
Abstract
Accumulating evidence has been obtained to understand the mechanisms of long non-coding RNAs (lncRNAs) in bladder cancer (BC). However, due to the recurrence and metastasis of BC, searching for lncRNAs that are related to prognosis and metastasis and exploring the pathogenesis of BC might provide new insights for the treatment of BC. In the present study, we used the TCGA and GEO databases and identified LINC02446 as associated with prognosis and differentially expressed in bladder cancer tissues and para-cancer tissues. Then, we found that LINC02446 could affect the proliferation, migration and invasion of BC cells. Additionally, we found that LINC02446 could bind to the EIF3G protein and regulate the protein stability of EIF3G and then inhibit the mTOR signalling pathway. In summary, all these findings show that LINC02446 might serve as a promising therapeutic target for BC intervention.
Similar content being viewed by others

Introduction
Bladder cancer (BC) is one of the most common malignancies of the genitourinary system. In 2018, the incidence of BC ranked 12th among malignancies worldwide, ranking 6th among males and 10th among females, and the incidence has been on the rise in recent years [1]. BC occurs more frequently in men over 65 years old, but in recent years, the onset age has tended to be younger, and the ratio between men and women is 3:1–4:1 [2]. In clinical practice, the most common pathological type of BC is urothelial carcinoma. Approximately 75% of patients with BC at the first diagnosis have superficial bladder cancer, and 25% have muscle-invasive bladder cancer (MIBC). Non-muscle–invasive BC has significantly different treatment regimens and prognoses from MIBC. The former is characterised by a high recurrence rate and low mortality, and the treatment is mainly assisted intravesical infusion of BCG (Bacillus Calmette-Guérin) or chemotherapy after urethral electrotomy. The latter is mainly treated with a combination of surgery, chemotherapy and radiotherapy, and ~50% of MIBC patients experience relapse within 2 years after radical cystectomy, depending on the pathological stage of the primary tumour and the status of lymph nodes. Local recurrence accounts for 30% of the recurrence, while distant metastasis is more common, with 10%–15% of MIBC patients already having metastasis at the time of the diagnosis of recurrence. The treatment of metastatic BC has always been a difficult problem in clinical practice. The median survival period is approximately 12 to 14 months, and the prognosis is far worse than that of kidney cancer and prostate cancer [3, 4].
As a kind of non-coding RNA, lncRNA refers to non-coding RNAs (ncRNAs) with a length of >200 nucleotides [5]. LncRNAs are involved in many biological functions, such as the regulation of cell development and differentiation, cell apoptosis and the cell cycle, as well as gene imprinting, splicing, epigenetic regulation, transcription and translation regulation [6,7,8]. With the continuous development of molecular biology technology and bioinformatics technology, it has been recognised that lncRNAs play important roles in various diseases, such as tumours [9,10,11,12,13]. The abnormal expression of lncRNAs is closely related to biological behaviours such as the cell proliferation, invasion and metastasis of malignant tumours [14,15,16]. Understanding each lncRNA, by identifying its cell function, dissecting its molecular mechanism, and linking it to its role in disease, is of profound significance for tapping the potential of lncRNAs in the application of cancer diagnosis and targeted therapy.
In the present study, we used The Cancer Genome Atlas (TCGA) database to search for differentially expressed lncRNAs associated with prognosis, and finally, we identified 38 differentially expressed lncRNAs. Then, the GEO database was used to identify differentially expressed lncRNAs associated with BC or para-cancer tissues and BC tissues with or without lymph node metastasis. Finally, we identified LINC02446 for further research and found that LINC02446 could affect proliferation, migration and invasion. Additionally, we found that LINC02446 could bind to EIF3G protein and regulate the protein stability of EIF3G and then inhibit the mTOR signalling pathway. All these findings may contribute new insights into the development of a novel therapeutic target for BC.
Methods and materials
Data acquisition and bioinformatics analysis
In this study, expression profiles and clinical characteristics were obtained from The Cancer Genome Atlas (TCGA) database (https://cancergenome.nih.gov/) and the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). The clinicopathological features of patients from the TCGA, GSE89006, and GSE121738 data sets were examined for a histological diagnosis of urothelial carcinoma. The TANRIC tool was used to obtain FPKM standardised data of BC lncRNA expression data. The linear fitting method was used to obtain the differentially expressed lncRNAs from samples of a specific survival period. Single-variable Cox regression was applied to screen the prognosis-related lncRNAs and to calculate prognosis-related lncRNA risk values from low to high. A LASSO prognostic model was also constructed for the accurate selection of prognosis-related lncRNAs. For predicting prognosis, Cox regression analysis was also performed and identified a specific 38-lncRNAs signature. The files downloaded from the GEO database were assessed with the R language analysis tool combined with the GCBI website (https://www.gcbi.com.cn/) to identify differentially expressed lncRNAs. The intersection of the identified lncRNAs from the two databases was obtained by Venn diagram.
Clinical samples
A collection of 40 BC samples was obtained from patients who underwent partial cystectomy or radical cystectomy at the First Hospital of China Medical University. Forty matched adjacent bladder epithelial tissues were acquired from a distance of more than 3 cm from the BC tissues. All tissues were processed to be examined histologically. The study was approved by the Medical Ethics Committee of the First Hospital of China Medical University, and written informed consent was obtained from each patient for surgical and research purposes. The patients’ clinical characteristics were list in Table 1.
