
Abstract
Modeling environmental spatial heterogeneity can improve the efficiency of forest tree genomic evaluation. Furthermore, genotyping costs can be lowered by reducing the number of markers needed. We investigated the impact on variance components, breeding value accuracy, and bias of two phenotypic data adjustments (experimental design and autoregressive spatial models), and a relationship matrix calculated from a subset of markers selected for their ability to infer ancestry. Using a multiple-trait multiple-site single-step Genomic Best Linear Unbiased Prediction (ssGBLUP) approach, four scenarios (2 phenotype adjustments × 2 marker sets) were applied to diameter at breast height (DBH), height (HT), and resistance to western gall rust (WGR) in four open-pollinated progeny trials of lodgepole pine, with 1490 (out of 11,188) trees genotyped with 25,099 SNPs. As a control, we fitted the conventional ABLUP model using pedigree information. The highest heritability estimates were achieved for the ABLUP followed closely by the ssGBLUP with the full marker set and using the spatial phenotype adjustments. The highest predictive ability was obtained by using a reduced marker subset (8000 SNPs) when either the spatial (DBH: 0.429, and WGR: 0.513) or design (HT: 0.467) phenotype corrections were used. No significant difference was detected in prediction bias among the six fitted models, and all values were close to 1 (0.918–1.014). Results demonstrated that selecting informative markers, such as those capturing ancestry, can improve the predictive ability. The use of spatial correlation structure increased traits’ heritability and reduced prediction bias, while increases in predictive ability were trait-dependent.
Similar content being viewed by others

Introduction
While Marker-Assisted-Selection (MAS) provides a frameworks for oligogenic traits (Muranty et al. 2014), Genomic Selection (GS) is a proven tool for predicting the genetic merit of complex polygenic traits of genotyped individuals by their genomic breeding value (BV) (Meuwissen et al. 2001). Genomic Best Linear Unbiased Prediction (GBLUP) is one of the most commonly used GS methods. The GBLUP uses the genomic realized relationship matrix (G-matrix) that describes the genetic relationships among individuals calculated from genetic markers such as single-nucleotide polymorphisms (SNPs) (Habier et al. 2013). While the GBLUP is the most widely used statistical method in GS for forest trees (e.g., Gamal El-Dien et al. 2015; Ratcliffe et al. 2015; Resende et al. 2017; Lenz et al. 2020; Mphahlele et al. 2020), its main limitation exists where evaluations (i.e., breeding values) are restricted only to the genotyped individuals (Lourenco et al. 2020).
In situations where not all trees in a population are genotyped, the single-step GBLUP GS approach (ssGBLUP) has been proposed as an effective analytical method (Legarra et al. 2009; Misztal et al. 2009; Aguilar et al. 2010; Christensen and Lund 2010). This approach considers both non-genotyped and genotyped individuals in a single genetic evaluation, combining the pedigree relationship A-matrix of the non-genotyped individuals with the G-matrix of the genotyped individuals in a blended relationship H-matrix. The effectiveness of this approach has been shown by improving the prediction of individual BVs through combining phenotypes, genotypes, and pedigree information. The ssGBLUP model has been successfully and routinely implemented in animal breeding genomic evaluation, producing accurate and less biased BV predictions compared to those relying on pedigree-based information alone (e.g., Legarra et al. 2014). This strategy has the added benefit of including historical phenotypic data without the concerns of the availability of DNA samples, making ssGBLUP attractive for forest tree breeding genetic evaluation where a substantial number of offspring is used (Cappa et al. 2017, 2018; Ratcliffe et al. 2017; Klápště et al. 2018, 2020; Thavamanikumar et al. 2020; Ukrainetz and Mansfield 2020a).
It should be stated that most of the above-referenced studies were limited to fitting models that accounted for classical experimental designs and did not consider a spatial correlation structure component at the test sites. Forest trees are large and long-lived organisms, occupying substantially larger areas compared to most cultivated crop species, and are generally planted on sites characterized by a high degree of heterogeneity (e.g., fertility, humidity, soil depth, aspect, or slope) (Cappa and Cantet 2007). The phenotypic measurements of trees grown in progeny trials can thus be spatially correlated due to microenvironment similarities. Given that the accuracy of BV predictions depends on the random effects’ covariance structure, the dispersion parameters specification should consider the existence of a positive spatial correlation in the environmental heterogeneity (Cappa et al. 2017). A common class of a posteriori spatial models for performing spatial adjustments in forest tree genetic evaluation trials is the first-order autoregressive (AR1) models (Gilmour et al. 1997). Several authors have employed pedigree-based spatial autoregressive individual-tree mixed models (e.g., Costa e Silva et al. 2001; Dutkowski et al. 2006; Cappa et al. 2012) which consistently increase the heritability and accuracy of predicted BV estimates, when comparing to the a priori model that only considers the experimental design effects (e.g., block effects), phenotype, and pedigree information. Since the ssGBLUP approach uses traditional BLUP mixed model equations, the extension to an individual-tree mixed model with environmental heterogeneity effects is straightforward (Cappa et al. 2017), and, therefore, phenotypic, genomic, pedigree, and spatial information can be integrated into the same analysis.
Since the ssGBLUP approach utilizes information from both genotyped (G-matrix) and non-genotyped (A-matrix) individuals (Misztal et al. 2009), the information produced by the former is greatly affected by the quality and informativeness of the SNPs used and their ability to accurately represent the true genetic relationships. As a result, the amount of identity-by-descent captured in the G-matrix is a determinant factor for the models’ predictability. This can be accomplished by effectively including only those SNPs that maximize the Ancestry Informativeness Coefficient (AIM) (Rosenberg et al. 2003). The AIM coefficient identifies SNPs with high information regarding population structure, thus a useful indicator of markers’ ability to infer identity-by-descent (Rosenberg et al. 2003). To explore the impact of a reduced number of selected SNPs on genetic parameter estimation, and consequently, genomic selection predictive ability, an optimized subset of SNPs can be selected according to their AIM coefficient considering the half-sib family structure of the present study population. To our knowledge, this is the first study to investigate the potential benefit of the AIM SNP selection on inferences (estimations of variance parameters) and on predictions in genomic forest tree breeding.
Here, we demonstrate the capability of the ssGBLUP approach, after accounting for the spatial correlation structure (i.e., environmental heterogeneity), and the utilization of a G-matrix calculated from an optimized subset of SNPs based on their AIM coefficient. We validate the effectiveness of ssGBLUP with four open-pollinated progeny trials of lodgepole pine (Pinus contorta var. latifolia Douglas ex Louden) of the Region C breeding program in central Alberta, Canada. Alberta has six lodgepole pine breeding regions (FGRMS 2016), with each representing similar ecological and climatic conditions. The Region C lies between 54.0°N and 55.3°N, 114.6°W and 117.0°W and encompasses an area of 1.2 M ha with an elevation band range between 800 and 1200 m. The program´s main objective is to increase volume gain of reforested stands while maintaining adequate genetic diversity and long-term adaptive capability. Secondary objectives include improved resistance to Endocronartium harknessii (western gall rust, WGR), good tree form, and undiminished wood quality. These four progeny trials are part of a large-scale tree genomic study (Thomas et al. 2019) and provided 1490 trees (out of 11,188) genotyped with 25,099 SNPs based on the genotyping-by-sequencing (GBS) platform. GBS is a low-cost, high-throughput genotyping technology that employs restriction enzymes to reduce genome complexity, thus does not require prior genomic information, making it suitable for non-model species such as forest trees (Ratcliffe et al. 2015). Chen et al. (2013) successfully demonstrated the suitability of GBS for SNP discovery in white spruce (Picea glauca (Moench) Voss) and lodgepole pine. In a recent study, Ukrainetz and Mansfield (2020b) used a fixed content SNP array for lodgepole pine and have experienced a low SNP recovery rate (19,584 out of 51,213 SNPs) caused by SNP calling difficulties while using the loblolly genome as a reference; even though they recovered a number of good quality SNPs that were adequate for their genomic selection study.
Overall, this study aims to provide evidence for the betterment of genetic variance component estimates, and consequently, improved genomic BV prediction of two growth attributes (total height and diameter at breast height) and resistance to WGR through an integrated approach that jointly considers spatial analysis and the informativeness in the genomic data. The main objectives of this study are to: (1) assess the performance of a multiple-trait multiple-site ssGBLUP approach for four large lodgepole pine open-pollinated progeny test trials, (2) evaluate the impact of utilizing phenotypes spatially adjusted by classical design versus autoregressive spatial analysis, and (3) produce and evaluate a G-matrix from a reduced number of SNPs that maximize ancestry information. Additionally, we compared the variance components and the efficiency, in terms of predictive ability and control of bias, of the conventional individual-tree model with pedigree-based relationship matrix (ABLUP) and the genomic ssGBLUP approach.
