
Abstract
During both development and adulthood, the human brain expresses many thousands of long noncoding RNAs (lncRNAs), and aberrant lncRNA expression has been associated with a wide range of neurological diseases. Although the biological significance of most lncRNAs remains to be discovered, it is now clear that certain lncRNAs carry out important functions in neurodevelopment, neural cell function, and perhaps even diseases of the human brain. Given the relatively inclusive definition of lncRNAs—transcripts longer than 200 nucleotides with essentially no protein coding potential—this class of noncoding transcript is both large and very diverse. Furthermore, emerging data indicate that lncRNA genes can act via multiple, non-mutually exclusive molecular mechanisms, and specific functions are difficult to predict from lncRNA expression or sequence alone. Thus, the different experimental approaches used to explore the role of a lncRNA might each shed light upon distinct facets of its overall molecular mechanism, and combining multiple approaches may be necessary to fully illuminate the function of any particular lncRNA. To understand how lncRNAs affect brain development and neurological disease, in vivo studies of lncRNA function are required. Thus, in this review, we focus our discussion upon a small set of neural lncRNAs that have been experimentally manipulated in mice. Together, these examples illustrate how studies of individual lncRNAs using multiple experimental approaches can help reveal the richness and complexity of lncRNA function in both neurodevelopment and diseases of the brain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The human genome produces tens of thousands of long noncoding RNAs (lncRNAs)—transcripts of greater than 200 nucleotides that lack evident protein coding potential (Djebali et al. 2012)—and it is now clear that certain lncRNAs can regulate important biological processes including those that underlie human disease (Rinn and Chang 2012; Lee 2012; Mercer and Mattick 2013; Batista and Chang 2013; Briggs et al. 2015). The developing and adult central nervous system (CNS) express a tremendous diversity of lncRNAs, many of which are brain-specific (Mercer et al. 2008; Derrien et al. 2012). Aberrant lncRNA expression has been associated with some of the most devastating neurological diseases including glioma (Ramos et al. 2016), schizophrenia (Barry et al. 2014), Alzheimer’s disease (Faghihi et al. 2008), developmental delay (Talkowski et al. 2012), and autism (Ziats and Rennert 2013). Although lncRNAs comprise an extensive class of noncoding transcripts, our general understanding of lncRNA function is still in its relative infancy. Nevertheless, the study of individual lncRNAs in the context of neural development and disease has provided fascinating and fundamentally important insights into the biological roles of this aspect of the noncoding genome.
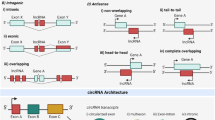
Like protein coding messenger RNAs (mRNAs), lncRNAs are transcribed from the genome by RNA Polymerase II, and many are also 5′ capped, spliced, and polyadenylated (Quinn and Chang 2015). lncRNA genes can have various genomic orientations (Fig. 1), with some overlapping protein-coding genes in the sense or antisense direction, others located between protein coding genes (intergenic), and a subset transcribed divergently from a neighboring gene through a shared promoter. While the genomic structure of lncRNAs can be further sub-classified (Mattick and Rinn 2015), the aforementioned basic differences in lncRNA gene location and orientation can provide some insight into potential mechanisms through which they might function.

Examples of possible genomic configurations of lncRNAs. a Antisense lncRNAs overlap other genes in the antisense direction, but lncRNAs can also overlap other genes in the sense direction. The overlapping regions may include exons, resulting in mature transcripts that share identical regions or have sequence complementarity, depending on the direction of overlap. b Non-overlapping lncRNAs can vary with respect to the distance to the nearest neighboring gene. Intergenic lncRNAs (lincRNAs) do not overlap known genes or regulatory elements, and may be transcribed in either direction relative to their nearest neighbors. A subset of intergenic lncRNAs known as divergent lncRNAs appear to share promoters (in yellow) with their closely neighboring genes, and are transcribed in the opposite direction
The currently known molecular mechanisms of lncRNA genes are very diverse, including the production of functional RNA transcripts (Rinn and Chang 2012), cis regulation of neighboring genes through lncRNA promoter activity and/or transcriptional elongation (Ørom et al. 2010; Kornienko et al. 2013; Li et al. 2013; Engreitz et al. 2016), and enhancer-like activity of lncRNA loci (Yin et al. 2015; Paralkar et al. 2016; Fulco et al. 2016). Furthermore, recent evidence indicates that certain transcripts currently annotated as lncRNAs can encode functional micropeptides (Andrews and Rothnagel 2014; Anderson et al. 2015; Nelson et al. 2016). Of note, these functions are not mutually exclusive, and any particular lncRNA gene might have multiple molecular roles. Thus, the use of different experimental approaches, such as knockdown of the lncRNA transcript versus genetic disruption of the lncRNA gene, may reveal different aspects of lncRNA function (Bassett et al. 2014) and could produce different phenotypes.
The expression of many lncRNAs is highly dynamic during CNS development and neural stem cell (NSC) differentiation (Mercer et al. 2010; Ramos et al. 2013), and in some cases can be regulated by neuronal activity (Lipovich et al. 2012; Barry et al. 2014). Furthermore, lncRNAs exhibit a high degree of cell-type specificity, even surpassing that of protein coding genes (Mercer et al. 2008; Cabili et al. 2011; Ramos et al. 2013; Liu et al. 2016). For instance, some lncRNAs are abundantly expressed in specific cell types, such as radial glia in the developing human brain (Liu et al. 2016). Large-scale screens reveal that essential lncRNA function appears to be surprisingly cell-type-specific, even for lncRNAs that are expressed across a wide range of cell types (Zhu et al. 2016; Liu et al. 2016). Therefore, experimental manipulations of lncRNAs through both large-scale screens and more in-depth individual analyses are key to advancing our understanding of the roles of lncRNAs in neural development and disease.
A growing number of lncRNAs have been found to regulate important biological functions in different neural cell types, and several recent reviews provide an excellent survey of such lncRNAs (Clark and Blackshaw 2014; Aprea and Calegari 2015; Briggs et al. 2015; Ramos et al. 2016; Hart and Goff 2016). In this review, we focus on a smaller subset of lncRNAs that have been experimentally manipulated in vivo in mice (Table 1). By discussing specific details of the experimental methods used to manipulate each of these lncRNAs, we hope to illustrate how studies of individual lncRNAs using multiple approaches can help reveal important insights regarding lncRNA function in both CNS development and diseases of the brain.
Overlapping, antisense lncRNAs
Numerous lncRNA genes overlap coding genes in an antisense direction (Fig. 1a). While some antisense lncRNAs seem to function primarily in cis—local to and dependent upon their site of transcription (Guil and Esteller 2012)—others appear to act in trans, independent from their site of transcription. Below, we review six antisense lncRNAs on a case-by-case basis with particular focus on the methods used to manipulate lncRNA expression. Despite these genes being classified together as antisense lncRNAs, the current data suggest that they function via distinct mechanisms.
-
BACE1-AS: potential disease significance through the stabilization of BACE1 mRNA.
The lncRNA BACE1-AS is transcribed antisense to β-site APP cleaving enzyme 1 (BACE1), a protease that plays a role in Alzheimer’s disease (AD) (Vassar et al. 1999; Vassar 2001) and is also required for multiple aspects of normal CNS function (Laird et al. 2005; Willem et al. 2006). In the human SH-SY5Y neuroblastoma cell line, BACE1-AS is detected in both the nucleus and cytoplasm (Faghihi et al. 2008). BACE1-AS knockdown (KD) with small interfering RNA (siRNA) decreases BACE1 mRNA and protein (Faghihi et al. 2008). Conversely, BACE1-AS overexpression (OE) increases BACE1 levels. Consistent with these results in human cell lines, continuous Bace1-AS siRNA infusion into the third ventricle of mice for 2 weeks reduces Bace1 mRNA and protein in the cortex, striatum, and hippocampus. Thus, BACE1-AS is a lncRNA that positively regulates its antisense partner, BACE1.
