
Abstract
Mitochondria are essential components of bioenergetic and biosynthetic pathways and are capable of making quick adjustments to accommodate the cell’s changing metabolic requirements. Mitochondria are dynamic organelles that conduct a variety of functions, including adenosine triphosphate synthesis, calcium homeostasis management, apoptosis, and population maintenance via fission and fusion. Mitochondrial malfunction manifested as oxidative stress and mutations can contribute to the aetiology of a variety of catastrophic disorders, including neurological disease, neuromuscular disease, ischemia-reperfusion damage, cancer, and the ageing process. This mitochondrial dysfunction prompted a thorough search for novel strategies that utilize critical facets of mitochondrial biogenesis, dynamics, mitophagy, redox metabolism, and the unfolded protein response to restore mitochondrial function. In the literature, from cardiovascular to neurodegenerative illnesses, targeting mitochondria as a therapeutic intervention to avoid cellular damage or cell death has been mentioned more frequently. For mitochondrial delivery currently, both passive and active targeting has been used. But due to the physical characteristics of mitochondria, passive targeting is challenging. In this review, we explore approaches and techniques for directing drugs and genetic materials to the mitochondria for mitochondrial dysfunction in the pathophysiology of various disorders associated with mitochondria.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Over a century ago, mitochondria were first identified and were initially termed as “bioblasts” by Richard Altmann, who described them as “elementary organisms” living inside cells and carrying out vital functions (Ernster and Schatz, 2009). Mitochondria, which are double membrane organelles having an intermembrane gap between their inner and outer membranes, are a crucial component of cellular metabolic processes. While the inner membrane functions as a tight diffusion barrier and is only permeable to tiny molecules to assist maintain the mitochondrial membrane potential across the membrane, the outer membrane is rich in lipids and is freely permeable. There are many cristae, which enhance the inner membrane surface area and are crucial for the mitochondria’s high respiratory activity (Zick et al., 2009). Mitochondria are susceptible to free radical damage because to their many copies of tiny (16.5 kb) circular DNA that lacks protective histones and their near proximity to ROS. The displacement-loop (D-loop), a 1.1 kb non-coding section of the DNA, comprises transcription and replication beginning sites for the mitochondria-specific polymerase-gamma (POLG) (Bebenek and Kunkel, 2004). 37 genes are encoded by mitochondrial DNA (mtDNA), comprising 22 tRNAs, two ribosomal RNAs (12S and 16S), and 13 electron transport chain (ETC) complex proteins. The cytosol produces the remaining hundreds of proteins needed for mitochondrial metabolism and maintenance, which are then transported into the mitochondria (Scarpulla, 2012). Mitochondrion, an membrane-bound organelle, hypothesized to be originated by endosymbiosis, are essential organelles for eukaryotes, performing key functions ranging from the generation of adenosine triphosphate (ATP) by oxidative phosphorylation (OXPHOS), generation and detoxification of reactive oxygen species (ROS) and thereby the regulation of cellular redox state; to the synthesis of nucleotides, Fe–S clusters, haem and amino acids, Fe2+/Ca2+, inflammation, and apoptosis (Malhi and Murthy, 2012; Osellame et al., 2012; Koliaki and Roden, 2016).
Mitochondrial illnesses are caused by inherited or spontaneous mutations in mtDNA or nuclear DNA (nDNA), which result in altered functioning of the proteins or RNA molecules that typically exist in mitochondria. Problems with mitochondrial function, on the other hand, may only impact certain tissues as a result of processes that occur throughout development and growth that we do not entirely comprehend. Even when tissue-specific isoforms of mitochondrial proteins are included, it is challenging to explain the clinically observed varied patterns of damaged organ systems in mitochondrial disease syndromes (Gorman et al., 2016; Zielonka et al., 2017). Genocopies are diseases that exhibit various symptoms yet result from the same mutation. On the other hand, illnesses are referred to be phenocopies when they have the same symptoms, but are brought on by mutations in separate genes. Leigh syndrome, which may be brought on by several distinct mutations, is an illustration of a phenocopy (Zori and Williams, 1991). Although the signs and symptoms of a mitochondrial illness can vary widely, they may include issues with vision or hearing, a loss of muscular coordination and weakness, neurodegenerative disease, learning impairments, etc. Parkinson’s disease, Alzheimer’s disease, bipolar disorder, schizophrenia, chronic fatigue syndrome, diabetes, neuromuscular disease, ischemia-reperfusion injury, cancer, as well as the aging process and other illnesses are also believed to entail some degree of mitochondrial malfunction (Mishra and Chan, 2016; Allouche et al., 2021). These intractable diseases have their roots in mtDNA defects have been increasing significantly and till now, more than 250 pathogenic mtDNA mutations have been identified. So, to treat these diseases, mitochondria have gained attention and are considered as the promising site for targeting various active compounds or genes. Mitochondria therapy can be obtained by targeting the carrier system containing a gene or drug to the mitochondria (Malhi and Murthy, 2012). The purpose of this review is to look over the possible and have an idea about the strategies to deliver drugs to mitochondria for treating mitochondrial associated diseases.
MITOCHONDRIAL DYSFUNCTION AND DISEASES
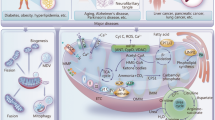
Human mitochondrial diseases result from mutations in either mtDNA genes or nDNA genes encoding the mitochondrial proteins required for aerobic ATP production, resulting in a change in the function of the proteins or RNA molecules in the matrix (Friedman and Nunnari, 2014). These mutations can either be inherited or spontaneous. Due to a germline genetic defect, the mitochondria become dysfunctional and are dispersed throughout the body. As a result, numerous cells and organs must endure the effects of decreased energy production (ATP), increased ROS production, lipid peroxidation, changes in membrane potential, and increased apoptotic signaling, which result in a number of disorders, as shown in Fig. 1. The cells that need high energy, like the heart, nerve, and muscle cells, suffer the most.

The underlying basic mechanism via which mitochondrial dysfunction results in several diseases.
The following section comes from the United Mitochondrial Disease Foundation:
(i) There are hundreds of distinct mitochondrial illnesses since mitochondria carry out various tasks in various organs. Because of the intricate interaction between the hundreds of genes and cells that must cooperate to keep our metabolic machinery running smoothly, it is a hallmark of mitochondrial diseases that identical mtDNA mutations may not produce similar conditions.
(ii) Numerous common polygenic diseases, such as cardiovascular, metabolic, neurodegenerative, neuromuscular diseases, and uncommon monogenic disorders, are all diseases caused by mitochondrial malfunction (Sorrentino et al., 2018).
(iii) If the germ line defect is sited in the pancreas, the β cells may slowly get destructed, resulting in low insulin production and ultimately the development of mitochondrial diabetes mellitus (mDM) (Karaa and The, 2014).
Some of the major diseases associated with mitochondrial dysfunction are mentioned below.
Cardiovascular Diseases
Cardiovascular disease (CVD) is the leading global cause of mortality in humans. Proper mitochondria functioning are mandatory to meet the heart’s high energy needs. Mitochondrial biogenesis and morphology are sensitive to the changing environment of the cardiac muscle cells and are controlled by fusion and fission proteins. Overexpression of fusion proteins, such as mitofusin 1 (Mfn1) and Mfn2 forms giant mitochondria that lead to structural changes in the mitochondria resulting in cardiac pathologies (Kraus and Cain, 1980). According to the annual (2019) report of the American Heart Association Research, adults in the US who had a CVD accounted for 121.5 million people. CVD, indexed as the basal cause of death, confers to around 840 678 deaths in the US in 2016, approximately 1 of every three deaths. Between 2013 and 2016, 121.5 million American adults lived with some form of CVD or the after-effects of stroke. Coronary Heart Disease was the leading contributor to CVD-related fatalities in 2016, accounting for 43.2% of cases in the US, followed by 16.9% of cases of stroke, 9.8% of High Blood Pressure, 9.3% of Heart Failure, 3.0% of arterial diseases, and 17.7% cases of other cardiovascular diseases (“Heart Disease and Stroke Statistics-2019 At-a-Glance,” 2019).
The etiology of CVD is complex and can lead to a progressive deterioration of mitochondria functions and structural integrity. Various metabolic abnormalities, excessive ROS production, energy depletion, autophagy deregulation, endoplasmic reticulum stress, and apoptosis activation are the consequences of CVD for functional abnormalities of cardiac mitochondria (Chistiakov et al., 2018). Deprivation of the mitochondrial membrane potential causes the activation of a number of signaling proteins that up-regulate many stress-responsive genes through retrograde signalling (Jazwinski, 2014). Excessive ROS are produced due to the uncoupling of the mitochondrial ETC from ATP production and causes extensive cell damage by oxidation of lipids and proteins. It also enhances the formation of fatty deposits in the arteries by boosting endothelial dysfunction, the pileup of oxidized low-density lipoprotein (LDL) in the arterial wall, vessel inflammation, development of initial lesions to plaque, and subsequent rupture of the plaque (Escribano-Lopez et al., 2009). In mitochondria, through ETC by OXPHOS mechanism synthesized the cellular ATP. The ATP content in the heart is relatively low because of immense ATP intake and rapid ATP output (Balaban and Kantor, 2016). During heart failure, the OXPHOS mechanism is impaired, and the ATP is further reduced, leading to poor cardiac performance (Ventura-Clapier et al., 2004). An alternative to glucose as a source of ATP production, lipids are used through the β-oxidation of fatty acids. Still, in heart failure, this insulin-dependent signaling process gets disrupted, resulting in hindrance in the β-oxidation process and subsequent compensation of energy (Neglia et al., 2020).
Cancer
The mtDNA mutation rate in cancer cells is an additional factor contributing to mitochondrial dysfunction (MDF). 90% of malignancies were found to include at least one point mutation that was absent in the matching normal cells, according to recent studies on the sequencing of mtDNA genomes. Numerous investigations found that mtDNA mutations had no appreciable effect on human patients with parathyroid, thyroid, glioblastoma, or adrenocortical carcinomas and that both healthy cells and malignant cells showed high levels of heteroplasmy. Numerous malignancies have been identified to contain somatic and germline mtDNA mutations. These include neuroblastomas and oncocytomas, renal adenocarcinoma, head and neck tumors, colon cancer cells, thyroid, ovarian, breast, breast, and astrocytic tumors (Brandon et al., 2006; Copeland et al., 2002). Oxidative stress (OS) and other endogenous mtDNA replication processes are a second component that links somatic mtDNA mutations to MDF. Given that mtDNA mutations can cause MDF, which leads to a build-up of additional ROS and the development of new mtDNA mutations, creating a vicious cycle (Lleonart et al., 2017). By transferring pathogenic or normal mtDNA to cancer cells, researchers have shown the importance of mtDNA in the disease, which results in modifications to the phenotypes of cancer cells (Petros et al., 2005; Ishikawa et al., 2008).