Materials and methods
Genetic material, trial description, and traits evaluated
We utilized the lodgepole pine Region C breeding program, initiated in the early 1980’s, and owned and managed by Blue Ridge Lumber (BRL), a Division of West Fraser Ltd., for study. The Region C breeding program consists of a commercial seed orchard producing seeds for reforestation and four progeny test sites planted in 1982 with open-pollinated (OP) seedlings. The test sites were chosen to represent a variety of site conditions present within the region. It is one of the most important lodgepole pine improvement program in the province in terms of seed use. The results of this study will inform the next generation of selections for the establishment of BRL’s second-generation seed orchard(s). The breeding program is composed of 224 OP families. Seed sources for these families were collected from phenotypically superior selected parents from five natural stands (i.e., provenances; Deer Mountain, Inverness River, Judy Creek, Swan Hills, and Virginia Hills) within Region C breeding region. Candidate parent trees were phenotypically selected for superior growth, stem straightness, health, branching, and crown traits (Dhir 1983). Additionally, this testing program included eight bulk seedlots collected from natural stands located within Regions B1, an adjacent low elevation lodgepole pine program, and C. Progeny testing trials were planted with 1-year-old containerized seedlings (Spencer-Lemaire Hillson container, volume 175 mL) on four test sites (Judy Creek: JUDY, Virginia Hills: VIRG, Swan Hills: SWAN, and Timeau: TIME) (Table 1). Each progeny trial was planted as a “set in reps” design with five replications (blocks), 21 sets per replication, and trees within sets were planted in 4-tree row plots at a 2.5 × 2.5 m spacing (John and Sadoway 2019 and Table 1). The sets (19 out of 21) are composed of two or three groups of four families originating from the same single stand. Two additional control sets consisting of four bulk seedlots each, were also included on each site. All trial sites were fenced to keep out ungulates and each trial had a single border row planted of lodgepole pine seedlings around the perimeter.
The entire Region C progeny trial population was assessed for two growth attributes (total height: HT and diameter at breast height: DBH) and one disease attribute, resistance to western gall rust (WGR). Both HT and DBH were assessed at age 30 years (DBH30 and HT30). In a previous study of lodgepole pine growing in Alberta, Rweyongeza (2016) reported high tree height age-age correlations between age 30 and long rotation ages of 50 (0.904) and 120 (0.805), demonstrating the robustness of early age height assessments. Resistance to WGR was assessed at age 36 (WGR36) using a qualitative scoring system with seven discrete categories ranging from no gall symptoms (category 1) to deceased (category 7). Given that there were very few trees in categories 3, 5, and 7 across trial sites, these categories were merged with the original categories 2, 4, and 6, respectively, resulting in four-category resistance ratings. The four-category WGR36 ratings were further transformed into a continuous normal score (NSWGR36) following Gianola and Norton (1981). After all data cleaning, 11,188 phenotyped trees were used in the analysis (Table 2).
Sample selection and genotyping by sequencing (GBS)
To sample the extent of genetic variability present in the progeny trial population (N = 224 OP families), 40 OP families were selected for SNP genotyping to represent the range in height variation. This selection was achieved using prior family rankings for tree height BVs. From these rankings, 15 families were chosen from high and low breeding value groups and 10 families were chosen from the medium breeding value group (Supplementary Fig. S1). The selection of families within these three breeding value groups, and individuals within families was random, however, consideration was also given to maintain a balanced experimental design (i.e., representation of families in all testing environments). Each site was represented by the same 40 OP families, with a target sampling of ~10 individual trees per OP family per site (n = 1600). An additional 35 potential forward selected trees, previously identified based on height BVs, were also included for sequencing. These 35 trees were from an additional 28 OP families, resulting in 1635 trees sequenced from a total of 68 OP families.
In the spring of 2017, freshly flushed needles were collected from the 1635 trees, kept in coolers and transported within 48 h to the University of Alberta for immediate storage at −80 °C until DNA extraction. After transport on dry-ice, genomic DNA was extracted at the Alberta Innovates (aka, InnoTech Alberta) facility in Vegreville, Alberta, using DNeasy 96 Plant Kit (Qiagen), and the quality of DNA extraction was assessed to ensure the required minimal 30 µl of DNA at 30-100 ng/µL for genotyping-by-sequencing (GBS). After extraction, DNA was shipped on dry-ice to the Institute of Biotechnology, Cornell University, for genotyping following Elshire et al. (2011) and Chen et al. (2013) with restriction enzyme Pst-1 (CTGCAG). Due to the lack of a lodgepole pine genome reference assembly, SNP determination was carried out with the reference-free UNEAK pipeline.
In total, 1554 samples from the 2017 field collection passed the DNA quality control and proceeded with GBS genotyping. After filtering for sequencing quality, 69.4% of the 6181 million sequencing raw reads were retained; and, a total of 9,133,021 read tags for Pst-1 were constructed using all the reads that passed quality control, with a minimum read count greater than 20. Aligning sequencing reads to the read tags generated 170,166 SNP markers. A final set of 1490 trees and 25,099 SNPs was obtained based on filtering the SNP data set for less than 30% missing data proportion and a minor allele frequency equal to or greater than 1%. Owing to the lack of a reference assembly genome and no genomic positions for the SNPs, missing data were then imputed using the mean observed allele at each locus.
Pedigree correction
Using the available 25,099 SNP markers for the sampled 68 OP families we validated the pedigree for records verification. Pedigree correction was done using a custom R-script and was based on the comparison of expected (pedigree) versus observed (molecular) additive genetic relationships. A customized R-script identified dubious pairs of samples by setting bounding thresholds on observed pairwise relationship coefficients for relationship groups for half-sibling of <0.10 or >0.375. For pedigree validation, the identified dubious pairs were then manually inspected, and the offending sample was reassigned to the appropriate maternal family. For paternal assignment, sample pairs with observed pairwise relationship coefficients >0.375 were identified and manually inspected, then paternal contribution was assigned appropriately in the pedigree. Supplementary Fig. S2 shows the comparison of observed pairwise relationship coefficients using the uncorrected and corrected pedigrees for the two SNP subsets (25K and 8K, see next SNP selection section).
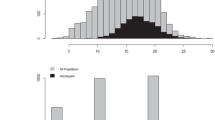
In total, pedigree records for 162 samples were modified or corrected. These changes mostly stemmed from the identification of 4 phantom mothers and 16 pollen donors (fathers). The corrected pedigree showed the 1490 trees originating from 83 parents (63 of the original mothers plus 4 new phantom mothers plus 16 new fathers) with a range of 1–41 genotyped trees per mother (and from 0 to 12 per site). The underrepresented families are those of the additional previously identified forward selections. The total number of phenotyped trees with at least one genotyped half-sib was 3467 (out of 11,188 total progenies tested in the four trials) (i.e., 30.99%) (see Table 2 for the summary). The phenotype distributions for the DBH30, HT30, and WGR36 traits of genotyped and non-genotyped trees are in Fig. 1. The genotyped trees follow the same distribution as non-genotyped trees for the three traits with similar mean and standard deviation (Table 2).

Frequency histograms of diameter at breast height (DBH30) and total height (HT30) at age 30 years, and western gall rust normal score at age 36 years (WGR36, based on the observed original scale 1–4) for the entire lodgepole pine population (gray, genotyped trees; black, non-genotyped trees).
SNP selection
Following Rosenberg et al. (2003) and with advice from Dr. Jaroslav Klápště (personal communication), a customized R-script was developed to calculate the Ancestry Informativeness Coefficient (AIM) (Eq. (4); Rosenberg et al. 2003) for each SNP using the half-sibling genetic family structure of the genotyped population and by including only families with at least 5 progeny. The R code used for the calculation of the ancestry informative coefficients is given as Supplementary Material (R_code S1).
A sensitivity analysis was then performed by adding increments of 1000 SNPs from 1000 to a maximum of 25,099 (e.g., 1000, 2000, 3000, …, 25,099) according to the descending AIM coefficient value. For each 1000 SNPs increment (n = 26 increments), the additive realized relationship matrix (G-matrix) (VanRaden 2008, see below) was estimated for all genotyped trees. A subset of 8000 SNPs (8K) was selected for use in this study based on minimizing the difference between the average pairwise half-sib relationships in G-matrix, according to the corrected pedigree (above), versus the expectation for an additive half-sib relationship (0.25). Results from this sensitivity analysis are illustrated in Supplementary Fig. S3.
Statistical analysis
Due to spatial heterogeneity within trials, as well as for reasons of computational efficiency, the statistical analyses were conducted in two stages. First, each trait was analyzed separately in each site using a pedigree-based classical a priori design model (Design) and an a posteriori spatial model with a first-order autoregressive residual (co)variance structure (AR1×AR1) (Cappa et al. 2019). The Akaike Information Criterion (AIC) was employed for model selection to determine the appropriateness of Design and AR1×AR1. The spatial effects from the Design and AR1×AR1 models for the traits DBH30, HT30, and NSWGR36 in the four studied sites, are illustrated in Supplementary Fig. S4 and the AIC is in Supplementary Table S1.
The single-trait single-site analysis was based on the following pedigree-based individual-tree mixed (Design) model:
where y is the vector of individual-tree observations, β is the vector of fixed effects for replicates and genetic groups formed according to provenance; s is the vector of random set nested within replications effects distributed as \(s\sim N(0,I\sigma _s^2)\), where \(\sigma _s^2\) is the set nested within replication variance; p is the vector of random plot effects distributed as \(p\sim N(0,I\sigma _p^2)\), where \(\sigma _p^2\) is the plot variance; b is the vector of random bulk seedlot effects distributed as \(b\sim N(0,I\sigma _b^2)\), where \(\sigma _b^2\) is the bulk seedlot variance; a is the vector of random effects that represents the genetic effects (or breeding values) distributed as \(a\sim N\left( {0,A\sigma _a^2} \right)\) where A is the average numerator relationship matrix derived from the pedigree information (Henderson 1984), and \(\sigma _a^2\) is the additive genetic variance. Finally, e is the vector of random residuals distributed as \(e\sim N(0,I\sigma _e^2)\)where I is the identity matrix, and \(\sigma _e^2\) is the residual variance. For the spatial model (AR1×AR1), the residual vector e was partitioned into spatially dependent (ξ) and independent (η) residuals. Therefore, the residual (co)variance matrix can be expressed as \(\sigma _\xi ^2\left[ {AR1(\rho _{col}) \sim AR1(\rho _{row})} \right] + \sigma _\eta ^2I\), where \(\sigma _\xi ^2\) is the spatially dependent variance, \(\sigma _\eta ^2\) is the spatially independent variance, \(AR1(\rho )\) is the first-order autoregressive correlation process, \(\rho _{col}\) and \(\rho _{row}\) are autocorrelations parameters for columns and rows, respectively, and ⨂ denotes the Kronecker product. The X, Zs, Zp, Zb and Za denote the incidence matrices for their respective effects.