Both of the BACE1-AS splice variants contain sequences complementary to BACE1 exons (Faghihi et al. 2008), which raises the possibility that the BACE1-AS lncRNA physically interacts with BACE1 mRNA. Consistent with this, the overlapping regions of the lncRNA and mRNA transcripts resist degradation in RNase protection assays, suggesting that they form an RNA duplex. Furthermore, KD of BACE1-AS reduces BACE1 mRNA half-life, while OE of BACE1-AS increases the half-life of the mRNA. Together, these data support a model in which BACE1-AS forms a duplex with BACE1 mRNA to increase its stability, leading to increased protein production (Faghihi et al. 2008).
BACE1 is a protease that cleaves amyloid precursor protein (APP), and aberrant cleavage is associated with AD (Vassar et al. 1999; Vassar 2001). The pathogenic APP cleavage products exhibit an intriguing functional relationship with BACE1-AS expression. In HEK-SW cells that harbor an AD-linked mutation (Su et al. 2003), KD of BACE1-AS decreases the APP cleavage products amyloid β (Aβ) 1–40 and Aβ 1–42 (Faghihi et al. 2008), suggesting that BACE1-AS normally promotes APP cleavage. Interestingly, cultured cells treated with Aβ 1–42 protein upregulate BACE1-AS expression (Tamagno et al. 2006; Faghihi et al. 2008). Collectively, these results suggest a positive feedback mechanism in which Aβ 1–42 induces elevated BACE1-AS expression, which increases BACE1 levels and thereby further promotes production of Aβ 1–42.
The expression levels of BACE1-AS also suggests a role for this lncRNA in AD. In patients with AD, BACE1-AS expression averaged across multiple brain regions is 2-fold higher as compared to controls (Faghihi et al. 2008), which is an increase larger than that observed for BACE1. Additionally, transgenic APP-tg19959 mice, which have elevated levels of Aβ 1–42 and are considered an AD model (Oltersdorf et al. 1990), also exhibit increased expression of Bace1-AS and Bace1 (Faghihi et al. 2008). From a translational standpoint, it will be very interesting to understand the effects of Bace1-AS KD on Bace1 expression and Aβ 1–42 biogenesis in this transgenic mouse model, as this could further implicate BACE1-AS in the regulation of these aspects of AD pathology.
-
BDNF-AS: a regulator of the growth factor BDNF.
BDNF-AS is a lncRNA transcribed antisense to brain-derived neurotrophic factor (BDNF) (Liu et al. 2005), a secreted neurotrophic polypeptide that regulates neuronal differentiation, maturation, and survival (Yoshimura et al. 2009; Chapleau et al. 2009; Hasbi et al. 2009). Knockdown of BDNF-AS through siRNA in several human and mouse cell lines induces the upregulation of BDNF (Modarresi et al. 2012; Lipovich et al. 2012). Consistent with the role of BDNF in promoting neuronal differentiation (Binder and Scharfman 2004), this resultant increase in BDNF upon KD of Bdnf-AS enhances neuronal outgrowth in mouse hippocampal neurosphere cultures (Modarresi et al. 2012). Moreover, in the mouse brain, Bdnf-AS KD achieved via intraventricular infusion of antisense oligonucleotides (ASOs) also increases BDNF and correlates with an increase in cell proliferation in the hippocampal dentate gyrus (Modarresi et al. 2012), a region of adult neurogenesis (Gonçalves et al. 2016). Thus, BDNF-AS appears to be a negative regulator of BDNF levels in multiple cell lines as well as in the adult mouse brain.
BDNF-AS splice variants all have exonic complementarity with BDNF mRNA (Modarresi et al. 2012), indicating the potential for lncRNA–mRNA duplex formation, as is observed with BACE1-AS and BACE1 transcripts (Faghihi et al. 2008). However, in HEK293T cells, BDNF-AS KD does not alter the half-life of BDNF mRNA (Modarresi et al. 2012), indicating that the increase in BDNF transcript levels observed with BDNF-AS KD does not relate to a change in BDNF transcript stability.
Certain lncRNA transcripts interact with chromatin-modifying proteins and can influence the chemical and/or structural state of chromatin at specific loci (Rinn and Chang 2012; Mercer and Mattick 2013). In HEK293T cells, BDNF-AS KD decreases the levels of histone 3 lysine 27 trimethylation (H3K27me3)—a histone modification associated with transcriptional repression (Aranda et al. 2015)—at the BDNF locus (Modarresi et al. 2012). This change is accompanied by a decrease in the enrichment of the H3K27-methyltransferase enhancer of zeste homolog 2 (EZH2) (Viré et al. 2005). While these data suggest a role for BDNF-AS in regulating the chromatin state at the BDNF locus, whether BDNF-AS interacts with EZH2 or other chromatin-modifying complexes has not been reported. It is also possible that the observed chromatin state changes at the BNDF locus simply reflect increased local transcriptional activity that results from increased levels of BDNF, which can promote transcription of its own gene via autoregulatory feedback (Bambah-Mukku et al. 2014). Precisely how BDNF-AS functions remains to be fully elucidated, but results from the siRNA- and ASO-mediated lncRNA KD experiments suggest that the BDNF-AS transcript itself negatively regulates the transcription and/or chromatin state of its antisense partner.
-
Evf2: neighboring gene regulation both in cis and in trans.
With its transcriptional start site (TSS) located within the Dlx5/6 bigene cluster, the lncRNA Evf2 is transcribed antisense to Dlx6 and contains the entire Dlx6 gene within a large intron (Fig. 2a) (Feng et al. 2006). Evf2 also contains the ultraconserved Dlx5/6 intergenic enhancer ei (Feng et al. 2006). In the developing mouse brain, Evf2 transcript is localized to the nucleus, and its pattern of expression overlaps that of Dlx5/6 and Dlx1/2 in the medial and lateral ganglionic eminences (MGE and LGE, respectively) (Porteus et al. 1994; Feng et al. 2006). Given that the Dlx family of transcription factors plays critical roles in GABAergic interneuron production (Anderson et al. 1997b; Pleasure et al. 2000; Cobos et al. 2005), it has been of great interest to determine whether this lncRNA modulates the expression and/or function of the Dlx genes. In this section, we describe how our understanding of Evf2 has evolved over time with the use of different experimental approaches.

The genomic configurations of Evf2 and Dlx1as, and the genetic mouse lines created to disrupt their transcription. a The lncRNA Evf2 exists within the Dlx5/6 bigene cluster. The ei and eii enhancer elements are shown in yellow. Evf2 overlaps the ei enhancer element and contains the entire Dlx6 gene within an intron. Evf2 is transcribed from the same strand as Dlx5 and the opposite strand from Dlx6. b The Evf2 TS allele includes a 3× polyA transcriptional stop (red) in the first exon of Evf2 to terminate transcription before the ei enhancer element. c Dlx1as is transcribed antisense to Dlx1 and contains exonic sequences that overlap with Dlx1 exons. d The Dlx1as 4xPA allele includes a 4x polyA transcriptional stop (red) inserted before the second exon to terminate transcription before overlap with Dlx1
In early experiments performed in cell culture, Evf2 was found to exhibit activity as a DLX2 transcriptional coactivator (Feng et al. 2006). Dlx1/2 is genetically required for the expression of the Dlx5/6 bigene cluster (Anderson et al. 1997a, b; Zerucha et al. 2000), and transient transfection of Dlx2 expression plasmids can activate reporter constructs containing Dlx5/6 enhancers, which are known targets of DLX2 (Zerucha et al. 2000; Zhou et al. 2004). When co-transfected with Dlx2 into the C17 and MN9D mouse neural cell lines, Evf2 increases Dlx2-dependent activation of these transcriptional reporters (Feng et al. 2006). Furthermore, immunoprecipitation of DLX proteins from cultured cells and rat branchial arches (Kohtz and Fishell 2004) enriches for Evf2 transcripts (Feng et al. 2006), indicating that DLX2 and Evf2 physically interact in vivo. Taken together, these results suggested that Evf2 transcripts can directly augment the ability of DLX2 protein to activate its downstream targets.