Neurodegenerative Diseases
Cell apoptosis can be triggered by mitochondrial oxidative stress damage caused by reactive oxygen species (ROS). Superoxide ion (\({\text{O}}_{2}^{ - }\)), hydrogen peroxide (H2O2), and hydroxyl radical (OH–) are three types of ROS that are primarily produced in the ETC when H+ escapes from complex I and complex III and interacts with dioxygen. The superoxide anions are catalyzed into oxygen and hydrogen peroxide by the superoxide dismutase (SODs) family, which includes the intracellular (SOD1) and manganese superoxide dismutase (MnSOD) or SOD2 or extracellular (SOD3) isoforms of copper/zinc SOD (Cu/ZnSOD). Then, glutathione peroxidase and catalase transform hydrogen peroxide into water, serving as an extra antioxidant defense (Murphy, 2009; Lubos et al., 2011). However, in conditions of significant ROS generation, the cell fails to protect itself and gradually acquires these toxic species (“Mighty Mitochondria and Neurodegenerative Diseases—Science in the News,” n.d.). It is one of the reasons for mitochondrial dysfunction and cell death, promoting the emergence and progression of neurological illnesses associated with aging. Damage from oxidative stress is brought on by the disparity between ROS and antioxidant defense systems (Wu et al., 2019). Parkinson’s disease (PD), Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), and Huntington’s disease (HD) are examples of neurodegenerative diseases that have links to mitochondrial dysfunction and mtDNA abnormalities (Sutong et al., 2021; Arun et al., 2015). Neurodegenerative illnesses can be treated by modulating the mitochondrial activity or altering the metabolites produced by mitochondria.
Diabetes
The production of ATP, carried out by mitochondria, is required for all living organisms. Additionally, mitochondria play a pivotal role in controlling how much insulin is secreted when glucose stimulates it in pancreatic β-cells (Wiederkehr and Wollheim, 2006). Over the last 20 years, a growing body of research has demonstrated how closely linked mitochondrial function is to various aspects of diabetes, including pancreatic β-cell failure, obesity, insulin resistance, and vascular implications of diabetes. In accordance with the WHO definition of diabetic Mellitus in 1980, the overwhelming majority of instances of diabetes fall into one of two groups of diabetes. Type 1 diabetes (T1D), also called insulin-dependent diabetes mellitus (IDDM), is the first group, and lack of insulin production characterizes it as the root cause of hyperglycemia. T1D’s complex etiology is caused by problems with beta cell control in addition to immune system dysregulation. The etiology of T1D may be influenced by mitochondrial dysfunction in beta cells and immune cells. The primary function of beta cells requires the generation of mitochondrial energy (insulin production in reaction to glucose). The generation of ROS occurs mainly in mitochondria. Mitochondrial ROS (mtROS) contributes to beta cell destruction during an immunological assault. Similarly, mitochondrial, morphology, physiology, and metabolism play a critical role in controlling T cell fate throughout immunological responses. The signaling process in the activation of antigen-specific T cells hinges on the generation of mtROS (Chen et al., 2018). The prevalence of this kind of diabetes in the general population is shown by the evidence of an autoimmune etiopathogenesis in pancreatic beta cells as the reason for reduced insulin output. From then, the trajectory continues downward until it reaches insulin insufficiency and, in the apparent absence of insulin therapy, a life-threatening and ketosis-prone condition.
The second type of diabetes, Type 2 diabetes (T2D) or Non-Insulin Dependent Diabetes Mellitus (NIDDM), is characterized by hyperlipidemia, hyperglycemia, and organismic insulin resistance. It is caused by the interaction of insulin resistance in insulin-sensitive tissues such as myocytes, hepatocytes, and adipocytes with pancreatic beta-cell dysfunction. This pathological alteration in circulating fuel levels and how central and peripheral tissues use energy substrates results in mitochondrial dysfunction throughout several organ systems. The International Diabetes Federation claims that type 2 diabetes mellitus (T2DM) now affects 451 million adults globally, ranging from 18 to 99 years old. If current trends continue, this number will rise to 693 million by 2045 (ID 2017). The significance of mitochondrial dysfunction is crucial in T2DM because of its documented relationship with insulin resistance (Fisher-Wellman and Neufer, 2012; Montgomery and Turner, 2014). ROS production, apoptosis, and the metabolism of energy substrates are all interconnected via the mitochondrion. The downstream deficiencies in critical processes, including hepatocyte metabolism, beta-cell insulin generation, cardiac output, skeletal muscle contraction, and neuronal health, are brought on by the imbalance of these systems in T2DM. The heap of mtDNA mutations and mtDNA copy count deficiency are aiding to explain the frequency of mitochondrial-related disorders like T2DM, even though mitochondria are famed to be vulnerable to a range of environmental and genetic insults. In addition, type 2 diabetes is a condition that worsens with time (US, 1998) (Sivitz and Yorek, 2010).
STRATEGIES TO TARGET MITOCHONDRIA
Current strategies for mitochondrial drug delivery include passive and active targeting. In the event of active targeting, certain interactions like ligand-receptor associations and antigen-antibody binding unfold at the sites of the mitochondria, which are further exploited and acquire utility of the affinity among the physiochemical properties of the carrier molecule (size, mass, hydrophilicity and electric charge) and the matrix of the mitochondria. Passive targeting of the mitochondria is challenging due to their physical characteristics; hence, active targeting procedures are currently being developed (Apostolova and Victor, 2015). Available strategies for mitochondrial targeting are as follows.
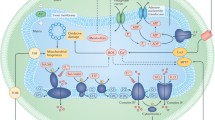
Targeting the Mitochondrial Electron Transport Chain
The mitochondrial ETC which is the fundamental facet in mitochondrial biological chemistry is ensconced in the inner mitochondrial membrane (IMM) comprising of four complexes that are concerned with OXPHOS (Yang et al., 2021). Certain substances, for instance, succinate which acquires increasingly positive redox potential besides NAD+/NADH have the ability to relocate electrons by dint of an individual complex (complex II). Coenzyme Q (ubiquinone) is adapted to disseminate across the membrane while cytochrome c is a highly water-soluble protein. The component (Fe–S) is extant in complexes I, II and III, and due to the Fe atom existing in the complexes aids in the transfer of the electron by deferring from Fe2+ to Fe3+ states, however, emanation of the electron may occur that results in the formation of superoxide (O2). In complex IV (cytochrome c oxidase) or cytochrome AA3, transfer of the electron takes place from cytochrome c to oxygen (O2) thereby, forming water and thus, help create a proton gradient which prompts the phosphorylation of adenosine diphosphate (ADP) (Kumari, 2018; Bhatti et al., 2021). The mitochondrial ETC besides the central component that generates the ATP and ROS, also render to be the central monitoring steps in apoptosis signalling (Trifunovic and Larsson, 2002; Qu et al., 2019). The deformity of complex I function in ETC implicated a sort of neurodegenerative disorders, inhibition of ATP productions, and thus generates extortionate ROS that leads to significant oxidative detriment (Bhatti et al., 2021). Various multisystem maladies in humans are attributable to mutations of mtDNA that intrude the function of one or more transfer RNA (tRNA) molecules that results in the mitochondrial translation impairment (Trifunovic and Larsson, 2002). As a consequence of this fact, proper functioning of the ETC is an obligatory as it stipulates energy in the regulation of cell death and survival processes (Bhatti et al., 2021). To retain ROS at adequate levels, cells generate antioxidant enzymes viz. Superoxide dismutase (SOD), catalase and glutathione peroxidase so as to avoid any damage of proteins, lipids or DNA, or oxidative injury (Pelicano et al., 2004). The obstruction of caspase actuation results in catalase degradation mediated by autophagy signifying the participation of autophagy in caspase-independent cell death, however, the function of ROS conceived from the mitochondrial ETC in autophagy-induced cell death remain uncharted. As per the study demonstrated by Chen et al., complex-I inhibitor rotenone and complex-II inhibitor thenoyl trifluoroacetone (TTFA) are capable of inducing autophagy, favourable for cell death in transformed and cancer cell lines which is arbitrated by ROS production. Contrarily, rotenone and TTFA failed to induce autophagy in primary mouse astrocytes, which alludes that the inhibition of mitochondrial complex I or complex II selectively induces autophagic cell death, in transformed and cancer cells mediated by ROS (Chen et al., 2007).
Targeting Transporters and Channels in Mitochondria
Transporters/channels in mitochondria serve as a key sensor and regulators for cellular redox signalling, the formation of ROS and nitrogen species in mitochondria, and reconciliation of cell survival and death. In both the outer and inner membranes of mitochondria, numbers of closely regulated ion channels are present. These pores due to their pharmacological modulation may promptly impact on the liberation of cytochrome c and thus emerge as a prominent oncological target. Apoptosis inducing chemotherapeutic drugs act by instigating detriment to DNA and stimulation of p53-dependent pathways that results to the relocation of pro-apoptotic Bcl-2 family members from the outer mitochondrial membrane (MOM) and subsequent MOM permeabilization (MOMP) resulting in the liberation of the cytochrome c from the intermembrane space of the mitochondria. However, by acting on the channels in mitochondria and particularly on the IMM create an opportunity to bypass p53-related events, hence, it is of importance to diverse classes of cancer develop chemo-resistance as a consequence of p53 mutations or anti-apoptotic protein upregulation or pro-apoptotic protein downregulation (Szabo et al., 2021). Besides targeting the inner and outer membrane, several proteins, ion channels, and transporters enclosed in the lipid membrane are targeted by the drugs as well. Some of the major drug targets are—lipophilic cations targeting the IMM (e.g., Rhodamine-123), carnitine palmitoyltransferase-1 (CPT-1) inhibitors (e.g., oxfenicine, perhexiline, and etomoxir), cardiolipin (CL) (e.g., 10-N-alkyl-arcine orange), Na+/Ca+2 exchanger regulators, IMM potassium channel regulators (e.g., glibencamide and diazoxide), B-cell lymphoma 2 (Bcl-2) protein inhibitors (e.g., Gossypol), and mitochondrial permeability transition MPT pore complex regulators (e.g., CsA). Some of the major drugs that are used for mitochondrial protein targets appertain to a group of proteins that mold the PTP complex over the MOM and IMM which is amenable for MPT and take the lead from both the survival and death signalling pathways. The actuation of MPT pore induces apoptosis and avert the demarcation of various tumour cells. Approach exhibited to prompt this effect typically imply activism detrimental to the MPT pore protein complex otherwise derivative action by impoverishing endogenous inhibitor of the complex MPT pore protein or augmenting calcium ions and ROS in the cytoplasm (Yang et al., 2021). On the contrary, CL, a negatively charged phospholipid, solely confined in the IMM asserts the architecture and membrane potential. However, diminution of CL content went hand in hand with mitochondrial detriment in myriad tissues, leading to assorted pathological conditions like—cardiac arrest, aging, and ischemia. Hence, with the purpose of overcoming the constraints, NAO (10-N-alkyl-arcine orange along with CL is pre-administered which is liberated responding to pro-apoptotic stimuli thus, abating the liberation of cytochrome c (Kumari, 2018; Bhatti et al., 2021).