Cell culture and transfection
The normal bladder epithelial cell line SV-HUC-1 and the bladder cancer cell lines 5637, RT4, SW780, TCCSUP, J82, T24 and UMUC3 were obtained from the Chinese Academy of Sciences Cell Bank (CASCB, China). The cells were cultured in RPMI 1640 medium (Gibco, USA) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Gibco, USA) at 37 °C in 5% CO2. LINC02446 and EIF3G overexpression plasmids and small-interfering RNAs (siRNAs) targeting LINC02446 were synthesised by GenePharma Co. (Shanghai, China). The cells were transfected with the above plasmids and siRNAs using Lipofectamine 3000 Reagent (Life Technologies Corporation, USA). The cells were transfected with 20 µM siRNA and 5 µg plasmid. The LINC02446 siRNA and negative control (NC) siRNA sequences used were as follows (5’–3’): siRNA, sense, UAUGGAAACAAAUGUACACUC and antisense, GUGUACAUUUGUUUCCAUAAA; and NC, sense, UUCUCCGAACGUGUCACGUTT and antisense, ACGUGACACGUUCGGAGAATT.
Quantitative real-time reverse transcription PCR (RT-PCR)
Total RNA was isolated with RNAiso Plus (Takara Biotechnology, Dalian, China). For RT-PCR, the treated RNA was directly reverse transcribed from 500 ng of RNA with random primers using Prime Script RT Master Mix (Takara, Dalian). To quantify the amount of LINC02446 and EIF3G mRNA, real‐time PCR analyses were performed using the SYBR® Premix Ex Taq™ Kit (Takara, Dalian). GAPDH was used as an internal control. All analyses were performed using the Thermal Cycler Dice™ Real-Time TP800 system (Takara, Kyoto, Japan). The 2-ΔΔCT method was used to calculate the relative expression of different genes. The primers are listed in Table 2.
Western blot analysis
Total protein was extracted by RIPA buffer. Protein quantification was performed using the Bradford method (Bio-Rad Co., USA). Equal amounts of proteins loaded on 4–20% sodium dodecyl sulfate polyacrylamide gel electrophoresis gels were subsequently transferred onto polyvinylidene fluoride membranes. Then, separated protein bands were blocked for one hour and incubated with primary anti-EIF3G (1:1000, ab192601, Abcam, MA, USA), anti-E-cadherin (1:1000, 14772, Cell Signaling Technology, MA, USA), anti-N-cadherin (1:1000, 13116, Cell Signaling Technology, MA, USA), anti-Vimentin (1:1000, 5741, Cell Signaling Technology, MA, USA), anti-mTOR (1:1000, 2972, Cell Signaling Technology, MA, USA), anti-p-mTOR (1:1000, 5536, Cell Signaling Technology, MA, USA) and anti-GAPDH (1:5000, sc-25778, Santa Cruz, CA, USA) antibodies overnight at 4 °C. The protein bands were washed with TBST three times after incubation with the secondary antibodies. Chemiluminescence detection was performed on a chemiluminescence system (Bio-Rad, CA, USA).
Real-time cell analysis (RTCA)
All the treated cells were dissociated with trypsin and then seeded in cell culture E-plates at a cell density of 3,500 cells per well and incubated in the incubator. The cell growth curves were automatically recorded on the xCELLigence System (Roche Applied Sciences) in real-time. The cell index was followed for 3 days.
Transwell assay
Transwell assay was performed with or without matrigel, respectively, of 2 × 104 BC cells suspended in 200 µL serum-free RPMI 1640 medium for invasion and migration. BC cells were incubated for 24 h at 37 °C. After incubation, the non-invaded cells on the upper membrane surface were removed with a cotton tip. Then, membrane was fixed and stained by violet crystalline.
RNA immunoprecipitation (RIP) and RNA pull-down assay
RIP and RNA pull-down assays were used to confirm the interaction between LINC02446 and EIF3G protein. According to the manufacturer’s recommendations, the RIP assay was performed via the EZ-Magna RIP RNA-binding Protein Immunoprecipitation Kit (Millipore, Billerica, MA, USA). T24 and UMUC3 cells were lysed and then incubated with RIP buffer containing magnetic beads conjugated with EIF3G and IgG antibodies. To identify the enrichment of LINC02446, purified RNAs were analysed using RT-PCR. An RNA pull-down assay was conducted to confirm the direct binding between LINC02446 and EIF3G protein.
Biotinylated RNAs were mixed with streptavidin magnetic beads (Life Technologies, USA) for 4 h. Total cell lysates were freshly prepared and added to each binding reaction with protease/phosphatase inhibitor cocktail and RNase inhibitor, and then the mixture was incubated with rotation for 1 h at 4 °C. After washing thoroughly three times, the RNA–protein binding mixture was detected by western blotting.
Statistical analysis
All data was expressed as the mean ± SD for three independent experiments. *p < 0.05, **p < 0.01 and ***p < 0.001. Data analyses were carried out using GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA, USA) and SPSS software ver. 20.0 (SPSS, Inc., Chicago, IL, USA). Probability (P) values < 0.05, as calculated using Student’s t-test as appropriate, was considered statistically significant. Comparisons of the expression of clinical sample data detected by RT-PCR and the LINC02446 levels between matched tumours and healthy adjacent specimens were performed by the non-parametric Wilcoxon test, while comparisons of tumour grades, stages and metastatic status were performed by the non-parametric Mann–Whitney test.