This initial understanding of Evf2 as a transcriptional co-activator was developed primarily from studies of Evf2 expressed from plasmids in cultured cells. To study the role of Evf2 transcribed from its endogenous genomic locus, genetic targeting was used to deplete this lncRNA in mice. To avoid removal of genomic DNA that could disrupt the known local transcriptional enhancers, a triple polyadenylation (polyA) signal was inserted into Evf2 exon 1 (Evf2 TS) to terminate lncRNA transcription before the ei enhancer (Bond et al. 2009) (Fig. 2b). This genetic strategy ablated full-length Evf2 transcripts in homozygous Evf2 TS/TS mice.
In the MGE of Evf2 TS/TS mice, the binding of DLX transcription factors at the Dlx5/6 enhancers is reduced (Bond et al. 2009). Surprisingly, however, the reduction of DLX at these enhancers correlates with increased levels of Dlx5 and Dlx6. While it possible that the insertion of polyA signals at this particular location disrupts uncharacterized DNA regulatory elements, siRNA-mediated Evf2 KD in vivo also increases Dlx5 expression in the MGE, suggesting that loss of the Evf2 transcript itself underlies the observed transcriptional changes. Moreover, Evf2 introduced in trans through electroporation can partially reverse the elevated levels of Dlx5 in Evf2 TS/TS mice. These in vivo genetic studies suggest that Evf2 transcripts normally inhibit Dlx5 expression in trans.
In contrast, Evf2 appears to regulate the expression of Dlx6 in cis. Evf2 expressed in trans via plasmid electroporation does not reduce Dlx6 expression (Bond et al. 2009), suggesting that the increased levels of Dlx6 in Evf2 TS/TS mice may result from a disruption of cis regulatory mechanisms and thus cannot be rescued in trans. This apparent cis regulation of Dlx6—but not Dlx5—perhaps relates to the genomic structure of this locus. Given that Dlx6 exists within an intron of Evf2 (Fig. 2a), active Evf2 transcription might normally inhibit Dlx6 expression from the same chromosome via transcriptional interference, wherein transcription along one DNA strand blocks transcription from the opposite strand (Shearwin et al. 2005; Cech and Steitz 2014). In this scenario, early termination of Evf2 elongation in the Evf2 TS/TS mice could alleviate transcriptional interference of Dlx6. Taken together, results from the Evf2 TS/TS mice indicate that Evf2 can regulate the Dlx5/6 bigene cluster via both cis and trans mechanisms.
DNA methylation and methyl CpG binding protein 2 (MECP2) have been investigated as mechanistic players underlying Evf2 function. MECP2 is a DNA methyl-binding protein associated with transcriptional repression (Nan et al. 1998) and is found at the Dlx5/6 locus (Horike et al. 2004). Mecp2-null mice exhibit increased Dlx5/6 expression (Berghoff et al. 2013), suggesting that Mecp2 normally represses this locus. In the MGE of Evf2 TS/TS mice, MECP2 enrichment at Dlx5/6 enhancers is decreased (Bond et al. 2009), correlating with the increased transcriptional activity. These data suggest that Evf2 is required for the localization of MECP2 at Dlx5/6 regulatory regions.
Interestingly, Evf2 TS/TS mice have increased levels of DNA methylation at the Dlx5/6 enhancers, and transgenic Evf2 expression reduces this excess methylation (Berghoff et al. 2013). Thus, while Evf2 is necessary for the enrichment of MECP2 at the Dlx5/6 enhancers, Evf2 also appears to inhibit DNA methylation of these enhancers in trans. Based on these and previous results (Anderson et al. 1997a, b; Zerucha et al. 2000; Bond et al. 2009), the authors propose that Evf2 inhibits DNA methylation at Dlx5/6 regulatory regions, facilitating antagonistic interactions between MECP2 and DLX proteins (Berghoff et al. 2013). How the proposed interactions enable differential control of Dlx5 and Dlx6 remains to be demonstrated.
Moreover, Evf2 also appears to mediate interactions between DLX1 and the SWI/SNF-related chromatin remodeler brahma-related gene 1 (BRG1) (Wang et al. 1996; Cajigas et al. 2015). DLX1 and BRG1 physically interact and co-localize at the Dlx5/6 ei enhancer (Cajigas et al. 2015). Furthermore, Evf2 can directly interact with BRG1, and loss of Evf2 reduces BRG1 levels at the Dlx5/6 enhancers. While Evf2 inhibits BRG1 ATPase activity in assays using recombinant proteins (Cajigas et al. 2015), the functional consequences of Evf2-BRG1 interactions in vivo remain to be tested. However, considering the previous work, one possibility is that Evf2 promotes interactions between BRG1 and DLX1/2, which serve to activate the Dlx5/6 locus and counteract repression by MECP2.
Given that the Dlx genes play crucial roles in the production of forebrain GABAergic interneurons (Anderson et al. 1997b; Pleasure et al. 2000; Cobos et al. 2005), disruption of Evf2 might be expected to influence this developmental process. Indeed, loss of Evf2 results in a 45–60% reduction of GAD1+ GABAergic interneurons in the hippocampus of early postnatal Evf2 TS/TS mice (Bond et al. 2009). This phenotype correlates with a 30% reduction in Gad1 expression in the mutant embryonic MGE, which can be partially restored by electroporation of Evf2. Surprisingly, the number of GAD1-expressing cells returns to normal in the adult Evf2 TS/TS hippocampus (Bond et al. 2009). However, impaired synaptic inhibition in the hippocampus persists throughout adulthood, demonstrating that the disruption of a neural lncRNA can lead to long-lasting changes in CNS function.
-
Dlx1as: a role in interneuron production.
Similar to Dlx5/6, the Dlx1/2 bigene cluster also contains a lncRNA gene, Dlx1as (McGuinness et al. 1996; Liu et al. 1997; Dinger et al. 2008; Jeong et al. 2008; Kraus et al. 2013). Transcribed antisense to Dlx1, Dlx1as overlaps the majority of the Dlx1 gene (Fig. 2c). However, unlike the Evf2-Dlx6 configuration, Dlx1as is not transcribed through the TSS of its coding gene partner Dlx1. To genetically disrupt Dlx1as expression in mice, four polyA signals were inserted between exons 1 and 2 (Dlx1as 4xPA) (Kraus et al. 2013) (Fig. 2d), promoting the termination of Dlx1as transcription before its overlap with Dlx1. However, despite the multiple polyA signals, Dlx1as expression is not completely ablated: in homozygous Dlx1as 4xPA/4xPA mice, Dlx1as is expressed at ~30% of wild-type levels.
Dlx1as is normally expressed in the embryonic ganglionic eminences (GEs) (Liu et al. 1997) in a pattern similar to that of Dlx1/2 (Porteus et al. 1994). In the GEs of Dlx1as 4xPA/4xPA embryos, Dlx1 transcript levels are moderately (~40%) increased (Kraus et al. 2013), suggesting that Dlx1as normally represses Dlx1 expression. Although postnatal Dlx1as 4xPA/4xPA mice have a 2-fold increase in the number of Dlx1+ cells in the hippocampus, the number of interneurons as assessed by Gad67 and Somatostatin expression was not altered in the mutant mice (Kraus et al. 2013). Given that overexpression of Dlx1 alone does not alter the number of GABAergic interneurons in the mouse brain (Hitoshi et al. 1991), it is perhaps not surprising that Dlx1as 4xPA/4xPA mice do not exhibit obvious changes in this population of neurons. Whether the development of other neuronal (and glial) cell types is affected by Dlx1as genetic disruption remains to be reported.