Superoxide Dismutase (SOD) as a Target in Mitochondria
Mitochondria are the vital source of ROS in most mammalian cells. ROS include the hydrogen peroxide, superoxide anion, hydroxyl radical, and single oxygen (Kowluru et al., 2006). SOD’s are classified among the chief antioxidant enzymes in eukaryotes that offers a defensive mechanism against toxic oxygen metabolites that are induced by free radicals, thereby, catalysed the dismutation of superoxide anions to oxygen and hydrogen peroxide (H2O2). Appertaining to the literature survey, three different SOD isoforms have been reported to be present in mammals—(i) The copper/zinc SOD (Cu/ZnSOD) or SOD1, which is coded by the Sod1 gene is a cytosolic enzyme, (ii) The SOD2, which is coded by the Sod2 gene is a key antioxidant enzyme in the mitochondria, and, (iii) Extracellular copper/zinc SOD or SOD3, which is coded by the Sod3 gene is minor SOD and is only revealed in a handful of tissues such as kidney, lungs and fat tissues (Barry Halliwell, 1989). SODs besides being the only enzymes that relate to superoxide also function in directing the levels of ROS and reactive nitrogen species (RNS), and acts as a key regulator of signalling. Apart from acting as signalling molecules, at elevated levels, these reactive species can become noxious (Wang et al., 2018). Oxidative stress emerges because of the subtle balance between ROS generation and ROS scavenging in the mitochondria results in the neutralization of endogenous antioxidant systems, uncoupling of oxidative phosphorylation, impairment of the electron transport and transformed membrane permeability. The primary antioxidative enzyme, Mn-SOD, is accountable for scavenging O2 in the mitochondrial matrix, thereby, contributing to surplus production of highly reactive oxidants, such as ONOO– and OH– as a consequence of which the mitochondrial dysfunction and disease development ensues (Kitada et al., 2020). SODs and other antioxidant enzymes serve to illuminate the signalling functions of H2O2 and superoxide. For instance, the SOD1 inhibitor tetrathiomolybdate (ATN-224) preserves PTP from oxidation and, thereby, the growth factor–mediated phosphorylation of ERK1/2 present in the tumour and endothelial cells extenuates by which H2O2 arbitrates such effect in lieu of superoxide. Notably, ATN-224 has been proven to mount O2 and also enhance the intracellular H2O2 levels that are rendered to hint an eccentric role of SOD1 (Juarez et al., 2008). Marinho et al., claimed that overexpression of the intrinsic gene (human SOD1) in mouse NIH 3T3 fibroblasts modulates vascular endothelial growth factor expression, which is conceivably restrained by overexpression of human catalase (indicative of the H2O2 and its role) (Marinho et al., 2014). Analogously, Connor et al., also affirmed that overexpression of the SOD2 gene in human fibrosarcoma mitochondria results in inactivating the phosphatase and tensin homolog (PTEN) and successive activation of PI3K-Akt signalling pathway, however, overexpression of catalase in mitochondria or cytosol are adequate for shielding the cells against oxidative stress induced apoptosis (Connor et al., 2005).
Mitochondrial KATP Channels Target for Therapy
Exploitation of the patch clamp technique set the existence of the mitoK channels across the inner mitochondrial membrane (IMM) (Wrzosek et al., 2022). Throughout the IMM various potassium channels have been marked namely—ATP-sensitive potassium channels (mitoKATP), small (mitoSKCa), intermediate (mitoIKCa) and large conductance calcium activated potassium channels (mitoBKCa), voltage-dependent potassium Channels (mitoKv1.3 and mitoKv7.4), and sodium dependent potassium channels (mitoSlo2) respectively (Wrzosek et al., 2022). In particular mitoKATP channel is reasoned the amplest of the K+ channels within the IMM. The reason is that trafficking of K+ channels across the plasmalemma performs a key role in a number of ways in conjunction with the supervision of cardiac function, neuronal excitability, muscle contraction, neurotransmitter release, epithelial electrolyte transport, insulin secretion, and cell proliferation. Consequently, the plasma membrane K+ channels have been acknowledged as potential therapeutic drug monitoring till date. For instance, voltage-regulated potassium channels render an opening for the discovery of new drugs in the context of assorted diseases—autoimmune disease, cancer, metabolic, neurological and cardiovascular diseases (Wrzosek et al., 2020). Furthermore, mitoKATP channels are hindered by ATP, thus, embroiled as latent mediator of cardioprotective mechanisms, particularly ischemic preconditioning (IPC), further mediated by mitigating matrix Ca2+ overload and by augmented mitochondrial ROS generation in the course of precondition, expeditiously resulting to the activation of protein kinase C (PKC) and attenuated ROS generation in the course of reperfusion. Also, the mitoKATP is systematized by a set of ligands (examples are—acetylcholine, adenosine, bradykinin, opioids) that tie up the sarcolemmal G-protein-coupled receptors, with succeeding activation of tyrosine protein kinases, calcium flux and, the PI3K/Akt pathway. According to reports published, drugs (like diazoxide and nicorandil) trigger the initiation of the mitochondrial ATP-sensitive potassium channel (mitoKATP) and simultaneously capable of inhibiting H2O2-induced apoptotic advancement in cardiomyocytes, which appeared to indicate that mitoKATP channels may as well be of central concern in mediating oxidative-stress signals in the mitochondrial apoptotic pathway (Marín-García, 2014), and a victim of potassium channel openers obvious to relate with plasma membrane KATP channels (Szewczyk and Marbán, 1999). As a result, mitoKATP are up and coming targets in view of developing modern pharmacological tools to regulate their functions. But the application of such tools fetches a grave threat that these compounds relate to disparate targets within the cell which may be conducive to redundant effect or creating difficulties to discern which effects are cognate with mitoKATP (Wrzosek et al., 2022).
Conjugates of Lipophilic Cations
As it is known that mitochondria confer to many facets of cell function and breakdown; for instance, mitochondria being—the chief source of superoxide; acquiescent to oxidative damage favorable for mitochondrial dysfunction and necrosis in a series of degenerative disorders (Murphy, 2008) hence, liberating various pro-apoptotic factors from the intermembrane space (IMS) of the mitochondria (Murphy and Smith, 2007). Consequently, reduced antioxidant molecules (tractable and small) are compelled which are selectively absorbed by mitochondria and propel to specific organs that are affected most by mitochondrial oxidative damage, and prevent the occurrence of further oxidative damage (Murphy, 2008). Interestingly, various mitochondrial targeting moieties such as—guanidinium, pyridinium, triethylammonium, triphenylphosphonium (TPP), 3-phenylsulfonylfuroxan, Rhodamine 19, Rhodamine 123, dimethylbenzothiazolium iodide and DQA are at hand; and these agents are usually lipophilic cations (Battogtokh et al., 2018). Two facets of lipophilic cations improve their efficiency at liberating the desiring antioxidants to mitochondria by transcending promptly via phospholipid bilayers without the need of special absorbing mechanism and thus, aggregating substantially throughout the mitochondria due to large membrane potential. The intent of antioxidants to mitochondria by conjugating to a lipophilic cation (e.g. Triphenylphosphonium) is a fundamental strategy to tackle this problem (Murphy and Smith, 2007). Triphenylphosphonium (TPP) besides having a favorable and toxicity profile, effortlessly resist the biological membrane without the necessity of a protein carrier and also chemically tractable making it simple to initiate synthetically within the molecule to be oriented. Another crucial element of the TPP targeting system is its positive charge and the substantial mitochondrial membrane potential that results in the aggregation of molecules into the energized mitochondria about ~1000-fold, as stated by the Nernst equation (Pala et al., 2020). Various antioxidants have been exploited to target the mitochondria by coupling to the TPP moiety, this involves TPP-conjugated derivatives of tocopherol, lipoic acid, ubiquinone, spin traps and the peroxidase mimetic Ebselen (Murphy, 2008). MitoE2 was the first studied mitochondria targeted antioxidant, it comprehends the α-tocopherol moiety (an effective chain-breaking antioxidant) of vitamin E, conjugated by the agency of two-carbon chain to a TPP cation. The presence of α-tocopherol moiety aids in preventing lipid peroxidation, which leads to the formation of α-tocopheroxyl radical and subsequently reclaim by the endogenous mitochondrial coenzyme Q pool. MitoE2 was rapidly absorbed by secluded mitochondria making it more efficient at impeding lipid peroxidation besides an untargeted α-tocopherol. Further, this approach has also been used to target the ubiquinol moiety of coenzyme Q to mitochondria. Ubiquinol is the pick due to its chain-breaking antioxidant properties which can be further reclaimed by the respiratory chain. In this approach Ubiquinol moiety was linked by a ten-carbon bridge to the TPP cation to produce MitoQ or MitoQ10 (Murphy and Smith, 2007; Graham et al., 2009). MitoQ is rapidly absorbed by the secluded mitochondria lead by Δψ, and almost all the gathered MitoQ within mitochondria adsorbs to the matrix surface of the inner membrane; and by dint of complex II, MitoQ is limited to the active ubiquinol antioxidant in the respiratory chain, however, the substrate is awful for the electron transfer flavoprotein-ubiquinone oxidoreductase or complex I. Owing to the reduced and feeble oxidized form by complex III, MitoQ fails to rehabilitate respiration in mitochondria lacking coenzyme Q; therefore, all traces of MitoQ are likely to be because of the aggregation of the antioxidant Ubiquinol form. In addition, the Ubiquinol form of MitoQ may also function as an antioxidant and gets oxidized to the ubiquinone form which is further promptly reduced by complex II thus, rejuvenating the antioxidant effectivity. It is therefore essential due to the reclamation of an antioxidant reverting to its active form after having been nullified a ROS is a key element of the efficacy of numerous antioxidants. MitoQ is also found to detoxify peroxynitrite and respond towards superoxide whilst with disparate Ubiquinols, its responsiveness remains insignificant with hydrogen peroxide (James et al., 2005; Kelso et al., 2001). A fascinating facet of the application of mitochondria-targeted antioxidants is that they can be practiced to a variety of afflictions and organs as mitochondrial oxidative damage conduces to a great deal of maladies. Additionally, they can be adapted to be used in various injuries like—acute injuries (ischemia-reperfusion injury during surgery), semi-acute injuries (liver damage from steatohepatitis) and, chronic degenerative disease (Friedreich’s ataxia, Parkinson’s disease, or type II diabetes). Also, perhaps, be feasible to oversee these compounds prophylactically (Murphy and Smith 2007).
