of allylic bromide and (L3-H)•CoBr complex (Fig. 5b, c) gave two linear curves (slope = 1.19 × 10−4 Ms−1; 6.79 × 10−3 Ms−1), which suggested a first-order rate dependence on allylic bromide and (L3-H)•CoBr complex. Similar kinetic studies on PMHS showed no change in kin within a certain concentrations range (Fig. 5d), indicating a zero-order rate dependence on hydrosilane. These quantitative kinetic studies suggests that the nucleophilic substitution of cobalt(II) alkenyl intermediate with allylic bromide could be the turnover-limiting step.

a A plot of kin vs alkyne concentrations. b A plot of kin vs allylic bromide concentrations. c A plot of kin vs catalyst concentrations. d A plot of kin vs PMHS concentrations.
Based on the experimental studies and previously reported literatures35,42,46,57,58,59, a possible mechanism is shown in Fig. 6. The cobalt hydride species C was obtained from the reaction of active intermediate (L3-H)•CoBr with LiOtBu and hydrosilane. The alkyne coordination with species C followed by the insertion of terminal alkyne into the cobalt hydride bond delivering majorly α-selective cobalt-alkenyl intermediate E. The quick isomerization balance between E and cobalt carbene zwitterion F led to the E/Z ratio variation based on the deuterium labeling experiment. The following SN2′ and SN2-like process of E with allylic electrophile generates species to deliver the corresponding skipped dienes. The hydrosilane and alkoxide might be likely responsible for the observed regioselectivity increase during the catalysis process.

A proposed mechanism of cobalt-hydride catalyzed electrophilic hydroallylation of terminal alkyne.
Discussion
In summary, we reported an efficient cobalt-hydride catalyzed branched selective electrophilic hydroallylation of terminal alkynes with allylic electrophile to access terminal skipped dienes with good regioselectivity and functional group tolerance under mild conditions. The reaction could be carried out on gram scale, and the TON was up to 1160. The primary mechanism of electrophilic allylation of α-selective cobalt alkenyl intermediate was proposed based on deuterium labeling experiment and kinetic studies. Various metal-hydride-catalyzed regioselective hydrofunctionalization of terminal alkynes will be further explored in our laboratory.
Methods
Materials
For NMR spectra of compounds in this manuscript, see Supplementary Information. For synthesis of ligands and substrates, see Supplementary Methods. For the optimization of reaction conditions, Supplementary Table 1. For isotopic labeling experiment, radical trapping experiment, and kinetic studies, see Supplementary Figs. 1–18 and Tables 2–9.
General procedure for hydroallylation of terminal alkynes
A 25 mL Schlenk flask equipped with a magnetic stirrer and a flanging rubber plug was dried with flame under vacuum. When cooled to ambient temperature, it was vacuumed and flushed with N2. This degassed procedure was repeated for three times. Then (L3-H)•CoBr (0.025 mmol, 5 mol%), THF (1.0 mL, 0.5 M), PMHS (0.75 mmol, 1.5 equiv.), terminal alkynes (1.0 mmol, 2 equiv.), allylic bromides (0.5 mmol, 1.0 equiv.), and LiOtBu (0.75 mmol, 1.5 equiv.) were added sequentially. The reaction was run at 50 °C for 30 min to 4 h. Then the resulting solution was quenched with 10 mL of PE and filtered through a pad of silica gel, washed with PE/EtOAc (5/1) (3 × 20 mL). The combined filtrate was concentrated under vacuum and the ratio of b/l was monitored by 1H NMR analysis. The mixture was purified by flash column chromatography to give the corresponding product.
Data availability
The authors declare that the data Supplementary the findings of this study are available within the paper and its Supplementary Information file. The experimental procedures and characterization of all new compounds are provided in the Supplementary Information.
References
Jie, M., Pasha, M. K. & Syed-Rahmatullah, M. S. K. Fatty acids, fatty acid analogues and their derivatives. Nat. Prod. Rep. 14, 163–189 (1997).
Macklin, T. K. & Micalizio, G. C. Convergent and Stereospecific Synthesis of Complex Skipped Polyenes and Polyunsaturated Fatty Acids. Nat. Chem. 2, 638–643 (2010).
McCammant, M. S., Liao, L. & Sigman, M. S. Palladium-Catalyzed 1,4-Difunctionalization of Butadiene to Form Skipped Polyenes. J. Am. Chem. Soc. 135, 4167–4170 (2013).
Zhurkin, F. E. & Hu, X. γ-Selective Allylation of (E)-Alkenylzinc Iodides Prepared by Reductive Coupling of Arylacetylenes with Alkyl Iodides. J. Org. Chem. 81, 5795–5802 (2016).
