
Abstract
The redox-active protein cytochrome c is a highly positively charged hemoglobin that regulates cell fate decisions of life and death. Under normal physiological conditions, cytochrome c is localized in the mitochondrial intermembrane space, and its distribution can extend to the cytosol, nucleus, and extracellular space under specific pathological or stress-induced conditions. In the mitochondria, cytochrome c acts as an electron carrier in the electron transport chain, facilitating adenosine triphosphate synthesis, regulating cardiolipin peroxidation, and influencing reactive oxygen species dynamics. Upon cellular stress, it can be released into the cytosol, where it interacts with apoptotic peptidase activator 1 (APAF1) to form the apoptosome, initiating caspase-dependent apoptotic cell death. Additionally, following exposure to pro-apoptotic compounds, cytochrome c contributes to the survival of drug-tolerant persister cells. When translocated to the nucleus, it can induce chromatin condensation and disrupt nucleosome assembly. Upon its release into the extracellular space, cytochrome c may act as an immune mediator during cell death processes, highlighting its multifaceted role in cellular biology. In this review, we explore the diverse structural and functional aspects of cytochrome c in physiological and pathological responses. We summarize how posttranslational modifications of cytochrome c (e.g., phosphorylation, acetylation, tyrosine nitration, and oxidation), binding proteins (e.g., HIGD1A, CHCHD2, ITPR1, and nucleophosmin), and mutations (e.g., G41S, Y48H, and A51V) affect its function. Furthermore, we provide an overview of the latest advanced technologies utilized for detecting cytochrome c, along with potential therapeutic approaches related to this protein. These strategies hold tremendous promise in personalized health care, presenting opportunities for targeted interventions in a wide range of conditions, including neurodegenerative disorders, cardiovascular diseases, and cancer.
Similar content being viewed by others
Facts
-
Cytochrome c acts beyond its traditional role in electron carrier in the electron transport chain and caspase-dependent apoptosis, engaging in chromatin remodeling, pyroptosis and the persister phenotype.
-
Post-translational modifications, including phosphorylation, acetylation, and nitration, intricately modulate its functions in electron transport, redox regulation, cell death pathways, and cell survival.
-
The translocation of cytochrome c to different cellular compartments, such as the cytosol, nucleus, and extracellular space, underscores its multifaceted role in cell death, cellular signaling, and immune responses.
Open questions
-
What are the precise molecular mechanisms through which cytochrome c interacts with other proteins in both apoptotic and non-apoptotic pathways to determine cell fate decisions?
-
How does the structure of cytochrome c contribute to its multifaceted roles in cellular biology across both physiological and pathological processes?
-
How do specific post-translational modifications of cytochrome c regulate its diverse function under both physiological conditions and in response to certain pathological contexts?
-
What are the current and emerging techniques for detecting cytochrome c, and how can these approaches contribute to understanding and addressing various diseases?
-
Could targeting the activity of cytochrome c offer therapeutic approaches for mitigating dysregulated cell death pathways in diseases such as neurodegenerative disorders, cardiovascular diseases, and cancer?
Introduction
Cytochromes were first described as respiratory pigments by Charles A. MacMunn in 1884 [1]. In the 1920s, David Keilin named these heme proteins “cytochromes” or “cellular pigments” [2]. Cytochromes are categorized into four major groups based on heme type and binding patterns: cytochrome a, cytochrome b group, cytochrome c group, and cytochrome d group [3]. Additionally, there are other types of cytochrome proteins such as cytochrome P450 and cytochrome f, each playing unique roles primarily in electron transport and metabolic reactions. Cytochrome c belongs to the cytochrome c group and possesses a unique heme c moiety that forms covalent bonds with cysteine residues of the protein scaffold [4]. The CYCS gene encodes its somatic form, essential for cellular respiration and cell death in most cells. The testis-specific variant, encoded by the CYCT gene, plays a unique role in spermatogenesis, adapting to the specific metabolic needs of sperm cells [5, 6]. Cytochrome c protein typically refers to the somatic form of the protein. However, humans only have a single ubiquitously expressed cytochrome c gene [7]. It is a highly conserved globular protein found in various organisms, including bacteria, plants, and animals, including humans. It is composed of 94–114 amino acids. In humans, cytochrome c is a single-chain protein consisting of 104 amino acid residues [8].
Under physiological conditions, cytochrome c primarily resides within the mitochondria, acting as an electron carrier in the electron transport chain. It facilitates electron transfer from coenzyme Q-cytochrome c reductase (complex III) to cytochrome c oxidase (complex IV), aiding in generating a proton gradient across the inner mitochondrial membrane, which is utilized for adenosine triphosphate (ATP) synthesis [9]. The indispensability of cytochrome c is underscored in cytochrome c-knockout mice studies, where global knockout leads to embryonic lethality during mid-gestation as the fetus transitions from glycolysis to aerobic energy production [10]. Additionally, selective cytochrome c elimination in adult forebrain neurons triggers severe behavioral abnormalities and premature death [11], highlighting its vital role in maintaining tissue homeostasis.
In response to numerous apoptotic stimuli, cytochrome c translocates from mitochondria to the cytosol primarily through mitochondrial outer membrane permeabilization (MOMP), a process largely controlled by the BCL2 family of proteins [12, 13]. Once in the cytosol, cytochrome c binds to apoptotic peptidase activator 1 [14] (APAF1; initially named apoptotic protease activator 1) and triggers the formation of an apoptosome, a multimeric protein complex composed of cytochrome c and APAF1, which mediates the activation of the initiator caspase, caspase 9 (CASP9), leading to activation of the effector CASP3/7 and ultimately apoptosis [15,16,17,18]. The CYCS K72A mutant, while retaining normal electron transfer function, fails to activate APAF1 [19, 20]. This mutant in knock-in mice exhibits embryonic or perinatal lethality due to central nervous system defects and disrupted lymphocyte homeostasis [21].
Upon stimuli that induce mitochondrial permeability transition (MPT), cytochrome c is released into the cytosol along with ATP, facilitating the assembly of pyroptosomes, comprising APAF1 and CASP4/11, and CASP4 activated in the APAF1 pyroptosome proceeds to cleave CASP3 and thereby GSDME to induce pyroptosis [22]. MPT refers to the abrupt loss of integrity of the inner mitochondrial membrane, which allows the free permeability of small molecules, leading to a loss of osmotic balance [23]. This process is mediated by the formation of the mitochondrial permeability transition pore (MPTP), a multiprotein complex pore whose composition, although still a matter of debate, includes adenine nucleotide translocator (ANT), cyclophilin D (CYPD), the voltage-dependent anion channel (VDAC), the mitochondrial phosphate carrier, and ATP synthase [24,25,26,27,28,29]. Mitochondrial F1FO (F)-ATP synthase dimers, monomers, or the c-subunit ring alone have also been implicated in MPTP formation, adding complexity to our understanding of MPTP’s structure and function [30].
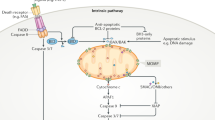
Cytochrome c, when translocated from the mitochondria to the cytosol in the context of sublethal MOMP induction, can enhance the survival of drug-resistant cells upon exposure to pro-apoptotic compounds known as BH3 mimetics [31]. Upon its translocation to the nucleus, cytochrome c contributes to chromatin assembly and remodeling, and it plays a role in regulating the accessibility of nucleolar proteins through liquid-liquid phase separation processes [32, 33]. In addition, upon release into the extracellular space, cytochrome c regulates immune responses [34, 35]. These findings underscore the central role of cytochrome c in maintaining homeostasis, governing cell death and survival pathways, and participating in immune responses (Fig. 1).

The timeline of cytochrome c research highlights significant milestones that have contributed to our current understanding of its function and biology. Many discoveries couldn’t be included here due to space limitations. These milestones, among others, have paved the way for further investigations. 1884 MacMunn describes “respiratory pigments” (cytochromes) [1]. 1925 Keilin gives cytochromes their modern name [2]. 1989 Nomenclature is created for electron transfer proteins [3]. 1996 Cytochrome c is found to induce apoptosis in cell-free extract [212]. 1999 Antioxidant functions of cytochrome c are discovered [95]. 2000 CYCS global knockout mice are developed [10]. 2003 Cytochrome c is found to bind with ITPR1 (inositol 1,4,5-trisphosphate receptor type) [133]. 2004 Discovery of cytochrome c nuclear translocation [142]. 2005 Electron transfer from cytochrome c to p66Shc triggering mitochondrial apoptosis via reactive oxygen species [98]. 2005 Apoptotic functions of cytochrome c are ablated in neurons [21]. 2006 First discovery and characterization of mammalian cytochrome c phosphorylation site [63]. 2008 CYCS G41S-mutant is found to be associated with thrombocytopenia [150]. 2017 Architectures of human mitochondria respiratory megacomplex is defined through cryo-electron microscopy [49]. 2020 Pyroptosomes consisting of APAF1/cytochrome c/CASP4/11 are discovered [22]. 2022 Sublethal cytochrome c is found to generate drug-tolerant persister cells [31]. 2022 Translocation of cytochrome c to the nucleus can regulate the liquid-liquid phase separation within the nucleolus, resulting in the release of proteins sequestered by nucleophosmin [32]. 2023 Lysine 39 acetylation of cytochrome c enhances porcine skeletal muscle’s cellular respiration and resilience to ischemia-reperfusion injury [78].
In this review, we provide an overview of the structure and function of mammalian cytochrome c, with an emphasis on human cytochrome c, and discuss its role in determining cell fate through posttranslational modifications, cellular localization, and interactions with multiple binding proteins. Moreover, we outline techniques for detecting cytochrome c and its aberrant alterations, highlighting its potential as a biomarker, along with methodologies for cytochrome c-targeted therapy. These strategies hold promise for personalized medicine in the realms of neurodegenerative disorders, cardiovascular diseases, and cancer.
Structures and functions of cytochrome c
The structural characteristics of cytochrome c have been elucidated since the 1960s [8, 36, 37]. Cytochrome c consists of five α-helices interconnected by extended omega (Ω) loops and a two-strand β-sheet structure [38, 39]. Fifteen amino acid residues of human cytochrome c (Fig. 2), have been conserved throughout evolution to support important functional roles [40]. Four highly conserved residues (Cys14, Cys17, His18 [constitutes a conserved cysteine-any-any-cysteine-histidine (CXXCH) motif], and Met80) are responsible for the tight binding of the heme molecule through covalent thioether linkages [41, 42]. His18 and Met80 act as a ligand to the heme iron and coordinate the heme axially, enabling redox potential and electron transfer to cytochrome c oxidase in the respiratory chain.

The structure of human cytochrome c exhibits conserved amino acid sites across various species. It is a heme protein that undergoes alkaline transition and has specific binding sites for ATP, APAF1, cytochrome c1, cytochrome c oxidase, and cardiolipin. In addition, cytochrome c can undergo modifications such as phosphorylation, acetylation, nitration, oxidation, and mutations associated with thrombocytopenia, including the variants G41S (PDB entry 3NWV), Y48H (PDB entry 5EXQ), and A51V (PDB entry 6DUJ). The 3D structure of cytochrome c, with highlighted residues, is displayed at the top (PDB entry 3ZCF).
A docking simulation of the cytochrome c-ATP complex demonstrated that the amine group and the phosphate groups of ATP bind with Glu62, Lys88, and Arg91 of cytochrome c through electrostatic interactions [43] (Fig. 2). Additionally, interactions between APAF1 and cytochrome c involve several residues of cytochrome c, including 7, 25, 39, 62–65, and 72 [19], further elucidating its apoptotic function. High-resolution X-ray crystal structures of human cytochrome c have been unveiled, providing detailed insights into its molecular architecture [44, 45]. Furthermore, the atomic structure of a complete mammalian apoptosome, resolved at a resolution of 3.8 Å through single-particle cryo-electron microscopy, revealed the mechanism by which cytochrome c facilitates the release of APAF1’s autoinhibition, primarily via interactions between specific amino acids (56, 72, 76, and 81) of cytochrome c and APAF1 [46] (Fig. 2). The key residues of cytochrome c important for binding to the respiratory complexes include lysines 8, 13, 27, 72, 86, and 87, which engage in electrostatic interactions to facilitate the formation of complexes with cytochrome c1 [47]. In the interaction with cytochrome c oxidase, Lys8, Gln12, Lys13, and Lys87 form critical hydrogen bonds and salt bridges near the exposed heme edge, facilitating electron transfer in the mitochondrial electron transport chain [48] (Fig. 2). In 2017, advancements in cryo-electron microscopy studies further expanded our understanding of cytochrome c’s role in cellular respiration. One study demonstrated the precise positioning of two cytochrome c molecules on the surface of the cytochrome c1 subunits of the complex III dimer in the human respiratory chain megacomplex [49]. It highlighted a difference in cytochrome c binding sites between reduced and oxidized states, with a structural shift bringing cytochrome c closer to the heme c1 in human complex III. The study challenges previous assumptions by demonstrating that both electron transfer pathways in the complex III homodimer are active, allowing simultaneous electron transfer from ubiquinol to two cytochrome c molecules. This underscores a complex interplay within the respiratory chain for efficient electron transport. All these structural studies highlight cytochrome c’s multifunctionality in both apoptosis and cellular respiration.