Mitochondria Targeted Peptides
As discussed above, targeting mitochondria has transpired out to be one of the novel therapeutic strategies. Another suitable technique for the mitochondrial specific delivery is the utilization of peptides which can penetrate the cell and the mitochondrial membrane, which have gained immense attention of the researchers can be classified on account of its numerous advantages. Based on the mode of action, MTPs may be sorted into four diverse groups comprising those that targets Bcl-2 family, elevated free radicals, overloaded calcium, and voltage-dependent anion channels (VDAC). Bcl-2 family proteins are crucial mitochondrial outer membrane permeabilization (MOMP) regulator and very critical in the intrinsic pathway of apoptosis. The Bcl-2 protein family comprises of pro-apoptotic or anti-apoptotic members of which pro-apoptotic members (specifically Bax and Bak) form pore to result in outer membrane permeabilization of the mitochondria and therapeutic targeting can be attained by using mimetic peptides (Malhi and Murthy, 2012; Farsinejad et al., 2015). Some of the anti-cancer peptides involves 99mTc-3PRGD2, NeuVax, Mistletoe, Gemcitabine, 177Lutetium-octreotate, which are under different phases of clinical trials (mostly Phase III and Phase IV Clinical trials (Chinnadrai et al., 2023).
Trans-Activator of Transcription (TAT) peptides obtained from HIV has a protein transduction domain (PTD) that has been utilized by Torchilin V. et al. for improved intracellular delivery of peptides, proteins and even drug carriers like 200-nm liposomes (Torchilin et al., 2001). However, there were certain obstruction in passing across the plasma membrane. So, this difficulty can be successfully countered through amalgamation PTD and mitochondrial targeting signal (MTS) to deliver exonuclease III protein, where the former helps in the cytoplasmic delivery and the latter helps in the mitochondrial intracellular trafficking (Shokolenko et al., 2005).
With further research in this aspect to make the delivery more efficient, many new findings were achieved. Few extremely cationic cell permeable peptides were found to respond to membrane potential (Horton et al., 2008). However, the process of cellular internalization and membrane permeation were difficult to understand (Malhi and Murthy, 2012). The mitochondria penetrating peptides (MPPs) constitute effective mitochondrial transporters that are convenient to make way across an organellar membrane that is laborious to penetrate (Wender et al., 2000). Improved cellular uptake and better proteolytic activity are achieved by certain modifications, such as changing the amide backbone for a peptide, ß-amide, carbamate, or alternative chemical scaffold (Malhi and Murthy, 2012).
Liposome-Based Carrier
Liposomes have gained immense interest as a novel drug carrier, having the potential to deliver both lipophilic and hydrophilic drugs, readily surface modifiable with ligands, biodegradable, biocompatible and their nontoxic nature. Mitochondrial specific drug targeting was first reported in 1998 to gene via liposomal cationic vesicles (DQAsomes) (Weissig et al., 1998).
Mitochondria-targeted liposomes may also consist of octaarginine (R8)-modified liposomes and cell-targeted liposomes are provided with a pH-dependent fusogenic peptide (Kakudo et al., 2004; Khalil et al., 2006).
An appropriate liposomal system for specific cell and mitochondrial targeting should preferentially consist of ligands for particular cell targeting, endosomal crossing and ultimately reaching the mitochondria. Enhanced endosomal escape can be attained on exposing the glutamic acid-alanine-leucine-alanine (GALA) peptide on the surface of the liposome. Also, the application of mitochondriotropic residues, such as TPP, on the surfaces of liposome can be their cause to be mitochondria specific (Boddapati et al., 2005, 2010). This lipophilic TPP cation furnish delocalized positive charge to the liposome, thereby reducing the free energy change for the liposome movement from hydrophilic to hydrophobic environment in regards to the mitochondrial membrane potential.
Yue C. and his team formulated IR-780 and Lonidamine encapsulated mitochondria-targeting thermosensitive liposomes (IL-TTSL). The IL-TTSL liposomes modified with triphenylphosphine were capable of targeting mitochondria and resulted in the decrease of the mitochondrial membrane potential because of the accumulation of liposomes in mitochondria. The stealth liposome showed promising results as a befitting technique for targeting mitochondria (Yue et al., 2017). Yanhong W. et al., in 2021 designed a novel dual functional liposomal system having potential extracellular charge reversal and also, specific mitochondrial targeting features to increase the accumulation of drug in mitochondria to activate cancer cell apoptosis. The charge reversion on Hyperoside loaded DSPE-Lys-DMA liposome system expedited the cellular internalization and mitochondrial accumulation for improved antitumor effect (Feng et al., 2021).
Mitochondria-Targeted Vesicles
Extracellular vesicles (EVs) are enclosed vesicles of phospholipid bilayers produced from all cell types in both pathological and physiological conditions and may be found in biological fluids such as blood, breast milk, cerebrospinal fluids, saliva and malignant ascites (Van Niel et al., 2018; Kalra et al., 2016; Igami et al., 2020; Amari and Germain, 2021). EVs have apparently become popular in the past few years as an intracellular conveying device for lipids, proteins, nucleic acids and even materials from other cellular compartments, which includes mitochondria (Chou et al., 2017; Shanmughapriya et al., 2020). There are various types of EVs, normally classified into three types depending on biogenesis: exosomes, micro-vesicles and apoptotic bodies (Igami et al., 2020). Exosomes were created at the time the limiting membrane of endosomes buds inward to form multivesicular bodies (MVBs). Exosomes are then discharged into the extracellular environment when MVBs fuse with the plasma membrane. Exosomes can interact with the extracellular matrix or provoke a reaction in cells in the microenvironment or at a distance after they are exempted from the cell surface (Alenquer and Amorim, 2015; Hessvik and Llorente, 2018). Micro-vesicles, on the other hand, are molded by the plasma membrane directly budding outward, resulting in a heterogeneous population of EVs. Apoptotic entities are also formed on the cell surface, but only dying cells releases them during cell fragmentation (Igami et al., 2020; Hessvik and Llorente, 2018; Bebelman et al., 2018). Exosomes are 40–120 nm in diameter, while micro-vesicles are 50–1000 nm in diameter (Zaborowski et al., 2015; Willms et al., 2018).
EVs transport a variety of biomolecules, including proteins, lipids, DNA, and a variety of RNA species (De Jong et al., 2019). EVs have characteristics that make them suitable for use as a therapeutic drug delivery system. EVs, for instance, transport and preserve a diverse range of nucleic acids and appear to be inherently capable of delivering them to target cells (Valadi et al., 2007). Another of their well-known characteristics is their capacity to escape the mononuclear phagocytic system by displaying the surface protein CD47 (De Jong et al., 2019). However, several contradictory findings have been noted, which could be associated with different EV sources and isolation methodologies (Elsharkasy et al., 2020).
Studies have demonstrated that EVs generated from particular progenitor cells carry biological cargo which facilitates angiogenesis, tissue healing, and immune function modulation (Turturici et al., 2014). As a result, these EVs seem to hold promise as a source of cellular treatment for a range of diseases, which may be taken advantage of by further modifying these EVs for therapeutic delivery. EVs have been demonstrated to readily traverse biological barriers, such as the blood-brain barrier (BBB), and produce functional alterations in target cells in multiple investigations (Cooper et al., 2014; Alvarez-Erviti et al., 2011; Liu et al., 2015; Lam et al., 2016).
Through a number of ligand/receptor interactions, EVs can relate with the plasma membrane on a cellular level (van Dongen et al., 2016). Consequently, EVs appear to be internalized more effectively than artificial and synthetic nanocarriers and use natural cellular endogenous processes, which may be helpful for drug delivery to intracellular sites (like mitochondrion) (Schindler et al., 2019; Millard et al., 2018). Furthermore, the basic mechanisms underlying EV intercellular transmission are yet unknown (Manickam, 2022).
Targeting through Mitochondria-Specific Bioactivation Reactions
An organelle-specific bioactivation reaction is another intriguing technique for selective mitochondrial transport of drugs. The availability of bioactivating enzymes in subcellular spaces like mitochondria that can be utilised to facilitate the transformation of a prodrug into a drug, underpins this technique. Mitochondrial monoamine oxidase B, cytochrome P450, and mitochondrial medium-chain acyl-CoA dehydrogenase are all promising candidates for regulated xenobiotic conversions. The mitochondrial β-oxidation enzymes catalyse the biotransformation of dietary and endogenously produced fatty acids, as well as a large list of xenobiotic alkanoic acids such tianeptine, chlorophenoxybutyric acid, and 5-hydroxydecanoic acid.
In the case of alkanoate-based prodrugs, mitochondrial targeting of antioxidants utilising mitochondrial β-oxidation has been investigated (Anders, 2011). The transporters and enzymes that make up the fatty acid ß-oxidation pathway for short and medium chain fatty acids are exclusively found in mitochondria, making the biotransformation of these compounds’ mitochondria-specific. Kurt S. Roser and his colleagues used the mitochondrial ß-oxidation pathway to deliver phenolic chain-breaking and thiol-based antioxidants to mitochondria (Roser et al., 2010).