Dlx1as is also expressed in the neurogenic lineage of the postnatal mouse ventricular-subventricular zone (V-SVZ) (Dinger et al. 2008; Ramos et al. 2013). In cultured V-SVZ neural stem cells, KD of Dlx1as with short hairpin RNA (shRNA) decreases the expression of Dlx1, Dlx2 and Dlx5 during differentiation (Ramos et al. 2013). These transcriptional changes correlate with a substantial decrease in the production of young neurons.
The contrasting effects upon gene expression in the embryonic brain of Dlx1as 4xPA/4xPA mice and V-SVZ cell cultures with Dlx1as KD could reflect potential cell type-specific functions of Dlx1as. In a recent genome-scale survey, lncRNA loci were found to have exquisitely cell type-specific functions for cell growth and proliferation (Liu et al. 2017). Alternatively, the differences observed may relate to the approaches used to reduce Dlx1as levels. In Dlx1as 4xPA/4xPA mice, transcriptional elongation of Dlx1as is abrogated, whereas, with Dlx1as KD, transcript levels are depleted without a direct effect upon transcription at the locus. Since Dlx1as overlaps Dlx1 (Fig. 2c), transcription of this lncRNA could potentially reduce expression of Dlx1 through transcriptional interference (Shearwin et al. 2005). Such repression in cis could be relieved by the transcriptional termination in Dlx1as 4xPA/4xPA mice but not by shRNA-mediated Dlx1as KD in V-SVZ NSCs. Additional approaches such as Dlx1as OE and attempted transgenic rescue of Dlx1as 4xPA/4xPA may be helpful in addressing these and other possibilities.
The mechanism by which Dlx1as regulates Dlx1 expression is still unclear, but, given that Evf2 can regulate its neighboring coding genes via both cis and trans mechanisms (Bond et al. 2009), it may be important to also address both possibilities for Dlx1as. Since Dlx1/2 are required for the expression of Evf2 (Anderson et al. 1997a; Zerucha et al. 2000), Dlx1as may indirectly affect Evf2 expression through regulating Dlx1. Interestingly, genetic deletion of Dlx5 or Dlx6 decreases Dlx1as expression in the developing branchial arches (Jeong et al. 2008). Given that Evf2 regulates the expression of both Dlx5 and Dlx6 (Bond et al. 2009), Evf2 may also indirectly regulate Dlx1as, adding an additional layer to the much-studied cross-talk between the Dlx1/2 and Dlx5/6 bigene clusters (McGuinness et al. 1996; Anderson et al. 1997b; Liu et al. 1997; Zerucha et al. 2000).
-
KCNA2-AS: implications for neuropathic pain.
KCNA2-AS is an antisense lncRNA that overlaps most of the KCNA2 gene, which encodes a voltage-gated potassium channel (Zhao et al. 2013). Decreased expression of KCNA2 correlates with neuropathic pain (Ishikawa et al. 1999; Kim et al. 2002), which can arise from damage to peripheral nerves (Campbell and Meyer 2006). While KCNA2-AS has not been studied in the context of the CNS, its role in the peripheral nervous system (PNS) highlights how a lncRNA might serve as a therapeutic target for an important neurological condition.
KCNA2-AS and KCNA2 are both expressed in the dorsal root ganglia (DRG) of rat, mouse, monkey, and human (Zhao et al. 2013). In the rat DRG, Kcna2-AS-positive neurons generally express low levels of Kcna2. Peripheral nerve injury caused by lumbar spinal nerve ligation (SNL) increases Kcna2-AS expression in neurons of the ipsilateral DRG, and these cells with elevated levels of Kcna2-AS exhibit decreased expression of Kcna2 (Zhao et al. 2013). These dynamic, reciprocal changes in Kcna2-AS and Kcna2 levels suggest a model in which expression of the Kcna2-AS lncRNA normally represses the expression of its antisense gene partner, Kcna2.
The transcription factor myeloid zinc finger 1 (MZF1) (Luo et al. 2009) binds to the Kcna2-AS promoter, and upon SNL, MZF1 levels increase and become further enriched at the Kcna2-AS promoter (Zhao et al. 2013). Moreover, MZF1 OE in cultured rat DRG neurons increases Kcna2-AS, while Kcna2 levels are decreased. Taken together, these data support a model in which peripheral nerve injury increases the expression of MZF1, which directly upregulates Kcna2-AS expression, thereby decreasing the expression of Kcna2. Consistent with this model, Kcna2-AS OE is sufficient to decrease Kcna2; however, the mechanism by which this lncRNA can downregulate its antisense coding gene partner in trans has not been reported.
In vivo, OE of Kcna2-AS in the rat DRG also decreases levels of Kcna2, resulting in altered voltage-gated potassium current density and resting membrane potentials (Zhao et al. 2013). These electrophysiological changes correspond to behavioral abnormalities associated with neuropathic pain, such as mechanical and cold hypersensitivity. Thus, increased levels of Kcna2-AS transcript appear to drive neuropathic pain via suppression of Kcna2 expression. Supporting this notion, KD of Kcna2-AS prior to SNL blocks the downregulation of Kcna2 normally observed after peripheral nerve injury. Importantly, Kcna2-AS KD also attenuates the neuropathic pain symptoms of mechanical, cold, and thermal hypersensitivity. These data raise the intriguing possibility that KD of Kcna2-AS can ameliorate—or perhaps even prevent—neuropathic pain symptoms that arise from injury to the peripheral nerves.
-
Ube3a-ATS: a role in imprinting with relevance to Angleman syndrome.
Some of the earliest-studied lncRNAs are involved in the regulation of gene dosage (Mohammad et al. 2009). The classic example is the lncRNA XIST, which is required for X-chromosome inactivation in female cells (Brown et al. 1992; Penny et al. 1996; Plath et al. 2002; Engreitz et al. 2013). Other lncRNAs play a role in imprinting, a process by which autosomal genes are expressed from a single allele in a parent of origin-specific manner (Bartolomei and Ferguson-Smith 2011). For instance, the lncRNA Air interacts with the histone methyltransferase G9a to enable imprinting of its neighboring genes in the mouse placenta (Nagano et al. 2008). Similarly, Kcnq1ot1 interacts with G9a as well as Polycomb repressive complex 2 (PRC2) to mediate imprinting (Pandey et al. 2008; Terranova et al. 2008). Investigating how lncRNAs regulate imprinting may be useful for understanding diseases caused by abnormal gene dosage, and could potentially lead to strategies for the reactivation of silenced alleles for therapeutic purposes.
Ube3a-ATS is a nuclear, neural-expressed antisense lncRNA that is involved in the repression of paternal Ube3a (Meng et al. 2012), which is imprinted specifically in neurons (Rougeulle et al. 1997; Vu and Hoffman 1997; Yamasaki et al. 2003). Ube3a-ATS is part of an extremely long transcript that extends over 1000 kb through multiple genes including the 3′ end of Ube3a. The relationship between Ube3a-ATS and Ube3a is of particular interest because of its implications for Angleman syndrome, a disorder characterized by neurological symptoms including seizures and intellectual disability. Angleman syndrome is caused by mutations in the maternal UBE3A allele, which results in complete loss of functional UBE3A protein in neurons due to silencing of the intact paternal copy (Kishino et al. 1997; Matsuura et al. 1997; Albrecht et al. 1997). Therefore, a potential therapeutic strategy is to reduce levels of UBE3A-ATS, thus relieving silencing of the normal paternal UBE3A (Meng et al. 2013, 2015).