Results
Identification of prognostic lncRNAs, establishment of the 38-lncRNA signature risk score and prediction of overall survival with the 38-lncRNA signature
We first downloaded BC data from the TCGA database, including the standardised expression data of second-generation sequencing FPKM, the clinical traits of patients and prognosis-related data. The expression data were matched with clinical data and prognostic data, and finally, we obtained relevant information for 400 patients with BC. All these data were used for the subsequent analysis. The limma linear fitting method was used to analyse samples with survival times of >3 years (a total of 69 samples) and samples with survival times of less than 1 year (a total of 77 samples). We then screened the differential lncRNAs with the thresholds of |logFC | >0.1 and P-value < 0.01 and identified 130 upregulated and 16 downregulated lncRNAs (Fig. 1A). Next, all 400 samples of the TCGA dataset were randomly divided into a training set of 300 samples and a test set of 100 samples. All samples from the training set were used for the next selection and model construction. The prognosis of the differential lncRNAs in the training set was analysed using the Cox model. A total of 116 prognostic lncRNAs were screened with P < 0.05 as the threshold. The 116 lncRNAs related to prognosis were input into the least absolute shrinkage and selection operator (LASSO) prognostic model, the best lambda was screened by 1000 cross validation, and the lambda value with the smallest validation error was selected as the best threshold. Finally, 38 lncRNAs were screened as key prognostic lncRNAs (Fig. 1B, C). Next, we designed a 38-lncRNA risk score based on the regression coefficients in single-variable Cox regression and the expression of the 38 prognosis-related lncRNAs. The 38-lncRNA risk score values of each patient were calculated, and then the patients were arranged from left to right according to the risk values from low to high. Next, all the patients were divided into two groups, the high-risk group (n = 150) and the low-risk group (n = 150), according to the median risk value of the training set as the cutoff. Then, the risk values, survival time and endpoint events of each patient and the expression levels of each lncRNA were marked (Fig. 1D–F). We then divided the patients into three risk groups (low risk, medium risk, and high risk) according to the prediction of each patient’s risk value of the 38-lncRNA signature by the Cox model. The results of the KM curves were compared by the log rank test. Survival analysis showed that the two models performed well in stratifying the three groups of patients in the training set and test set (Fig. 1G). Then, the two models for the prediction of overall survival time were assessed by the AUC. The AUC results showed that the model performed well in terms of the prediction effect (Fig. 1H).

A Volcano plot showing the differentially expressed lncRNAs in samples with survival times of more than 3 years (a total of 69 samples) and survival times of less than 1 year (a total of 77 samples) with adjusted |logFC | >0.1 and P-value < 0.01. B The tuning parameter (λ) selection in the LASSO model used 1000‐fold cross‐validation via minimum criteria. The vertical dashed line indicates the minimum partial likelihood deviation. C LASSO coefficient profiles of the 116 prognosis-related lncRNAs. D The 38 prognosis-related lncRNA signature risk score distributions. E Four-hundred patients’ survival time distribution according to the risk score. F Heatmap showing the 38 prognosis-related lncRNA expression profiles. The blue dotted line represents the median lncRNA risk score cutoff dividing the patients into low-risk and high-risk groups. G Kaplan–Meier curves of overall survival in the training (n = 300) and testing (n = 100) sets stratifying the three groups of patients with low risk, medium risk and high risk. H The AUC results showed the prediction of overall survival time in the training (n = 300) and testing (n = 100) sets.
Prognostic value of the 38- lncRNA signature, identification of 38-lncRNA signature correlated biological pathways and co-expression network
In this study, univariate and multivariate Cox regression analyses were performed to predict the prognosis related to some clinical characteristics and specific lncRNAs in BC. A nomogram was constructed to improve the convenience of the application of our findings in clinical practice (Fig. 2A). Some clinical characteristics (age, gender, lymph node count, lymph nodes positive, response, new tumour event, new tumour type, metastasis, neoplasm grade, tumour stage and pathologic stage), as well as the 38-lncRNA risk score were assessed by a Cox test to examine the independent risk factors in BC. By comparing the hazard ratio of these factors, some clinical characteristics, including positive lymph nodes, response, new tumour event, metastasis, tumour stage and pathologic stage, and the 38-lncRNA risk score were identified as covariates in the analysis of the dataset. In Fig. 2B, the width of the horizontal line represents the 95% confidence interval. Additionally, receiver operating characteristic (ROC) analysis was performed to assess the sensitivity and specificity of various models of survival prediction, including the 38-lncRNA risk score and some significant clinical characteristics obtained in the previous steps. In the ROC analysis, 3-year and 5-year survival predictions were tested in the training set and test set. Compared with the current clinical indicators, our 38-lncRNA signature exhibited a higher accuracy in predicting the 3-year and 5-year overall survival of BC patients (Fig. 2C, D). Predicting functions and relevant pathways can provide a more comprehensive and deeper understanding of the pathogenesis of the relevant factors. First, the ssGSEA method was used to score the pathway functions of each patient based on the gene expression information of the full transcriptome, and the 2199 functional gene set of canonical pathways (CP) in the MSigDB database was selected to score each patient. Then, according to the risk score, the patients were divided into the low-risk group and the high-risk group, and the limma linear fitting method was used for functional difference analysis. Finally, a total of 50 pathways with significant differences in function between the two risk groups were obtained with |logFC | >0.8 and FDR < 0.05 as the threshold (Fig. 2E). In addition, we also used the WGCNA co-expression network to analyse the co-expression of the 38 lncRNAs with miRNAs and mRNAs (Fig. 2F).

A A nomogram was constructed to improve the convenience of the application of our findings in clinical practice. B Forest plot represents the prognostic characteristics in the multivariable Cox proportional hazards regression analysis. C ROC analysis for predicting 3-year survival in the training and test sets. D ROC analysis for predicting 5-year survival in the training and test sets. E Heatmap showing the top 50 significantly different functional pathways between the two risk groups with |logFC | >0.8 and FDR < 0.05 as the threshold. F The co-expression of the 38 lncRNAs with miRNAs and mRNAs using the co-expression network.