Investigations of the biotransformation of x-(phenoxy)alkanoates, 3-(phenoxy)acrylates, and x-(1-methyl-1H-imidazol-2-ylthio)alkanoates are used to examine the possibilities of thia- and oxaalkanoate-based prodrugs. In a hypoxia reoxygenation paradigm of rat cardiomyocytes, 3- and 5-(1-methyl-1H-imidazol-2-ylthio)alkanoates were demonstrated to undergo fast biotransformation further into antioxidant methimazole and to provide a cytoprotective effect (Roser et al., 2010). 3-(2,6-Dimethylphenoxy)propanoic acid and 3-(2,6-dimethylphenoxy)acrylic acid similarly provided cytoprotectant in the same model. Etomoxir, an inhibitor of carnitine palmitoyl transferase I, the mitochondrial enzyme that allows these chemicals to enter the mitochondria, reduced the protective effect of these compounds. Importantly, albeit being described as an effective and adaptable ROS scavenger (Kim et al., 2001) methimazole ended in failure to provide cardiomyocyte cytoprotectant when provided alone under hypoxia-reoxygenation circumstances. This information is backed with the assumption that antioxidants must be supplied precisely to mitochondria in order to achieve the required “in situ” level to be active (Heidari et al., 2014). To confirm the therapeutic potential and associated chemicals in vivo, more study in this field is required.
Mn Porphyrin-Based Targeting
The class of compounds known as porphyrins consists of tetrapyrrole macrocycles with a 1D, 2D, or rarely 3D skeleton. The substitution of nitrogen atoms and other alternatives for the α, β and other carbon atoms of porphyrins can significantly change the properties of porphyrins as well as their biological applications (Tovmasyan et al., 2013). Initially created as potent mimics of the SOD family of enzymes, Mn(III) substituted pyridylporphyrins (collectively referred to as MnPs) were introduced. The porphyrin ligand’s macrocyclic structure ensures the integrity of the metal centre, where all reactions transpired. For this reason, nature has used the porphyrin ligand in a variety of proteins and enzymes, including myoglobin, haemoglobin, nitric oxide synthases and the cytochrome P450 family of enzymes. The porphyrin ligand possesses redox chemistry that is beyond the range that would have enabled it to react with biomolecules; also, the porphyrin ligand is a potent photosensitizer (Batinic-Haberle et al., 2021). The low molecular weight synthetic nonpeptides known as SOD mimics, or redox-active manganese (Mn) porphyrins, have demonstrated extraordinary therapeutic results in spinal cord, kidney and brain ischemia/reperfusion injuries, disorders that share oxidative stress with SCD. Interestingly, these cationic substances can pass the plasma membrane and function largely in the intracellular compartment because they are non-immunogenic. Mn porphyrins are not only superoxide dismutation scavengers but also participate in thiol signalling (Thamilarasan et al., 2020).
The majority of mammalian cells’ energy source is produced by mitochondria, which are essential organelles in mammalian cell organelles. They are also crucial in apoptosis and other forms of programmed cell death, and they are readily harmed by ROS production. The potential of the mitochondrial membrane elevated as the mitochondria became cancerous, leading to greater accumulation and persistence of cationic mitochondrial targeting agents in cancer cells as opposed to healthy cells (Tokarz and Blasiak, 2014). Therefore, the introduction of the lipophilic cations, such as triphenylphosphine, to the photosensitizer may be necessary to deliver it to the mitochondria of cancer cells as they can cause it to undergo passive diffusion and take advantage of the electrochemical potential existing between the inner and outer layers of the cell membrane. The cations’ ability to diffuse is made easier by the electrochemical potential energy, which improves the cations’ absorption by cells. Consequently, conjugating a photodynamic agent with a cationic mitochondrial targeting agent might hasten the destruction of cancer cells, enhance therapeutic results, and minimise needless side effects (Yang et al., 2019).
Clinical studies are now being conducted with two Mn(III) porphyrin analogues. It has been investigated if MnTnBuOE-2-PyP5+ (BMX-001) is effective for radio-protecting healthy tissue during the treatment of cancers of the head and neck (NCT02990468), anal (NCT0338650) and glioma (NCT02655601), as well as the normal brain in cancer patients with brain metastases (NCT03608020). It is significant to highlight that BMX-001 functions as a tumour radio- and chemosensitizer but does not shield cancer cells or malignant tissue. MnTE-2-PyP5+ (AEOL10113, BMX-010), a second porphyrin analogue, is being studied for a non-cancer use, a topical dermatitis and itch (NCT02457858) (Batinic-Haberle and Tome, 2019).
CONCLUSIONS
Cellular metabolic processes depend heavily on mitochondria. Devastating illnesses can arise as a result of mitochondrial malfunction, which compromises their structure and function, compromises ETC functioning, and increases ROS production. Therefore, mitochondrial malfunction, primarily in their metabolic functions, has been linked to a variety of human disorders, including cancer, ischemia-reperfusion damage, neurodegenerative diseases, neuromuscular diseases, and metabolic diseases. The distinct structural and functional properties of mitochondria allow for the targeted delivery of medications intended to change this organelle’s functionality for therapeutic purposes. Finding the best ways to attack mitochondria has advanced significantly. We have discussed various potential therapeutic targets for mitochondria. Despite the variety of potential strategies discussed in this article, the complexity of the mitochondrial homeostatic mechanisms highlights the fact that a scientifically sound approach to mitochondrial therapy is still in its infancy.
REFERENCES
Alenquer, M. and Amorim, M.J., Exosome biogenesis, regulation, and function in viral infection, Viruses, 2015, vol. 7, no. 9, pp. 5066–5083. https://doi.org/10.3390/v7092862
Allouche, S., Schaeffer, S., and Chapon, F., Mitochondrial diseases in adults: an update, Rev. Med. Intern., 2021, vol. 42, no. 8, pp. 541–557. https://doi.org/10.1016/j.revmed.2020.12.002
Alvarez-Erviti, L., Seow, Y., Yin, H., Betts, C., Lakhal, S., and Wood, M.J.A., Delivery of SiRNA to the mouse brain by systemic injection of targeted exos, Nat. Biotechnol., 2011, vol. 29, no. 4, pp. 341–345. https://doi.org/10.1038/nbt.1807
Amari, L. and Germain, M., Mitochondrial extracellular vesicles—origins and roles, Front. Mol. Neurosci., 2021, vol. 14, pp. 1–7. https://doi.org/10.3389/fnmol.2021.767219
Anders, M.W., Putting bioactivation reactions to work: targeting antioxidants to mitochondria, Chem.-Biol. Interact., 2011, vol. 192, no. 1–2, pp. 8–13. https://doi.org/10.1016/j.cbi.2010.10.004
Apostolova, N. and Victor, V.M., Molecular strategies for targeting antioxidants to mitochondria: therapeutic implications, Antioxid. Redox Signal., 2015, vol. 22, no. 8, pp. 686–729. https://doi.org/10.1089/ars.2014.5952
Arun, S., Liu, L., and Donmez, G., Mitochondrial biology and neurological diseases, Curr. Neuropharmacol., 2015, vol. 14, no. 2, pp. 143–154. https://doi.org/10.2174/1570159x13666150703154541
Batinic-Haberle, I. and Tome, M.E., Thiol regulation by Mn porphyrins, commonly known as SOD mimics, Redox Biol., 2019, vol. 25, p. 101139. https://doi.org/10.1016/j.redox.2019.101139
Batinic-Haberle, I., Tovmasyan, A., Huang, Z., Duan, W., Du Li, Siamakpour-Reihani, S., Cao, Z., Sheng, H., Spasojevic, I., and Alvarez, A. 2nd, H2O2-Driven anticancer activity of Mn porphyrins and the underlying molecular pathways, Oxid. Med. Cell. Longevity, 2021, pp. 1–25. https://doi.org/10.1155/2021/6653790
Battogtokh, G., Choi, Y.S., Kang, D.S., Park, S.J., Shim, M.S., Huh, K.M., Cho, Y.-Y., Lee, J.Y., Lee, H.S., and Kang, H.C., Mitochondria-targeting drug conjugates for cytotoxic, anti-oxidizing and sensing purposes: current strategies and future perspectives, Acta Pharm. Sin. B, 2018, vol. 8, no. 6, pp. 862–880. https://doi.org/10.1016/j.apsb.2018.05.006
Bebelman, M.P., Smit, M.J., Pegtel, D.M., and Baglio, S.R., Biogenesis and function of extracellular vesicles in cancer, Pharmacol. Ther., 2018, vol. 188, pp. 1–11. https://doi.org/10.1016/j.pharmthera.2018.02.013
Bebenek, K. and Kunkel, T.A., Functions of DNA polymerases, in DNA Repair and Replication, vol. 69, Advances in Protein Chemistry, Academic Press, 2004, pp. 137–165.
Bhatti, J.S., Pahwa, P., Reddy, P.H., and Bhatti, G.K., Impaired mitochondrial bioenergetics and signaling pathways, in Clinical Bioenergetics, Ostojic, S.B.T.-C.B., Ed., Elsevier, 2021, pp. 61–79.
Boddapati, S.V., Tongcharoensirikul, P., Hanson, R.N., D’Souza, G.G.M., Torchilin, V.P., and Weissig, V., Mitochondriotropic liposomes, J. Liposome Res., 2005, vol. 15, nos. 1–2, pp. 49–58. https://doi.org/10.1081/LPR-64958
Boddapati, S.V., D’Souza, G.G.M., and Weissig, V., Liposomes for drug delivery to mitochondria, Methods Mol. Biol. (Clifton, N.J.), 2010, vol. 605, pp. 295–303.