In a mouse model of Angleman syndrome, genetic and pharmacological reduction of Ube3a-ATS expression has shown therapeutic promise. Premature termination of Ube3a-ATS via the insertion of a triple polyA cassette relieves silencing of the paternal Ube3a allele, rescuing neurological deficits associated with Angleman syndrome (Meng et al. 2013). Moreover, a single intraventricular administration of ASOs targeting Ube3a-ATS reduces levels of this lncRNA in the brain, partially de-repressing paternal Ube3a for 4 months (Meng et al. 2015). This ASO treatment even ameliorates certain Angleman syndrome-associated phenotypes, such as aberrant contextual fear behavior. Given the sustained lncRNA KD achieved with a single intraventricular injection of ASOs, the targeting of neural lncRNAs with ASOs may be clinically feasible, at least for those lncRNAs that are expressed in non-dividing neurons.
Non-overlapping lncRNAs
Non-overlapping lncRNAs can vary greatly in terms of their distance from their nearest neighboring gene, with some only a few bases apart and others separated by megabases (Cabili et al. 2011) (Fig. 1b). A subset of lncRNAs can carry out enhancer-like roles (Ørom et al. 2010), and the act of transcription of some lncRNAs appears to facilitate the expression of their close gene neighbors (Engreitz et al. 2016). However, even in these cases where non-overlapping lncRNAs have roles in cis, it is still possible that the lncRNA transcript itself has additional functions, and work from mouse ES cells suggests that many lncRNAs function in trans (Guttman et al. 2011).
-
Six3OS: a mediator of retinal progenitor cell differentiation.
The lncRNA Six3OS (originally Rncr1) is transcribed divergently from Six3, a transcription factor that is critical for eye development (Zhu et al. 2002). Six3OS is largely co-expressed with its coding gene partner in cells of the developing mouse retina (Blackshaw et al. 2004; Alfano et al. 2005; Geng et al. 2007; Rapicavoli et al. 2011). In situ hybridization (ISH) reveals Six3OS transcripts in both the nucleus and cytoplasm of embryonic retinal progenitor cells (RPCs) (Rapicavoli et al. 2011).
There are multiple isoforms of Six3OS, and OE of the most abundant isoform in perinatal RPCs via in vivo plasmid electroporation indicates a functional relationship between this lncRNA and Six3. RPCs are multipotent, giving rise to rod cells, bipolar cells, amacrine cells, and Müller glia (Cepko 2014). Six3OS OE in RPCs has a relatively modest effect, reducing the proportion of syntaxin (syn)-expressing amacrine cells (Rapicavoli et al. 2011). In contrast, as previously reported (Zhu et al. 2002), Six3 OE in RPCs produces a much broader range of phenotypes, affecting the rod bipolar cells and rod photoreceptors, in addition to increasing the proportion of syn+amacrine cells (Rapicavoli et al. 2011). Interestingly, when Six3OS is co-electroporated with Six3, these Six3-induced phenotypes are not observed. These results indicate that, at least in the context of overexpression, Six3OS can counteract the effects of Six3.
Interestingly, shRNA-mediated KD of Six3 and Six3OS generates results that would not have been predicted by the phenotypes of their OE (Rapicavoli et al. 2011). Despite the opposing effects of Six3OS and Six3 in the context of overexpression, Six3OS KD produces phenotypes similar to those observed with Six3 KD. Furthermore, combined KD of Six3OS and Six3 reveals complex functional relationships between this lncRNA and its coding gene neighbor. For example, some retinal cell populations that are not affected by KD of either Six3OS or Six3 alone are altered upon their combined KD (Rapicavoli et al. 2011). Surprisingly, in other cell populations in which the single KDs result in similar phenotypes, the combined KD actually rescues these effects. These complex, non-additive phenotypes suggest an epistatic association between these two genes and demonstrate the elaborate relationship that a lncRNA can share with its neighbor.
How Six3OS functionally interacts with Six3 is still not clear. Genetic deletion of Six3 does not alter expression of Six3OS (Geng et al. 2007), indicating that neither an intact Six3 locus nor SIX3 protein are required for expression of the Six3OS lncRNA. Given that neither KD nor OE of Six3OS transcript affects SIX3 protein abundance (Rapicavoli et al. 2011), it appears that Six3OS can modulate the function of Six3 without changing the levels of SIX3 protein. It remains to be tested whether the Six3OS locus itself can regulate Six3 expression in cis, potentially via enhancer-like activity (Groff et al. 2016).
While Six3OS was not found to directly interact with SIX3 protein, both RNA immunoprecipitation and protein microarray analyses indicate that Six3OS binds to the EYA family of proteins as well as EZH2 (Rapicavoli et al. 2011). EYA family proteins are important transcriptional co-regulators in retinal development (Bonini et al. 1993; Pignoni et al. 1997) and can interact directly with proteins of the SIX family (Jemc and Rebay 2007). However, it remains to be determined how the Six3OS interactions with EYA and EZH2 might influence cell fate outcomes.
Six3OS is also expressed in V-SVZ NSCs and is downregulated during neuronal differentiation (Ramos et al. 2013). In V-SVZ cultures, shRNA-mediated KD of Six3OS results in 2-fold fewer neuroblasts and 3-fold fewer oligodendrocyte lineage cells, accompanied by a corresponding increase in the proportion of glial fibrillary acidic protein (GFAP)-expressing glial cells. Thus, similar to the studies from the retina (Rapicavoli et al. 2011), these results suggest that Six3OS can regulate cell fate decisions in neural precursor cells of the CNS.
-
Pnky: a regulator of neurogenesis from postnatal and embryonic neural stem cells.
The intergenic lncRNA Pnky neighbors the gene Brn2 (Pou3f2) (Ramos et al. 2013, 2015), which encodes a transcription factor with key roles in neocortical development (Sugitani et al. 2002) and direct neuronal reprogramming (Vierbuchen et al. 2010). Pnky and Brn2 are separated by 2.2 kb and are transcribed in divergent directions, and therefore do not share any overlapping regions (Ramos et al. 2015).
Pnky is neural-specific, and is enriched in the nucleus of V-SVZ NSCs (Ramos et al. 2013, 2015). Throughout postnatal and adult life, mouse V-SVZ NSCs give rise to transit-amplifying cells that divide two to three times before generating neuroblasts (Doetsch et al. 1999; Lim and Alvarez-Buylla 2014). These neuroblasts migrate through the rostral migratory stream to the olfactory bulb where they differentiate into interneurons (Lim and Alvarez-Buylla 2014). Over this course of V-SVZ neuronal differentiation, levels of Pnky transcript decrease (Ramos et al. 2013, 2015).
In V-SVZ NSC cultures, shRNA-mediated KD of Pnky increases neuroblast production 3- to 4-fold, indicating that Pnky influences V-SVZ neurogenesis (Ramos et al. 2015). Time-lapse microscopy analysis of individual V-SVZ NSCs reveals that Pnky regulates multiple aspects of V-SVZ neurogenesis. Firstly, Pnky KD increases neurogenic commitment by ~50% (Ramos et al. 2015). Furthermore, with Pnky KD, the transit-amplifying population undergoes additional rounds of cell division. Combined, these effects greatly increase the number of neuroblasts produced from this population of postnatal NSCs.
Pnky also regulates neurogenesis from embryonic NSCs (Ramos et al. 2015). The cortical ventricular zone—where embryonic NSCs are located—is enriched for Pnky transcripts in both the mouse and human. Pnky KD in mouse embryonic ventricular zone cells via in utero electroporation of shRNA constructs promotes neurogenic differentiation in vivo, increasing the proportion of SATB2+ young neurons while decreasing the SOX2+ NSC population. Thus, reducing Pnky transcript levels in NSCs from both the embryonic and postnatal brain promotes neuronal differentiation.