At physiological pH, cytochrome c carries a total charge of +8 (isoelectric point around 10) [50, 51], facilitating its interaction with negatively charged molecules such as cardiolipin at specific sequence positions, namely 72, 73, 86, and 87 [52]. Oxidized cardiolipin has a reduced affinity for cytochrome c, leading to an increase of free cytochrome c in the intermembrane space. This accumulation facilitates cytochrome c release into the cytoplasm upon MOMP [51].
The alkaline transition phenomenon refers to pH-dependent ligand exchange, involving the deprotonation of alternative ligands and the breaking of the iron-methionine ligation bond [53]. This transition is influenced by the flexibility of the Ω loops (70–85 and 40–57, with a particular emphasis on the epsilon-amino groups of Lys79 and Lys73) and the axial ligands (Met80 and Tyr67), which impact electron transfer and conformational changes in cytochrome c, thereby regulating its functions [54, 55]. These alterations can lead to the disruption of the iron-Met80 bond and increased accessibility of H2O2 to the heme center, thereby enhancing cytochrome c’s peroxidase activity. This increased peroxidase activity is indicative of cytochrome c’s altered state [51], and once MOMP occurs, triggered by various pro-apoptotic signals and events, it allows the nonmembrane-associated cytochrome c to be released into the cytosol. In the cytosol, cytochrome c can then participate in apoptosome formation and the activation of downstream caspases, leading to apoptosis.
In addition, the functional diversity of cytochrome c is influenced by various posttranslational modifications, which will be further discussed below.
Posttranslational modifications of cytochrome c
Posttranslational modifications of cytochrome c, such as phosphorylation, acetylation, glycosylation, glycation, deamidation, homocysteinylation, carbonylation, nitration, and sulfoxidation of specific amino acids, have been identified in various species and reviewed elsewhere [56,57,58]. In this section, we will primarily focus on discussing the posttranslational modifications observed in mammalian cytochrome c (Table 1 and Table 2).
Nitration
Nitration impacts cytochrome c’s five tyrosine residues (Tyr46, Tyr48, Tyr67, Tyr74, Tyr97). Cytochrome c can act as a catalyst and a target for nitrite-hydrogen peroxide–dependent protein nitration [59]. In general, in vitro nitration lowers cytochrome c’s midpoint redox potential and hinders caspase activation. While nitration of Tyr46 and Tyr48 does not affect electron transfer, it obstructs apoptosome formation [60]. Nitration of Tyr46 and Tyr48 might trigger cytochrome c degradation when heme iron state shifts to a high spin state [61]. Nitration of Tyr74 boosts peroxidase activity and inhibits apoptosis [62]. Notably, in vivo, only nitration of Tyr67, Tyr74, and Tyr97 has been observed, hinting at potential discrepancies in cytochrome c nitration effects between in vivo and in vitro conditions [57]. Although nitration sites can be mapped via sensitive methods such as mass spectrometry, no study has yet quantitatively shown any significant in vivo nitration of cytochrome c affecting a significant part of the cytochrome c pool to elicit biological effects.
Phosphorylation
Mass spectrometry identified five phosphorylation sites in mammalian cytochrome c. Tyr97, first found in bovine heart [63] and inducible by insulin treatment in the brain [64], was the initial site recognized and functionally analyzed. Its phosphomimetic mutant Y97-pCMF (p-carboxy-methyl-l-phenylalanine) impairs mitochondrial ROS production and reduces CASP3 activation activity without altering cytochrome c’s overall structure [65]. The second site, Tyr48 from bovine liver [66], inhibits mitochondria respiration and CASP3 activity [67], disrupts electron transfer, and alters electron transport chain flux [68], switching cytochrome c’s function from apoptotic to anti-apoptotic [67, 69]. The phosphomimetic mutants Y48E or Y48pCMF lower alkaline transition and midpoint redox potential while compromising caspase activation [69, 70].
Later, cytochrome c Thr28 and Ser47 phosphorylations were found in human skeletal muscle and brain tissue respectively but weren’t functionally characterized [71]. Phosphorylation of Ser47, identified in the brain, was then shown to be mediated by Akt, downregulating respiration and inhibiting apoptosis [72]. This phosphorylation is lost during ischemia and its loss was proposed to drive cell death during reperfusion [73]. Thr28 phosphorylation was identified in the kidney, mediated by AMPK, partially inhibits cytochrome c oxidase reaction without affecting CASP3 activity [74]. Thr58 was reported in rat kidney [75].
These modifications regulate cytochrome c functions across different tissues and reflect each tissue’s unique metabolic demands and regulatory needs. All identified phosphorylations inhibit respiration, with some also interfering with apoptosome formation and caspase activation. Under physiological conditions, cytochrome c phosphorylations modulate electron transport chain flux, preventing excessive mitochondrial membrane potentials and ROS generation [76]. During stress conditions like ischemic stroke, loss of cytochrome c phosphorylations triggers ROS burst during reperfusion, leading to cell death [77].
As discussed above, in most publications, quantitation of a given cytochrome c posttranslational modification is not provided, and the modifications may often occur at very low levels, such as in the case of nitration, questioning their biological importance. Future work should take this point into consideration because one can argue that a significant fraction of a protein must be modified to have a functional effect. Notably, for some cytochrome c phosphorylation, quantitative information is available and indicates that a significant cytochrome c fraction carries the modification. Reported numbers range from 35% for S47 phosphorylation in the brain [72] to 83% for T28 phosphorylation in kidney [74], or the number was indirectly inferred based on functional studies, indicating that cytochrome c from liver to be at least 50% phosphorylated on Y48 [66]. It should also be noted that depending on the method used, contrasting results can be obtained, as was the case for K39 acetylation discussed below, where mass spectrometry suggested low occupancy while X-ray crystallography and functional experiments suggested that the major fraction of the protein carried the modification [78].
Tissue-specific cytochrome c phosphorylation seems highly complex but may be interpreted in light of the energy demand and susceptibility to apoptosis of a tissue or organ. For example, we propose that Tyr 48 phosphorylation maintains an optimal intermediate mitochondrial membrane potential by partially inhibiting electron transport chain flux. In addition, this modification blocks apoptosis, protecting an organ that is tasked with the function to detoxify compounds an organism takes up as food or inhales through the lung. In contrast, Tyr97 phosphorylation in the heart only shows small kinetic differences in the reaction with cytochrome c oxidase compared to the unphosphorylated protein, likely because the heart is constantly working and fully dependent on aerobic energy production. Similar consideration may apply to other posttranslational modifications, including acetylation.
Acetylation
Recent studies have explored cytochrome c acetylation. Initially, a proteomics study reported Lys8 acetylation [79], which, although not functionally characterized, was later found to reduce cytochrome c oxidase activity [80] similar to phosphorylation, when studied using lysine to acetylmimetic glutamine replacement. Lys53 acetylation, discovered in human prostate cancer specimens, also inhibits cytochrome c oxidase activity and apoptosis, providing a potential mechanism that drives Warburg metabolism and apoptosis evasion in cancer [81]. In contrast to cytochrome c phosphorylation, which is lost during ischemia, lysine 39 acetylation is the first example of a posttranslational modification that is gained during ischemia. It was identified in ischemic porcine tibialis anterior skeletal muscle, enhanced cytochrome c oxidase activity and inhibited apoptosis [78]. It was then proposed that the gain of this posttranslational modification allows skeletal muscle to meet increased energy demand while at the same time providing the tissue with effective resilience to ischemia-reperfusion injury. Additionally, acetylations on lysines 27, 79, and 86 were found in both control and ischemic samples [78].
Oxidation
Cytochrome c can undergo autooxidation at the Met80 residue through the formation of oxygen groups leading to the cleavage of the Met80-heme iron bond. A human cytochrome c mutant (Delta83/84 cytochrome c) was created by removing two amino acids (Val83 and Gly84) from the loop, and it was observed that Met80 of Delta83/84 cytochrome c was site-specifically modified to methionine sulfoxide. This modification may impact the peroxidase activity of cytochrome c, highlighting a feedback mechanism between cytochrome c peroxidase activity and oxidative stress during cell death [82].
Subcellular compartment-dependent functions of cytochrome c
Cytochrome c exhibits a diverse range of functions depending on its cellular localization and interaction partners (Table 3). Considering its electrostatic properties, caution is needed when interpreting cytochrome c interactions in experimental buffers with varying ionic strength, as inadequate low ionic strength may promote nonspecific binding [83]. To prevent this, it is beneficial to use physiological ionic strength in buffers and validate observed interactions for their physiological relevance. In the subsequent sections (Fig. 3), we summarize and discuss how the localization of cytochrome c determines its function and the associated regulatory mechanisms.

Cytochrome c exhibit distinct functions depending on its subcellular localization. In mitochondria, cytochrome c serves as a vital component of the electron transport chain, facilitating electron transfer between complexes III and IV in the mitochondrial intermembrane space. The interaction between cytochrome c and HIGD1A (hypoxia-inducible domain family member 1A) is pivotal in regulating mitochondrial oxidative phosphorylation and the cellular stress response. Cytochrome c also plays a role in scavenging reactive oxygen species (ROS), particularly through the detoxification of hydrogen peroxide (H2O2). Moreover, when cytochrome c interacts with cardiolipin, it demonstrates peroxidase activity, playing a role in the initiation of apoptosis. It can further amplify ROS generation and trigger apoptosis via its interaction with p66Shc. Cytochrome c also interacts with other proteins, such as the growth hormone-inducible transmembrane protein (GHITM, also known as MICS1). Alongside GHITM, coiled-coil-helix-coiled-coil-helix domain containing 2 (CHCHD2) binds to cytochrome c. These interactions influence mitochondrial morphology or the release of cytochrome c. Members of the BCL-2 family, including BCL2 and BCL2L1, can inhibit the release of cytochrome c. A Under cellular stress, BH3-only proteins, such as Bim and Bid, become activated, bind and inactivate the anti-apoptotic proteins, such as BCL2 and BCL2L1, whose primary function is to restrain the effectors Bax and Bak. Once the anti-apoptotic BCL2 proteins are neutralized by the BH3-only proteins, Bax and Bak undergo spontaneous activation, homo-oligomerize, and form pores on the mitochondrial outer membrane, allowing the escape of cytochrome c and other apoptogeneic factors from the intermembrane space. After cytochrome c is translocated to the cytosol, it interacts with APAF1, initiating apoptosome assembly and subsequently activating CASP9 and CASP3. B It can interact with the inositol 1,4,5-trisphosphate receptor type 1 (ITPR1) on the endoplasmic reticulum membranes, triggering calcium ion (Ca2+) release. In addition, cytochrome c can bind to 14-3-3epsilon and block 14-3-3epsilon–mediated inhibition of APAF1, thus acting as an indirect activator of CASP9/3. Heat shock proteins have been identified as inhibitors of cytochrome c release or apoptosome formation. C Mitochondrial permeability transition (MPT) induces the assembly of the pyroptosome, which consists of APAF1, cytochrome c, and CASP4. This complex cleaves CASP3 and triggers the activation of gasdermin E (GSDME), leading to pyroptosis. D In the presence of cardiolipin, O2, and H2O2, cytochrome c oxidizes the plasmalogen vinyl ether linkage, facilitating its hydrolytic cleavage leading to lipid peroxidation. E BH3 mimetics can induce sublethal cytochrome c release, which activates heme-regulated inhibitor kinase (HRI or EIF2AK1) and enables the translation of activating transcription factor 4 (ATF4), resulting in a persister phenotype. Furthermore, cytochrome c can enter the nucleus, where it directly binds and inhibits histone chaperone SET/TAF-Iβ during DNA damage, and thus hinders SET/TAF-Iβ nucleosome assembly activity. Cytochrome c can directly bind to nucleolar nucleophosmin (NPM) by triggering a conformational change, driving alternative reading frame (ARF) release followed by the activation of p53 pathway in response to DNA damage. NPM engages in liquid-liquid phase separation. Finally, cytochrome c can be released into the extracellular space from damaged cells and taken up by astrocytes or immune cells in a toll-like receptor 4 (TLR4)-dependent manner. This uptake leads to the secretion of cytokines such as IL-1β, IL-12, and GM-CSF that trigger immune responses.