Gene expression microarray analyses in the GEO database and hub lncRNA selection
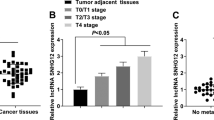
Currently, the Gene Expression Omnibus (GEO) database is the largest and most comprehensive international public gene expression data resource. LncRNAs associated with prognosis are usually differentially expressed in tumour tissues and corresponding normal or lymph node tissues. Therefore, in the present study, we first compared human bladder carcinoma tissues with matched para-carcinoma tissues in a GEO dataset (GSE89006). Differential gene expression analysis screened the top 5000 differentially expressed lncRNAs by fold change > 2 with p ≤ 0.01 (Fig. 3A, B). Next, we used another GEO dataset (GSE121738) to compare differentially expressed lncRNAs in BC with or without lymph node metastasis. A total of 8427 DEGs were found by fold change > 2 with p ≤ 0.01 (Fig. 3C, D). The differential lncRNA analysis from the original chip data was performed by using the R language analysis tool from the GCBI website. Heat maps and volcano plots were applied to visualise the distribution of the expressed lncRNAs between the two groups. To identify the hub lncRNAs that might have an important influence on the occurrence and prognosis of BC, a three-way Venn diagram of the 38 prognosis-related lncRNAs and the GSE89006 and GSE121738 data sets was constructed and revealed that two explicit lncRNAs were commonly changed (Fig. 3E). The two explicit lncRNAs were HCG27 and LINC02446. HCG27 has been reported in many diseases. Finally, we selected LINC02446, which has not been reported before, for further research. According to gene expression profiling interactive analysis (GEPIA) (http://gepia.cancer-pku.cn/), low levels of LINC02446 indicated poor prognosis (Fig. 3F).

A Representative images of the heatmap showing the differentially expressed lncRNAs in human bladder carcinoma tissues and matched para-carcinoma tissues from the GEO dataset (GSE89006). B Representative images of the volcano plot showing the differentially expressed lncRNAs in human bladder carcinoma tissues and matched para-carcinoma tissues from the GEO dataset (GSE89006). C Representative images of the heatmap map showing the differentially expressed lncRNAs in BC tissues with or without lymph node metastasis from the GEO dataset (GSE89006). D Representative images of the volcano plot showing the differentially expressed lncRNAs in BC tissues with or without lymph node metastasis from the GEO dataset (GSE89006). E Analysis of the common differentially expressed lncRNAs of the 38 prognosis-related lncRNAs and the GSE89006 and GSE121738 data sets by Venn diagram. F Kaplan–Meier analysis of OS for the patients with different LINC02446 expression.
LINC02446 expression is downregulated and acts as a tumour suppressor in BC
As described above, LINC02446 was downregulated in BC tissues compared with adjacent normal tissues, was downregulated in BC tissues with lymph node metastasis compared with BC tissues without lymph node metastasis and was negatively correlated with prognosis. Therefore, we first reconfirmed whether the expression of LINC02446 was downregulated in BC tissues and BC cell lines. A total of 40 pairs of BC tissues and matched para-cancer tissues were compared by qRT-PCR to examine the expression of LINC02446. Compared with that in matched para-cancer tissues, LINC02446 expression was downregulated in BC tissues (Fig. 4A). Among the 40 BC tissues, LINC02446 expression was downregulated in 14 high-grade BC samples compared with 26 low-grade BC samples (Fig. 4B). The expression of LINC02446 was also downregulated in 15 invasive BC samples compared with 25 non-invasive BC samples (Fig. 4C). Additionally, LINC02446 expression was downregulated in 10 BC tissues with metastasis compared with 20 BC tissues without metastasis (Fig. 4D). After confirming the LINC02446 expression level in tissues, we also found that LINC02446 showed low expression in 5 BC cell lines (5637, TCCSUP, J82, T24, and UMUC3) compared to the immortalised normal urothelial cell line, SV-HUC-1 (SV) (Fig. 4E). All the above findings urged us to explore the functional performance of LINC02446. Thus, we transfected T24 and UMUC3 cells with LINC02446 knockdown (LINC02446 si) and LINC02446 overexpression plasmids (LINC02446-OE). The relative expression of LINC02446 was assessed using qRT-PCR (Fig. 4F). Then, an RTCA assay was performed to evaluate the proliferation of T24 and UMUC3 BC cells, and LINC02446 expression revealed an inverse correlation with cell proliferation (Fig. 4G). Next, we used a transwell assay to assess the effect of LINC02446 on the migration and invasion abilities of T24 and UMUC3 BC cells, and an inversely correlated relationship was found between LINC02446 expression and migration and invasion abilities (Fig. 4H). In addition, as epithelial-mesenchymal transition (EMT) plays an essential role in invasion and metastasis in cancer, we also explored the expression of EMT-associated markers, including E-cadherin, N-cadherin and Vimentin, using western blotting. In this study, the western blot assay showed that E-cadherin expression was positively correlated with LINC02446 expression, while N-cadherin and Vimentin expression was inversely correlated with LINC02446 expression (Fig. 4I). Moreover, we found that LINC02446 could inhibit the activation of the mTOR signalling pathway via western blotting (Fig. 4J).