In the second stage, the phenotypic data adjusted with the design and spatial effects are obtained for each tree and trait and at each site by subtracting the estimated replication, set, and plot effects (design phenotype adjustment). The spatial phenotype adjustment also included the autoregressive residual effects (ξ, model (2)). After adjusting phenotypes, the pedigree information was used in the ABLUP analyses. Further, the genotypic data from the full SNP set (25K) and the subset of SNPs (8K) jointly with the adjusted phenotypes (design and spatial) and the pedigree information, were used with the single-step GBLUP (ssGBLUP) analyses. Therefore, the following multiple-trait multiple-site individual-tree ABLUP was employed:
where y is the vector of design or spatially adjusted phenotypes for each i trait (i = DBH30, HT30, and NSWGR36) and j site (j = JUDY, VIRG, SWAN, TIME); β is the vector of fixed effects for provenance for each trait-site combination; a is the random vector of genetic effects distributed as \(a\sim N\left( {0,\Sigma _0 \otimes A} \right)\), where \(\Sigma _0\) is the (co)variance matrix of genetic effects for each combination of the three traits (i.e., DBH30, HT30, and NSWGR36) and the four sites (JUDY, VIRG, SWAN, TIME) with dimension 12 × 12, and A is the numerator relationship matrix (Henderson 1984) containing the additive relationships among all trees: 242 parents without records plus 11,188 offspring with data in y. Finally, e is the vector of random residuals distributed as \(e\sim N(0,R_0 \otimes I)\) where R0 is the residual (co)variance matrix for each combination for the three traits and four sites with dimension 12 × 12. We assumed an unstructured (co)variance matrix for the genetic effects (\(\Sigma _0\)). However, for the residual (co)variances (R0), we assumed residual (co)variance between traits within site, but given that the sites were assessed separately, residual (co)variance between traits across sites is assumed to be zero. The matrices \(X_{ij}\) and \(Z_{ij}\) related the design or spatial adjusted phenotypes to the means of the provenances \(\left[ {\beta _{ij}^\prime } \right]\), and the genetic effects in \(\left[ {a_{ij}^\prime } \right]\). The symbol ‘ indicates the transpose operation.
In the ssGBLUP approach, the A-matrix of the previous mixed model (2) was replaced by the combined pedigree- and marker-based relationship matrix H of the same dimension as the pedigree-based matrix. The inverse of the relationship matrix that combines pedigree and genomic information (H−1) was derived by Legarra et al. (2009), Misztal et al. (2009), Aguilar et al. (2010), and Christensen and Lund (2010), and calculated for each G-matrix as:
where λ scales the differences between genomic and pedigree-based information, G−1 is the inverse of the genomic-based relationship matrices, and \(A_{22}^{ - 1}\) is the inverse of the pedigree-based relationship matrix for the genotyped individuals only (the subscript 2 represent the genotyped individuals). The weighting factor λ was set to 0.95 for all ssGBLUP models. The G-matrix was estimated following the first method proposed by VanRaden (2008):
where, W is the centered matrix of SNP covariates, and \(p_k\) is the current (or observed) allele frequency of the genotyped trees for marker k. The G-matrix was scaled to have the same diagonal and off-diagonal averages as the corresponding A22-matrix, according to Christensen et al. (2012) (Eq. (4)).
The narrow-sense individual heritability, \(h^2\), was estimated as: \(\hat h^2 = \hat \sigma _{a_{ij}}^2/\left( {\hat \sigma _{a_{ij}}^2 + \hat \sigma _{e_{ij}}^2} \right)\), where \(\hat \sigma _{a_{ij}}^2\) is the estimated genetic variance for the ith trait and jth site, and \(\hat \sigma _{e_{ij}}^2\) the estimated residual variance for the ith trait and jth site from the multiple-trait multiple-site model (2). The genetic correlation, r, was estimated as: \(\hat r = \hat \sigma_{a_{ij}}/\sqrt {\hat \sigma_{a_{ii}}^2\hat \sigma_{a_{jj}}^2}\), where \(\hat \sigma _{a_{ii}}^2\) and \(\hat \sigma _{a_{jj}}^2\) are the genetic variances for the traits or sites i and j, respectively, and \(\hat \sigma _{a_{ij}}\) is the estimated covariance between traits or sites i and j from the multiple-trait multiple-site model (2).
Heat map of the pair-wise relationship coefficients among the 1490 genotyped lodgepole pine trees (Supplementary Fig. S5) and estimation of provenance fixed effects (i.e., BLUE) for the three studied traits and the ABLUP and ssGBLUP evaluation models fitted (results no shown), revealed that differences attributable to provenance origins were not important. Family structure presented by the small squares around the diagonal elements with ~40 individual trees (from 29 to 41) is clearly demonstrated in the Supplementary Fig. S5.
The two ABLUP and the four ssGBLUP evaluation models (details in Table 3) were fitted in R (www.r-project.org) with the function remlf90 from the package breedR (Muñoz and Sanchez 2020), using the Expectation-Maximization (EM) algorithm followed by one iteration with the Average Information (AI) algorithm to compute the approximated standard errors of the variance components (Chateigner et al. 2020). The remlf90 function in the R-package breedR is based in the REMLF90 (for the EM algorithm) and AIREMLF90 (for the AI algorithm) of the BLUPF90 family (Misztal et al. 2018). The program preGSf90, also from the BLUPF90 family (Misztal et al. 2018), was used to create the inverse of the different H-matrices from the respective G-matrices calculated with the full 25K and the 8K subset of SNP markers, and then used to fit the ssGBLUP models (2) with the breedR package. The linkage disequilibrium (LD) between pairs of GBS-derived SNPs were calculated as the squared correlation statistic using also the program preGSf90 from the BLUPF90 family (Misztal et al. 2018).
Model comparisons
We evaluated the two ABLUP and the four ssGBLUP models for both predictive ability and prediction bias by 10-fold cross-validation, where one subsample was used as the validation set, and the remaining nine samples as the training set. All trees with phenotypic data were in the training population at least once. The variance components were fixed to the respective variance components calculated with all the available trees with phenotypic data in the cross-validation analysis. The predictive ability was determined as the Pearson correlation between the observed breeding values from the full data set (i.e., using all the available 11,188 trees with phenotypes) and those breeding values of the validation set predicted from the respective ABLUP and ssGBLUP evaluation method (Table 3), multiplied by the square root of the narrow-sense heritability of each trait calculated using the pedigree-based information for all available trees (Legarra et al. 2008).
The prediction bias was calculated by the regression coefficient between the observed breeding values from the full data set and those predicted with each ABLUP and ssGBLUP model (Table 3). A regression coefficient equal to one is considered to have no bias, while a regression coefficient greater or smaller than one indicates deflated or inflated predictions, respectively.
Predictive ability and prediction bias are only reported for the genotyped trees from the validation set for all site-trait combinations examined. An analysis of variance (ANOVA) using a linear model with fixed effect for site and Least Significant Difference (LSD) multiple comparison tests were performed at a significance level α = 0.05, to test the significance of the difference in predictive ability and prediction bias between the ABLUP and ssGBLUP approaches performed for each trait.
Programs from the BLUPF90 family (Misztal et al. 2018) were used for cross-validation analyses. A custom R-script was written to automate the cross-validation analysis.
Results
Model fit and variance components
Our evaluation of the model fit revealed that the ssGBLUP model using 25K SNPs had a slightly lower AIC (i.e., better fit) (66,700 for design (ssGBLUPd_25K) and 65,200 for spatial phenotype adjustments (ssGBLUPs_25K)) than the ssGBLUP model with 8K SNPs (66,708 for design (ssGBLUPd_8K) and 65,204 for spatial phenotype adjustments (ssGBLUPs_8K)). However, the ABLUP models showed the lowest AIC values for block (66,673) and design (65,171) phenotype adjustments.
Overall, for the three studied traits, the ABLUP produced slightly higher heritability estimates compared to the ssGBLUP (0.24 vs. 0.23 for DBH30, 0.30 vs. 0.29 for HT30 and 0.51 vs. 0.49 for NSWGR36) (Table 4). The DBH30 trait showed heritability estimates ranging from 0.22 to 0.28 for ABLUP and from 0.19 to 0.26 for ssGBLUP. While, slightly higher heritability estimates were obtained for HT30 across sites, with values ranging from 0.22 to 0.40 for both ABLUP and ssGBLUP models. However, the heritability estimates for NSWGR36 were highest for both ABLUP (range: 0.45–0.57) and ssGBLUP (range: 0.39–0.55) models.