Mitochondrial cytochrome c
Mitochondrial cytochrome c interacts with multiple proteins, thereby playing an important role in determining its function within the mitochondria (Table 3). Mitochondrial cytochrome c generally performs four major functions.
1) Electron transport. Cytochrome c is a peripheral membrane protein located between the inner and outer mitochondrial membranes. It undergoes oxidation and reduction as its iron atom transitions between the ferrous and ferric forms and forms complexes with several redox partner proteins, including cytochrome bc1, cytochrome c oxidase. Cytochrome bc1, also called complex III, is a multi-subunit complex involved in mitochondrial electron transport, while cytochrome c1 is a specific subunit within this complex that transfers electrons to cytochrome c. Electron transfer from cytochrome c1 to cytochrome c is energetically favored within the immobilized cytochrome c1-cytochrome c complex [84].The function of cytochrome c oxidase can be regulated by a respiratory supercomplex factor hypoxia-inducible domain family member 1A (HIGD1A). The interaction between cytochrome c and HIGD1A regulates mitochondrial oxidative phosphorylation and response to cellular stress [65] (Fig. 3). High-resolution structural models of yeast respiratory supercomplexes have revealed that the diffusion of cytochrome c enhances electron transport [85]. The axial bond formed by Met80 with heme-iron in cytochrome c contributes to its high redox potential, enabling its functional role in the respiratory chain [86].
2) Peroxidase activity. Cardiolipin is an anionic phospholipid that constitutes about 20% of the inner mitochondrial membrane, and can move from the inner to the outer leaflet of the inner mitochondrial membrane, a process facilitating cardiolipin remodeling [87]. Cytochrome c engages with cardiolipin, exhibiting peroxidase activity that aids in the initiation of apoptosis. [88] (Fig. 3). Membrane curvature affects peripheral cytochrome c-cardiolipin interactions, leads to the conformational change of cytochrome c that switches on its peroxidase activity [89]. In addition, a decrease in pH at the inner mitochondrial membrane interface enhances cytochrome c’s peroxidase activity [90]. The binding of the ferrous state of cytochrome c and cardiolipin induces higher peroxidase activity of cytochrome c and higher permeability of the cardiolipin membrane [91], which may play a pivotal role in apoptosis initiation. The mitochondria-targeted antioxidant SkQ1 (plastoquinonyl-decyl-triphenylphosphonium, also known as visomitin), has been shown to inhibit the peroxidase activity of the cytochrome c-cardiolipin complex [92]. SkQ1 has also been reported to reduce inflammation following hemorrhagic shock by protecting myocardial mitochondria [93].
3) Dual role of cytochrome c in redox regulation. The redox states of cytochrome c are involved in the detoxification of hydrogen peroxide (H2O2) [94, 95]. Particularly, the ferric state of cytochrome c is instrumental in safeguarding mitochondrial cytochrome c oxidase from oxidative damage [96]. In addition, cytochrome c effectively processes free fatty acid hydroperoxides (FFA-OOH) in mitochondria, mitigating toxicity and regulating mitochondrial functions [97]. It should be noted that cytochrome c can also amplify ROS generation and trigger apoptosis through interaction with p66Shc [98] (Fig. 3).
4) Regulation of mitochondrial physiological functions. Cytochrome c regulates several mitochondrial physiological functions via protein interactions. It is associated with coiled-coil-helix-coiled-coil-helix domain containing 2 (CHCHD2), important for mitochondrial cristae structure and oxygen consumption. Mutations in CHCHD2 have been identified in patients with Parkinson’s disease (PD). Introduction of CHCHD2 has been shown to alleviate PD-associated pathological phenotypes [99] (Fig. 3). Cytochrome c also interacts with other proteins like growth hormone-inducible transmembrane protein (GHITM, also known as MICS1) [100] (Fig. 3), transient receptor potential cation channel subfamily V member 4 (TRPV4) [101], affecting mitochondrial morphology, or cytochrome c release, and caspase activation. The BCL2 family, such as BCL2 and BCL2L1 [102], can inhibit the release of cytochrome c (Fig. 3). BCL2L1 has been shown to interact directly with cytochrome c in response to ionizing radiation and genotoxic stress [103]. However, there is debate over whether this interaction is direct [104]. BCL2 does not appear to have the same activity [104]. Direct binding between BCL2L1 and cytochrome c was observed by co-immunoprecipitation, size-exclusion chromatography [105], and mass spectrometry [106]. Nevertheless, these studies lacked a mutational analysis using an inactive BCL2L1 mutant in the cellular context, which is necessary for determining the functional significance of this interaction. Moreover, it remains unclear whether the BCL2L1-cytochrome c interaction is relevant for the regulation of apoptosis in vivo. Cytochrome c is also vital for redox-coupled protein import to the mitochondrial intermembrane space via the Mia40/Erv1 electron relay pathway in yeast [107]. In human cells, augmenter of liver regeneration (ALR) is notably more effective in transferring electrons to cytochrome c than to oxygen [108]. These findings demonstrate the involvement of cytochrome c in the regulation of multiple mitochondrial physiological functions.
Cytochrome c translocation to the cytoplasm
Proteomic analysis of human cancer cells suggests the interactions between cytochrome c and several pro-survival and apoptotic proteins in the cytoplasm [109]. Four major functions have been demonstrated following the release of cytochrome c into the cytosol.
1) Pro-apoptosis function. The translocation of cytochrome c into the cytosol following MOMP is essential for caspase-dependent intrinsic apoptosis [110, 111]. MOMP is considered the point of no return in the commitment to cell death. The release of cytochrome c is pivotal for the subsequent activation of initiator and effector apoptotic caspases. However, if caspase activity is inhibited, cell death can proceed through alternative, caspase-independent pathways once MOMP has occurred [112, 113], cell death can proceed through alternative, caspase-independent pathways once MOMP has occurred [112, 113]. MOMP is primarily and precisely regulated by various members of the BCL2 family, which consists of anti-apoptotic proteins (BCL2, BCL2-like 1 [BCL2L1, also known as Bcl-XL], etc.) [114], effector proteins (BCL2-associated X [Bax], BCL2 antagonist/killer 1 [Bak], and BCL2-related ovarian killer [Bok]), and the BH3-only proteins [110, 115]. Under cellular stress, BH3-only proteins, such as Bim (also called BCL2L11) [116] and BH3 interacting domain death agonist (Bid) [117, 118], become activated and bind and inactivate the anti-apoptotic proteins, such as BCL2 and BCL2L1, whose primary function is to restrain the effectors Bax and Bak [119, 120]. Once the anti-apoptotic BCL2 proteins are neutralized by the BH3-only proteins, Bax and Bak undergo spontaneous activation, homo-oligomerize, and form pores on the mitochondrial outer membrane, allowing the escape of cytochrome c and other apoptogeneic factors from the intermembrane space [121,122,123]. Some BH3-only proteins, such as truncated Bid (tBid) and Bim, may also directly bind and activate Bax and Bak [13, 110, 124], although such interaction appears to be dispensable for Bax/Bak-mediated apoptosis [125, 126]. It is worth noting that tBid was shown to remodel mitochondrial structure and open up the junction between mitochondrial cristae and the intermembrane space, allowing maximal release of cytochrome c [127]. Cytochrome c release appears to require a two-step process [128]. Specifically, solubilization of cytochrome c involves disrupting the electrostatic interactions it maintains with cardiolipin. Following this disruption, MOMP facilitates the release of cytochrome c into the cytosol. Once in the cytosol, cytochrome c tightly binds to its cytoplasmic receptor, APAF1, and in the presence of ATP/deoxyadenosine triphosphate (dATP), triggers the formation of the ∼1.4 mDa protein assembly known as an apoptosome. Both dATP and ATP are capable of facilitating the formation of the apoptosome. Historically, research has emphasized the role of dATP, based on early cell-free system studies where dATP seemed more effective. However, later research has shown that ATP, which is far more abundant in living cells, can also enable apoptosome formation [129]. Cryogenic electron microscopy studies revealed that an apoptosome is a wheel-shaped, multimeric, 7-fold symmetric protein complex composed of 7 cytochrome c and 7 APAF1 molecules [130, 131]. Once formed in the cytosol, the apoptosome efficiently recruits pro-CASP9 and causes its activation, leading to the activation of the effector CASP3 and CASP7 [46] (Fig. 3). Moreover, cytochrome c can bind to inositol 1,4,5-trisphosphate receptor type 1 (ITPR1, also called IP3R), a calcium channel located on the outer membrane of the endoplasmic reticulum, releasing calcium that triggers a feedback loop activating CASP9/3 [132, 133] (Fig. 3). Studies using NMR spectroscopy, site mutagenesis, and computational calculations have demonstrated that cytochrome c can bind to 14–3–3epsilon (a direct inhibitor of APAF1) and block 14-3-3epsilon-mediated inhibition of APAF1, thus acting as an indirect activator of CASP9/3. Additionally, heat shock proteins have been identified as inhibitors of cytochrome c release or apoptosome formation [134, 135] (Fig. 3).
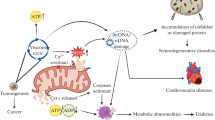
2) Pro-pyroptosis function. Several lines of evidence suggest that cytochrome c is involved in regulating pyroptosis, a form of inflammasome-associated cell death, mediated by caspase cleavage of the gasdermin family proteins at certain circumstances. First, iron-mediated ROS production triggers pyroptosis through the TOM20-BAX-cytochrome c-CASPs-gasdermin E (GSDME) pathway as was shown in melanoma cells [136]. Second, lobaplatin-induced ROS production and JNK phosphorylation result in BAX-dependent cytochrome c release, followed by CASP9/3 activation and GSDME cleavage, leading to pyroptosis [137]. CASP3-mediated production of N-terminal-GSDME induces MOMP, leading to cytochrome c release and apoptosis [138]. In certain cases, cytochrome c-mediated apoptosis activation may restrict inflammasome activation [139]. These studies highlight crosstalk between pyroptosis and apoptosis. Finally, excessive calcium, the activation of adenine nucleotide translocator 1 (ANT, also called SLC25A4), oxidative stress, and high bile acid levels lead to MPT, resulting in a pyroptosome formation with APAF1, cytochrome c, and CASP4/11 (not CASP9), causing CASP3-dependent GSDME cleavage and pyroptosis [22] (Fig. 4). These results are from a study revealing that while inhibitors of cyclophilin D and the mitochondrial phosphate carrier did not prevent cell death, bongkrekic acid — by inhibiting ANT — significantly protected cells from both MPT and subsequent cell death. Moreover, lonidamine, which specifically targets ANT1, prompts the swift release of ATP from mitochondria to the cytoplasm. This rapid ATP release is necessary for assembling the pyroptosome in the cytoplasm, facilitating bile acid-induced MPT and leading to pyroptotic cell death. The findings indicate that pyroptosis demands significantly stronger stimuli and a higher threshold than other cell death mechanisms. Remarkably, MPT-induced pyroptosis occurs within a much narrower time frame ( < 4 h) compared to MOMP-induced apoptosis, which usually unfolds over more than 18 h. This suggests that the APAF1 pyroptosome assembly serves as an immediate response mechanism, enabling cells to quickly react to severe threats by facilitating the removal of dying cells. Additionally, the recruitment of CASP4 to APAF1 necessitates considerably higher ATP levels than CASP9, and CASP4 has been shown to competitively inhibit CASP9 activation in high-ATP conditions, aligning with the time course of enzyme activities [22]. However, the mechanism behind the cellular decision between apoptosis and pyroptosis remains unclear.

Cytochrome c plays a critical role in various cellular processes, including apoptosis, pyroptosis, and the development of a persister phenotype. a In apoptosis, stressful stimuli such as chemotherapy or low doses of bile acids trigger MOMP. This leads to the release of cytochrome c into the cytosol, where it interacts with APAF1, initiating apoptosome assembly. Activated caspase-9 and caspase-3 are then recruited, resulting in apoptosis. b Under specific stimuli like bile acids, calcium overload, or activators of ANT, mitochondrial MPT promotes the assembly of a pyroptosome. The pyroptosome consists of APAF1, cytochrome c, and CASP4/11 (a general sensor of MPT) but not CASP9. This assembly leads to the cleavage of CASP3 and GSDME, inducing pyroptosis. c In the context of BH3-mimetic treatment, a sublethal release of cytochrome c enables cancer cells to acquire a persister phenotype. This phenotype is driven by the cytochrome c-EIF2AK1/HRI-ATF4 pathway. It allows cancer cells to persist despite treatment and contributes to treatment resistance. ANT adenine nucleotide translocator, MOMP mitochondrial outer membrane permeabilization, MPT mitochondrial permeability transition, ATF4 activating transcription factor 4, HRI heme-regulated inhibitor/EIF2AK1, eukaryotic translation initiation factor 2 alpha kinase 1.