A Relative expression level of LINC02446 in BC tissues and matched para-carcinoma tissues using qRT-PCR. B Relative expression level of LINC02446 in high-grade BC tissues and low-grade BC tissues using qRT-PCR. C Relative expression level of LINC02446 in non-invasive BC tissues and invasive BC tissues using qRT-PCR. D Relative expression level of LINC02446 in non-metastatic BC tissues and metastatic BC tissues using qRT-PCR. E LINC02446 expression in seven bladder cancer cell lines and SV-HUC-1 (SV) cells according to qRT-PCR. F Relative expression level of LINC02446 in T24 and UMUC3 cells using qRT-PCR. G Proliferation in treated T24 and UMUC3 cells was detected by RTCA. H Cell migration and invasion in treated T24 and UMUC3 cells was performed by transwell assays with or without Matrigel, respectively. I Relative expression levels of cell adhesion molecules (E-cadherin, N-cadherin and Vimentin) in treated T24 and UMUC3 cells using western blotting. J Relative expression of mTOR and p-mTOR in treated T24 and UMUC3 cells evaluated using western blotting.
LINC02446 binds with EIF3G and regulates the protein stability of EIF3G in BC cells
After understanding the biological functions of LINC02446, we intended to further explore the pathogenesis of LINC02446 in BC. In recent years, many studies have shown that lncRNAs can bind to specific proteins and then mediate protein modification [17, 18], so we aimed to identify the specific proteins that can bind to LINC02446. Using a bioinformatics programme, catRAPID, we identified EIF3G as the putative target of LINC02446 (Fig. 5A). The expression of EIF3G mRNA was not significantly different in BC and adjacent normal tissues according to the GEPIA database (Fig. 5B). EIF3G protein was overexpressed in BC tissues compared with matched adjacent normal tissues (Fig. 5C). Next, we attempted to investigate the regulatory effect of LINC02446 on EIF3G, and EIF3G mRNA and protein levels were measured by qRT-PCR and western blot analysis. The results showed no significant difference in EIF3G mRNA expression with the change in LINC02446 compared with that in the corresponding control group (Fig. 5D). EIF3G protein expression was negatively regulated by LINC02446 compared with that in the corresponding control group (Fig. 5E). To confirm the binding relationship between LINC02446 and EIF3G, we performed an RNA pull-down assay and confirmed that the full-length LINC02446 probe pulled down EIF3G (Fig. 5F). In addition, we also conducted a RIP assay to further provide proof of the binding relationship between LINC02446 and EIF3G (Fig. 5G). To further explore the regulatory mode of LINC02446 on the EIF3G protein, we adopted the bioinformatics method PhosphoSitePlus (https://www.phosphosite.org/) to explore the molecular structure characteristics of the EIF3G protein and its modification sites. Interestingly, we found that EIF3G protein contains the RPM1 domain, which is an RNA-binding domain that is known to bind single-stranded RNAs, at the position of 241 to 310 amino acids. Additionally, near the RPM1 domain, there may be a ubiquitination site at the 180th lysine of the EIF3G protein (Fig. 5H). All these findings indicated that LINC02446 regulates the biological process of BC by binding to EIF3G protein and then affecting the stability of EIF3G protein. With this assumption, to determine the effect of LINC02446 on the half-life of EIF3G protein, we first transfected T24 and UMUC3 cells with vector- and LINC02446-overexpressing plasmids, and then all the cells were treated with 100 µg/mL cycloheximide (CHX) for the indicated times. Western blot analysis revealed that the half-life of EIF3G was reduced following the overexpression of LINC02446 (Fig. 5I). Then, we used DMSO or 10 µmol/L MG-132 to treat T24 and UMUC3 cells, which were transfected with vector- and LINC02446-overexpressing plasmids to determine the effect of LINC02446 on the protein stability of EIF3G. The results showed that overexpression of LINC02446 decreased EIF3G protein expression, which could be restored after MG-132 treatment (Fig. 5J). To investigate the ubiquitination site at the 180th lysine of the EIF3G protein on the half-life and proteasome-dependent degradation of EIF3G, plasmids with a substitution of the lysine residues (K180) of EIF3G with arginine residues (K180R) were constructed separately. Western blot analysis revealed that the half-life of EIF3G was more stable following the mutation of EIF3G with arginine (Fig. 5K). In addition, the cells with EIF3G K180 and K180R transfected with vector and LINC02446 overexpression plasmids were used to determine the effect of LINC02446 on the protein stability of the EIF3G K180 site, and the results demonstrated that overexpression of LINC02446 could destroy the stability of EIF3G protein with K180R (Fig. 5L).

A Identification of putative proteins for binding with LINC02446 using a bioinformatics programme. B EIF3G mRNA expression between BC tissues and adjacent normal tissues from the GEPIA database. C Relative EIF3G protein expression between BC tissues and matched adjacent normal tissues according to western blotting. D T24 and UMUC3 cells were transfected with LINC02446 siRNA or LINC02446 overexpression plasmid. The relative expression of EIF3G was evaluated using qRT-PCR. E T24 and UMUC3 cells were transfected with LINC02446 siRNA or LINC02446 overexpression plasmid. The relative expression of EIF3G was evaluated using western blotting. F The binding relationship between LINC02446 and EIF3G was determined by an RNA pull-down assay. G The interaction relationship between LINC02446 and EIF3G was detected by a RIP assay. H Molecular structure characteristics of EIF3G protein and its modification sites by a bioinformatics programme. I T24 and UMUC3 cells were transfected with vector or LINC02446 overexpression plasmid and treated with cycloheximide (100 µg/mL). The relative expression of EIF3G was evaluated using western blotting. J T24 and UMUC3 cells were transfected with vector or LINC02446 overexpression plasmid and treated with MG-132 (10 µmol/L). The relative expression of EIF3G was evaluated using western blotting. K T24 and UMUC3 cells were transfected with WT (K180) and K180R-mutant (K180R) EIF3G plasmids and treated with cycloheximide (100 μg/mL). The relative expression of EIF3G was evaluated using western blotting. L T24 and UMUC3 cells were transfected with vector and LINC02446 overexpression plasmids and transfected with WT (K180) and K180R-mutant (K180R) EIF3G plasmids. The relative expression of EIF3G was evaluated using western blotting.