The phenotype data adjustment by autoregressive spatial effects generally displayed a consistent reduction in the residual variance compared to the design adjustment, with the most obvious reduction found in the trait with the stronger spatial pattern of environmental variation (Supplementary Table S1 and Supplementary Fig. S4), HT30 (averaged across sites 0.83 vs. 0.95 for the full set of 25K SNPs and 0.85 vs. 0.97 for the 8K subset, Table 4). However, both phenotype adjustments, spatial and design, resulted in very similar estimates of additive variances. Consequently, the narrow-sense heritability within sites estimated from the spatial phenotype adjustment were higher than those estimated from the design adjustment for both sets of markers (Fig. 2a and Table 4). Averaging across sites, the highest increase in heritability (10.28%) for the spatial phenotype adjustment respect to the design, was achieved for HT30 from the 8K SNP subset (0.30 vs. 0.27, respectively) follow by 25K SNP set (0.31 vs. 0.29, respectively, (8.70%)) (Table 4). The lowest increase in heritability (0.51%) was achieved for the NSWGR36 trait with 25K SNP set (0.498 vs. 0.495, respectively for the spatial and design phenotype adjustments). There was only one exception to this, for NSWGR36 and the 8K SNP subset, where the heritability from the spatial adjustment showed slightly lower estimates compared to the design adjustment (averaged across sites 0.47 vs. 0.48, respectively, (3.11%)).

Average relative variation in the narrow-sense heritability from the ssGBLUP models with spatial phenotype adjustment over those estimated with the design adjustment for the full SNP set (Full SNP set (25K)) and highest ancestry-informative SNP subset (Ancestry-Informative SNP subset (8K)) (a), and with a G-matrix calculated using the highest ancestry-informative SNP subset over those estimated with the full SNP set for the design (Design) and spatial (Spatial) phenotype adjustments (b) for the three traits studied. See text for traits´ abbreviations.
Regarding the impact of the number of SNP markers used to calculate the realized genomic relationships, we found that the ssGBLUP model with the full set of 25K SNPs showed higher heritability estimates with respect to the ssGBLUP model obtained from the AIM analysis (8K), for the two phenotype adjustment methods (Fig. 2b and Table 4). For instance, averaging across sites, the highest increase in heritability (6.96%) for the full set of SNPs (25K) respect to the 8K subset, was achieved for the trait HT30 with the design phenotype adjustment (0.29 vs. 0.27, respectively), while the lowest increase (2.53%) was achieved for the NSWGR36 trait with the design phenotype adjustment (0.50 vs. 0.48, respectively). These higher heritability estimates are a result of higher additive genetic variance and lower error variance of the ssGBLUP model using all available SNPs compared with those based on a subset of 8K SNPs (Table 4).
Genetic correlations
Within sites, genetic correlation estimated between DBH30 and HT30 using the ssGBLUP model were high (range: 0.435–0.717) (Supplementary Table S2). In contrast, negative genetic correlations were found between growth traits (DBH30 and HT30) and NSWGR36, with an average of −0.388 for DBH30 and −0.294 for HT30 (Supplementary Table S2). Our results suggest that selection for faster-growing trees could potentially improve WGR genetic resistance, or selection for pest resistant trees does not seem to adversely affect growth. Furthermore, between sites genetic correlations estimated with ssGBLUP were high for DBH30 (range: 0.678–0.891) and NSWGR36 (range: 0.813–0.955), and moderate to high for HT30 (range: 0.556–0.788) (Supplementary Table S3) with only slightly differences with those estimated from the ABLUP models (range: 0.606–0.837 for DBH30, 0.849–0.950 for NSWGR36, and 0.574–0.779 for HT30), indicative of nonsignificant genotype by environmental interactions across sites for these traits. The lowest genetic correlation between sites (average across models equal to 0.647) were observed for the HT30 trait with those pairs involving the JUDY site, probably caused for the high mortality in this site (survival at 30 years 58%) due to early competition for insufficient weed control.
Predictive ability and genomic breeding values bias
Overall, the results showed that the maximum predictive ability (PA) was reached using the G-matrix from the 8K subset of ancestry informative SNPs for HT30 using the design adjustment, and for DBH30 and NSWGR36 with the spatial phenotype adjustment (Table 5). The lowest PA was found when all the SNPs (25K) were used compared to the 8K, with either design (0.414 vs. 0.423 for DBH30) or spatial (0.451 vs. 0.464 for HT30 and 0.492 vs. 0.513 for NSWGR36) phenotype adjustment. With respect to the lowest PA obtained (i.e., when all the 25K SNPs were used), the maximum improvement in the PA values were statistically significant and in the order of 3.71% for DBH30 (0.414 for ssGBLUPd_25K vs. 0.429 for ssGBLUPs_8K), 3.43% for HT30 (0.451 for ssGBLUPs_25K vs. 0.467 for ssGBLUPd_8K), and 6.81% for NSWGR36 (0.481 for ssGBLUPd_25K vs. 0.513 for ssGBLUPs_8K). Although not statistically significant, the phenotypic data with a spatial adjustment provided higher PA than with the design adjustment (0.418 vs. 0.414 for the 25K SNPs and 0.429 vs. 0.423 for the 8K SNPs for DBH30, and 0.492 vs. 0.481 for the 25K SNPs and 0.513 vs. 0.509 for the 8K SNPs for NSWGR36), except for HT30 where a slight reduction in PA was observed (0.451 vs. 0.453 for the 25K SNPs and 0.464 vs. 0.467 for the 8K SNPs) (Table 5). Averaging across sites, all 8K SNPs ssGBLUP models and three studied traits had higher predictive ability than those estimated with the pedigree-based ABLUP model for the respective design and spatial phenotype adjustments (Table 5).
The regression coefficients of genomic breeding values were used as a measure of prediction bias (PB) for ABLUP and ssGBLUP models. Table 5 shows that all calculated PB values were close to one (range: 0.918–1.014), indicative of unbiased predictions of our four ssGBLUP models. For example, the regression coefficients averaged across the four sites and the four ssGBLUP approaches ranged from 0.970 to 0.987 (DBH30), from 0.997 to 1.014 (HT30), and from 0.918 to 0.965 (NSWGR36). Although no evidence of significant bias was detected, the ssGBLUP models using 8K SNP showed a higher bias, except for DBH30 (Table 5). In general, the spatial correction had less bias than the prediction from the design phenotype adjustment (across trait and SNP sets the averaged absolute deviation from 1 was equal to 0.027 and 0.036, respectively). Therefore, the spatial phenotype adjustment reduced the bias of the genomic ssGBLUP approaches. The ssGBLUP model with the least bias was ssGBLUPs_25K (across sites and trait the averaged absolute deviation from 1 was equal to 0.022). However, the PB values were still lower for the ABLUP model with design (ABLUPd) and spatial (ABLUPs) phenotype adjustments (across sites and trait the averaged absolute deviation from 1 was equal to 0.010 and 0.016, respectively, Table 5).
Discussion
The present study aimed to jointly utilize spatial analysis and SNP selection with the ssGBLUP approach, to improve the (co)variance component estimation and predictive ability and to reduce the genomic prediction bias of three economically important traits in lodgepole pine. As a control, the performance of the ssGBLUP models was also compared to the conventional pedigree-based ABLUP model. We found that the highest heritability estimates, associated with the highest genetic and lowest residual variances, were achieved for the ABLUP model using the spatial phenotype adjustments followed closely by the ssGBLUP model based on the full SNP set (25K) and also using the spatial phenotype adjustments. Meanwhile, the highest predictive ability was obtained by using the 8K SNP subset when either the spatial (DBH30 and NSWGR36) or design (HT30) phenotype corrections were used. Additionally, no significant difference was detected in the prediction bias among the six models fitted as these values were all close to 1, indicating an unbiased prediction.
Recent studies on forest tree species revised by Grattapaglia et al. (2018) have shown that GBLUP and ssGBLUP GS approaches were successfully applied in many breeding programs focused on improving wood quality and growth traits, such as total height and diameter at breast height assessed in this lodgepole pine population, which are seen as complex Mendelian traits controlled by a large number of genes of small-effects. However, compared to BLUP-based GS approaches, Bayesian predictive GS models such as BayesCπ were more accurate in predicting simple Mendelian traits (e.g., resistance to biotic stresses) regulated by few genes of large-effects (i.e., oligogenic traits). For instance, Ukrainetz and Mansfield (2020b) found comparable predictions accuracies between BayesC and GBLUP in lodgepole pine for different polygenic growth (tree height) and wood quality traits when there was relatedness (0.81 for Bayes C and 0.83 for GBLUP) or unrelatedness (0.27 for BayesC and 0.29 for GBLUP) between the training and validation populations. However, Bayesian-based GS approaches were effective in predicting fusiform rust disease incidence with high accuracy in Pinus taeda L (loblolly pine), a trait controlled by a few major quantitative trait loci (Resende et al. 2012; Shalizi et al. 2021). In contrast, in Picea abies (Norway spruce), Lenz et al. (2020) showed that BayesCπ did not lead to improvement of the predictive ability compared with methods assuming that all genes had small effects (GBLUP) for white pine weevil (Pissodes strobi) highlighted the evidence of a polygenic control of this insect resistance trait. Polygenic control seems also reasonable for WGR given that it is influenced by many physical as well as chemical factors, but efforts to examine whether some larger gene effects are detectable for WGR need to be realized. The development of modern genomic tools such as machine learning algorithms could be capable to converge all these different GS techniques (Cortés et al. 2020).
For oligogenic traits, marker Assisted Selection (MAS) offers an alternative to GS approaches, i.e., when small number of QTLs of large effect that account for a large percentage of the variation in the selected trait (e.g., 30% or more; Muranty et al. 2014). However, there are only very few examples of MAS application in forest trees (Bernatzky and Mulcahy 1992). Recently, Shalizi et al. (2021) identified three SNP loci with large effects on fusiform rust disease outcome in a set of clonally replicated full-sib families of Pinus taeda, explaining 54% of the trait´s genetic variation. However, as the authors stated, these markers should be further validated in an independent study to be considered for MAS. Moreover, given that MAS generally introduces one gene at the time, it is not suitable for simultaneously managing the recombination of many genes (Muranty et al. 2014). Consequently, the rare application of MAS in forest tree species is associated with the polygenic nature of most quantitative genetic traits (as the analyzed traits of this lodgepole pine population), highlight considering the GS approaches, which are based on capturing the whole-genome effect, as a realistic way in lodgepole pine and other tree species.