Hamilton, J. Y., Sarlah, D. & Carreira, E. M. Iridium-Catalyzed Enantioselective Allylic Vinylation. J. Am. Chem. Soc. 135, 994–997 (2016).
Kabalka, G. W. & Al-Masum, M. Microwave-Enhanced Palladium-Catalyzed Cross-Coupling Reactions of Potassium Vinyltrifluoroborates and Allyl Acetates: A New Route to 1,4-Pentadienes. Org. Lett. 8, 11–13 (2013).
Ilies, L., Yoshida, T. & Nakamura, E. Iron-Catalyzed Chemo- and Stereoselective Hydromagnesiation of Diarylalkynes and Diynes. J. Am. Chem. Soc. 134, 16951–16954 (2012).
Yadav, J. S., Reddy, B. V. S., Reddy, P. M. K. & Gupta, M. K. Zn/[bmim]PF6-mediated Markovnikov Allylation of Unactivated Terminal Alkynes. Tetrahedron Lett. 46, 8411–8413 (2005).
Ranu, B. C. & Majee, A. Indium-mediated Regioselective Markovnikov Allylation of Unactivated Terminal Alkynes. Chem. Commun. 1997, 1225–1226 (1997).
Fujiwara, N. & Yamamoto, Y. Allyl- and Benzylindium Reagents. Carboindation of Carbon−Carbon and Carbon−Nitrogen Triple Bonds. J. Org. Chem. 64, 4095–4101 (1999).
Lee, P. H., Heo, Y., Seomoon, D., Kim, S. & Lee, K. Regioselective Allylgallation of Terminal Alkynes. Chem. Commun. 2005, 1874–1876 (2005).
Yasui, H., Nishikawa, T., Yorimitsu, H. & Oshima, K. Cobalt-Catalyzed Allylzincations of Internal Alkynes. Bull. Chem. Soc. Jpn. 79, 1271–1274 (2006).
Snider, B. B. Lewis-Acid-Catalyzed Ene Reactions. Acc. Chem. Res. 13, 426–432 (1980).
Hoffmann, H. M. R. The Ene Reaction. Angew. Chem., Int. Ed. 8, 556–577 (1969).
Lee, Y., Akiyama, K., Gillingham, D. G., Brown, M. K. & Hoveyda, A. H. Highly Site- and Enantioselective Cu-Catalyzed Allylic Alkylation Reactions with Easily Accessible Vinylaluminum Reagents. J. Am. Chem. Soc. 130, 446–447 (2008).
Gao, F., Carr, J. L. & Hoveyda, A. H. Copper-Catalyzed Enantioselective Allylic Substitution with Readily Accessible Carbonyl- and Acetal-Containing Vinylboron Reagents. Angew. Chem., Int. Ed. 51, 6613–6617 (2012).
Trost, B. M., Probst, G. D. & Schoop, A. Ruthenium-Catalyzed Alder Ene Type Reactions. A Formal Synthesis of Alternaric Acid. J. Am. Chem. Soc. 120, 9228–9236 (1998).
Trost, B. M., Pinkerton, A. B., Toste, F. D. & Sperrle, M. Synthesis of 1,1-Disubstituted Alkenes via a Ru-Catalyzed Addition. J. Am. Chem. Soc. 123, 12504–12509 (2001).
Trost, B. M., Papillon, J. P. N. & Nussbaumer, T. Ru-Catalyzed Alkene−Alkyne Coupling. Total Synthesis of Amphidinolide P. J. Am. Chem. Soc. 127, 17921–17937 (2005).
Trost, B. M. & James, J. C. Ruthenium-Catalyzed Alkene–Alkyne Coupling of Disubstituted Olefins: Application to the Stereoselective Synthesis of Trisubstituted Enecarbamates. J. Am. Chem. Soc. 137, 620–623 (2015).
Trost, B. M. & Gholami, H. Propene as an Atom-Economical Linchpin for Concise Total Synthesis of Polyenes: Piericidin A. J. Am. Chem. Soc. 140, 11623–11626 (2018).
Hilt, G. & Treutwein, J. Cobalt-Catalyzed Alder-Ene Reaction. Angew. Chem., Int. Ed. 46, 8500–8502 (2007).
Hilt, G., Vogler, T., Hess, W. & Galbiati, F. A Simple Cobalt Catalyst System for the Efficient and Regioselective Cyclotrimerisation of Alkynes. Chem. Commun. 2005, 1474–1475 (2005).
Hilt, G., Hess, W., Vogler, T. & Hengst, C. Ligand and Solvent Effects on Cobalt(I)-Catalysed Reactions: Alkyne Dimerisation Versus [2+2+2]-Cyclotrimerisation Versus Diels-Alder Reaction Versus [4+2+2]-Cycloaddition. J. Organomet. Chem. 690, 5170–5181 (2005).