3) Plasmalogenase activity. Plasmalogens, phospholipids with a vinyl ether bond at the sn-1 position, are vital for cellular signaling. In the presence of cardiolipin, O2, and H2O2, cytochrome c oxidizes the plasmalogen vinyl ether linkage, facilitating its hydrolytic cleavage leading to lipid peroxidation [140, 141]. Cytochrome c, when released from myocardial mitochondria, can cleave the vinyl ether bond of plasmenylcholine and plasmenylethanolamine in membrane bilayers [140, 141]. Inhibiting cytochrome c-mediated plasmalogen degradation using a specific monoclonal antibody targeting cytochrome c has been proposed as a potential Alzheimer’s disease therapy [140]. Further research is to determine whether cytochrome c functions as a plasmalogenase to a significant degree under normal physiological conditions or in the context of excessive stress. Additionally, the exploration of any potential therapeutic applications indicated by this study will necessitate more in-depth investigations.
4) Pro-survival functions in drug-persisting cells. The translocation of cytochrome c to the cytosol can also result in a pro-survival signal, assisting cancer cells in developing a persister phenotype during treatment with BH3 mimetics, both in vitro and in vivo. This process is triggered by sublethal cytochrome c release, which activates heme-regulated inhibitor kinase (HRI or eukaryotic translation initiation factor 2 alpha kinase 1 [EIF2AK1]) through physical interaction, initiates the integrated stress response (ISR), and induces translational reprogramming. This reprogramming suppresses global translation while selectively enabling the translation of activating transcription factor 4 (ATF4), a key driver of the persister phenotype (Fig. 4) [31]. Additional research is required to uncover the extent and importance of this effect.
Cytochrome c translocation to the nucleus
Mitochondrial cytochrome c translocates into the nucleus upon DNA damage [142], initiating cellular responses like chromatin condensation, chromosome reorganization, and gene transcription regulation. This effect was independent of caspase activation [142]. Nuclear cytochrome c directly binds and inhibits histone chaperone SET/template-activating factor-Iβ (TAF-Iβ; Table 3) during DNA damage, and thus hinders SET/TAF-Iβ nucleosome assembly activity [143]. Cytochrome c can directly bind to nucleolar nucleophosmin (NPM) by triggering a conformational change, driving alternative reading frame (ARF) release in response to DNA damage. NPM engages in liquid-liquid phase separation through heterotypic interactions with lysine-rich motifs of cytochrome c [33]. These motifs facilitate the formation of nucleolar-like droplets when combined with NPM. As a result, this intricate complex regulates the movement and accessibility of nucleolar proteins upon DNA damage [32]. Nuclear cytochrome c has a cytotoxicity threshold; low-level DNA damage promotes nuclear translocation for enhanced nucleosome assembly and DNA repair, while persistent damage surpasses this threshold, causing cytochrome c to obstruct DNA remodeling [144].
Cytochrome c release into the extracellular space
Cytochrome c can be released extracellularly by damaged or dying cells and acts as a signaling molecule possibly alerting nearby cells to tissue damage. Elevated serum cytochrome c levels have been noted in various diseases including inflammatory arthritis, myocardial infarction, and liver diseases [145]. It’s also a potential marker of mitochondrial injury post heart failure resuscitation, snakebite envenomation, and chemotherapy [146,147,148]. The released cytochrome c can trigger immune responses in astrocytes via toll-like receptor 4 (TLR4) interaction [34, 35] (Fig. 3), suggesting that inhibiting the cytochrome c-TLR4 pathway could mitigate inflammation caused by cell death. It should be noted that the opposite way of action has also been reported. Intravenous cytochrome c injection in septic mice restored cytochrome c oxidase activity in failing myocardium, significantly improving survival [149].
Role of cytochrome c in diseases
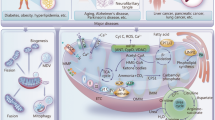
In addition to its physiological functions, abnormal cytochrome c signaling is implicated in various pathological conditions and diseases. Dysregulated cytochrome c expression, release, and mutations are closely associated with human diseases (Fig. 5A).

A Cytochrome c has been implicated in various human diseases, including neurodegenerative diseases, cardiovascular diseases, inflammatory diseases, and cancer. It is also relevant to treatments such as anti-retroviral therapy and cancer treatment. Furthermore, methodologies have been developed for detecting cytochrome c in serum, aiding in disease diagnosis and monitoring. Traditional techniques such as enzyme-linked immunosorbent assays (ELISAs), western blots, high-performance liquid chromatography (HPLC), immunocytochemistry (IC), flow cytometry (FCM), surface plasmon resonance (SPR) and biosensors could potentially be used to detect and analyze cytochrome c. B Biosensors offer a valuable tool for monitoring mitochondria damage or therapeutic interventions that induce cell death in tumors, resulting in increased levels of cytochrome c release that can be found in the bloodstream. These biosensors consist of three main components: (a) Bioreceptor: The bioreceptor employs capture probes such as aptamers, enzymes, or antibodies specifically designed for sensing cytochrome c. The electrode surfaces are modified with gold nanoparticles (AuNPs), carbon nanotubes (CNTs), quantum dots, trypsin, enzymes (e.g., cytochrome c oxidase [CcO] and cytochrome c reductase [CcR]), antibodies, or aptamers. (b) Detector Element or Transducer System: This component of the biosensor utilizes various techniques such as square wave voltammetry, electrochemistry, fluorescence, differential pulse voltammetry, interferometric reflectance spectroscopy, or surface-enhanced Raman scattering (SERS) to detect and measure the presence of cytochrome c. (c) Reader Device: The biosensor is accompanied by a reader device that enables the analysis and interpretation of the detected signals. The specifications of these biosensors include high sensitivity, rapid response times, low cost, multiplexing capability (i.e., simultaneous detection of multiple targets), and miniaturization for portability and ease of use. By incorporating these components and meeting the specified requirements, cytochrome c biosensors provide a valuable analytical tool for sensitive, rapid, and cost-effective detection and quantification of cytochrome c in serum. C The strategies involve specific property designs to enhance targeted delivery and tailor the platform for cytochrome c delivery and include: (a) Incorporation of targeting molecules on the NP surface: Antibodies (e.g., HER-2 antibody), toxins, peptides, aptamers, and ligands (e.g., transferrin) can be attached to the NP surface, facilitating specific and efficient NP uptake by cancer cells. (b) Utilization of therapeutic delivery systems: NP-based delivery systems coated with ligands that recognize cancer cells through surface markers. cytochrome c can be encapsulated inside the NP/nanogel or bound to its surface. (c) Implementation of linkers: A linker is employed to connect the cytochrome c-conjugated protein or peptide to the NP. For example, a redox-sensitive linker containing a disulfide bond can be used, as the disulfide bonds are cleaved within the reductive chemical environment of the cancer cell cytoplasm, leading to the release of cytochrome c. (d) Enhancement of cytochrome c stability: Methods such as PEGylation, ionic liquids, B-DNA, or conjugation with proteins or peptides can be employed to improve the stability of cytochrome c. PEGylation involves the conjugation of poly(ethylene glycol) (PEG) to drugs or nanoparticles, increasing their circulation time and reducing unwanted host responses. By incorporating these strategies, precision NPs can be developed to effectively deliver cytochrome c to cancer cells, enabling targeted therapeutic interventions.
Mutations of CYCS in human thrombocytopenia
Mutations in CYCS have been identified in thrombocytopenia, which is a heterogeneous group of inherited diseases characterized by low platelet counts. Three specific CYCS mutations have been described: G41S [150], Y48H [151], and A51V [152, 153]. Another newly identified CYCS mutation, Lys101del, is associated with bleeding tendencies and platelet aggregation defects [154]. Large-scale sequencing studies have also identified multiple previously unknown CYCS mutations, with potential clinical significance for the mutation of Asn52 based on its interaction with cardiolipin [58, 155].
There are contradictory reports suggesting that the G41S mutant exhibits elevated peroxidase activity without affecting the strength of the Met80-Fe bond [45, 156]. Despite the increased peroxidase activity, the mutant does not display heightened affinity for cardiolipin or increased release to the cytoplasm during apoptosis stimulation [157]. The Y48H variant of CYCS in thrombocytopenia exhibits a 30%-40% reduction in oxygen consumption and an increase in apoptotic activity. In vitro studies have shown that megakaryocytes from patients with thrombocytopenia can produce proplatelets and release platelets with normal microtubule coil formation [151]. The A51V variant of CYCS in thrombocytopenia displays increased accessibility to non-native conformers and exhibits 6- to 15-fold higher peroxidase activity [153]. So far, there is no explanation as to why these mutations cause such a specific yet mild phenotype and why there are so few known variants. This may be due to the essential roles of cytochrome c in the electron transport chain and apoptosis initiation, together with a high degree of early evolutionary optimization and conservation, likely restricting the occurrence of a broader range of mutations. The impact of CYCS mutations on other human diseases remains unknown.
Cytochrome c as a biomarker in diseases
Upon release into extracellular space, cytochrome c serves as a novel in vivo marker for mitochondrial injury following heart failure and chemotherapy. Circulating cytochrome c is associated with anti-retroviral-induced toxicity in HIV patients [158, 159]. Elevated serum cytochrome c levels are significantly related to the occurrence of nephropathy in myocardial infarction patients treated with percutaneous coronary intervention [160]. In patients with advanced non–small-cell lung cancer (NSCLC), significantly lower serum cytochrome c levels were observed, which correlates with poor survival outcomes. In contrast, patients with higher serum cytochrome c levels showed better responses to chemotherapy, indicating that serum cytochrome c levels may serve as an indicator of severity and prognosis in NSCLC patients [161]. Similarly, breast cancer patients exhibit increased serum cytochrome c levels in response to chemotherapeutic agents [162]. Intracellular cytochrome c deficiency leads to apoptosome and mitochondrial dysfunction in African American men with prostate cancer [163]. Furthermore, a study established three gene models (mRNA of T-box transcription factor 21 [TBX21], TGFB-induced factor homeobox 2 [TGIF2], and cytochrome c) to classify breast cancer patients and predict recurrence. The study showed that mortality was negatively correlated with the expression value of TBX21 and TGIF2 but positively correlated with cytochrome c, emphasizing the high possibility of recurrence with high cytochrome c expression [164].
Cytochrome c detection in diseases
Traditional techniques such as enzyme-linked immunosorbent assays (ELISAs) [161], western blots, high-performance liquid chromatography (HPLC), immunocytochemistry (IC) [165], and flow cytometry (FCM) [166] have been used to detect intracellular and extracellular levels of cytochrome c (Fig. 5). Researchers are focusing on the development of sensitive and cost-effective biosensors for medical technologies that can diagnose clinical abnormalities. A biosensor for cytochrome c detection and quantification in human serum consists of three components (Fig. 5B). 1) The bioreceptor utilizes capture probes such as aptamers, enzymes, and antibodies specifically designed for sensing cytochrome c [167, 168]. The electrode surfaces are modified with gold nanoparticles (AuNPs), carbon nanotubes (CNTs), quantum dots, trypsin, enzymes, antibodies, or aptamers [169,170,171,172]. 2) The detector element or transducer system employs various techniques including square wave voltammetry, electrochemistry, fluorescence, differential pulse voltammetry, interferometric reflectance spectroscopy, and surface-enhanced Raman scattering (SERS) to facilitate detection and quantification [168, 173, 174]. 3) The reader device is responsible for capturing and analyzing the signals generated by the biosensor system. In addition, quantitative photo-crosslinking mass spectrometry or photon counting histograms can distinguish different conformers of cytochrome c [175, 176]. These technologies also provide a viable platform for cell-based screening of compounds that induces cytochrome c release [177].
Targeting cytochrome c in therapeutics
1) Inhibiting cytochrome c-mediated apoptosis to limit mitochondria injury and therapy toxicity. Several approaches have been explored to inhibit cytochrome c-mediated apoptosis. HIV protease inhibitors (nelfinavir and ritonavir) prevent apoptosis in photoreceptor cells by inhibiting MOMP and subsequent cytochrome c release [178]. Inhibiting MPT by cyclosporin A or decylubiquinone also reduces cytochrome c-mediated apoptosis, which can be beneficial for patients with ischemic respiratory depression [179, 180]. Synuclein prevents neuronal apoptosis by covalently hetero-oligomerization with cytochrome c in Parkinson’s disease [181]. Additionally, blocking the interaction between cytochrome c and ITPR1 shows promise in treating apoptosis-associated disorders [182].