The ssGBLUP GS method extends the genomic enabled predictive ability to all pedigree connected trees across trials (Grattapaglia et al. 2018), even to those trials with few genetic connections (Callister et al. 2021), this is particularly relevant advantage for large progeny trial networks commonly deployed in forest tree breeding (Paludeto et al. 2021). The ssGBLUP GS approach has been applied in forest trees to improve the theoretical accuracy or predictive ability of breeding values using the single-trait (Cappa et al. 2017; Paludeto et al. 2021) and multiple-trait (Ratcliffe et al. 2017; Cappa et al. 2018; Mphahlele et al. 2021) models. Furthermore, using a multiple-site ssGBLUP model, Ukrainetz and Mansfield (2020a) showed an improved predictive ability within environments for individual growth and wood quality traits in a lodgepole pine population. Callister et al. (2021) also concluded that the multiple-site ssGBLUP model significantly improved the prediction accuracy of parents and genotyped trees for steam volume data (and wood quality) in a Eucalyptus globulus population conformed by 48 (and 20) sites spread across three regions of southern Australia. Multiple-traits multiple-sites GBLUP and ssGBLUP models simultaneously use the information from multiple traits and environments to capture the correlations among traits (attributable in part to pleiotropy) and their interrelationships with environmental factors, and allows for an extension for dominance and epistatic interaction genetic effects. The role in predicting phenotypes of the pleiotropic and epistatic interactions has been recognized in forest trees species (Holliday et al. 2013; Gamal El-Dien et al. 2016; Chhetri et al. 2019; Klápště et al. 2020); however, these studies are still scarce. Specifically, pleiotropic and epistatic interactions for growth and disease traits are not well known in forest tree species. However, a parallel study, using the same lodgepole pine population assessed for 15 productivity and climate-adaptability traits that include the three traits of the present study, showed that a multi-trait genome-wide association (GWA) analysis identified many more significant associations than single-trait GWA analysis for growth and WGR resistant traits, potentially revealing pleiotropic effects of individual genes. The current study is one of the first to address the applicability of GS in forestry that simultaneously consider multiple-traits and multiple-sites in an individual tree mixed ssGBLUP model. Although the application of GS approaches in lodgepole pine is in its infancy (Ukrainetz and Mansfield 2020a, 2020b), our results showed that environmental heterogeneity and genetic covariance among traits and test environments, used for the estimation of variance components and phenotype prediction, can be accounted for concurrently.
The advantage of using spatial autoregressive models has been seen in a number of studies in forestry. With using only phenotypes and pedigree (ABLUP model), these studies found a consistent reduction in the error variance and an increase in heritability using the classical, a priori design models (e.g., Ye and Jayawickrama 2008; Cappa et al. 2019b). To elucidate, in a first-generation Douglas-fir (Pseudotsuga menziesii (Mirb.) Franco) progeny trial, Ye and Jayawickrama (2008) showed that, on average, 14–34% of the residual variance could be attributed to spatial heterogeneity, and accounting for such variance resulted in an increase in the heritability estimates. Similarly, in the current study, our results support the use of jointly applying spatial adjustment with pedigree-based (ABLUP) and GS approaches (ssGBLUP) for long rotation lodgepole pine growing in highly heterogeneous environments in central Alberta’s boreal forests (70–90 years) (Supplementary Fig. S4).
Our results generally showed slightly lower heritability estimates from the ssGBLUP compared to ABLUP (Table 4), and confirm previous studies that suggested inflated heritability estimates from the pedigree-based ABLUP analyses caused by an overestimation of additive variance. For instance, Ratcliffe et al. (2017) using an open-pollinated Picea glauca (Moench) Voss population, and utilizing a varying proportion of genotyped trees in the ssGBLUP, showed higher heritability estimates for total height and wood density from ssGBLUP than those estimates from ABLUP. Ukrainetz and Mansfield (2020a) using an open-pollinated lodgepole pine population, showed that ABLUP had the highest heritability estimates for wood density compared to ssGBLUP. Mphahlele et al. (2021) also studying an open-pollinated population of Eucalyptus grandis, observed higher heritability estimates from ABLUP for dimeter and tolerance to Leptocybe invasa gall wasp and fungal stem diseases Botryosphaeria dothidea and Teratosphaeria zuluensis, compared to those estimates from ssGBLUP.
In addition to the betterment of heritability estimates identified (Table 4), we investigated the effectiveness of spatial corrections on the phenotypes for predictive ability (PA). In the two SNP datasets evaluated, the ssGBLUP approaches with the spatial autoregressive structure to correct the phenotypic data, resulted in a modest, albeit non-significant, increase in the PA of DBH30 and NSWGR36 compared to the simplest design adjustment, but a marginal reduction (also non-significant) for HT30 (Table 5). Previous studies applying GS in crop breeding have proposed adjusting continuous spatial effects to increase PA by fitting spatial coordinates expressed as either classification variables, covariables (Lado et al. 2013), or first-order autoregressive covariance structures (Bernal-Vasquez et al. 2014; Ward et al. 2019; Mao et al. 2020; Tsai et al. 2020). However, these studies used only genotyped individuals (i.e., GBLUP models). Lado et al. (2013) used different row-column and moving-means models to adjust a spatial trend in phenotypic data of 384 wheat genotypes and concluded that correction of spatial variation is essential to increasing the PA in genomic selection models. Comparably, in Tsai et al. (2020), the PA for wheat and barley yield improved by combining a multi-trait response and spatial effects in the model. On the contrary, although corrections for spatial variation increased across-environment trait heritability estimates by 25%, Ward et al. (2019) found little effect on model PA in wheat yields. Mao et al. (2020), using two empirical datasets of maize and wheat and a simulation, recently showed adjusting for spatial effects improved genotypically superior plants’ selection. Nonetheless, prior to our current research, only two studies have directly investigated the efficiency of including spatial analysis in genomic prediction with ssGBLUP in forest trees (Cappa et al. 2017; Klápště et al. 2020), where Cappa et al. (2017) brought forth the first study that applied competition or spatial analysis using ssGBLUP and showed increased heritability estimates and theoretical accuracies of predicted breeding values. However, these two previous studies considered the theoretical accuracies of predicted breeding values of trees with phenotype information and from the current generation (Cappa et al. 2017), and they did not include any comparison with the conventional phenotype adjustment of the field experimental design (Klápště et al. 2020).
Our results showed, for the two phenotype adjustments and the three studied traits, that using the ancestry informative SNP subset (8K) provided higher PA than using the full 25K SNP set (Table 5). Moreover, when we compared the PA obtained from the 8K subset selected according to their ancestry information with other three 8K SNP subsets selected according to the population statistic Fst scores (Ling et al. 2021) and two association statistics from a single-step multiple-trait multiple-site genome-wide association analysis (ssGWAS): SNPs ranked by the largest to the smallest effects in absolute value averaged across the four sites, and the smallest to largest p values averaged across the four sites, the results showed higher PA for the ancestry-informative SNP subset than those 8K SNPs subsets selected according to their population and association statistics (Fig. 3 and Supplementary Table S4). Therefore, these results showed the higher ability of the ancestry-informative 8K SNP subset to capture realized relatedness than those 8K SNP selected using population (Fst scores) or association statistics (absolute marker effects or p values). As shown Thistlethwaite et al. (2020) in Douglas-fir, these findings also support the fact that the predictive performance is mainly driven by the markers ability to capture realized relatedness and not the linkage disequilibrium (LD) between SNP marker loci and putative QTLs underlying the trait. Based on these empirical results, we propose that it is possible to concurrently reduce the number of SNP markers while improving the PA for complex traits in a long-lived tree species. In summary, we suggest that strategies like AIM SNP selection can be used when establishing the genotyping platform required for the broad adoption of genomic selection. Specifically, the choice of SNP markers to include on genotyping arrays requires careful consideration since the genotyping cost per sample is proportional to not only the number of SNPs on the array, but also to how much genetic relatedness the selected SNP could potentially capture (Meuwissen et al. 2013). The SNPs identified by the AIM selection can be used as a starting point to identify high-quality SNPs that accurately capture individual genealogical relationships as well as population structure, while also accounting for Mendelian variation between siblings, and delivering high predictive ability for complex traits in non-model organisms.

Black lines indicate the average of the predictive ability for the respective DBH30, HT30, and NSWGR36 traits, and estimated from the 8K subset of markers selected for their ability to infer ancestry (solid line, ssGBLUPs_8K model), from the full marker set (25K) (dotted line, ssGBLUPs_25K model) and from the conventional pedigree-based model (dashed line, ABLUPs). See text for traits´ abbreviations, and Table 3 for models´ abbreviations.
Several forest tree breeding studies have previously documented the benefits of using selected marker subsets to improve PA in GS analyses for the genotyped trees (i.e., GBLUP) (Resende et al. 2012; Tan et al. 2017; Chen et al. 2018). Resende et al. (2012) reduced the number of SNPs using the marker effect ranked in decreasing order by their absolute values in a Pinus taeda (loblolly pine) population. Two interesting scenarios about predictive ability emerged from Resende et al. (2012). For 13 out of the studied 17 traits, the correlation reached an asymptote with a subset from 820 to 4790 SNPs; however, maximum PA was reached with smaller marker subsets (110–590 SNPs) and decreased with the addition of more markers for disease-resistance-related traits, wood density, and crown width, lending evidence for a simpler genetic architecture for these traits. Likewise, Tan et al. (2017) increased the number of SNPs sampled at random and also showed that only a slight improvement in the PA was achieved when more than 5K SNPs (out of 41K) were used for all growth and wood quality traits studied in their Eucalyptus hybrid population. Further, Chen et al. (2018), using the two sampling strategies (i.e., randomly and largest positive SNP marker effects), showed the predictive ability reached a plateau with the top 4K SNPs (out of 100K) that had the highest positive marker effects for height in Norway spruce (Picea abies).