LINC02446 affects cell function and inhibits the mTOR signalling pathway in an EIF3G-mediated manner in BC
To further determine whether the oncogenic role of EIF3G in BC depends on LINC02446, we transfected cells with LINC02446-overexpressing plasmids or EIF3G-overexpressing plasmids or co-transfected them with both. RTCA assays for cell proliferation and transwell assays for cell migration and invasion were performed. All the results revealed that overexpression of EIF3G significantly promoted proliferation, migration and invasion, while overexpression of LINC02446 depleted this promotion (Fig. 6A, B). Next, we also explored the expression of EMT-associated markers, including E-cadherin, N-cadherin and Vimentin, using western blotting. The results showed that E-cadherin expression was negatively correlated with EIF3G expression, and N-cadherin and Vimentin expression was positively correlated with EIF3G expression, while overexpression of LINC02446 reversed this effect (Fig. 6C). As EIF3G could affect the mTOR signalling pathway in colorectal cancer, we sought to directly test whether overexpression of EIF3G could result in pathogenic mTOR activation and whether LINC02446 drives mTOR activity in an EIF3G-dependent manner. Ultimately, according to western blot analysis, we found that overexpression of EIF3G could significantly activate the mTOR signalling pathway, while LINC02446 overexpression could rescue the abnormal activation of the mTOR signalling pathway in EIF3G-overexpressing BC cells (Fig. 6D). The flow chart of the study procedure is presented in supplementary Fig. 1.

A The proliferation of treated T24 and UMUC3 cells was detected by RTCA. B The cell migration and invasion of treated T24 and UMUC3 cells was assessed by transwell assays with or without Matrigel, respectively. C The relative expression levels of cell adhesion molecules (E-cadherin, N-cadherin and Vimentin) in treated T24 and UMUC3 cells were assessed by western blotting. D T24 and UMUC3 cells were transfected with LINC02446 overexpression plasmid, EIF3G overexpression plasmid or both. The relative expression of mTOR and p-mTOR was evaluated using western blotting.
Discussion
Although much progress has been made in the research of BC in recent years, due to the recurrence and metastasis of BC, the current treatment of BC is still limited to surgery and chemotherapy, and molecular targeted therapy drugs targeting specific targets have not yet been used in clinical practice [19, 20]. Therefore, the discovery of specific molecular targets related to the occurrence and development of BC, the identification of pathogenic mechanisms and the search for key molecular signalling pathways are important for the future treatment of BC.
With the development of microarray chip technology, especially gene chip technology, it has been found that non-coding RNAs play significant roles in the normal life activities of the organism, as well as in the occurrence and development of diseases [21, 22]. LncRNAs, a type of ncRNA, have attracted more attention recently and become a hot topic in current research. LncRNAs have various and complex functions in the body and can regulate gene expression at the transcription, post-transcription and epigenetic levels, participate in genomic modification, transcriptional activation, transcriptional interference, chromosome precipitation and other processes, and play an important role in the occurrence and development of various diseases, including tumours [23,24,25,26]. The study of the intrinsic mechanisms of lncRNAs can help us to obtain a deeper understanding of tumours, and lncRNAs are expected to be used as molecular markers for the diagnosis and prognosis of tumours in clinical practice, thus providing a new target for the clinical treatment of tumours.
The first step of our research was to search for differentially expressed lncRNAs, to find the specific lncRNAs associated with prognosis and to search for independent predictors that may influence the progression of BC by using the TCGA database, which is a large public resource database [27]. After downloading BC data from the TCGA database, collecting the clinical and prognostic data of patients, applying the limma linear fitting method to analyse the difference in lncRNAs of patients with a specific survival period, and further using the Cox model to analyse the survival prognosis of the different lncRNAs, we applied the LASSO prognosis model and finally selected 38 lncRNAs related to prognosis for further analysis. A Cox model was constructed with the 38 specific lncRNAs, and the risk score was calculated for each patient. Then, the patients were divided into low-risk, medium-risk and high-risk groups according to the risk score in the training set and test set, and their overall survival was analysed using Kaplan–Meier analysis. Finally, we found that the 38-lncRNA signature could be used to predict the survival of patients with BC. Further analysis indicated that the prognostic value of the 38-lncRNA signature was independent of lymph node metastasis, new tumour event, metastasis, tumour stage and pathologic stage. In addition, the independent predictors of the 38-lncRNA signature were also assessed by ROC curve analysis and evaluated for predicting 3-year and 5-year survival. All these findings showed that the 38-lncRNA signature had good stability and accuracy in predicting patient prognosis.
By using the GEO database to search for lncRNAs that were differentially expressed in BC and corresponding adjacent tissues, we further screened lncRNAs that were differentially expressed in BC tissues with or without lymph node metastasis. Based on the results of the TCGA database analysis, we finally selected LINC02446 as the final study object and speculated that LINC02446 may play an important role in the occurrence, development, and prognosis of BC and may become an ideal molecular target for the diagnosis and treatment of BC. Further exploration of the functional annotation of prognosis-related LINC02446 might provide a better understanding of its molecular mechanisms. Subsequent experiments showed that LINC02446 expression in tumour tissues was lower than that in adjacent tissues. Compared with that in low-grade tumour tissues, LINC02446 expression was lower in high-grade tumour tissues. Compared with that in non-invasive tumour tissues, LINC02446 expression was lower in invasive tumour tissues. Compared with that in non-metastatic tumour tissues, LINC02446 expression was lower in metastatic tumour tissues. Moreover, functional experiments showed that LINC02446 negatively regulated the proliferation, migration and invasion of BC cells.