Tsuji, Y. & Fujihara, T. Copper-Catalyzed Transformations Using Cu–H, Cu–B, and Cu–Si as Active Catalyst Species. Chem. Rec. 16, 2294–2313 (2016).
Suess, A. M. & Lalic, G. Copper-Catalyzed Hydrofunctionalization of Alkynes. Synlett 27, 1165–1174 (2016).
Jordan, A. J., Lalic, G. & Sadighi, J. P. Coinage Metal Hydrides: Synthesis, Characterization, and Reactivity. Chem. Rev. 116, 8318–8372 (2016).
Chen, J., Guo, J. & Lu, Z. Recent Advances in Hydrometallation of Alkenes and Alkynes via the First Row Transition Metal Catalysis. Chin. J. Chem. 36, 1075–1109 (2018).
Wei, D. & Darcel, C. Iron Catalysis in Reduction and Hydrometalation Reactions. Chem. Rev. 119, 2550–2610 (2019).
Ai, W., Zhong, R., Liu, X. & Liu, Q. Hydride Transfer Reactions Catalyzed by Cobalt Complexes. Chem. Rev. 119, 2876–2953 (2019).
Nájera, C., Beletskaya, I. P. & Yus, M. Metal-catalyzed Regiodivergent Organic Reaction. Chem. Soc. Rev. 48, 4515–4618 (2019).
Uehling, M. R., Suess, A. M. & Lalic, G. Copper-Catalyzed Hydroalkylation of Terminal Alkynes. J. Am. Chem. Soc. 137, 1424–1427 (2015).
Cheung, C. W., Zhurkin, F. E. & Hu, X. Z-Selective Olefin Synthesis via Iron-Catalyzed Reductive Coupling of Alkyl Halides with Terminal Arylalkynes. J. Am. Chem. Soc. 137, 4932–4935 (2015).
Lu, X. et al. 1,1-Disubstituted Olefin Synthesis via Ni-Catalyzed Markovnikov Hydroalkylation of Alkynes with Alkyl Halides. Chem. Commun. 52, 5324–5327 (2016).
Mailig, M., Hazra, A., Armstrong, M. K. & Lalic, G. Catalytic Anti-Markovnikov Hydroallylation of Terminal and Functionalized Internal Alkynes: Synthesis of Skipped Dienes and Trisubstituted Alkenes. J. Am. Chem. Soc. 139, 6969–6977 (2017).
Xu, G. et al. Ligand-Controlled Regiodivergent and Enantioselective Copper-Catalyzed Hydroallylation of Alkynes. Angew. Chem., Int. Ed. 56, 13130–13134 (2017).
Li, Y. et al. Cobalt-catalysed enantioselective C(sp3)–C(sp3) coupling. Nat. Catal. 4, 901 (2021).
Zhang, D., Li, M., Li, J., Lin, A. & Yao, Z. Rhodium-catalyzed intermolecular enantioselective Alder–ene type reaction of cyclopentenes with silylacetylenes. Nat. Commun. 12, 6627 (2021).
Guo, J., Cheng, Z., Chen, J., Chen, X. & Lu, Z. Iron- and Cobalt-Catalyzed Asymmetric Hydrofunctionalization of Alkenes and Alkynes. Acc. Chem. Res. 54, 2701–2716 (2021).
Chen, X., Cheng, Z., Guo, J. & Lu, Z. Asymmetric remote C-H borylation of internal alkenes via alkene isomerization. Nat. Commun. 9, 3939 (2018).
Shen, X. et al. Ligand-promoted Cobalt-catalyzed Radical Hydroamination of Alkenes. Nat. Commun. 11, 783 (2020).
Chen, J., Shen, X. & Lu, Z. Cobalt-Catalyzed Markovnikov Selective Sequential Hydrogenation/Hydrohydrazidation of Aliphatic Terminal Alkynes. J. Am. Chem. Soc. 142, 14455–14460 (2020).
Cheng, Z. et al. Regio-controllable Cobalt-Catalyzed Sequential Hydrosilylation/Hydroboration of Arylacetylenes. Angew. Chem., Int. Ed. 60, 22454–22460 (2021).
Lu, P. et al. Iron-Catalyzed Highly Enantioselective Hydrogenation of Alkenes. J. Am. Chem. Soc. 143, 12433–12438 (2021).
Sun, Y., Guo, J., Shen, X. & Lu, Z. Ligand relay catalysis for cobalt-catalyzed sequential hydrosilylation and hydrohydrazidation of terminal alkynes. Nat. Commun. 13, 650 (2022).