Brandon, M., Baldi, P., and Wallace, D.C., Mitochondrial mutations in cancer, Oncogene, 2006, vol. 25, no. 34, pp. 4647–4662. https://doi.org/10.1038/sj.onc.1209607
Chen, J., Stimpson, S.E., Fernandez-Bueno, G.A., and Mathews, C.E., Mitochondrial reactive oxygen species and type 1 diabetes, Antioxid. Redox Signal., 2018, vol. 29, no. 14, pp. 1361–1372. https://doi.org/10.1089/ars.2017.7346
Chen, Y., McMillan-Ward, E., Kong, J., Israels, S.J., and Gibson, S.B., Mitochondrial electron-transport-chain inhibitors of complexes I and II induce autophagic cell death mediated by reactive oxygen species, J. Cell Sci., 2007, vol. 120, no. 23, pp. 4155–4166. https://doi.org/10.1242/jcs.011163
Chinnadurai, R.K., Khan, N., Meghwanshi, G.K., Ponne, S., Althobiti, M., and Kumar, R., Current research status of anti-cancer peptides: mechanism of action, production, and clinical applications, Biomed. Pharmacother., 2023, vol. 164, p. 114996. https://doi.org/10.1016/j.biopha.2023.114996
Chistiakov, D.A., Shkurat, T.P., Melnichenko, A.A., Grechko, A.V., and Orekhov, A.N., The role of mitochondrial dysfunction in cardiovascular disease: a brief review, Ann. Med., 2018, vol. 50, no. 2, pp. 121–127. https://doi.org/10.1080/07853890.2017.1417631
Chou, S.H., Jing Lan, Y., Esposito, E., Ning, M.M., Balaj, L., Ji, X., Lo, E.H., and Hayakawa, K., Extracellular mitochondria in cerebrospinal fluid and neurological recovery after subarachnoid hemorrhage, Stroke, 2017, vol. 48, no. 8, pp. 2231–2237. https://doi.org/10.1161/STROKEAHA.117.017758
Connor, K.M., Subbaram, S., Regan, K.J., Nelson, K.K., Mazurkiewicz, J.E., Bartholomew, P.J., Aplin, A.E., Tai, Y.-T., Aguirre-Ghiso, J., Flores, S.C., and Andres Melendez, J., Mitochondrial H2O2 regulates the angiogenic phenotype via PTEN oxidation, J. Biol. Chem., 2005, vol. 280, no. 17, pp. 16916–16924. https://doi.org/10.1074/jbc.M410690200
Cooper, J.M., Wiklander, M P.B.O., Nordin, J.Z., Al-Shawi, R., Wood, M.J., Vithlani, M., Schapira, A.H.V., Simons, J.P., El-Andaloussi, S., and Alvarez-Erviti, L., Systemic Exosomal SiRNA Delivery Reduced Alpha-Synuclein Aggregates in Brains of Transgenic Mice, Movement Disord., 2014, vol. 29, no. 12, pp. 1476–1485. https://doi.org/10.1002/mds.25978
Copeland, W.C., Wachsman, J.T., Johnson, F.M., and Penta, J.S., Mitochondrial DNA alterations in cancer, Cancer Invest., 2002, vol. 20, no. 4, pp. 557–569. https://doi.org/10.1081/cnv-120002155
De, G., Ko, J.K., Tan, T., Zhu, H., Li, H., and Ma, J., Amphipathic tail-anchoring peptide is a promising therapeutic agent for prostate cancer treatment, Oncotarget, 2014, vol. 5, no. 17, pp. 7734–7747. https://doi.org/10.18632/oncotarget.2301
van Dongen, H.M., Masoumi, N., Witwer, K.W., and Pegtel, D.M., Extracellular vesicles exploit viral entry routes for cargo delivery, Microbiol. Mol. Biol. Rev., 2016, vol. 80, no. 2, pp. 369–386. https://doi.org/10.1128/mmbr.00063-15
Elsharkasy, O.M., Nordin, J.Z., Hagey, D.W., de Jong, O.G., Schiffelers, R.M., Andaloussi, S.E.L., and Vader, P., Extracellular vesicles as drug delivery systems: why and how?, Adv. Drug Delivery Rev., 2020, vol. 159, pp. 332–343. https://doi.org/10.1016/j.addr.2020.04.004
Ernster, L. and Schatz, G., A historical review mitochondria, Cell, 2009, vol. 91, no. 3, part 2, pp. 227s–255s.
Escribano-Lopez, I., Diaz-Morales, N., Rovira-Llopis, S., Bañuls, C., Lopez-Domenech, S., Castelló, R., Falcón, R., et al., Oxidative stress and mitochondrial dysfunction in atherosclerosis: mitochondria-targeted antioxidants as potential therapy, Curr. Med. Chem., 2009, vol. 16, no. 35, pp. 4654–4667. https://doi.org/10.2174/9781681081755116080005
Farsinejad, S., Gheisary, Z., Samani, S.E., and Alizadeh, A.M., Mitochondrial targeted peptides for cancer therapy, Tumor Biol., 2015, vol. 36, no. 8, pp. 5715–5725. https://doi.org/10.1007/s13277-015-3719-1
Feng, Y., Qin, G., Chang, S., Jing, Z., Zhang, Y., and Wang, Y., Antitumor effect of hyperoside loaded in charge reversed and mitochondria-targeted liposomes, Int. J. Nanomed., 2021, vol. 16, pp. 3073–3089. https://doi.org/10.2147/IJN.S297716
Fisher-Wellman, K.H. and Neufer, P.D., Linking mitochondrial bioenergetics to insulin resistance via redox biology, Trends Endocrinol. Metab., 2012, vol. 23, no. 3, pp. 142–153. https://doi.org/10.1016/j.tem.2011.12.008
Friedman, J.R. and Nunnari, J., Mitochondrial form and function, Nature, 2014, vol. 505, no. 7483, pp. 335–343. https://doi.org/10.1038/nature12985
Gorman, G.S., Chinnery, P.F., DiMauro, S., Hirano, M., Koga, Y., McFarland, R., Suomalainen, A., Thorburn, D.R., Zeviani, M., and Turnbull, D.M., Mitochondrial diseases, Nat. Rev. Dis. Primers, 2016, vol. 2, pp. 16–36. https://doi.org/10.1038/nrdp.2016.80
Graham, D., Huynh, N.N., Hamilton, C.A., Beattie, E., Smith, R.A.J., Cochemé, H.M., Murphy, M.P., and Dominiczak, A.F., Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy, Hypertension, 2009, vol. 54, no. 2, pp. 322–328. https://doi.org/10.1161/HYPERTENSIONAHA.109.130351
Halliwell, B. and Gutteridge, J.M.C., Protection against oxidants in biological systems : the superoxide theory of oxygen toxicity, Free Radical Biol. Med., 1989, pp. 86–123.
Heart Disease and Stroke Statistics-2019 at-a-Glance, 2019.
Heidari, R., Niknahad, H., Jamshidzadeh, A., and Abdoli, N., Factors affecting drug-induced liver injury: antithyroid drugs as instances, Clin. Mol. Hepatol., 2014, vol. 20, no. 3, pp. 237–248. https://doi.org/10.3350/cmh.2014.20.3.237
Hessvik, N.P. and Llorente, A., Current knowledge on exosome biogenesis and release, Cell. Mol. Life Sci., 2018, vol. 75, no. 2, pp. 193–208. https://doi.org/10.1007/s00018-017-2595-9
Horton, K.L., Stewart, K.M., Fonseca, S.B., Guo, Q., and Kelley, S.O., Mitochondria-penetrating peptides, Chem. Biol., 2008, vol. 15, no. 4, pp. 375–382. https://doi.org/10.1016/J.CHEMBIOL.2008.03.015
ID Federation, IDF Diabetes Atlas, Brussels: International Diabetes Federation, 2017, vol. 4, 8th ed., pp. 905–911.
Igami, K., Uchiumi, T., Ueda, S., Kamioka, K., Setoyama, D., Gotoh, K., Akimoto, M., Matsumoto, S., and Kang, D., Characterization and function of medium and large extracellular vesicles from plasma and urine by surface antigens and annexin V, PeerJ Anal. Chem., 2020, vol. 2, pp. 1–23. https://doi.org/10.7717/peerj-achem.4
Ishikawa, K., Takenaga, K., Akimoto, M., Koshikawa, N., Yamaguchi, A., Imanishi, H., Nakada, K., Honma, Y., and Hayashi, J.-I., ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis, Science, 2008, vol. 320, no. 5876, pp. 661–664. https://doi.org/10.1126/science.1156906
James, A.M., Cochemé, H.M., Smith, R.A.J., and Murphy, M.P., Interactions of mitochondria-targeted and untargeted ubiquinones with the mitochondrial respiratory chain and reactive oxygen species. Implications for the use of exogenous ubiquinones as therapies and experimental tools, J. Biol. Chem., 2005, vol. 280, no. 22, pp. 21295–21312. https://doi.org/10.1074/jbc.M501527200
Jazwinski, S.M., The retrograde response: when mitochondrial quality control is not enough, Biochim. Biophys. Acta, 2014, vol. 1833, no. 2, pp. 400–409. https://doi.org/10.1016/j.bbamcr.2012.02.010
Juarez, J.C., Manuia, M., Burnett, M.E., Betancourt, O., Boivin, B., Shaw, D.E., Tonks, N.K., Mazar, A.P., and Doñate, F., Superoxide dismutase 1 (SOD1) is essential for H2O2-mediated oxidation and inactivation of phosphatases in growth factor signaling, Proc. Natl. Acad. Sci. U. S. A., 2008, vol. 105, no. 20, pp. 7147–7152. https://doi.org/10.1073/pnas.0709451105
Kakudo, T., Chaki, S., Futaki, S., Nakase, I., Akaji, K., Kawakami, T., Maruyama, K., Kamiya, H., and Harashima, H., Transferrin-modified liposomes equipped with a pH-sensitive fusogenic peptide: an artificial viral-like delivery system, Biochemistry, 2004, vol. 43, no. 19, pp. 5618–5628. https://doi.org/10.1021/bi035802w
Kalra, H., Drummen, G., and Mathivanan, S., Focus on extracellular vesicles: introducing the next small big thing, Int. J. Mol. Sci. 2016, vol. 17, no. 2, pp. 1–30. https://doi.org/10.3390/ijms17020170
Karaa, A., and Goldstein, A., The spectrum of clinical presentation, diagnosis, and management of mitochondrial forms of diabetes, Pediatr. Diabetes, 2014, pp. 1–9. https://doi.org/10.1111/pedi.12223
Kelso, G.F., Porteous, C.M., Coulter, C.V., Hughes, G., Porteous, W.K., Ledgerwood, E.C., Smith, R.A., and Murphy, M.P., Selective targeting of a redox-active ubiquinone to mitochondria within cells: antioxidant and antiapoptotic properties, J. Biol. Chem., 2001, vol. 276, no. 7, pp. 4588–4596. https://doi.org/10.1074/jbc.M009093200
Khalil, I.A., Kogure, K., Futaki, S., and Harashima, H., High density of octaarginine stimulates macropinocytosis leading to efficient intracellular trafficking for gene expression, J. Biol. Chem., 2006, vol. 281, no. 6, pp. 3544–3551. https://doi.org/10.1074/jbc.M503202200
Kim, Ho, Lee, T.H., Hwang, Y.S., Bang, M.A., Kim, K.H., Suh, J.M., Chung, H.K., Yu, D.Y., Lee, K.K., Kwon, O.Y., Ro, H.K., and Shong, M., Methimazole as an antioxidant and immunomodulator in thyroid cells: mechanisms involving interferon-γ signaling and H2O2 scavenging, Mol. Pharmacol., 2001, vol. 60, no. 5, pp. 972–980. https://doi.org/10.1124/mol.60.5.972
Kitada, M., Xu, J., Ogura, Y., Monno, I., and Koya, D., Manganese superoxide dismutase dysfunction and the pathogenesis of kidney disease, Front. Physiol., 2020, vol. 11, p. 755. https://doi.org/10.3389/fphys.2020.00755
Koliaki, C. and Roden, M., Alterations of mitochondrial function and insulin sensitivity in human obesity and diabetes mellitus, Annu. Rev. Nutr., 2016, pp. 1–31. https://doi.org/10.1146/annurev-nutr-071715-050656
Kowluru, R.A., Atasi, L., and Ho, Y.-S., Role of mitochondrial superoxide dismutase in the development of diabetic retinopathy, Invest. Ophthalmol. Visual Sci., 2006, vol. 47, no. 4, pp. 1594–1599. https://doi.org/10.1167/iovs.05-1276
Kraus, B. and Cain, H., Giant mitochondria in the human mycardium—morphogenesis and fate, Virchows Arch. B Cell Pathol., 1980, vol. 89, pp. 77–89.