EIF3 is an integral part of eukaryotic translation initiation factors (EIFs) and participates in protein synthesis at the level of translation initiation [28]. Recent research has revealed that EIF3, which contains 13 subunits named EIF3a to EIF3m, is a key regulator in meditating the interaction and formation of the 43S pre-initiation complex and EIF4f-bound mRNA clients [29]. In addition to promoting the translation initiation of protein synthesis, EIFs have been shown to be involved in a variety of pathological and physiological processes. The aberrant expression of EIFs has been reported in many diseases, including narcolepsy, neurodegenerative disorders, infection and especially cancers [30,31,32]. As an essential part of the subunits of EIFs, EIF3G could bind to the EIF3 holo-complex via EIF3E, which is a necessary component to facilitate the initiation of mRNA transcription. Recent studies have shown that EIF3G is overexpressed in several tumours, including colorectal cancer, breast cancer and prostate cancer [33,34,35]. The aberrant expression of EIF3G may play an oncogenic role and may implicate the occurrence, development and progression of tumours by mediating apoptosis, drug resistance and other molecular mechanisms [36, 37]. As EIF3G acts as an important physiological regulator of transcription initiation and has a pathologic molecular mechanism that promotes the progression of many diseases, it is essential to explore its expression and pathogenesis in BC. In the present study, we observed that EIF3G was overexpressed at the protein level in BC but not at the mRNA expression level. Mechanistically, we applied bioinformatics methods and demonstrated that LINC02446 could bind with EIF3G protein and regulate the protein stability of EIF3G. We also discovered that the LINC02446/EIF3G axis could activate the mTOR signalling pathway in BC. In conclusion, all these findings might provide a deeper understanding of the molecular mechanisms of BC and aid in the development of new target-related therapeutic approaches for clinical practice. However, there are still some limitations in this study. First, this paper only screened prognostic lncRNAs from the perspective of bioinformatics and did not perform further analysis on the survival prognosis related to this lncRNA. Second, there were few clinical data samples applied to assess the expression levels of the studied factors in this paper, which need to be further supplemented in the future. In addition, this paper only carried out bioinformatics analysis of the studied factors, and the corresponding gene sequencing verification and subsequent experimental verification were few, so further research is needed for further exploration.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Chen WQ, Li H, Sun KX, Zheng RS, Zhang SW, Zeng HM, et al. [Report of Cancer Incidence and Mortality in China, 2014]. Zhonghua Zhong Liu Za Zhi. 2018;40:5–13.
Babjuk M, Burger M, Compérat EM, Gontero P, Mostafid AH, Palou J, et al. European Association of Urology Guidelines on non-muscle-invasive bladder cancer (TaT1 and Carcinoma In Situ)−2019 update. Eur Urol. 2019;76:639–57.
Ucpinar B, Erbin A, Ayranci A, Caglar U, Alis D, Basal S, et al. Prediction of recurrence in non-muscle invasive bladder cancer patients. Do patient characterisics matter?. J Buon. 2019;24:1659–65.
St LG, Wahlestedt C, Kapranov P. The landscape of long noncoding RNA classification. Trends Genet. 2015;31:239–51.
Schmitz SU, Grote P, Herrmann BG. Mechanisms of long noncoding RNA function in development and disease. Cell Mol Life Sci. 2016;73:2491–509.
Chen LL. Linking long noncoding RNA localization and function. Trends Biochem Sci. 2016;41:761–72.
Bhan A, Soleimani M, Mandal SS. Long noncoding RNA and cancer: a new paradigm. Cancer Res. 2017;77:3965–81.
Salamon I, Saccani JG, Condorelli G. The long noncoding RNA landscape in cardiovascular disease: a brief update. Curr Opin Cardiol. 2018;33:282–9.
Luo Q, Chen Y. Long noncoding RNAs and Alzheimer’s disease. Clin Interv Aging. 2016;11:867–72.
Yang Z, Jiang S, Shang J, Jiang Y, Dai Y, Xu B, et al. LncRNA: Shedding light on mechanisms and opportunities in fibrosis and aging. Ageing Res Rev. 2019;52:17–31.
Delás MJ, Sabin LR, Dolzhenko E, Knott SR, Munera ME, Jackson BT, et al. lncRNA requirements for mouse acute myeloid leukemia and normal differentiation. Elife. 2017;6:e25607.
Peng WX, Koirala P, Mo YY. LncRNA-mediated regulation of cell signaling in cancer. Oncogene. 2017;36:5661–7.
Wei GH, Wang X. lncRNA MEG3 inhibit proliferation and metastasis of gastric cancer via p53 signaling pathway. Eur Rev Med Pharm Sci. 2017;21:3850–6.
Dong J, Teng F, Guo W, Yang J, Ding G, Fu Z. lncRNA SNHG8 promotes the tumorigenesis and metastasis by sponging miR-149-5p and predicts tumor recurrence in hepatocellular carcinoma. Cell Physiol Biochem. 2018;51:2262–74.
Wang LX, Wan C, Dong ZB, Wang BH, Liu HY, Li Y. Integrative analysis of long noncoding RNA (lncRNA), microRNA (miRNA) and mRNA expression and construction of a competing endogenous RNA (ceRNA) network in metastatic melanoma. Med Sci Monit. 2019;25:2896–907.