2) Limiting cytochrome c-mediated oxidative stress in oxidative-associated disease. Cytochrome c administration has shown potential in limiting oxidative stress and lipid peroxidation in preclinical models. In rat resuscitation and hemorrhagic shock models, cytochrome c injection has been found to prevent liver oxidative damage and reduce oxidative stress and acidosis [183]. On the other hand, cytochrome c can cleave plasmenylcholine in membrane bilayers, a process that may be targeted with specific antibodies to benefit patients with conditions like Alzheimer’s disease [140]. In addition, artificial cytochrome c mimics have been developed for reducing highly toxic pollutants by utilizing cytochrome c’s bioreduction ability [184].
3) Promoting cytochrome c-mediated apoptosis in cancer therapy. Selective induction of apoptosis in tumor cells while sparing normal cells is a key strategy in cancer treatment. Various approaches have been explored, including small-molecule drugs, natural compounds, and nanocarrier-mediated delivery of multiple anticancer drugs [185]. For example, artemisinin and berberine can induce cytochrome c-mediated apoptosis to suppress cancer growth [186, 187]. Precision nanoparticles have been engineered for targeted delivery of cytochrome c in cancer cells (Fig. 5C) and four specific properties have been designed to tailor this platform: a) utilizing targeting molecules [188,189,190], such as antibodies [185, 189], prodrugs [174, 188], peptides, aptamers, and ligands (e.g., transferrin [191, 192]); b) different therapeutic delivery systems [193, 194] through cytochrome c encapsulation or binding to nanoparticles/nanogels [190, 195]; c) the linker systems, such as redox-sensitive linker, whose disulfide bonds are cleaved within the reductive or ROS-rich microenvironment, or a pH-sensitive agent in acidic lysosome microenvironment, or thermo-responsive NGs [195, 196]; and d) improving cytochrome c stability using PEGylation [195], ionic liquids, B-DNA, conjugated peptides, or glycosylation that can increase cytochrome c stability and bioavailability [197, 198].
Conclusion and perspective
Over the past three decades, research on the structure and function of cytochrome c has generated continuous interest in understanding its roles in various physiological and pathological processes. The diverse functions of cytochrome c in cell death and redox modulation are intricately regulated by posttranslational modifications, interacting proteins as well as its localization in response to various stimuli. Despite advances in cytochrome c research, challenges remain. Key issues include identifying alternative cytochrome c conformations and to study their in vivo functions, understanding how cytochrome c mutations affect disease onset and progression, and establishing the link between cytochrome c mutations, apoptosis deficiency, and tumorigenesis. Additionally, distinguishing cytochrome c’s anti-oxidation and pro-oxidation functions, pinpointing the balance between its pro-survival and pro-death effects during treatment, and developing reliable biomarkers to evaluate mitochondrial cytochrome c release’s antagonistic functions are. Further exploring cytochrome c functional diversity may aid in creating specific inhibitors or activators for disease treatment.
References
Munn CAM, Foster MVI. Researches on myohamatin and the histohæmatins. Philos Trans R Soc Lond. 1886;177:267–98.
Keilin D. On cytochrome, a respiratory pigment, common to animals, yeast, and higher plants. Proc R Soc Lond B. 1925;98:312–39.
Nomenclature Committee of the International Union of Biochemistry (NC-IUB). Nomenclature of electron-transfer proteins. Recommendations 1989. J Biol Chem. 1992;267:665–77.
Reedy CJ, Gibney BR. Heme protein assemblies. Chem Rev. 2004;104:617–49.
Narisawa S, Hecht NB, Goldberg E, Boatright KM, Reed JC, Millán JL. Testis-specific cytochrome c-null mice produce functional sperm but undergo early testicular atrophy. Mol Cell Biol. 2002;22:5554–62.
Vitale I, Pietrocola F, Guilbaud E, Aaronson SA, Abrams JM, Adam D, et al. Apoptotic cell death in disease-Current understanding of the NCCD 2023. Cell Death Differ. 2023;30:1097–154.
Hüttemann M, Jaradat S, Grossman LI. Cytochrome c oxidase of mammals contains a testes-specific isoform of subunit VIb–the counterpart to testes-specific cytochrome c? Mol Reprod Dev. 2003;66:8–16.
Margoliash E. Primary structure and evolution of Cytochrome C. Proc Natl Acad Sci USA. 1963;50:672–9.
Scott RA. Cytochrome c: A multidisciplinary approach. Sausalito, Calif: AGM; 1996.
Li K, Li Y, Shelton JM, Richardson JA, Spencer E, Chen ZJ, et al. Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell. 2000;101:389–99.
Pinto M, Vempati UD, Diaz F, Peralta S, Moraes CT. Ablation of Cytochrome c in adult forebrain neurons impairs oxidative phosphorylation without detectable apoptosis. Mol Neurobiol. 2019;56:3722–35.
Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18:157–64.
Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63.
Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–13.
Wang X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001;15:2922–33.
Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–19.
Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem. 2004;73:87–106.
King LE, Hohorst L, Garcia-Saez AJ. Expanding roles of BCL-2 proteins in apoptosis execution and beyond. J Cell Sci. 2023;136:jcs260790.
Yu T, Wang X, Purring-Koch C, Wei Y, McLendon GL. A mutational epitope for cytochrome C binding to the apoptosis protease activation factor-1. J Biol Chem. 2001;276:13034–8.
Kluck RM, Ellerby LM, Ellerby HM, Naiem S, Yaffe MP, Margoliash E, et al. Determinants of cytochrome c pro-apoptotic activity. The role of lysine 72 trimethylation. J Biol Chem. 2000;275:16127–33.
Hao Z, Duncan GS, Chang CC, Elia A, Fang M, Wakeham A, et al. Specific ablation of the apoptotic functions of cytochrome C reveals a differential requirement for cytochrome C and Apaf-1 in apoptosis. Cell. 2005;121:579–91.
Xu W, Che Y, Zhang Q, Huang H, Ding C, Wang Y, et al. Apaf-1 Pyroptosome Senses mitochondrial permeability transition. Cell Metab. 2021;33:424–36.e410.
Izzo V, Bravo-San Pedro JM, Sica V, Kroemer G, Galluzzi L. Mitochondrial permeability transition: new findings and persisting uncertainties. Trends Cell Biol. 2016;26:655–67.
Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–62.
Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–8.
Javadov S, Jang S, Parodi-Rullán R, Khuchua Z, Kuznetsov AV. Mitochondrial permeability transition in cardiac ischemia–reperfusion: whether cyclophilin D is a viable target for cardioprotection? Cell Mol Life Sci. 2017;74:2795–813.
Karch J, Molkentin JD. Identifying the components of the elusive mitochondrial permeability transition pore. Proc Natl Acad Sci USA. 2014;111:10396–7.
Zhivotovsky B, Galluzzi L, Kepp O, Kroemer G. Adenine nucleotide translocase: a component of the phylogenetically conserved cell death machinery. Cell Death Differ. 2009;16:1419–25.
Carrer A, Tommasin L, Sileikyte J, Ciscato F, Filadi R, Urbani A, et al. Defining the molecular mechanisms of the mitochondrial permeability transition through genetic manipulation of F-ATP synthase. Nat Commun. 2021;12:4835.
Bernardi P, Gerle C, Halestrap AP, Jonas EA, Karch J, Mnatsakanyan N, et al. Identity, structure, and function of the mitochondrial permeability transition pore: controversies, consensus, recent advances, and future directions. Cell Death Differ. 2023;30:1869–85.
Kalkavan H, Chen MJ, Crawford JC, Quarato G, Fitzgerald P, Tait SWG, et al. Sublethal cytochrome c release generates drug-tolerant persister cells. Cell. 2022;185:3356–74.e3322.
González-Arzola K, Díaz-Quintana A, Bernardo-García N, Martínez-Fábregas J, Rivero-Rodríguez F, Casado-Combreras M, et al. Nucleus-translocated mitochondrial cytochrome c liberates nucleophosmin-sequestered ARF tumor suppressor by changing nucleolar liquid-liquid phase separation. Nat Struct Mol Biol. 2022;29:1024–36.
González-Arzola K, Guerra-Castellano A, Rivero-Rodríguez F, Casado-Combreras M, Pérez-Mejías G, Díaz-Quintana A, et al. Mitochondrial cytochrome c shot towards histone chaperone condensates in the nucleus. FEBS Open Bio. 2021;11:2418–40.
Gouveia A, Bajwa E, Klegeris A. Extracellular cytochrome c as an intercellular signaling molecule regulating microglial functions. Biochim Biophys Acta Gen Subj. 2017;1861:2274–81.
Wenzel TJ, Bajwa E, Klegeris A. Cytochrome c can be released into extracellular space and modulate functions of human astrocytes in a toll-like receptor 4-dependent manner. Biochim Biophys Acta Gen Subj. 2019;1863:129400.
Dickerson RE, Takano T, Eisenberg D, Kallai OB, Samson L, Cooper A, et al. Ferricytochrome c. I. General features of the horse and bonito proteins at 2.8 A resolution. J Biol Chem. 1971;246:1511–35.
Margoliash E, Smith EL. Amino-Acid Sequence of Horse Heart Cytochrome C : Peptides released by Digestion with Chymotrypsin. Nature. 1961;192:1121–3.
Imai M, Saio T, Kumeta H, Uchida T, Inagaki F, Ishimori K. Investigation of the redox-dependent modulation of structure and dynamics in human cytochrome c. Biochem Biophys Res Commun. 2016;469:978–84.
Stelter M, Melo AM, Pereira MM, Gomes CM, Hreggvidsson GO, Hjorleifsdottir S, et al. A novel type of monoheme cytochrome c: biochemical and structural characterization at 1.23 A resolution of rhodothermus marinus cytochrome c. Biochemistry. 2008;47:11953–63.
Zaidi S, Hassan MI, Islam A, Ahmad F. The role of key residues in structure, function, and stability of cytochrome-c. Cell Mol Life Sci. 2014;71:229–55.
Mavridou DA, Ferguson SJ, Stevens JM. Cytochrome c assembly. IUBMB Life. 2013;65:209–16.
Ferguson SJ, Stevens JM, Allen JW, Robertson IB. Cytochrome c assembly: a tale of ever increasing variation and mystery? Biochim Biophys Acta. 2008;1777:980–4.
Sinibaldi F, Mei G, Polticelli F, Piro MC, Howes BD, Smulevich G, et al. ATP specifically drives refolding of non-native conformations of cytochrome c. Protein Sci. 2005;14:1049–58.
Rajagopal BS, Edzuma AN, Hough MA, Blundell KL, Kagan VE, Kapralov AA, et al. The hydrogen-peroxide-induced radical behaviour in human cytochrome c-phospholipid complexes: implications for the enhanced pro-apoptotic activity of the G41S mutant. Biochem J. 2013;456:441–52.
Liptak MD, Fagerlund RD, Ledgerwood EC, Wilbanks SM, Bren KL. The Proapoptotic G41S mutation to human Cytochrome c alters the heme electronic structure and increases the electron self-exchange rate. J Am Chem Soc. 2011;133:1153–5.
Zhou M, Li Y, Hu Q, Bai XC, Huang W, Yan C, et al. Atomic structure of the apoptosome: mechanism of cytochrome c- and dATP-mediated activation of Apaf-1. Genes Dev. 2015;29:2349–61.
Kokhan O, Wraight CA, Tajkhorshid E. The binding interface of cytochrome c and cytochrome c(1) in the bc(1) complex: rationalizing the role of key residues. Biophys J. 2010;99:2647–56.
Shimada S, Shinzawa-Itoh K, Baba J, Aoe S, Shimada A, Yamashita E, et al. Complex structure of cytochrome c–cytochrome c oxidase reveals a novel protein–protein interaction mode. EMBO J. 2017;36:291–300.
Guo R, Zong S, Wu M, Gu J, Yang M. Architecture of human mitochondrial respiratory megacomplex I(2)III(2)IV(2). Cell. 2017;170:1247–57.e1212.
Koppenol WH, Margoliash E. The asymmetric distribution of charges on the surface of horse cytochrome c. Functional implications. J Biol Chem. 1982;257:4426–37.
Hannibal L, Tomasina F, Capdevila DA, Demicheli V, Tórtora V, Alvarez-Paggi D, et al. Alternative conformations of Cytochrome c: Structure, function, and detection. Biochemistry. 2016;55:407–28.