Kumari, A., Electron transport chain, in Sweet Biochemistry, Kumari, A.B.T.-S.B., Ed., Elsevier, 2018, pp. 13–16.
Lam, S.J., O’Brien-Simpson, N.M., Pantarat, N., Sulistio, A., Wong, E.H.H., Chen, Y.-Y., Lenzo, J.C., Holden, J., Blencowe, A., Reynolds, E.C., and Qiao, G.G., Combating multidrug-resistant gram-negative bacteria with structurally nanoengineered antimicrobial peptide polymers, Nat. Microbiol., 2016, vol. 1, no. 11, p. 16162. https://doi.org/10.1038/nmicrobiol.2016.162
Liu, Y., Li, D., Liu, Z, Zhou, Y., Chu, D., Li, Z., Jiang, X., Hou, D., Chen, X., Chen, Y., Yang, Z., Jin, L., Jiang, W., Tian, C., Zhou, G., Zen, K., Zhang, J., Zhang, Y., Li, J., and Zhang, C.Y., Targeted exosome-mediated delivery of opioid receptor mu SiRNA for the treatment of morphine relapse, Sci. Rep., 2015, vol. 5, pp. 1–10. https://doi.org/10.1038/srep17543
Lleonart, M.E., Grodzicki, R., Graifer, D.M., and Lyakhovich, A., Mitochondrial dysfunction and potential anticancer therapy, Med. Res. Rev., 2017, vol. 37, no. 6, pp. 1275–1298. https://doi.org/10.1002/med.21459
Lubos, E., Kelly, N.J., Oldebeken, S.R., Leopold, J.A., Zhang, Y.-Y., Loscalzo, J., and Handy, D.E., Glutathione peroxidase-1 deficiency augments proinflammatory cytokine-induced redox signaling and human endothelial cell activation, J. Biol. Chem., 2011, vol. 286, no. 41, pp. 35407–35417. https://doi.org/10.1074/jbc.M110.205708
Malhi, S.S. and Ramachandr Murthy, R.S., Delivery to mitochondria: a narrower approach for broader therapeutics, Expert Opin. Drug Delivery, 2012, vol. 9, no. 8, pp. 909–935. https://doi.org/10.1517/17425247.2012.694864
Manickam, D.S., Delivery of mitochondria via extracellular vesicles—a new horizon in drug delivery, J. Controlled Release, 2022, vol. 343, pp. 400–407. https://doi.org/10.1016/J.JCONREL.2022.01.045
Marín-García, J., Post-genomics cardiovascular signaling pathways, in Post Genomic Cardiology, J. B. T.-P.-G. C., and Marín-García, E. Eds., Boston: Academic, 2014, Ch. 3, 2nd ed, pp. 57–112.
Marinho, H.S., Real, C., Cyrne, L., Soares, H., and Antunes, F., Hydrogen peroxide sensing, signaling and regulation of transcription factors, Redox Biol., 2014, vol. 2, pp. 535–562. https://doi.org/10.1016/j.redox.2014.02.006
Mighty mitochondria and neurodegenerative diseases—science in the news, n.d.
Millard, M., Yakavetsa, I., Piffoux, M., Brun, A., Gazeau, F., Guigner, J. M., Jasniewski, J., Lassalle, H.P., Wilhelm, C., and Bezdetnaya, L., MTHPC-loaded extracellular vesicles outperform liposomal and free MTHPC formulations by an increased stability drug delivery efficiency and cytotoxic effect in tridimensional model of tumors, Drug Delivery, 2018, vol. 25, no. 1, pp. 1790–1801. https://doi.org/10.1080/10717544.2018.1513609
Mishra, P. and Chan, D.C., Metabolic regulation of mitochondrial dynamics, J. Cell Biol., 2016, vol. 212, no. 4, pp. 379–387. https://doi.org/10.1083/jcb.201511036
Montgomery, M.K. and Turner, N., Mitochondrial dysfunction and insulin resistance : an update, Endocrine Connections, 2014, vol. 4, no. 1, pp. R1–R2. https://doi.org/10.1530/EC-14-0092
Murphy, M.P., Targeting lipophilic cations to mitochondria, Biochim. Biophys. Acta, Bioenergetics 2008, vol. 1777, no. 7, рр. 1028–1031. https://doi.org/https://doi.org/10.1016/j.bbabio.2008.03.029
Murphy, M.P., How mitochondria produce reactive oxygen species, Biochem. J., 2009, vol. 417, no. 1, pp. 1–13. https://doi.org/10.1042/BJ20081386
Murphy, M.P. and Smith, R.A.J., Targeting antioxidants to mitochondria by conjugation to lipophilic cations, Annu. Rev. Pharmacol. Toxicol., 2007, vol. 47, no. 1, pp. 629–656. https://doi.org/10.1146/annurev.pharmtox.47.120505.105110
Neglia, D., De Caterina, A., Marraccini, P., Natali, A., Ciardetti, M., Vecoli, C., Gastaldelli, A., et al., Impaired myocardial metabolic reserve and substrate selection flexibility during stress in patients with idiopathic dilated cardiomyopathy, Am. J. Physiol., Heart Circ. Physiol., 2020, vol. 3270–3278. https://doi.org/10.1152/ajpheart.00887.2007
Osellame, L.D., Blacker, T.S., and Duchen, M.R., Cellular and molecular mechanisms of mitochondrial function, Best Pract. Res. Clin. Endocrinol. Metab., 2012, vol. 26, no. 6, pp. 711–723. https://doi.org/10.1016/j.beem.2012.05.003
Pala, L., Senn, H.M., Caldwell, S.T., Prime, T.A., Warrington, S., Bright, T.P., Prag, H.A., Wilson, C., Murphy, M.P., and Hartley, R.C., Enhancing the mitochondrial uptake of phosphonium cations by carboxylic acid incorporation, Front. Chem., 2020, vol. 8.
Pelicano, H., Carney, D., and Huang, P., ROS stress in cancer cells and therapeutic implications, Drug Resist. Updates: Rev. Comment. Antimicrob. Anticancer Chemother., 2004, vol. 7, no. 2, pp. 97–110. https://doi.org/10.1016/j.drup.2004.01.004
Petros, J.A., Baumann, A.K., Ruiz-Pesini, E., Amin, M.B., Sun, K.Q., Hall, J., Lim, S.D., et al., MtDNA mutations increase tumorigenicity in prostate cancer, Proc. Natl. Acad. Sci. U. S. A., 2005, vol. 102, no. 3, pp. 719–724. https://doi.org/10.1073/pnas.0408894102
Qu, C., Zhang, S., Wang, W., Li, M., Wang, Y., and Van Der Heijde-Mulder, M., Mitochondrial electron transport chain complex III sustains hepatitis E virus replication and represents an antiviral target, FASEB J., 2019, vol. 33, no. 1, pp. 1008–1019. https://doi.org/10.1096/fj.201800620R
Roser, K.S., Brookes, P.S., Wojtovich, A.P., Olson, L.P., Shojaie, J., Parton, R.L., and Anders, M.W., Mitochondrial biotransformation of ω-(phenoxy)alkanoic acids, 3-(phenoxy)acrylic acids, and ω-(1-methyl-1H-imidazol-2-ylthio)alkanoic acids: a prodrug strategy for targeting cytoprotective antioxidants to mitochondria, Bioorg. Med. Chem., 2010, vol. 18, no. 4, pp. 1441–1448. https://doi.org/10.1016/j.bmc.2010.01.019
Roser, K.S., Brookes, P.S., Wojtovich, A.P., Olson, L.P., Shojaie, J., Parton, R.L., and Anders, M.W., Mitochondrial biotransformation of ω-(phenoxy)alkanoic acids, 3-(phenoxy)acrylic acids, and ω-(1-methyl-1h-imidazol-2-ylthio)alkanoic acids: a prodrug strategy for targeting cytoprotective antioxidants to mitochondria, Bioorg. Med. Chem., 2010, vol. 18, no. 4, pp. 1441–1448. https://doi.org/10.1016/j.bmc.2010.01.019
Balaban, R.S., Kantor, H.L., Katz, L.A., and Briggs, R.W., Relation between work and phosphate metabolite in the in vivo paced mammalian heart, Am. Assoc. Adv. Sci., 2016, vol. 232.