This increase in neurogenesis observed with Pnky KD suggests a role for Pnky that is distinct from several other lncRNAs known to have neurodevelopmental function. For instance, KD of lncRNAs megamind (Lin et al. 2014), RMST (Ng et al. 2012, 2013), Six3OS (Rapicavoli et al. 2011; Ramos et al. 2013) and Dlx1as (Ramos et al. 2013) can all decrease neurogenesis, suggesting that these other lncRNAs are required to positively regulate neuronal differentiation. Similarly, genetic deletion of Linc-Brn1b appears to decrease the proliferation of embryonic cortical intermediate progenitors, reducing upper layer neurogenesis (Sauvageau et al. 2013). As noted above, mice lacking Evf2 have defective interneuron production (Bond et al. 2009). Thus, unlike the aforementioned lncRNAs that appear to potentiate neuronal production, Pnky appears to restrain neurogenesis from NSCs, perhaps serving to control their long-term self-renewal and/or rate of neuronal production.
While Pnky is relatively close to its neighbor Brn2, both genes appear to have their own promoters: each TSS region has a separate conserved CpG island that exhibits dynamic chromatin-state changes characteristic of gene promoters (Ramos et al. 2013), including the H3K4me3 modification associated with transcriptional activity as well as the repression-associated H3K27me3 (Venkatesh and Workman 2015) (Fig. 3). Furthermore, while Pnky expression decreases during neurogenesis, Brn2 transcript levels increase in transit-amplifying cells (Ramos et al. 2013), suggesting that these gene neighbors are not always co-expressed.

The lncRNA Pnky is transcribed in a diverging manner from its nearest neighboring gene, Pou3f2 (Brn2). While Pnky and Pou3f2 are relatively close, they appear to have independent promoters as indicated by separate conserved CpG islands that exhibit dynamic chromatin modifications. In embryonic stem cells (ESC), these promoters are enriched with both the H3K4me3 modification, which correlates with active transcription, and the repression-associated H3K27me3. In V-SVZ NSCs, only H3K4me3 is enriched, demonstrating cell type-specific chromatin modifications at these regions
Given that some lncRNAs can regulate the expression of gene neighbors in cis, one possibility is that Pnky regulates Brn2 expression. However, KD of Pnky in V-SVZ cultures does not significantly alter expression of Brn2 or any of the other genes within a 5-MB window, suggesting that the Pnky transcript does not regulate gene expression in cis, at least in this experimental context (Ramos et al. 2015). Whether the act of Pnky transcription (and/or the Pnky locus itself) can regulate expression of Brn2 or other neighboring genes remains to be shown.
Pnky transcripts are located in multiple foci throughout the nucleus of neural cells (Ramos et al. 2015). The presence of Pnky transcripts in more than two spatially disparate locations indicates that this lncRNA can localize in regions of the nucleus that are far from the two Pnky loci, suggesting that this lncRNA has trans roles. Many lncRNAs with trans function regulate gene expression through interaction with specific proteins (Yang et al. 2015). Biotinylated-Pnky pulldown as well as RNA immunoprecipitation analysis reveal that polypyrimidine tract-binding protein 1 (PTBP1) (Keppetipola et al. 2012) interacts with Pnky in cultured V-SVZ NSCs (Ramos et al. 2015). Interestingly, the lncRNAs megamind (Lin et al. 2014), MEG3 (Zhang et al. 2017), and XIST (Maenner et al. 2010) also interact with PTBP1, but the potential significance of these other lncRNA–PTBP1 interactions has not been reported.
PTBP1 is localized in the nucleus of NSCs and appears to function as a repressor of neuronal differentiation. In the embryonic brain, genetic deletion of Ptbp1 results in precocious neuronal differentiation (Shibasaki et al. 2013), and KD of PTBP1 alone in fibroblasts leads to direct neuronal trans-differentiation (Xue et al. 2013). In V-SVZ NSC cultures, PTBP1 KD increases the size of neurogenic colonies, which is a phenotype similar to that of Pnky KD (Ramos et al. 2015). The individual KDs of either PTBP1 or Pnky produce gene expression changes as well as mRNA splicing changes that are highly similar, suggesting that this splicing factor and lncRNA regulate a common set of transcripts in NSCs. Furthermore, double KD of PTBP1 and Pnky does not have a synergistic or additive effect upon transcriptional changes observed with the single KDs, suggesting that Pnky and PTBP1 function in the same molecular pathway. Taken together, these data indicate that Pnky and PTBP1 physically and genetically interact and regulate proper splicing and gene expression in V-SVZ NSCs.
-
Gomafu: from retinal development to schizophrenia.
Gomafu (also known as MIAT or Rncr2) was identified as a lncRNA that is dynamically expressed during retinal development (along with Six3OS, see above) (Blackshaw et al. 2004). Gomafu is intergenic, located 20 kb away from its nearest protein-coding neighbor (Blackshaw et al. 2004), and is expressed widely in the developing nervous system, localizing to multiple foci within the nucleus (Sone et al. 2007).
In the developing mouse retina, Gomafu is expressed in a large proportion of the progenitor cells, with its expression decreasing postnatally and becoming undetectable by adulthood (Rapicavoli et al. 2010). In vivo electroporation experiments indicate developmental roles for Gomafu in RPCs (Rapicavoli et al. 2010). While OE of Gomafu does not produce any overt phenotype, shRNA-mediated KD of this lncRNA increases the production of Müller glia and amacrine cells, suggesting that Gomafu normally represses these neural cell fates.
Given that the Gomafu lncRNA is normally found in the nucleus, the mis-localization of Gomafu transcripts to the cytoplasm might be expected to have a dominant-negative effect via the sequestration of its nuclear binding partners in the cytoplasmic compartment (Zhang et al. 2004; Miyazaki et al. 2007). Indeed, Gomafu transcripts fused to IRES-GFP sequences localize in the cytoplasm, and OE of this Gomafu-IRES-GFP construct in the developing retina causes phenotypes similar to those observed with Gomafu KD (Rapicavoli et al. 2010). As OE of Gomafu alone does not have an observable phenotype, the effect of Gomafu-IRES-GFP OE has been attributed to this mis-localization of Gomafu sequences. Transfection of Gomafu-IRES-GFP fusion constructs also enabled the dissection of distinct functional domains of Gomafu. For instance, when fused to IRES-GFP, OE of the 5′ end increases the proportion of amacrine cells while the 3′ end increases the Müller glial cell population (Rapicavoli et al. 2010). Thus, distinct regions of the Gomafu transcript appear to regulate different aspects of RPC fate determination, perhaps through interactions with unique binding partners.
Since lncRNAs exhibit relatively low evolutionary conservation at the primary sequence level (Johnsson et al. 2014; Ulitsky 2016), any regions of conservation may provide insights into lncRNA function. Gomafu orthologs identified in mouse, human, chicken, and Xenopus all contain multiple consensus recognition sites for the splicing factor quaking (QKI) (Rapicavoli et al. 2010). Furthermore, Gomafu physically interacts with QKI (Barry et al. 2014) and other splicing factors including serine/arginine-rich splicing factor 1 (SRSF1) (Barry et al. 2014) and splicing factor 1 (SF1) (Tsuiji et al. 2011).
Since QKI has been implicated in schizophrenia (Aberg et al. 2006a, b), an interaction between GOMAFU and QKI could be relevant for this disorder. Interestingly, KD of GOMAFU in human iPSC-derived neurons increases the levels of the DISC1 and ERBB4 splice variants that have been associated with schizophrenia (Morikawa and Manabe 2010; Barry et al. 2014), while GOMAFU OE in these cells has the opposite effect (Barry et al. 2014). In schizophrenia patient samples, GOMAFU levels are reduced, further suggesting a connection between GOMAFU dysregulation and aberrant RNA splicing in schizophrenia (Barry et al. 2014). Additionally, GOMAFU expression is regulated by neuronal activity, becoming significantly decreased with depolarization (Barry et al. 2014). Thus, while GOMAFU downregulation may be important for splicing changes in the context of normal neuronal activity, its constitutive downregulation could be relevant to the neuropathology underlying schizophrenia.