Elmer-Dixon MM, Bowler BE. Electrostatic constituents of the interaction of Cardiolipin with Site A of Cytochrome c. Biochemistry. 2018;57:5683–95.
Blouin C, Guillemette JG, Wallace CJ. Resolving the individual components of a pH-induced conformational change. Biophys J. 2001;81:2331–8.
Oviedo-Rouco S, Perez-Bertoldi JM, Spedalieri C, Castro MA, Tomasina F, Tortora V, et al. Electron transfer and conformational transitions of cytochrome c are modulated by the same dynamical features. Arch Biochem Biophys. 2020;680:108243.
Maity H, Rumbley JN, Englander SW. Functional role of a protein foldon–an Omega-loop foldon controls the alkaline transition in ferricytochrome c. Proteins. 2006;63:349–55.
Guerra-Castellano A, Márquez I, Pérez-Mejías G, Díaz-Quintana A, De la Rosa MA, Díaz-Moreno I. Post-translational modifications of cytochrome c in cell life and disease. Int J Mol Sci. 2020;21:8483.
Kalpage HA, Bazylianska V, Recanati MA, Fite A, Liu J, Wan J, et al. Tissue-specific regulation of cytochrome c by post-translational modifications: respiration, the mitochondrial membrane potential, ROS, and apoptosis. FASEB J. 2019;33:1540–53.
Alvarez-Paggi D, Hannibal L, Castro MA, Oviedo-Rouco S, Demicheli V, Tortora V, et al. Multifunctional Cytochrome c: Learning new tricks from an old dog. Chem Rev. 2017;117:13382–460.
Castro L, Eiserich JP, Sweeney S, Radi R, Freeman BA. Cytochrome c: a catalyst and target of nitrite-hydrogen peroxide-dependent protein nitration. Arch Biochem Biophys. 2004;421:99–107.
Garcia-Heredia JM, Diaz-Moreno I, Diaz-Quintana A, Orzaez M, Navarro JA, Hervas M, et al. Specific nitration of tyrosines 46 and 48 makes cytochrome c assemble a non-functional apoptosome. FEBS Lett. 2012;586:154–8.
Diaz-Moreno I, Garcia-Heredia JM, Diaz-Quintana A, Teixeira M, De la Rosa MA. Nitration of tyrosines 46 and 48 induces the specific degradation of cytochrome c upon change of the heme iron state to high-spin. Biochim Biophys Acta. 2011;1807:1616–23.
Garcia-Heredia JM, Diaz-Moreno I, Nieto PM, Orzaez M, Kocanis S, Teixeira M, et al. Nitration of tyrosine 74 prevents human cytochrome c to play a key role in apoptosis signaling by blocking caspase-9 activation. Biochim Biophys Acta. 2010;1797:981–93.
Lee I, Salomon AR, Yu K, Doan JW, Grossman LI, Hüttemann M. New prospects for an old enzyme: mammalian cytochrome c is tyrosine-phosphorylated in vivo. Biochemistry. 2006;45:9121–8.
Sanderson TH, Mahapatra G, Pecina P, Ji Q, Yu K, Sinkler C, et al. Cytochrome C is tyrosine 97 phosphorylated by neuroprotective insulin treatment. PLoS One. 2013;8:e78627.
Guerra-Castellano A, Diaz-Quintana A, Perez-Mejias G, Elena-Real CA, Gonzalez-Arzola K, Garcia-Maurino SM, et al. Oxidative stress is tightly regulated by cytochrome c phosphorylation and respirasome factors in mitochondria. Proc Natl Acad Sci USA. 2018;115:7955–60.
Yu H, Lee I, Salomon AR, Yu K, Hüttemann M. Mammalian liver cytochrome c is tyrosine-48 phosphorylated in vivo, inhibiting mitochondrial respiration. Biochim Biophys Acta. 2008;1777:1066–71.
Pecina P, Borisenko GG, Belikova NA, Tyurina YY, Pecinova A, Lee I, et al. Phosphomimetic substitution of cytochrome C tyrosine 48 decreases respiration and binding to cardiolipin and abolishes ability to trigger downstream caspase activation. Biochemistry. 2010;49:6705–14.
Gomila AMJ, Pérez-Mejías G, Nin-Hill A, Guerra-Castellano A, Casas-Ferrer L, Ortiz-Tescari S, et al. Phosphorylation disrupts long-distance electron transport in cytochrome c. Nat Commun. 2022;13:7100.
Garcia-Heredia JM, Diaz-Quintana A, Salzano M, Orzaez M, Perez-Paya E, Teixeira M, et al. Tyrosine phosphorylation turns alkaline transition into a biologically relevant process and makes human cytochrome c behave as an anti-apoptotic switch. J Biol Inorg Chem. 2011;16:1155–68.
Moreno-Beltran B, Guerra-Castellano A, Diaz-Quintana A, Del Conte R, Garcia-Maurino SM, Diaz-Moreno S, et al. Structural basis of mitochondrial dysfunction in response to cytochrome c phosphorylation at tyrosine 48. Proc Natl Acad Sci USA. 2017;114:E3041–E3050.
Zhao X, León IR, Bak S, Mogensen M, Wrzesinski K, Højlund K, et al. Phosphoproteome analysis of functional mitochondria isolated from resting human muscle reveals extensive phosphorylation of inner membrane protein complexes and enzymes. Mol Cell Proteom. 2011;10:M110.000299.
Kalpage HA, Wan J, Morse PT, Lee I, Hüttemann M. Brain-specific Serine-47 modification of Cytochrome c regulates Cytochrome c Oxidase activity attenuating ROS Production and Cell Death: Implications for Ischemia/Reperfusion Injury and Akt Signaling. Cells. 2020;9:1843.
Kalpage HA, Vaishnav A, Liu J, Varughese A, Wan J, Turner AA, et al. Serine-47 phosphorylation of cytochrome c in the mammalian brain regulates cytochrome c oxidase and caspase-3 activity. FASEB J. 2019;33:13503–14.
Mahapatra G, Varughese A, Ji Q, Lee I, Liu J, Vaishnav A, et al. Phosphorylation of Cytochrome c Threonine 28 regulates electron transport chain activity in kidney: implications for AMP kinase. J Biol Chem. 2017;292:64–79.
Wan J, Kalpage HA, Vaishnav A, Liu J, Lee I, Mahapatra G, et al. Regulation of respiration and apoptosis by Cytochrome c Threonine 58 phosphorylation. Sci Rep. 2019;9:15815.
Kalpage HA, Wan J, Morse PT, Zurek MP, Turner AA, Khobeir A, et al. Cytochrome c phosphorylation: Control of mitochondrial electron transport chain flux and apoptosis. Int J Biochem Cell Biol. 2020;121:105704.
Morse PT, Wan J, Bell J, Lee I, Goebel DJ, Malek MH, et al. Sometimes less is more: inhibitory infrared light during early reperfusion calms hyperactive mitochondria and suppresses reperfusion injury. Biochem Soc Trans. 2022;50:1377–88.
Morse PT, Perez-Mejias G, Wan J, Turner AA, Marquez I, Kalpage HA, et al. Cytochrome c lysine acetylation regulates cellular respiration and cell death in ischemic skeletal muscle. Nat Commun. 2023;14:4166.
Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–18.
Marquez I, Perez-Mejias G, Guerra-Castellano A, Olloqui-Sariego JL, Andreu R, Calvente JJ, et al. Structural and functional insights into lysine acetylation of cytochrome c using mimetic point mutants. FEBS Open Biol. 2021;11:3304–23.
Bazylianska V, Kalpage HA, Wan J, Vaishnav A, Mahapatra G, Turner AA, et al. Lysine 53 Acetylation of Cytochrome c in prostate cancer: warburg metabolism and evasion of apoptosis. Cells. 2021;10:802.
Wang Z, Ando Y, Nugraheni AD, Ren C, Nagao S, Hirota S. Self-oxidation of cytochrome c at methionine80 with molecular oxygen induced by cleavage of the Met-heme iron bond. Mol Biosyst. 2014;10:3130–7.
Uren RT, Dewson G, Bonzon C, Lithgow T, Newmeyer DD, Kluck RM. Mitochondrial release of pro-apoptotic proteins: electrostatic interactions can hold cytochrome c but not Smac/DIABLO to mitochondrial membranes. J Biol Chem. 2005;280:2266–74.
Pérez-Mejías G, Olloqui-Sariego JL, Guerra-Castellano A, Díaz-Quintana A, Calvente JJ, Andreu R, et al. Physical contact between cytochrome c(1) and cytochrome c increases the driving force for electron transfer. Biochim Biophys Acta Bioenerg. 2020;1861:148277.
Berndtsson J, Aufschnaiter A, Rathore S, Marin-Buera L, Dawitz H, Diessl J, et al. Respiratory supercomplexes enhance electron transport by decreasing cytochrome c diffusion distance. EMBO Rep. 2020;21:e51015.
Ferri T, Poscia A, Ascoli F, Santucci R. Direct electrochemical evidence for an equilibrium intermediate in the guanidine-induced unfolding of cytochrome c. Biochim Biophys Acta. 1996;1298:102–8.
Elmer-Dixon MM, Hoody J, Steele HBB, Becht DC, Bowler BE. Cardiolipin preferentially partitions to the inner leaflet of mixed lipid large unilamellar vesicles. J Phys Chem B. 2019;123:9111–22.
Kapralov AA, Yanamala N, Tyurina YY, Castro L, Samhan-Arias A, Vladimirov YA, et al. Topography of tyrosine residues and their involvement in peroxidation of polyunsaturated cardiolipin in cytochrome c/cardiolipin peroxidase complexes. Biochim Biophys Acta. 2011;1808:2147–55.
Elmer-Dixon MM, Xie Z, Alverson JB, Priestley ND, Bowler BE. Curvature-dependent binding of Cytochrome c to Cardiolipin. J Am Chem Soc. 2020;142:19532–9.
Parui PP, Sarakar Y, Majumder R, Das S, Yang H, Yasuhara K, et al. Determination of proton concentration at cardiolipin-containing membrane interfaces and its relation with the peroxidase activity of cytochrome c. Chem Sci. 2019;10:9140–51.
Zhu J, Jiang M, Ma H, Zhang H, Cheng W, Li J, et al. Redox-state-mediated regulation of Cytochrome c Release in apoptosis revealed by surface-enhanced raman scattering on nickel substrates. Angew Chem Int Ed Engl. 2019;58:16499–503.
Firsov AM, Kotova EA, Orlov VN, Antonenko YN, Skulachev VP. A mitochondria-targeted antioxidant can inhibit peroxidase activity of cytochrome c by detachment of the protein from liposomes. FEBS Lett. 2016;590:2836–43.
Jia B, Ye J, Gan L, Li R, Zhang M, Sun D, et al. Mitochondrial antioxidant SkQ1 decreases inflammation following hemorrhagic shock by protecting myocardial mitochondria. Front Physiol. 2022;13:1047909.
Velayutham M, Hemann C, Zweier JL. Removal of H2O2 and generation of superoxide radical: role of cytochrome c and NADH. Free Radic Biol Med. 2011;51:160–70.
Korshunov SS, Krasnikov BF, Pereverzev MO, Skulachev VP. The antioxidant functions of cytochrome c. FEBS Lett. 1999;462:192–8.
Sedlak E, Fabian M, Robinson NC, Musatov A. Ferricytochrome c protects mitochondrial cytochrome c oxidase against hydrogen peroxide-induced oxidative damage. Free Radic Biol Med. 2010;49:1574–81.
Belikova NA, Tyurina YY, Borisenko G, Tyurin V, Samhan Arias AK, Yanamala N, et al. Heterolytic reduction of fatty acid hydroperoxides by cytochrome c/cardiolipin complexes: antioxidant function in mitochondria. J Am Chem Soc. 2009;131:11288–9.
Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–33.
Meng H, Yamashita C, Shiba-Fukushima K, Inoshita T, Funayama M, Sato S, et al. Loss of Parkinson’s disease-associated protein CHCHD2 affects mitochondrial crista structure and destabilizes cytochrome c. Nat Commun. 2017;8:15500.
Oka T, Sayano T, Tamai S, Yokota S, Kato H, Fujii G, et al. Identification of a novel protein MICS1 that is involved in maintenance of mitochondrial morphology and apoptotic release of cytochrome c. Mol Biol Cell. 2008;19:2597–608.
Das R, Kumar A, Dalai R, Goswami C. Cytochrome C interacts with the pathogenic mutational hotspot region of TRPV4 and forms complexes that differ in mutation and metal ion-sensitive manner. Biochem Biophys Res Commun. 2022;611:172–8.