Scarpulla, R.C., Nucleus-encoded regulators of mitochondrial function: integration of respiratory chain expression, nutrient sensing and metabolic stress, Biochim. Biophys. Acta, 2012, nos. 9–10, pp. 1088–1097. https://doi.org/10.1016/j.bbagrm.2011.10.011
Schindler, C., Collinson, A., Matthews, C., Pointon, A., Jenkinson, L., Minter, R.R., Vaughan, T.J., and Tigue, N.J., Exosomal delivery of doxorubicin enables rapid cell entry and enhanced in vitro potency, PLoS One, 2019, vol. 14, no. 3, pp. 1–19. https://doi.org/10.1371/journal.pone.0214545
Shanmughapriya, S., Langford, D., and Natarajaseenivasan, K., Inter and intracellular mitochondrial trafficking in health and disease, Ageing Res. Rev., 2020, vol. 62, pp. 101–128. https://doi.org/10.1016/j.arr.2020.101128
Shokolenko, I.N., Alexeyev, M.F., Ledoux, S.P., and Wilson, G.L., TAT-mediated protein transduction and targeted delivery of fusion proteins into mitochondria of breast cancer cells, DNA Repair, 2005, vol. 4, no. 4, pp. 511–518. https://doi.org/10.1016/j.dnarep.2004.11.009
Sivitz, W.I. and Yorek, M.A., Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities, Antioxid. Redox Signal., 2010, vol. 12, pp. 537–577.
Sorrentino, V., Menzies, K.J., and Auwerx, J., Repairing mitochondrial dysfunction in disease, Annu. Rev. Pharmacol. Toxicol., 2018, vol. 58, no. 1, pp. 353–389. https://doi.org/10.1146/annurev-pharmtox-010716-104908
Sutong, X., Zhang, X., Liu, C., Liu, Q., Chai, H., Luo, Y., and Li, S., Role of mitochondrial in neurodegenerative diseases: from an epigenetic perspective, Front. Cell Dev. Biol., 2021, vol. 9, pp. 1–16. https://doi.org/10.3389/fcell.2021.688789
Szabo, I., Zoratti, M., and Biasutto, L., Targeting mitochondrial ion channels for cancer therapy, Redox Biol., 2021, vol. 42, p. 101846. https://doi.org/10.1016/j.redox.2020.101846
Szewczyk, A. and Marbán, E., Mitochondria: a new target for k channel openers?, Trends Pharm. Sci., 1999, vol. 20, no. 4, pp. 157–161. https://doi.org/10.1016/s0165-6147(99)01301-2
Thamilarasan, M., Estupinan, R., Batinic-Haberle, I., and Zennadi, R., Mn porphyrins as a novel treatment targeting sickle cell NOXs to reverse and prevent acute vaso-occlusion in vivo, Blood Adv., 2020, vol. 4, no. 11, pp. 2372–2386. https://doi.org/10.1182/bloodadvances.2020001642
Tokarz, P. and Blasiak, J., Role of mitochondria in carcinogenesis, Acta Biochim. Pol., 2014, vol. 61, no. 4, pp. 671–678.
Torchilin, V.P., Rammohan, R., Weissig, V., and Levchenko, T.S., TAT peptide on the surface of liposomes, Proc. Natl. Acad. Sci. U. S. ., 2001, vol. 98, no. 15, pp. 8786–8791.
Tovmasyan, A., Sheng, H., Weitner, T., Arulpragasam, A., Lu, M., Warner, D.S., Vujaskovic, Z., Spasojevic, I., and Batinic-Haberle, I., Design, mechanism of action, bioavailability and therapeutic effects of Mn porphyrin-based redox modulators, Med. Principles Pract., 2013, vol. 22, no. 2, pp. 103–130. https://doi.org/10.1159/000341715
Trifunovic, A. and Larsson, N.-G., Tissue-specific knockout model for study of mitochondrial DNA mutation disorders, in Redox Cell Biology and Genetics Part B, Sen, C.K. and Packer, L.B.T.-M.E., Academic Press, 2002, vol. 353, pp. 409–421.
Turturici, G., Tinnirello, R., Sconzo, G., and Geraci, F., Extracellular membrane vesicles as a mechanism of cell-to-cell communication: advantages and disadvantages, Am. J. Physiol., Cell Physiol., 2014, vol. 306, no. 7, pp. 621–633. https://doi.org/10.1152/ajpcell.00228.2013
US Group, Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group, Lancet, 1998, vol. 352, pp. 837–853.
Valadi, H., Ekström, K., Bossios, A., Sjöstrand, M., Lee, J.J., and Lötvall, J.O., Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells, Nat. Cell Biol., 2007, vol. 9, no. 6, pp. 654–659. https://doi.org/10.1038/ncb1596
Van Niel, G., D’Angelo, G., and Raposo, G., Shedding light on the cell biology of extracellular vesicles, Nat. Rev. Mol. Cell Biol., 2018, vol. 19, no. 4, pp. 213–228. https://doi.org/10.1038/nrm.2017.125
Ventura-Clapier, R., Garnier, A., and Veksler, V., Energy metabolism in heart failure, J. Physiol., 2004, vol. 555, no. 1, pp. 1–13. https://doi.org/10.1113/jphysiol.2003.055095
Wang, Y., Branicky, R., Noë, A., and Hekimi, S., Superoxide dismutases: dual roles in controlling ROS damage and regulating ROS signaling, J. Cell Biol., 2018, vol. 217, no. 6, pp. 1915–1928. https://doi.org/10.1083/jcb.201708007
Weissig, V., Lasch, J., Erdos, G., Meyer, H.W., Rowe, T.C., and Hughes, J., DQAsomes: a novel potential drug and gene delivery system made from DequaliniumTM, Pharm. Res., 1998, vol. 15, no. 2, pp. 334–337. https://doi.org/10.1023/A:1011991307631
Wender, P.A., Mitchell, D.J., Pattabiraman, K., Pelkey, E.T., Steinman, L., and Rothbard, J.B., The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters, Proc. Natl. Acad. Sci. U. S. A., 2000, vol. 97, no. 24, pp. 13003–13008. https://doi.org/10.1073/pnas.97.24.13003
Wiederkehr, A., and Wollheim, C.B., Minireview: implication of mitochondria in insulin secretion and action, Endocrinology, 2006, vol. 147, no. 6, pp. 2643–2649. https://doi.org/10.1210/en.2006-0057
Willms, E., Cabañas, C., Mäger, I., Wood, M.J.A., and Vader, P., Extracellular vesicle heterogeneity: subpopulations, isolation techniques, and diverse functions in cancer progression, Front. Immunol., 2018, vol. 9, no. 738, pp. 1–17. https://doi.org/10.3389/fimmu.2018.00738
Wrzosek, A., Augustynek, B., Żochowska, M., and Szewczyk, A., Mitochondrial potassium channels as druggable targets, Biomolecules, 2020, vol. 10, no. 8, p. 1200. https://doi.org/10.3390/biom10081200
Wrzosek, A., Gałecka, S., Żochowska, M., Olszewska, A., and Kulawiak, B., Alternative targets for modulators of mitochondrial potassium channels, Molecules, 2022, vol. 27, no. 1, p. 299.
Wu, Y., Chen, M., and Jiang, J., Mitochondrion mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling, Mitochondrion, 2019, vol. 49, pp. 35–45.
Yang, M., Deng, J., Guo, D., Sun, Q., Wang, Z., Wang, K., and Wu, F., Mitochondria-targeting Pt/Mn porphyrins as efficient photosensitizers for magnetic resonance imaging and photodynamic therapy, Dyes Pigments, 2019, vol. 166, pp. 189–195. https://doi.org/10.1016/J.DYEPIG.2019.03.048
Yang, Q., Wang, L., Liu, J., Cao, W., Pan, Q., and Li, M., Targeting the complex I and III of mitochondrial electron transport chain as a potentially viable option in liver cancer management, Cell Death Discovery, 2021, vol. 7, no. 1, p. 293. https://doi.org/10.1038/s41420-021-00675-x
Yue, C., Yang, Y., Song, J., Alfranca, G., Zhang, C., Zhang, Q., Yin, T., Pan, F., De La Fuente, J.M., and Cui, D., Mitochondria-targeting near-infrared light-triggered thermosensitive liposomes for localized photothermal and photodynamic ablation of tumors combined with chemotherapy, Nanoscale, 2017, vol. 9, no. 31, pp. 11103–11118. https://doi.org/10.1039/c7nr02193c
Zaborowski, M.P., Balaj, L., Breakefield, X.O., and Lai, C.P., Extracellular vesicles: composition, biological relevance, and methods of study, BioScience, 2015, vol. 65, no. 8, pp. 783–797. https://doi.org/10.1093/biosci/biv084
Zick, M., Rabl, R., and Reichert, A.S., Cristae formation-linking ultrastructure and function of mitochondria, Biochim. Biophys. Acta, 2009, vol. 1793, no. 1, pp. 5–19. https://doi.org/10.1016/j.bbamcr.2008.06.013
Zielonka, J., Joseph, J., Sikora, A., Hardy, M., Ouari, O., Vasquez-Vivar, J., Cheng, G., Lopez, M., and Kalyanaraman, B., Mitochondria-targeted triphenylphosphonium-based compounds: syntheses, mechanisms of action, and therapeutic and diagnostic applications, Chem. Rev., 2017, vol. 117, no. 15, pp. 10043–10120. https://doi.org/10.1021/acs.chemrev.7b00042
Zori, R.T. and Williams, C.A., Phenocopy versus genocopy, Am. J. Med. Genet., 1991, vol. 40, no. 2, pp. 248–249.
ACKNOWLEDGMENTS
Not applicable.
Funding
No funding organization in the public, private, or nonprofit sectors provided a particular grant for this work.
Author information
Authors and Affiliations
Contributions
Dr. H.K Sharma conceptualized the idea, synthesized the review technique, and reviewed the manuscript. El Bethel Lalthavel Hmar and Sujata Paul contributed to the content significantly and edited the paper. Rofiqul Islam contributed partially to the targeting strategies of the mitochondria section.
Corresponding author
Ethics declarations
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
This work does not contain any studies involving human and animal subjects.
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors of this work declare that they have no conflicts of interest.
CONSENT STATEMENT
Not applicable.
Additional information
Publisher’s Note.
Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Hmar, E.B., Paul, S., Islam, R. et al. An Insight into the Approach Taken to Appurtenances Disorders Linked to Mitochondria. Biol Bull Russ Acad Sci 51, 271–285 (2024). https://doi.org/10.1134/S1062359023604962
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1062359023604962