Given the results described above, further consideration of trait-specific SNP selection could be recommended to supplement the SNP selection in addition to the AIM marker selection. Preliminary results using additional SNPs, i.e., over the ancestry-informative 8K SNP subset (e.g., 11K and 14K), and ranked according to their p values and explained variance based in a ssGWAS analysis, suggests that no improvement was detected in PA when more markers with a large effect and variance were included, once the highly informative markers for ancestry inference are already included in the SNP subset. For example, for DBH30, the averaged PA of the ssGBLUP model using spatial adjustment and either the 11K or 14K SNP subsets were 0.428 and 0.427, respectively, and were not statistically different from the 8K subset (0.429).
In addition, the impact of use lower number of SNPs (<8K) on the predictive ability was studied using five SNPs subsets (500, 2000, 4000, 6000, and 8000) and the three different strategies of SNPs selection based on population (Fst scores) and associations (absolute marker effects and their p values) statistics. When the number of SNPs ranked according to their Fst scores, absolute marker effects or p values decreasing from 8K to 0.5K, the PA of the genomic breeding values increased by ~8.2% for the growth traits and 22.2% for NSWGR36 (average across selection methods) (Fig. 3 and Supplementary Table S4). As discussed previously, several studies in forest trees species have showed that lower numbers of SNPs provided a PA of genomic breeding values greater (Resende et al. 2012) or equivalent to those observed using all SNPs available (Lenz et al. 2017; Tan et al. 2017; Chen et al. 2018). As discussed Lenz et al. (2017), the fact that higher PA of predicted genomic breeding values can be obtained by using a reduced number of SNP with largest effects, compared to GS models estimated with all markers, could imply that part of the short-range LD could be picked-up when a reduced numbers of large-effect markers are utilized to build the G-matrices. Alternatively, these small number of markers could have the ability to better account for the realized relationship between related (and unrelated) trees. Despite that the 8K ancestry-informative SNP subset capturing the historical pedigree much better than other 8K SNPs selected by population and associations statistics, other selection strategies to optimize SNP sets could be potentially useful to reduce the genotyping efforts and GS cost in this lodgepole pine population and should be evaluated further.
The current study employed a reference-free SNP calling method, with the main research objective focusing on genetic parameter estimation and predictive analyses. Although the current SNP build might not provide the needed capacity for estimating LD at the physical level, we studied the relationships between the markers’ interdependence and predictability through genetic correlations of SNPs. For the entire 25,099 GBS-derived SNP set, the percentage of pairs with LD > 0.9 was 3.08% (Fig. 4). However, when conducting variable selection for most predictable SNPs, a much greater proportion of SNP markers were found in correlation (e.g., 57.5% of SNPs with significant marker effects found in LD > 0.9; details, see Fig. 5). It is, therefore, notable that SNPs with greater predictability might be in correlation, statistically. Whether or not they underlie the variations within the same pathways and determine the importance of population genetic and demographic parameters that govern trait variability, remains to be addressed when a genome assembly is available.

Distribution of pairwise linkage disequilibrium (LD) values greater than 0.10 between pairs of the 25,099 (25K) SNP set calculated as the squared correlation statistic.

Distribution of pairwise linkage disequilibrium LD values greater than 0.10 between pairs of the 500 (0.5K) selected SNPs based on the Fst scores (Fst), the largest absolute estimated effects (Marker_effect), and the lowest p-values (p-value) calculated as the squared correlation statistic.
For the 500 selected SNPs based on the largest absolute estimated effects, the SNP subset and method with the highest PA (Fig. 3 and Supplementary Table S4), we examine the impact of the training/validation (TS/VS) size on PA. Following Tan et al. (2017), we divided all 1490 trees into six different size groups with a TS/VS ratio of 1:1, 2:1, 3:1, 4:1, 5:1, and 9:1, with the corresponding size of 745/745, 993/497, 1118/373, 1192/298, 1242/248 and 1341/149 trees. Our results showed that the PA improved with the increasing sizes of the TS for all the studied traits (Fig. 6). The maximum PA values were achieved with TS/VS ratio of 9:1, indicating that this 9:1 ratio is the optimal for this lodgepole pine population. However, given that the PA increased without reaching a plateau, we expect further increase of the PA of genomic predictions with much larger training population size. The increment of PA as the size of the training set increased supports earlier GBLUP studies using either simulated (Grattapaglia and Resende 2011) and empirical (Tan et al. 2017; Calleja-Rodriguez et al. 2020) data sets in forest tree species. For larger training population, larger diversity is captured and more robust prediction would be obtained (Arenas et al. 2021).

Estimates were obtained across six training/validation sets (TS/VS) sizes in numbers of individuals (x-axis): 745/745, 993/497, 1118/373, 1192/298, 1242/248 and 1341/149. See text for traits´ abbreviations.
Although ssGBLUP approaches that included all available marker information (25K) increased the proportion of variance explained, i.e., higher individual-tree heritability estimates, ssGBLUP based on the ancestry-informative 8K SNP subset showed a greater PA. That is, the estimated heritability did not provide a good indication of what one would expect for the prediction of PA. Using simulation and real human data from related and unrelated individuals, de los Campos et al. (2013) also reported that the estimates of genomic heritability did not follow the same patterns as those of PA. These authors suggested that this lack of concurrence might be due to a contrasting situation in the entries of the derived G-matrix: some of the elements (related off-diagonal elements and diagonal elements) accurately represent the true covariance function, while others from off-diagonal elements (distant relatives and pairs of unrelated individuals) do not describe the patterns of realized genetic relationships at causal loci well. In addition, as they pointed out, variance components estimators are functions of diagonal and off-diagonal elements of G-matrix whereas the predictive ability is mainly determined by the off-diagonal entries of G-matrix and, therefore, these two tasks (inference vs. prediction) are driven, in part, by different forces (de los Campos et al. 2013). Our contrasting results on heritability estimates and PA reported here for this lodgepole pine population could be explained by these factors, especially when considering that the proportion of pairs of expected unrelated genotyped trees is very large (1,083,733) compared to the half-sib related trees (25,572); i.e., only 2.36% of all pairwise relationship coefficients for the genotyped trees are half-sib.
Conclusion
Despite best efforts in the original design of these progeny trials installed over 30-years ago, analytical tools available today can greatly enhance the genetic parameter estimation and breeding value predictions that can be gleaned from these efforts through the combined analysis of phenotypic, spatial, and genomic information. Mature progeny trials are necessary to develop robust genomic selection models, and their importance cannot be underestimated in moving adoption of these new technologies forward. This study showed that using of an autoregressive structure for correcting spatial variability led to increases in trait heritability estimates (from 0.51 to 10.28%). Though slightly decreased in heritability estimates (from 2.53 to 6.96%), the use of the strategically selected ancestry-informative 8K SNP subset can significantly improve predictive ability with either the design or spatial phenotype adjustment. For example, combining spatial adjustment and the 8K SNP subset analysis resulted in a 3.71% and 6.81% increase in the predictive ability for DBH30, and NSWGR36, respectively, compared to the use of design phenotype adjustment and the full 25K SNP set. These empirical results suggest incorporating SNP selection with AIM coefficients when considering enabling technologies, such as the establishment of a species-wide SNP array or more cost-effective target SNP genotyping approaches (Nagano et al. 2020; Zhang et al. 2020). Further, although the use of spatial autoregressive structure increased trait heritability and reduced prediction bias, the increases of predictive ability depend on the trait analyzed.
Covering ~30% of Earth’s land surface, forest ecosystem provides innumerable agricultural, ecological and societal benefits, while playing a critical role in global biogeochemical cycles and climate regulation (Bonan 2008). However, climate-induced forest mortality has become an emerging global phenomenon (Anderegg et al. 2015). And as an example, the ongoing outbreak of mountain pine beetle that began in 2001 has killed over 10 million hectares of mature lodgepole pine in Alberta and British Columbia (Westfall and Ebata 2012). Fueled by the ongoing drought pressure, species range of lodgepole pine is expected to shrink to only 17% of its current distribution in about 60 years (Coops and Waring 2011). The slow reaction time of 80–120 years to develop “genetic options” has made traditional trait-driven tree improvement an unfeasible solution. Our results attest to this powerful transition to genomics technologies (Bohra et al. 2020; Varshney et al. 2021). To this end, we reckon immediate adoption of agrigenomics approaches in forestry and in its regulatory agencies to manage the genomic adaptation in the face of climate challenges (Qaim 2020).
Data availability
Genotyping-by-sequencing (GBS) raw reads used in this study have been deposited in NCBI SRA BioProject - PRJNA715165. Information of the lodgepole pine trials including pedigree and phenotypic data are available in the GitHub repository: https://github.com/RESFOR/quantitative_genetics_R/blob/main/data_multi-trait_multi-site.txt.