Carthy CM, Yanagawa B, Luo H, Granville DJ, Yang D, Cheung P, et al. Bcl-2 and Bcl-xL overexpression inhibits cytochrome c release, activation of multiple caspases, and virus release following coxsackievirus B3 infection. Virology. 2003;313:147–57.
Kharbanda S, Pandey P, Schofield L, Israels S, Roncinske R, Yoshida K, et al. Role for Bcl-xL as an inhibitor of cytosolic cytochrome C accumulation in DNA damage-induced apoptosis. Proc Natl Acad Sci USA. 1997;94:6939–42.
Mahajan NP, Linder K, Berry G, Gordon GW, Heim R, Herman B. Bcl-2 and Bax interactions in mitochondria probed with green fluorescent protein and fluorescence resonance energy transfer. Nat Biotechnol. 1998;16:547–52.
Yadaiah M, Rao PN, Harish P, Bhuyan AK. High affinity binding of Bcl-xL to cytochrome c: possible relevance for interception of translocated cytochrome c in apoptosis. Biochim Biophys Acta. 2007;1774:1370–9.
Zhang W, Zhang T, Chen Y. Simultaneous quantification of Cyt c interactions with HSP27 and Bcl-xL using molecularly imprinted polymers (MIPs) coupled with liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based targeted proteomics. J Proteom. 2019;192:188–95.
Bihlmaier K, Mesecke N, Terziyska N, Bien M, Hell K, Herrmann JM. The disulfide relay system of mitochondria is connected to the respiratory chain. J Cell Biol. 2007;179:389–95.
Farrell SR, Thorpe C. Augmenter of liver regeneration: a flavin-dependent sulfhydryl oxidase with cytochrome c reductase activity. Biochemistry. 2005;44:1532–41.
Martinez-Fabregas J, Diaz-Moreno I, Gonzalez-Arzola K, Janocha S, Navarro JA, Hervas M, et al. Structural and functional analysis of novel human cytochrome C targets in apoptosis. Mol Cell Proteom. 2014;13:1439–56.
Czabotar PE, Garcia-Saez AJ. Mechanisms of BCL-2 family proteins in mitochondrial apoptosis. Nat Rev Mol Cell Biol. 2023;24:732–48.
Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of Caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–90.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541.
Lartigue L, Kushnareva Y, Seong Y, Lin H, Faustin B, Newmeyer DD. Caspase-independent mitochondrial cell death results from loss of respiration, not cytotoxic protein release. Mol Biol Cell. 2009;20:4871–84.
Song X, Kim S-Y, Lee YJ. The role of Bcl-xL in synergistic induction of apoptosis by Mapatumumab and Oxaliplatin in combination with hyperthermia on human colon cancer. Mol Cancer Res. 2012;10:1567–79.
Moldoveanu T, Czabotar PE. BAX, BAK, and BOK: A coming of age for the BCL-2 family effector proteins. Cold Spring Harb Perspect Biol. 2020;12:a036319.
Puthalakath H, O’Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–49.
Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501.
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–90.
Fletcher JI, Meusburger S, Hawkins CJ, Riglar DT, Lee EF, Fairlie WD, et al. Apoptosis is triggered when prosurvival Bcl-2 proteins cannot restrain Bax. Proc Natl Acad Sci USA. 2008;105:18081–7.
Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–305.
Newton K, Strasser A, Kayagaki N, Dixit VM. Cell death. Cell. 2024;187:235–56.
O’Neill KL, Huang K, Zhang J, Chen Y, Luo X. Inactivation of prosurvival Bcl-2 proteins activates Bax/Bak through the outer mitochondrial membrane. Genes Dev. 2016;30:973–88.
Luo X, O’Neill KL, Huang K. The third model of Bax/Bak activation: a Bcl-2 family feud finally resolved? F1000Res. 2020;9:F1000.
Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, et al. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013;152:519–31.
Huang K, O’Neill KL, Li J, Zhou W, Han N, Pang X, et al. BH3-only proteins target BCL-xL/MCL-1, not BAX/BAK, to initiate apoptosis. Cell Res. 2019;29:942–52.
Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–9.
Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, et al. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67.
Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA. 2002;99:1259–63.
Riedl SJ, Salvesen GS. The apoptosome: signalling platform of cell death. Nat Rev Mol Cell Biol. 2007;8:405–13.
Cheng TC, Hong C, Akey IV, Yuan S, Akey CW. A near atomic structure of the active human apoptosome. Elife. 2016;5:e17755.
Yu X, Acehan D, Menetret JF, Booth CR, Ludtke SJ, Riedl SJ, et al. A structure of the human apoptosome at 12.8 A resolution provides insights into this cell death platform. Structure. 2005;13:1725–35.
Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ. 2006;13:1423–33.
Boehning D, Patterson RL, Sedaghat L, Glebova NO, Kurosaki T, Snyder SH. Cytochrome c binds to inositol (1,4,5) trisphosphate receptors, amplifying calcium-dependent apoptosis. Nat Cell Biol. 2003;5:1051–61.
Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, et al. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–75.
Gorman AM, Szegezdi E, Quigney DJ, Samali A. Hsp27 inhibits 6-hydroxydopamine-induced cytochrome c release and apoptosis in PC12 cells. Biochem Biophys Res Commun. 2005;327:801–10.
Zhou B, Zhang JY, Liu XS, Chen HZ, Ai YL, Cheng K, et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018;28:1171–85.
Yu J, Li S, Qi J, Chen Z, Wu Y, Guo J, et al. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019;10:193.
Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10:1689.
Shi CS, Kehrl JH. Cytochrome c Negatively Regulates NLRP3 Inflammasomes. PLoS One. 2016;11:e0167636.
Jenkins CM, Yang K, Liu G, Moon SH, Dilthey BG, Gross RW. Cytochrome c is an oxidative stress-activated plasmalogenase that cleaves plasmenylcholine and plasmenylethanolamine at the sn-1 vinyl ether linkage. J Biol Chem. 2018;293:8693–709.
Goldfine H. Cytochrome c takes on plasmalogen catabolism. J Biol Chem. 2018;293:8710–1.
Nur EKA, Gross SR, Pan Z, Balklava Z, Ma J, Liu LF. Nuclear translocation of cytochrome c during apoptosis. J Biol Chem. 2004;279:24911–4.
Gonzalez-Arzola K, Diaz-Moreno I, Cano-Gonzalez A, Diaz-Quintana A, Velazquez-Campoy A, Moreno-Beltran B, et al. Structural basis for inhibition of the histone chaperone activity of SET/TAF-Ibeta by cytochrome c. Proc Natl Acad Sci USA. 2015;112:9908–13.
Diaz-Moreno I, Velazquez-Cruz A, Curran-French S, Diaz-Quintana A, De la Rosa MA. Nuclear cytochrome c - a mitochondrial visitor regulating damaged chromatin dynamics. FEBS Lett. 2018;592:172–8.
Chimenti MS, Sunzini F, Fiorucci L, Botti E, Fonti GL, Conigliaro P, et al. Potential role of Cytochrome c and Tryptase in Psoriasis and Psoriatic Arthritis Pathogenesis: Focus on resistance to apoptosis and oxidative stress. Front Immunol. 2018;9:2363.
Au AK, Aneja RK, Bell MJ, Bayir H, Feldman K, Adelson PD, et al. Cerebrospinal fluid levels of high-mobility group box 1 and cytochrome C predict outcome after pediatric traumatic brain injury. J Neurotrauma. 2012;29:2013–21.
Zornetta I, Caccin P, Fernandez J, Lomonte B, Gutierrez JM, Montecucco C. Envenomations by Bothrops and Crotalus snakes induce the release of mitochondrial alarmins. PLoS Negl Trop Dis. 2012;6:e1526.
Adachi N, Hirota M, Hamaguchi M, Okamoto K, Watanabe K, Endo F. Serum cytochrome c level as a prognostic indicator in patients with systemic inflammatory response syndrome. Clin Chim Acta. 2004;342:127–36.
Piel DA, Deutschman CS, Levy RJ. Exogenous cytochrome C restores myocardial cytochrome oxidase activity into the late phase of sepsis. Shock. 2008;29:612–6.
Morison IM, Cramer Borde EM, Cheesman EJ, Cheong PL, Holyoake AJ, Fichelson S, et al. A mutation of human cytochrome c enhances the intrinsic apoptotic pathway but causes only thrombocytopenia. Nat Genet. 2008;40:387–9.
De Rocco D, Cerqua C, Goffrini P, Russo G, Pastore A, Meloni F, et al. Mutations of cytochrome c identified in patients with thrombocytopenia THC4 affect both apoptosis and cellular bioenergetics. Biochim Biophys Acta. 2014;1842:269–74.
Johnson B, Lowe GC, Futterer J, Lordkipanidzé M, MacDonald D, Simpson MA, et al. Whole exome sequencing identifies genetic variants in inherited thrombocytopenia with secondary qualitative function defects. Haematologica. 2016;101:1170–9.
Lei H, Bowler BE. Naturally occurring A51V variant of human Cytochrome c destabilizes the native state and enhances peroxidase activity. J Phys Chem B. 2019;123:8939–53.
Uchiyama Y, Yanagisawa K, Kunishima S, Shiina M, Ogawa Y, Nakashima M, et al. A novel CYCS mutation in the alpha-helix of the CYCS C-terminal domain causes non-syndromic thrombocytopenia. Clin Genet. 2018;94:548–53.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Karsisiotis AI, Deacon OM, Wilson MT, Macdonald C, Blumenschein TM, Moore GR, et al. Increased dynamics in the 40-57 Omega-loop of the G41S variant of human cytochrome c promote its pro-apoptotic conformation. Sci Rep. 2016;6:30447.
Josephs TM, Morison IM, Day CL, Wilbanks SM, Ledgerwood EC. Enhancing the peroxidase activity of cytochrome c by mutation of residue 41: implications for the peroxidase mechanism and cytochrome c release. Biochem J. 2014;458:259–65.
Langs-Barlow A, Selvaraj S, Ogbuagu O, Shabanova V, Shapiro ED, Paintsil E. Association of circulating cytochrome c with clinical manifestations of antiretroviral-induced toxicity. Mitochondrion. 2015;20:71–4.
Mensah EA, Sarfo B, Bonney EY, Parbie PK, Ocloo A. Symptoms of toxicity and plasma cytochrome c levels in human immunodeficiency virus-infected patients receiving anti-retroviral therapy in Ghana: A cross-sectional study. Infect Disord Drug Targets. 2020;20:88–97.
Tang C, Hou J, Yan G, Qiao Y, Wang D, Zhu B, et al. Effects of Serum Cytochrome c on contrast-induced nephropathy in patients with ST-elevation myocardial infarction undergoing percutaneous coronary intervention. Biomed Res Int. 2019;2019:9357203.
Javid J, Mir R, Julka PK, Ray PC, Saxena A. Extracellular cytochrome c as a biomarker for monitoring therapeutic efficacy and prognosis of non-small cell lung cancer patients. Tumour Biol. 2015;36:4253–60.
Kadam CY, Abhang SA. Serum levels of soluble Fas ligand, granzyme B and cytochrome c during adjuvant chemotherapy of breast cancer. Clin Chim Acta. 2015;438:98–102.
Kumar R, Bhat TA, Walsh EM, Chaudhary AK, O’Malley J, Rhim JS, et al. Cytochrome c deficiency confers Apoptosome and mitochondrial dysfunction in African-American men with prostate cancer. Cancer Res. 2019;79:1353–68.
Zhao S, Shen W, Du R, Luo X, Yu J, Zhou W, et al. Three inflammation-related genes could predict risk in prognosis and metastasis of patients with breast cancer. Cancer Med. 2019;8:593–605.
Crowley LC, Marfell BJ, Scott AP, Waterhouse NJ. Analysis of Cytochrome c release by immunocytochemistry. Cold Spring Harb Protoc. 2016;2016. https://doi.org/10.1101/pdb.prot087338.
Christensen ME, Jansen ES, Sanchez W, Waterhouse NJ. Flow cytometry based assays for the measurement of apoptosis-associated mitochondrial membrane depolarisation and cytochrome c release. Methods. 2013;61:138–45.
Pessoa J. Cytochrome c in cancer therapy and prognosis. Biosci Rep. 2022;42:BSR20222171.
Zhu L, Luo F, Li Z, Dai G, He P, Wang Q, et al. Selective detection of cytochrome C by microchip electrophoresis based on an aptamer strategy. Electrophoresis. 2019;40:1331–6.