Gomafu has also been implicated in anxiety-related behaviors in mice (Spadaro et al. 2015). Upon fear conditioning, Gomafu is significantly downregulated in the medial prefrontal cortex. Moreover, Gomafu KD through ASO infusion into the prefrontal cortex enhances freezing behavior during fear conditioning. This in vivo KD also increases behaviors related to anxiety, such as stereotypic grooming and avoidance of an open field. In primary cortical neuron cultures, KD of Gomafu upregulates Crybb1 (Spadaro et al. 2015), a chaperone protein that has been associated with schizophrenia (Gill et al. 1996; Takahashi et al. 2003). Interestingly, in these cultures, Gomafu physically interacts with the PRC1 complex member BMI1 (Haupt et al. 1991; Meng et al. 2010), and KD of Gomafu reduces BMI1 occupancy at the Crybb1 promoter (Spadaro et al. 2015). Therefore, Gomafu may directly regulate Crybb1 expression through a physical interaction with the repressor BMI1. Taken together, these results demonstrate that Gomafu can modulate neuronal function and potentially impact neuropsychiatric disorders through multiple molecular mechanisms.
Recently, a Gomafu knockout mouse has been reported (Ip et al. 2016). Surprisingly, loss of Gomafu did not result in any overt developmental phenotypes in the hippocampus, despite the high level of Gomafu expression reported in hippocampal CA1 neurons (Sone et al. 2007). This could reflect different requirements for Gomafu in different cell types, or developmental compensation by parallel genetic pathways. Alternatively, this could be due to differences resulting from constitutive deletion of the entire locus as opposed to acute knockdown of the transcript. Nevertheless, these mice did present moderate behavioral abnormalities, including a hyperactivity phenotype that could be exacerbated by treatment with the psychostimulant methamphetamine (Ip et al. 2016). Given that these mice exhibit behavioral defects without any obvious developmental aberrations, it is possible that a relatively subtle effect on neurodevelopment and/or change in neuronal function underlies the behavioral abnormalities. These considerations may be particularly important for the study of complex neuropsychiatric diseases that are difficult to fully model in mice.
Concluding remarks
From this survey of the data regarding the function of a small set of neural lncRNAs, it is apparent that this class of noncoding transcripts is extremely diverse in terms of biological roles and molecular mechanisms. This diversity, while intriguing, presents a challenge for determining the function of lncRNAs, as it is currently difficult to predict whether a particular lncRNA has important cellular functions, much less how that lncRNA might operate at the level of molecular mechanism. Further complicating these issues, it appears that certain lncRNAs—despite being expressed in many different cell types—have biological functions that are exquisitely cell-type-specific (Liu et al. 2017). Thus, manipulating lncRNA expression can lead to complex phenotypes that vary with the cell type being analyzed and the timing of the lncRNA perturbation. Using a combination of different, complementary strategies may therefore prove especially important for revealing the functions of lncRNAs.
Given the extraordinary diversity of lncRNA structure and function, it can be useful to sub-set this class of noncoding transcripts based on certain aspects of their genomic structure. Many antisense lncRNAs affect the expression of their partner genes transcribed from the opposite strand. However, despite this functional similarity, the mechanisms by which such regulation is achieved can be quite distinct. For example, Evf2 regulates the expression of its gene neighbors, Dlx5 and Dlx6, via both cis and trans mechanisms (Bond et al. 2009), while BACE1-AS transcripts interact directly with the BACE1 mRNA, influencing mRNA stability (Faghihi et al. 2008). Thus, while it might be reasonable to hypothesize that antisense lncRNAs regulate their partner genes, it is currently not possible to predict how they might do so.
Non-overlapping lncRNAs are perhaps even more challenging to decipher, particularly when they do not include any known genomic regulatory elements. Even when they are close to another gene, lncRNAs may not have any regulatory relationship with that neighbor. Given the many different possibilities, determining the molecular mechanism through which an intergenic lncRNA functions can be quite difficult. Therefore, an initial genetic characterization of whether the lncRNA acts in cis or in trans (or both) may provide a crucial foundation for developing any mechanistic understanding of how the lncRNA functions.
In this review, we have focused on a small set of lncRNAs in order to provide a more in-depth discussion of their neural functions. However, there are several other lncRNAs that also have important roles in the nervous system. For instance, Taurine Upregulated Gene 1 (Tug1) is required for proper photoreceptor differentiation (Young et al. 2005) and interacts with the PRC2 complex to repress p53-dependent cell cycle regulation (Khalil et al. 2009). LncOL1 also interacts with components of the PRC2 complex to promote oligodendrocyte maturation and is required for proper myelination as well as remyelination following injury (He et al. 2017). Notch signaling in the developing human cortex is mediated by LncND, which sequesters the microRNA miR-143-3p to regulate the expression of Notch receptors (Rani et al. 2016). RMST influences the differentiation of human embryonic stem cells into dopaminergic neurons through interacting with SOX2 and enabling its binding to specific target promoters (Ng et al. 2012, 2013). Cyrano and megamind (also known as TUNA) are required for proper neural development in zebrafish, and their loss can be rescued by their human or mouse orthologs (Ulitsky et al. 2011). Moreover, megamind expression is altered in Huntington’s disease, with megamind levels exhibiting a negative correlation with disease grade (Lin et al. 2014). Additionally, a set of knockout mice for 18 intergenic lncRNAs has been reported (Sauvageau et al. 2013). This set includes Linc-Brn1b, which is expressed in the neural progenitors of the VZ and SVZ in the developing embryonic cortex. Linc-Brn1b appears to positively regulate the expression of its neighboring gene Brn1, a neurogenic transcription factor, and loss of Linc-Brn1b reduces the proliferation of intermediate progenitors and decreases the production of upper layer neurons (Sauvageau et al. 2013). When this set of knockout mice is further analyzed, additional lncRNAs with neural functions may be identified.
As the lncRNA field continues to mature, additional approaches will be used to manipulate lncRNA expression. For some lncRNAs, a conditional knockout allele will allow targeting of a particular subset of cells with precise control over the timing of deletion. For example, given that Pnky appears to function in trans and does not overlap known genes or enhancers (Ramos et al. 2015), this lncRNA may be suitable for conditional knockout methods. For other lncRNAs, this traditional genetic approach may not be possible due to their complex genomic configurations, such as overlapping important coding genes and/or enhancers. In these cases, perhaps CRISPR interference-based approaches can be used to affect the expression of the lncRNA while avoiding any alterations to the underlying DNA (Liu et al. 2017). Methods that specifically target the lncRNA transcript, such as KD through shRNA or ASOs, may assist in distinguishing the function of the transcript from potential regulatory roles of the locus itself. BAC transgenics could also be used to assess lncRNA overexpression or to attempt to rescue loss of the endogenous lncRNA. Combining multiple complementary approaches will provide important insights into how lncRNAs carry out their roles. Moving forward, a precise understanding of the intricate molecular mechanisms through which lncRNAs function will be essential for revealing how specific lncRNAs can play important roles in neural development and disease.