References
Aguilar I, Misztal I, Johnson DL, Legarra A, Tsuruta S, Lawlor TJ (2010) Hot topic: a unified approach to utilize phenotypic, full pedigree, and genomic information for genetic evaluation of Holstein final score. J Dairy Sci 93:743–752
Anderegg WRL, Hicke JA, Fisher RA, Allen CD, Aukema J, Bentz B et al. (2015) Tree mortality from drought, insects, and their interactions in a changing climate. N Phytol 208:674–683
Arenas S, Cortés AJ, Mastretta-Yanes A, Jaramillo-Correa JP (2021) Evaluating the accuracy of genomic prediction for the management and conservation of relictual natural tree populations. Tree Genet Genomes 17:12
Bernal-Vasquez A-M, Möhring J, Schmidt M, Schönleben M, Schön C-C, Piepho H-P (2014) The importance of phenotypic data analysis for genomic prediction-a case study comparing different spatial models in rye. BMC Genomics 15:646
Bernatzky R, Mulcahy DL (1992) Marker-aided selection in a backcross breeding program for resistance to chestnut blight in the American chestnut. Can J Res 22:1031–1035
Bohra A, Chand Jha U, Godwin ID, Kumar Varshney R (2020) Genomic interventions for sustainable agriculture. Plant Biotechnol J 18:2388–2405
Bonan GB (2008) Forests and climate change: forcings, feedbacks, and the climate benefits of forests. Science 320:1444–1449
Calleja-Rodriguez A, Pan J, Funda T, Chen Z, Baison J, Isik F et al. (2020) Evaluation of the efficiency of genomic versus pedigree predictions for growth and wood quality traits in Scots pine. BMC Genomics 21:796
Callister AN, Bradshaw BP, Elms S, Gillies RAW, Sasse JM, Brawner JT (2021) Single-step genomic BLUP enables joint analysis of disconnected breeding programs: an example with Eucalyptus globulus Labill. G3 Genes|Genomes|Genetics 11:jkab253
Cappa EP, Cantet RJC (2007) Bayesian estimation of a surface to account for a spatial trend using penalized splines in an individual-tree mixed model. Can J For Res 2677–2688
Cappa EP, El-Kassaby YA, Muñoz F, Garcia MN, Villalba PV, Klápště J et al. (2017) Improving accuracy of breeding values by incorporating genomic information in spatial-competition mixed models. Mol Breed 37:125
Cappa EP, El-Kassaby YA, Muñoz F, Garcia MN, Villalba PV, Klápště J, et al. (2018) Genomic-based multiple-trait evaluation in Eucalyptus grandis using dominant DArT markers. Plant Sci 271:27–33
Cappa EP, de Lima BM, da Silva-Junior OB, Garcia CC, Mansfield SD, Grattapaglia D (2019) Improving genomic prediction of growth and wood traits in Eucalyptus using phenotypes from non-genotyped trees by single-step GBLUP. Plant Sci 284:9–15
Cappa EP, Muñoz F, Sanchez L (2019) Performance of alternative spatial models in empirical Douglas-fir and simulated datasets. Ann For Sci 76:16
Cappa EP, Yanchuk AD, Cartwright CV (2012) Bayesian inference for multi-environment spatial individual-tree models with additive and full-sib family genetic effects for large forest genetic trials. Ann For Sci 69:627–640
Chateigner A, Lesage-Descauses MC, Rogier O, Jorge V, Leplé JC, Brunaud V et al. (2020) Gene expression predictions and networks in natural populations supports the omnigenic theory. BMC Genomics 21:416
Chen ZQ, Baison J, Pan J, Karlsson B, Gull BA, Westin J, et al. (2018) Accuracy of genomic selection for growth and wood quality traits in two control - pollinated progeny trials using exome capture as genotyping platform in Norway spruce. BMC Genom 19:946
Chen C, Mitchell SE, Elshire RJ, Buckler ES, El-Kassaby YA (2013) Mining conifers’ mega-genome using rapid and efficient multiplexed high-throughput genotyping-by-sequencing (GBS) SNP discovery platform. Tree Genet Genomes 9:1537–1544
Chhetri HB, Macaya-Sanz D, Kainer D, Biswal AK, Evans LM, Chen J-G et al. (2019) Multitrait genome-wide association analysis of Populus trichocarpa identifies key polymorphisms controlling morphological and physiological traits. N Phytol 223:293–309
Christensen OF, Lund MS (2010) Genomic prediction when some animals are not genotyped. Genet Sel Evol 42:2
Christensen OF, Madsen P, Nielsen B, Ostersen T, Su G (2012) Single-step methods for genomic evaluation in pigs. Animal 6:1565–1571
Coops NC, Waring RH (2011) A process-based approach to estimate lodgepole pine (Pinus contorta Dougl.) distribution in the Pacific Northwest under climate change. Clim Change 105:313–328
Cortés AJ, Restrepo-Montoya M, Bedoya-Canas LE (2020) Modern strategies to assess and breed forest tree adaptation to changing climate. Front Plant Sci 11:1606
Costa e Silva J, Dutkowski GW, Gilmour AR (2001) Analysis of early tree height in forest genetic trials is enhanced by including a spatially correlated residual. Can J Res 31:1887–1893
Dhir NK (1983) Development of genetically improved strains of lodgepole pine seed for reforestation in Alberta. In: USDA Forest Service (ed) Lodgepole pine: regeneration and management., General Technical Report, PNW-157: 20–22, p 20
Dutkowski GW, Costa E, Silva J, Gilmour AR, Wellendorf H, Aguiar A (2006) Spatial analysis enhances modelling of a wide variety of traits in forest genetic trials. Can J Res 36:1851–1870
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES et al. (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6:1–10
FGRMS (2016) Alberta forest genetic resource management and conservation standards. Alberta Agriculture and Forestry, Government of Alberta, Edmonton, Alberta, 158
Gamal El-Dien O, Ratcliffe B, Klapste J, Chen C, Porth I, El-Kassaby YA et al. (2015) Prediction accuracies for growth and wood attributes of interior spruce in space using genotyping-by-sequencing. BMC Genomics 16:370
Gamal El-Dien O, Ratcliffe B, Klápště J, Porth I, Chen C, El-Kassaby YA (2016) Implementation of the realized genomic relationship matrix to open-pollinated white spruce family testing for disentangling additive from nonadditive genetic effects. G3; Genes|Genomes|Genet 6:743–753
Gianola D, Norton HW (1981) Scaling threshold characters. Genetics 99:357–364
Gilmour AR, Cullis BR, Verbyla AP, Verbyla AP (1997) Accounting for natural and extraneous variation in the analysis of field experiments. J Agric Biol Environ Stat 2:269
Grattapaglia D, Resende MDV (2011) Genomic selection in forest tree breeding. Tree Genet Genomes 7:241–255
Grattapaglia D, Silva-Junior OB, Resende RT, Cappa EP, Müller BSF, Tan B et al. (2018) Quantitative genetics and genomics converge to accelerate forest tree breeding. Front Plant Sci 9:1–10
Habier D, Fernando RL, Garrick DJ (2013) Genomic BLUP decoded: a look into the black box of genomic prediction. Genetics 194:597–607
Henderson CR (1984) Applications of linear models in animal breeding. University of Guelph, Guelph
Holliday JA, Wang T, Aitken S (2013) Predicting adaptive phenotypes from multilocus genotypes in sitka spruce (Picea sitchensis) using random forest. G3#58; Genes|Genomes|Genet 2:1085–1093
John S, Sadoway S (2019) Region C lodgepole pine controlled parentage program plan seed orchards G284 and G827. West Fraser Mills Ltd, Blue Ridge Lumber Inc. Alberta, Canada
Klápště J, Dungey HS, Graham NJ, Telfer EJ (2020) Effect of trait’s expression level on single-step genomic evaluation of resistance to Dothistroma needle blight. BMC Plant Biol 20:205
Klápště J, Dungey HS, Telfer EJ, Suontama M, Graham NJ, Li Y et al. (2020) Marker selection in multivariate genomic prediction improves accuracy of low heritability traits. Front Genet 11:1240
Klápště J, Suontama M, Dungey HS, Telfer EJ, Graham NJ, Low CB et al. (2018) Effect of hidden relatedness on single-step genetic evaluation in an advanced open-pollinated breeding program. J Hered 109:802–810
Lado B, Matus I, Rodriguez A, Inostroza L, Poland J, Belzile F et al. (2013) Increased genomic prediction accuracy in wheat breeding through spatial adjustment of field trial data. G3 Genes, Genomes, Genet 3:2105–2114
Legarra A, Aguilar I, Misztal I (2009) A relationship matrix including full pedigree and genomic information. J Dairy Sci 92:4656–4663
Legarra A, Christensen OF, Aguilar I, Misztal I (2014) Single Step, a general approach for genomic selection. Livest Sci 166:54–65
Legarra A, Robert-Granié C, Manfredi E, Elsen JM (2008) Performance of genomic selection in mice. Genetics 180:611–618
Lenz PRN, Beaulieu J, Mansfield SD, Clément S, Desponts M, Bousquet J (2017) Factors affecting the accuracy of genomic selection for growth and wood quality traits in an advanced-breeding population of black spruce (Picea mariana). BMC Genomics 18:335
Lenz PRN, Nadeau S, Mottet MJ, Perron M, Isabel N, Beaulieu J et al. (2020) Multi-trait genomic selection for weevil resistance, growth, and wood quality in Norway spruce. Evol Appl 13:76–94
Ling AS, Hay EH, Aggrey SE, Rekaya R (2021) Dissection of the impact of prioritized QTL-linked and -unlinked SNP markers on the accuracy of genomic selection(1). BMC Genom data 22:26
de los Campos G, Vazquez AI, Fernando R, Klimentidis YC, Sorensen D (2013) Prediction of complex human traits using the genomic best linear unbiased predictor. PLoS Genet 9:e1003608