Shamsipur M, Chabok A, Molaabasi F, Seyfoori A, Hajipour-Verdom B, Shojaedin-Givi B, et al. Label free phosphate functionalized semiconducting polymer dots for detection of iron(III) and cytochrome c with application to apoptosis imaging. Biosens Bioelectron. 2019;141:111337.
Ghayyem S, Faridbod F. A fluorescent aptamer/carbon dots based assay for Cytochrome c protein detection as a biomarker of cell apoptosis. Methods Appl Fluoresc. 2018;7:015005.
Zhu J, Chu H, Shen J, Wang C, Wei Y. Nitrogen and fluorine co-doped green fluorescence carbon dots as a label-free probe for determination of cytochrome c in serum and temperature sensing. J Colloid Interface Sci. 2021;586:683–91.
Amouzadeh Tabrizi M, Ferré-Borrull J, Marsal LF. Highly sensitive IRS based biosensor for the determination of cytochrome c as a cancer marker by using nanoporous anodic alumina modified with trypsin. Biosens Bioelectron. 2020;149:111828.
Manickam P, Kaushik A, Karunakaran C, Bhansali S. Recent advances in cytochrome c biosensing technologies. Biosens Bioelectron. 2017;87:654–68.
Han C, Xu X, Zhang C, Yan D, Liao S, Zhang C, et al. Cytochrome c light-up graphene oxide nanosensor for the targeted self-monitoring of mitochondria-mediated tumor cell death. Biosens Bioelectron. 2020;173:112791.
Muller F, Graziadei A, Rappsilber J. Quantitative photo-crosslinking Mass Spectrometry revealing protein structure response to environmental changes. Anal Chem. 2019;91:9041–8.
Perroud TD, Bokoch MP, Zare RN. Cytochrome c conformations resolved by the photon counting histogram: watching the alkaline transition with single-molecule sensitivity. Proc Natl Acad Sci USA. 2005;102:17570–5.
Zhang H, Zhang B, Di C, Ali MC, Chen J, Li Z, et al. Label-free fluorescence imaging of cytochrome c in living systems and anti-cancer drug screening with nitrogen doped carbon quantum dots. Nanoscale. 2018;10:5342–9.
Hisatomi T, Nakazawa T, Noda K, Almulki L, Miyahara S, Nakao S, et al. HIV protease inhibitors provide neuroprotection through inhibition of mitochondrial apoptosis in mice. J Clin Invest. 2008;118:2025–38.
Borutaite V, Jekabsone A, Morkuniene R, Brown GC. Inhibition of mitochondrial permeability transition prevents mitochondrial dysfunction, cytochrome c release and apoptosis induced by heart ischemia. J Mol Cell Cardiol. 2003;35:357–66.
Giacomotto J, Brouilly N, Walter L, Mariol MC, Berger J, Segalat L, et al. Chemical genetics unveils a key role of mitochondrial dynamics, cytochrome c release and IP3R activity in muscular dystrophy. Hum Mol Genet. 2013;22:4562–78.
Bayir H, Kapralov AA, Jiang J, Huang Z, Tyurina YY, Tyurin VA, et al. Peroxidase mechanism of lipid-dependent cross-linking of synuclein with cytochrome C: protection against apoptosis versus delayed oxidative stress in Parkinson disease. J Biol Chem. 2009;284:15951–69.
Boehning D, van Rossum DB, Patterson RL, Snyder SH. A peptide inhibitor of cytochrome c/inositol 1,4,5-trisphosphate receptor binding blocks intrinsic and extrinsic cell death pathways. Proc Natl Acad Sci USA. 2005;102:1466–71.
Powell RD, Goodenow DA, Mixer HV, McKillop IH, Evans SL. Cytochrome c limits oxidative stress and decreases acidosis in a rat model of hemorrhagic shock and reperfusion injury. J Trauma Acute Care Surg. 2017;82:35–41.
Chen Z, Liu X, Huang C, Li J, Shen X. Artificial Cytochrome c Mimics: Graphene Oxide-Fe(III) complex-coated molecularly imprinted colloidosomes for selective photoreduction of highly toxic pollutants. ACS Appl Mater Interfaces. 2020;12:6615–26.
Morales-Cruz M, Cruz-Montanez A, Figueroa CM, Gonzalez-Robles T, Davila J, Inyushin M, et al. Combining stimulus-triggered release and active targeting strategies improves cytotoxicity of cytochrome c nanoparticles in tumor cells. Mol Pharm. 2016;13:2844–54.
Laleve A, Panozzo C, Kuhl I, Bourand-Plantefol A, Ostojic J, Sissoko A, et al. Artemisinin and its derivatives target mitochondrial c-type cytochromes in yeast and human cells. Biochim Biophys Acta Mol Cell Res. 2020;1867:118661.
Zhao Y, Jing Z, Lv J, Zhang Z, Lin J, Cao X, et al. Berberine activates caspase-9/cytochrome c-mediated apoptosis to suppress triple-negative breast cancer cells in vitro and in vivo. Biomed Pharmacother. 2017;95:18–24.
Pei Y, Li M, Hou Y, Hu Y, Chu G, Dai L, et al. An autonomous tumor-targeted nanoprodrug for reactive oxygen species-activatable dual-cytochrome c/doxorubicin antitumor therapy. Nanoscale. 2018;10:11418–29.
Kucheryavykh YV, Davila J, Ortiz-Rivera J, Inyushin M, Almodovar L, Mayol M, et al. Targeted delivery of nanoparticulate Cytochrome C into Glioma cells through the proton-coupled targeted delivery of nanoparticulate Cytochrome C. Biomolecules. 2019;9:154.
Chen X, Zhu Q, Xu X, Shen S, Zhang Y, Mo R. Sequentially site-specific delivery of apoptotic protein and tumor-suppressor gene for combination cancer therapy. Small. 2019;15:e1902998.
Saxena M, Delgado Y, Sharma RK, Sharma S, Guzman S, Tinoco AD, et al. Inducing cell death in vitro in cancer cells by targeted delivery of cytochrome c via a transferrin conjugate. PLoS One. 2018;13:e0195542.
Macone A, Masciarelli S, Palombarini F, Quaglio D, Boffi A, Trabuco MC, et al. Ferritin nanovehicle for targeted delivery of cytochrome C to cancer cells. Sci Rep. 2019;9:11749.
Tollefson EJ, Allen CR, Chong G, Zhang X, Rozanov ND, Bautista A, et al. Preferential binding of Cytochrome c to anionic ligand-coated gold nanoparticles: a complementary computational and experimental approach. ACS Nano. 2019;13:6856–66.
Choi E, Lim DK, Kim S. Hydrolytic surface erosion of mesoporous silica nanoparticles for efficient intracellular delivery of cytochrome c. J Colloid Interface Sci. 2020;560:416–25.
Guo C, Zhang Y, Li Y, Zhang L, Jiang H, Tao J, et al. Gold nanoparticle-guarded large-pore mesoporous silica nanocomposites for delivery and controlled release of cytochrome c. J Colloid Interface Sci. 2021;589:34–44.
Miceli E, Wedepohl S, Osorio Blanco ER, Rimondino GN, Martinelli M, Strumia M, et al. Semi-interpenetrated, dendritic, dual-responsive nanogels with cytochrome c corona induce controlled apoptosis in HeLa cells. Eur J Pharm Biopharm. 2018;130:115–22.
Delgado Y, Morales-Cruz M, Hernandez-Roman J, Martinez Y, Griebenow K. Chemical glycosylation of cytochrome c improves physical and chemical protein stability. BMC Biochem. 2014;15:16.
Mendez J, Morales Cruz M, Delgado Y, Figueroa CM, Orellano EA, Morales M, et al. Delivery of chemically glycosylated cytochrome c immobilized in mesoporous silica nanoparticles induces apoptosis in HeLa cancer cells. Mol Pharm. 2014;11:102–11.
Rodriguez-Roldan V, Garcia-Heredia JM, Navarro JA, De la Rosa MA, Hervas M. Effect of nitration on the physicochemical and kinetic features of wild-type and monotyrosine mutants of human respiratory cytochrome c. Biochemistry. 2008;47:12371–9.
Karsisiotis AI, Deacon OM, Rajagopal BS, Macdonald C, Blumenschein TM, Moore GR, et al. Backbone resonance assignments of ferric human cytochrome c and the pro-apoptotic G41S mutant in the ferric and ferrous states. Biomol NMR Assign. 2015;9:415–9.
Godoy LC, Munoz-Pinedo C, Castro L, Cardaci S, Schonhoff CM, King M, et al. Disruption of the M80-Fe ligation stimulates the translocation of cytochrome c to the cytoplasm and nucleus in nonapoptotic cells. Proc Natl Acad Sci USA. 2009;106:2653–8.
Hirota S, Yamashiro N, Wang Z, Nagao S. Effect of methionine80 heme coordination on domain swapping of cytochrome c. J Biol Inorg Chem. 2017;22:705–12.
Yadav N, Gogada R, O’Malley J, Gundampati RK, Jayanthi S, Hashmi S, et al. Molecular insights on cytochrome c and nucleotide regulation of apoptosome function and its implication in cancer. Biochim Biophys Acta Mol Cell Res. 2020;1867:118573.
Chertkova RV, Sharonov GV, Feofanov AV, Bocharova OV, Latypov RF, Chernyak BV, et al. Proapoptotic activity of cytochrome c in living cells: effect of K72 substitutions and species differences. Mol Cell Biochem. 2008;314:85–93.
Nold SM, Lei H, Mou TC, Bowler BE. Effect of a K72A mutation on the structure, stability, dynamics, and peroxidase activity of human Cytochrome c. Biochemistry. 2017;56:3358–68.
Lei H, Nold SM, Motta LJ, Bowler BE. Effect of V83G and I81A substitutions to human Cytochrome c on acid unfolding and peroxidase activity below a neutral pH. Biochemistry. 2019;58:2921–33.
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89.
Boehning D, Patterson RL, Snyder SH. Apoptosis and calcium: new roles for cytochrome c and inositol 1,4,5-trisphosphate. Cell Cycle. 2004;3:252–4.
Saelens X, Kalai M, Vandenabeele P. Translation inhibition in apoptosis: caspase-dependent PKR activation and eIF2-alpha phosphorylation. J Biol Chem. 2001;276:41620–8.
Elena-Real CA, Diaz-Quintana A, Gonzalez-Arzola K, Velazquez-Campoy A, Orzaez M, Lopez-Rivas A, et al. Cytochrome c speeds up caspase cascade activation by blocking 14-3-3epsilon-dependent Apaf-1 inhibition. Cell Death Dis. 2018;9:365.
Duncan AM, Ozawa T, Suzuki H, Rozen R. Assignment of the gene for the cytochrome c1 subunit of the mitochondrial cytochrome bc1 complex (CYC1) to human chromosome 8q24.3. Genomics. 1994;19:400–1.
Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–57.
Acknowledgements
We thank Dave Primm (Department of Surgery, University of Texas Southwestern Medical Center) for his critical reading of the manuscript. We are thankful to Dr. Douglas Green (St. Jude Hospital) for his critical review and invaluable contributions to the manuscript.
Funding
Research by X.S. was partly supported by an American Cancer Society grant (IRG-21-142-16), Simmons Comprehensive Cancer Center support grant (P30CA142543), and the Elsa U. Pardee Foundation (project ID: 21005817). Research by D.T. and R.K. was supported by grants from the National Institutes of Health (NIH; R01CA160417, R01CA229275, and R01CA211070). Research by Y.L. was supported by the NIH (R01CA265827 and R21CA259243), and Department of Defense (RA210084). Research by M.H. was supported by the Office of the Assistant Secretary of Defense for Health Affairs through the Prostate Cancer Research Program (HT94252410073), NIH (R01NS120322 and U44NS125160), the Michigan Prostate SPORE Developmental Research Program, the National Science Foundation (MCB-2329629), and the Michigan Translational Research and Commercialization (MTRAC) Award. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the funding agencies, including the Department of Defense and NIH.
Author information
Authors and Affiliations
Contributions
ZZ, DT, and XS were involved in the conceptualization. All authors contributed to writing the manuscript. DT, MH, and XS further edited, reviewed, and approved the final manuscript for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhou, Z., Arroum, T., Luo, X. et al. Diverse functions of cytochrome c in cell death and disease. Cell Death Differ 31, 387–404 (2024). https://doi.org/10.1038/s41418-024-01284-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-024-01284-8
- Springer Nature Limited
This article is cited by
-
Comparative proteomics of oral squamous cell carcinoma tissue in consumers and non-consumers of tobacco
Journal of Proteins and Proteomics (2024)