Ferrè F, Colantoni A, Helmer-Citterich M. Revealing protein-lncRNA interaction. Brief Bioinform. 2016;17:106–16.
Cheng S, Zhang Y, Chen S, Zhou Y. LncRNA Hotair Participates in microglia activation and inflammatory factor release by regulating the ubiquitination of MYD88 in traumatic brain injury. J Mol Neurosci. 2021;71:169–77.
Rouprêt M, Colin P, Xylinas E, Compérat E, Dubosq F, Houédé N, et al. [CCAFU french national guidelines 2016–2018 on upper tract tumors]. Prog Urol. 2016;27(Suppl 1):S55–S66.
Bahrami S, Kazemi B, Zali H, Black PC, Basiri A, Bandehpour M, et al. Discovering therapeutic protein targets for bladder cancer using proteomic data analysis. Curr Mol Pharmacol. 2020;13:150–72.
Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer. 2018;18:5–18.
Hombach S, Kretz M. Non-coding RNAs: classification, biology and functioning. Adv Exp Med Biol. 2016;937:3–17.
Jarroux J, Morillon A, Pinskaya M. History, discovery, and classification of lncRNAs. Adv Exp Med Biol. 2017;1008:1–46.
Andersen RE, Lim DA. Forging our understanding of lncRNAs in the brain. Cell Tissue Res. 2018;371:55–71.
Darbellay F, Necsulea A. Comparative transcriptomics analyses across species, organs, and developmental stages reveal functionally constrained lncRNAs. Mol Biol Evol. 2020;37:240–59.
Kazimierczyk M, Kasprowicz MK, Kasprzyk ME, Wrzesinski J. Human long noncoding RNA interactome: detection, characterization and function. Int J Mol Sci. 2020;21:1207.
Tomczak K, Czerwińska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol (Pozn). 2015;19(1A):A68–A77.
Beznosková P, Wagner S, Jansen ME, von der Haar T, Valášek LS. Translation initiation factor eIF3 promotes programmed stop codon readthrough. Nucleic Acids Res. 2015;43:5099–111.
Dong Z, Zhang JT. Initiation factor eIF3 and regulation of mRNA translation, cell growth, and cancer. Crit Rev Oncol Hematol. 2006;59:169–80.
Holm A, Lin L, Faraco J, Mostafavi S, Battle A, Zhu X, et al. EIF3G is associated with narcolepsy across ethnicities. Eur J Hum Genet. 2015;23:1573–80.
Gomes-Duarte A, Lacerda R, Menezes J, Romão L. eIF3: a factor for human health and disease. RNA Biol. 2018;15:26–34.
Yin Y, Long J, Sun Y, Li H, Jiang E, Zeng C, et al. The function and clinical significance of eIF3 in cancer. Gene. 2018;673:130–3.
Yang C, Zhang Y, Du W, Cheng H, Li C. Eukaryotic translation initiation factor 3 subunit G promotes human colorectal cancer. Am J Transl Res. 2019;11:612–23.
Zheng Q, Liu H, Ye J, Zhang H, Jia Z, Cao J. Nuclear distribution of eIF3g and its interacting nuclear proteins in breast cancer cells. Mol Med Rep. 2016;13:2973–80.
Nupponen NN, Isola J, Visakorpi T. Mapping the amplification of EIF3S3 in breast and prostate cancer. Genes Chromosome Cancer. 2000;28:203–10.
Yang C, Liu X, Li C, Li S, Du W, Yang D. Eukaryotic translation initiation factor 3 subunit G (EIF3G) resensitized HCT116/5-Fu to 5-fluorouracil (5-Fu) via inhibition of MRP and MDR1. Onco Target Ther. 2018;11:5315–24.
Dalton HM, Curran SP. Hypodermal responses to protein synthesis inhibition induce systemic developmental arrest and AMPK-dependent survival in Caenorhabditis elegans. PLOS Genet. 2018;14:e1007520.
Acknowledgements
This work was supported by National Natural Science Fund [Grant No. 81672525], the Project of Liaoning Distinguished Professor (Grant No. [2012]145), Liaoning Natural Science Fund [Grant No. 201602830], Shenyang Plan Project of Science and Technology (Grant No. F17-230-9-08) and Shenyang clinical medicine research centre (Grant No. [2017]76), China Medical University’s 2017 discipline promotion programme (Grant No. 2017XK08), China Medical University’s 2018 discipline promotion programme. Funding agency did not participate in the design of the study and collection, analysis and interpretation of data and in writing the manuscript.
Funding
This work was supported by National Natural Science Fund [Grant No. 81672525], the Project of Liaoning Distinguished Professor (Grant No. [2012]145), Liaoning Natural Science Fund [Grant No. 201602830], Shenyang Plan Project of Science and Technology (Grant No. F17-230-9-08) and Shenyang clinical medicine research centre (Grant No. [2017]76), China Medical University’s 2017 discipline promotion programme (Grant No. 2017XK08), China Medical University’s 2018 discipline promotion programme.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors gave consent for the publication of the manuscript. The authors declare no competing financial interests. All authors read and approved the final manuscript.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Zhang, X., Zhang, J., Zhao, W. et al. Long non-coding RNA LINC02446 suppresses the proliferation and metastasis of bladder cancer cells by binding with EIF3G and regulating the mTOR signalling pathway. Cancer Gene Ther 28, 1376–1389 (2021). https://doi.org/10.1038/s41417-020-00285-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-020-00285-2
- Springer Nature America, Inc.