The OPQC•CoBr was identified, see, Chen, J., Shen, X. & Lu, Z. Cobalt-Catalyzed Markovnikov-Type Selective Hydroboration of Terminal Alkynes. Angew. Chem., Int. Ed. 60, 690–694 (2021).
Zhao, X., Zhu, S., Qing, F. & Chu, L. Reductive Hydrobenzylation of Terminal Alkynes via Photoredox and Nickel Dual Catalysis. Chem. Commun. 57, 9414–9417 (2021).
Wang, F., Zhu, S. & Chu, L. Synergistic Catalysis for Stereodivergent Synthesis of trans- and cis-Skipped Dienes. Synlett 31, 1741–1746 (2020).
Shin, K., Joung, S., Kim, Y. & Chang, S. Selective Synthesis of Silacycles by Borane-Catalyzed Domino Hydrosilylation of Proximal Unsaturated Bonds: Tunable Approach to 1,n-Diols. Adv. Synth. Catal. 359, 3428–3436 (2017).
Chen, J., Cheng, B., Cao, M. & Lu, Z. Iron-Catalyzed Asymmetric Hydrosilylation of 1,1-Disubstituted Alkenes. Angew. Chem., Int. Ed. 54, 4661–4664 (2015).
Wu, X. et al. Nickel-Catalyzed Hydrosilylation of Terminal Alkenes with Primary Silanes via Electrophilic Silicon–Hydrogen Bond Activation. Org. Lett. 23, 1434–1439 (2021).
Lu, S., Qi, L. & Li, Z. Cobalt-Catalyzed Alkylation-Peroxidation of Alkenes with 1,3-Dicarbonyl Compounds and T-Hydro. Asian J. Org. Chem. 6, 313–321 (2017).
Zhao, J., Cheng, B., Chen, C. & Lu, Z. Cobalt-Catalyzed Migrational Isomerization of Styrenes. Org. Lett. 22, 837–841 (2020).
Zhang, S. et al. Cobalt(II)-Catalyzed Stereoselective Olefin Isomerization: Facile Access to Acyclic Trisubstituted Alkenes. J. Am. Chem. Soc. 142, 8910–8917 (2020).
Li, J. et al. Supramolecular Materials Based on AIE Luminogens (AIEgens): Construction and Applications. Chem. Soc. Rev. 49, 1144–1172 (2020).
Xu, J., Wu, X., Guo, J., Zhao, Z. & Tang, B. Sky-blue Delayed Fluorescence Molecules Based on Pyridine-substituted Acridone for Efficient Organic Light-emitting Diodes. J. Mater. Chem. C. 9, 15505–15510 (2021).
Wang, Y. & Buchwald, S. L. Enantioselective CuH-Catalyzed Hydroallylation of Vinylarenes. J. Am. Chem. Soc. 138, 5024–5027 (2016).
Han, J., Guo, G., Zhang, X., Liao, J. & Ye, K. Recent Advances in Cobalt-catalyzed Allylic Functionalization. Org. Biomol. Chem. 18, 7740–7750 (2020).
Blasius, C., Vasilenko, V., Matveeva, R., Wadepohl, H. & Gade, H. Reaction Pathways and Redox States in α-Selective Cobalt-Catalyzed Hydroborations of Alkynes. Angew. Chem., Int. Ed. 59, 23010–23014 (2020).
Acknowledgements
Financial support was provided by National Key R&D Program of China (2021YFF0701603 and 2021YFA1500200), NSFC (21922107), Zhejiang Provincial Natural Science Foundation of China (LR19B020001), the Fundamental Research Funds for the Central Universities (226-2022-00224), and Center of Chemistry for Frontier Technologies. We thank the fund of the excellent doctoral dissertation funding from Zhejiang University.
Author information
Authors and Affiliations
Contributions
Z.L. proposed this project. J.C. provided initial results. J.C. performed the experiments. Z.L. and J.C. prepared the manuscript. J.C. and J.Y. prepared the Supplementary Information.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, J., Ying, J. & Lu, Z. Cobalt-catalyzed branched selective hydroallylation of terminal alkynes. Nat Commun 13, 4518 (2022). https://doi.org/10.1038/s41467-022-32291-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-32291-3
- Springer Nature Limited
This article is cited by
-
Cobalt-catalyzed hydrothiolation of alkynes for the diverse synthesis of branched alkenyl sulfides
Nature Communications (2024)
-
Regio‐ and enantioselective nickel-alkyl catalyzed hydroalkylation of alkynes
Nature Communications (2024)
-
NNN pincer palladium(II) complexes with N-(2-(1H-pyrazol-1-yl)phenyl)-picolinamide ligands: synthesis, characterization, and application to heck coupling reaction
Transition Metal Chemistry (2024)