Lourenco D, Legarra A, Tsuruta S, Masuda Y, Aguilar I, Misztal I (2020) Single-step genomic evaluations from theory to practice: using snp chips and sequence data in blupf90. Genes (Basel) 11:1–32
Mao X, Dutta S, Wong RKW, Nettleton D (2020) Adjusting for spatial effects in genomic prediction. J Agric Biol Environ Stat 25:699–718
Meuwissen THE, Hayes BJ, Goddard ME (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Meuwissen T, Hayes B, Goddard M (2013) Accelerating improvement of livestock with genomic selection. Annu Rev Anim Biosci 1:221–237
Misztal I, Legarra A, Aguilar I (2009) Computing procedures for genetic evaluation including phenotypic, full pedigree, and genomic information. J Dairy Sci 92:4648–4655
Misztal I, Tsuruta S, Lourenco D, Aguilar I, Legarra A, Vitezica Z (2018) Manual for BLUPF90 family of programs. University of Georgia, Athens, USA, 125
Mphahlele MM, Isik F, Hodge GR, Myburg AA (2021) Genomic breeding for diameter growth and tolerance to leptocybe gall wasp and botryosphaeria/teratosphaeria fungal disease complex in Eucalyptus grandis. Front Plant Sci 12:228
Mphahlele MM, Isik F, Mostert-O’Neill MM, Reynolds SM, Hodge GR, Myburg AA (2020) Expected benefits of genomic selection for growth and wood quality traits in Eucalyptus grandis. Tree Genet Genomes 16:1–12
Muñoz F, Sanchez L (2020) breedR: Statistical methods for forest genetic resources analysts. R package version 0.12–4. https://github.com/famuvie/breedR
Muranty H, Jorge V, Bastien C, Lepoittevin C, Bouffier L, Sanchez L (2014) Potential for marker-assisted selection for forest tree breeding: lessons from 20 years of MAS in crops. Tree Genet Genomes 10:1491–1510
Nagano S, Hirao T, Takashima Y, Matsushita M, Mishima K, Takahashi M, et al. (2020) SNP genotyping with target amplicon sequencing using a multiplexed primer panel and its application to genomic prediction in Japanese cedar, Cryptomeria japonica (L.f.) D.Don. Forests 11:898
Paludeto JGZ, Grattapaglia D, Estopa RA, Tambarussi EV (2021) Genomic relationship–based genetic parameters and prospects of genomic selection for growth and wood quality traits in Eucalyptus benthamii. Tree Genet Genomes 17:38
Qaim M (2020) Role of new plant breeding technologies for food security and sustainable agricultural development. Appl Econ Perspect Policy 42:129–150
Ratcliffe B, El-Dien OG, Klápště J, Porth I, Chen C, Jaquish B et al. (2015) A comparison of genomic selection models across time in interior spruce (Picea engelmannii × glauca) using unordered SNP imputation methods. Heredity (Edinb) 115:547–555
Ratcliffe B, Gamal El-Dien O, Cappa EP, Porth I, Klapste J, Chen C, et al. (2017) Single-step BLUP with varying genotyping effort in open-pollinated picea glauca. G3 Genes|Genomes|Genetics 7:935–942
Resende MFR, Munoz P, Resende MD, Garrick DJ, Fernando RL, Davis JM et al. (2012) Accuracy of genomic selection methods in a standard data set of loblolly pine (Pinus taeda L.). Genetics 190:1503–1510
Resende RT, Resende MDV, Silva FF, Azevedo CF, Takahashi EK, Silva-Junior OB, et al. (2017) Assessing the expected response to genomic selection of individuals and families in Eucalyptus breeding with an additive-dominant model. Heredity 119:245–255
Rosenberg NA, Li LM, Ward R, Pritchard JK (2003) Informativeness of genetic markers for inference of ancestry. Am J Hum Genet 73:1402–1422
Rweyongeza DM (2016) A new approach to prediction of the age-age correlation for use in tree breeding. Ann Sci 73:1099–1111
Shalizi MN, Cumbie WP, Isik F (2021) Genomic prediction for fusiform rust disease incidence in a large cloned population of Pinus taeda. G3 Genes|Genomes|Genetics 11:jkab235
Tan B, Grattapaglia D, Martins GS, Ferreira KZ, Sundberg B, Ingvarsson PK (2017) Evaluating the accuracy of genomic prediction of growth and wood traits in two Eucalyptus species and their F1 hybrids. BMC Plant Biol 17:110
Thavamanikumar S, Arnold RJ, Luo J, Thumma BR (2020) Genomic studies reveal substantial dominant effects and improved genomic predictions in an open-pollinated breeding population of Eucalyptus pellita. Genes|Genomes|Genet 10:g3.401601.2020
Thistlethwaite FR, El-Dien OG, Ratcliffe B, Klápště J, Porth I, Chen C et al. (2020) Linkage disequilibrium vs. pedigree: Genomic selection prediction accuracy in conifer species. PLoS One 15:1–14
Thomas B, El-Kassaby Y, Cappa E, Klutsch J, Ullah A, Erbilgin N (2019) Genome Canada’s RES-FOR project: Genomic selection for white spruce and lodgepole pine – linking phenotypes and genotypes. In: Genomic Selection for white spruce and lodgepole pine - linking phenotypes and genotypes, Quebec, Canada
Tsai HY, Cericola F, Edriss V, Andersen JR, Orabi J, Jensen JD et al. (2020) Use of multiple traits genomic prediction, genotype by environment interactions and spatial effect to improve prediction accuracy in yield data. PLoS One 15:1–14
Ukrainetz NK, Mansfield SD (2020a) Prediction accuracy of single-step BLUP for growth and wood quality traits in the lodgepole pine breeding program in British Columbia. Tree Genet Genomes 16:1–13
Ukrainetz NK, Mansfield SD (2020b) Assessing the sensitivities of genomic selection for growth and wood quality traits in lodgepole pine using Bayesian models. Tree Genet Genomes 16:14
VanRaden PM (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91:4414–4423
Varshney RK, Bohra A, Yu J, Graner A, Zhang Q, Sorrells ME (2021) Designing future crops: genomics-assisted breeding comes of age. Trends Plant Sci 26:631–649
Ward BP, Brown-Guedira G, Tyagi P, Kolb FL, van Sanford DA, Sneller CH et al. (2019) Multienvironment and multitrait genomic selection models in unbalanced early-generation wheat yield trials. Crop Sci 59:491–507
Westfall J, Ebata T (2012) Summary of forest health conditions in British Columbia. Victoria, British Columbia
Ye TZ, Jayawickrama KJS (2008) Efficiency of using spatial analysis in first-generation coastal Douglas-fir progeny tests in the US Pacific Northwest. Tree Genet Genomes 4:677–692
Zhang J, Yang J, Zhang L, Luo J, Zhao H, Zhang J et al. (2020) A new SNP genotyping technology target SNP-seq and its application in genetic analysis of cucumber varieties. Sci Rep 10:5623
Acknowledgements
We thank Dr. Jaroslav Klápště for his advice in implementing the AIM coefficient for SNP selection. The authors also would like to thank three anonymous referees for their constructive comments on an earlier version of the manuscript.
Funding
We acknowledge cash funding for this research from Genome Canada, Genome Alberta through Alberta Economic Trade and Development, Genome British Columbia, the University of Alberta, and the University of Calgary. Further cash funding has been provided by Alberta Innovates BioSolutions, the Forest Resource Improvement Association of Alberta, and the Forest Resource Improvement Program through West Fraser Ltd. (Blue Ridge Lumber and Hinton Wood Products) and Weyerhaeuser Timberlands (Grande Prairie and Pembina). In-kind funding has been provided by Alberta Agriculture and Forestry, Blue Ridge Lumber West Fraser, Weyerhaeuser Timberlands Grande Prairie, and the Thomas, Wishart, and Erbilgin labs in support of the Resilient Forests (RES-FOR): Climate, Pests & Policy – Genomic Applications LSARP project. This work was completed utilizing the High-Performance Computing Center facilities of Oklahoma State University (NSF MRI-1531128), and also in part by the Extreme Science Foundation Environment (XSEDE, NSF ACI-1548562). Specifically, it used the Bridges system, which was supported by NSF award number ACI-14456606, at the Pittsburgh Supercomputing Center (PSC). The allocation is under the MCB180177 of Charles Chen.
Author information
Authors and Affiliations
Contributions
Conceptualization: EPC and YAE-K; Data Curation and Resources: BRT, CC, BR, JGK, XW, YL, and JS-A; Genomic Data Resources: CC, and BR; Field Trials Resources: AB, SS, BRT, JGK, and NE; Methodology and Formal Analysis: EPC; Software and Writing-Original Draft Preparation: EPC and BR; Writing-Reviewing and Editing, Visualization: YAE-K, CC, NE, BRT, BR, and AB; Funding acquisition and Project Supervision: BRT, NE, and YAE-K.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Cappa, E.P., Ratcliffe, B., Chen, C. et al. Improving lodgepole pine genomic evaluation using spatial correlation structure and SNP selection with single-step GBLUP. Heredity 128, 209–224 (2022). https://doi.org/10.1038/s41437-022-00508-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-022-00508-2
- Springer Nature Switzerland AG
This article is cited by
-
Genomic evaluation for breeding and genetic management in Cordia africana, a multipurpose tropical tree species
BMC Genomics (2024)
-
Preselection of QTL markers enhances accuracy of genomic selection in Norway spruce
BMC Genomics (2023)
-
Genome-wide association studies: an intuitive solution for SNP identification and gene mapping in trees
Functional & Integrative Genomics (2023)
-
Multiple-trait analyses improved the accuracy of genomic prediction and the power of genome-wide association of productivity and climate change-adaptive traits in lodgepole pine
BMC Genomics (2022)