References
Aberg K, Saetre P, Jareborg N, Jazin E (2006a) Human QKI, a potential regulator of mRNA expression of human oligodendrocyte-related genes involved in schizophrenia. Proc Natl Acad Sci U S A 103:7482–7487
Aberg K, Saetre P, Lindholm E, Ekholm B, Pettersson U, Adolfsson R, Jazin E (2006b) Human QKI, a new candidate gene for schizophrenia involved in myelination. Am J Med Genet Part B Neuropsychiatr Genet 141B:84–90
Albrecht U, Sutcliffe JS, Cattanach BM, Beechey CV, Armstrong D, Eichele G, Beaudet AL (1997) Imprinted expression of the murine Angelman syndrome gene, Ube3a, in hippocampal and Purkinje neurons. Nat Genet 17:75–78
Alfano G, Vitiello C, Caccioppoli C, Caramico T, Carola A, Szego MJ, McInnes RR, Auricchio A, Banfi S (2005) Natural antisense transcripts associated with genes involved in eye development. Hum Mol Genet 14:913–923
Anderson SA, Eisenstat DD, Shi L, Rubenstein JL (1997a) Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science 278:474–476
Anderson SA, Qiu M, Bulfone A, Eisenstat DD, Meneses J, Pedersen R, Rubenstein JL (1997b) Mutations of the homeobox genes Dlx-1 and Dlx-2 disrupt the striatal subventricular zone and differentiation of late born striatal neurons. Neuron 19:27–37
Anderson DM, Anderson KM, Chang C-L, Makarewich CA, Nelson BR, McAnally JR, Kasaragod P, Shelton JM, Liou J, Bassel-Duby R, Olson EN (2015) A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 160:595–606
Andrews SJ, Rothnagel JA (2014) Emerging evidence for functional peptides encoded by short open reading frames. Nat Rev Genet 15:193–204
Aprea J, Calegari F (2015) Long non-coding RNAs in corticogenesis: deciphering the non-coding code of the brain. EMBO J 34:2865–2884
Aranda S, Mas G, Di Croce L (2015) Regulation of gene transcription by Polycomb proteins. Sci Adv 1:e1500737–e1500737
Bambah-Mukku D, Travaglia A, Chen DY, Pollonini G, Alberini CM (2014) A positive autoregulatory BDNF feedback loop via C/EBPβ mediates hippocampal memory consolidation. J Neurosci 34:12547–12559
Barry G, Briggs JA, Vanichkina DP, Poth EM, Beveridge NJ, Ratnu VS, Nayler SP, Nones K, Hu J, Bredy TW, Nakagawa S, Rigo F, Taft RJ, Cairns MJ, Blackshaw S, Wolvetang EJ, Mattick JS (2014) The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol Psychiatry 19:486–494
Bartolomei MS, Ferguson-Smith AC (2011) Mammalian genomic imprinting. Cold Spring Harb Perspect Biol 3:a002592–a002592
Bassett AR, Akhtar A, Barlow DP, Bird AP, Brockdorff N, Duboule D, Ephrussi A, Ferguson-Smith AC, Gingeras TR, Haerty W, Higgs DR, Miska EA, Ponting CP (2014) Considerations when investigating lncRNA function in vivo. elife 3:e03058
Batista PJ, Chang HY (2013) Long noncoding RNAs: cellular address codes in development and disease. Cell 152:1298–1307
Berghoff EG, Clark MF, Chen S, Cajigas I, Leib DE, Kohtz JD (2013) Evf2 (Dlx6as) lncRNA regulates ultraconserved enhancer methylation and the differential transcriptional control of adjacent genes. Development 140:4407–4416
Binder DK, Scharfman HE (2004) Brain-derived neurotrophic factor. Growth Factors 22:123–131
Blackshaw S, Harpavat S, Trimarchi J, Cai L, Huang H, Kuo WP, Weber G, Lee K, Fraioli RE, Cho S-H, Yung R, Asch E, Ohno-Machado L, Wong WH, Cepko CL (2004) Genomic analysis of mouse retinal development. PLoS Biol 2:e247
Bond AM, Vangompel MJW, Sametsky EA, Clark MF, Savage JC, Disterhoft JF, Kohtz JD (2009) Balanced gene regulation by an embryonic brain ncRNA is critical for adult hippocampal GABA circuitry. Nat Neurosci 12:1020–1027
Bonini NM, Leiserson WM, Benzer S (1993) The eyes absent gene: genetic control of cell survival and differentiation in the developing drosophila eye. Cell 72:379–395
Briggs JA, Wolvetang EJ, Mattick JS, Rinn JL, Barry G (2015) Mechanisms of long non-coding RNAs in mammalian nervous system development, plasticity, disease, and evolution. Neuron 88:861–877
Brown CJ, Hendrich BD, Rupert JL, Lafrenière RG, Xing Y, Lawrence J, Willard HF (1992) The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 71:527–542
Cabili MN, Trapnell C, Goff L, Koziol M, Tazon-Vega B, Regev A, Rinn JL (2011) Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25:1915–1927
Cajigas I, Leib DE, Cochrane J, Luo H, Swyter KR, Chen S, Clark BS, Thompson J, Yates JR, Kingston RE, Kohtz JD (2015) Evf2 lncRNA/BRG1/DLX1 interactions reveal RNA-dependent inhibition of chromatin remodeling. Development 142:2641–2652
Campbell JN, Meyer RA (2006) Mechanisms of neuropathic pain. Neuron 52:77–92
Cech TR, Steitz JA (2014) The noncoding RNA revolution — trashing old rules to forge new ones. Cell 157:77–94
Cepko C (2014) Intrinsically different retinal progenitor cells produce specific types of progeny. Nat Rev Neurosci 15:615–627
Chapleau CA, Larimore JL, Theibert A, Pozzo-Miller L (2009) Modulation of dendritic spine development and plasticity by BDNF and vesicular trafficking: fundamental roles in neurodevelopmental disorders associated with mental retardation and autism. J Neurodev Disord 1:185–196
Clark BS, Blackshaw S (2014) Long non-coding RNA-dependent transcriptional regulation in neuronal development and disease. Front Genet 5:164
Cobos I, Calcagnotto ME, Vilaythong AJ, Thwin MT, Noebels JL, Baraban SC, Rubenstein JLR (2005) Mice lacking Dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat Neurosci 8:1059–1068
Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, Ruan Y, Lassmann T, Carninci P, Brown JB, Lipovich L, Gonzalez JM, Thomas M, Davis CA, Shiekhattar R, Gingeras TR, Hubbard TJ, Notredame C, Harrow J, Guigo R (2012) The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22:1775–1789
Dinger ME, Amaral PP, Mercer TR, Pang KC, Bruce SJ, Gardiner BB, Askarian-Amiri ME, Ru K, Solda G, Simons C, Sunkin SM, Crowe ML, Grimmond SM, Perkins AC, Mattick JS (2008) Long noncoding RNAs in mouse embryonic stem cell pluripotency and differentiation. Genome Res 18:1433–1445
Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Röder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Bar NS, Batut P, Bell K, Bell I, Chakrabortty S, Chen X, Chrast J, Curado J, Derrien T, Drenkow J, Dumais E, Dumais J, Duttagupta R, Falconnet E, Fastuca M, Fejes-Toth K, Ferreira P, Foissac S, Fullwood MJ, Gao H, Gonzalez D, Gordon A, Gunawardena H, Howald C, Jha S, Johnson R, Kapranov P, King B, Kingswood C, Luo OJ, Park E, Persaud K, Preall JB, Ribeca P, Risk B, Robyr D, Sammeth M, Schaffer L, See L-H, Shahab A, Skancke J, Suzuki AM, Takahashi H, Tilgner H, Trout D, Walters N, Wang H, Wrobel J, Yu Y, Ruan X, Hayashizaki Y, Harrow J, Gerstein M, Hubbard T, Reymond A, Antonarakis SE, Hannon G, Giddings MC, Ruan Y, Wold B, Carninci P, Guigó R, Gingeras TR (2012) Landscape of transcription in human cells. Nature 489:101–108
Doetsch F, Caillé I, Lim DA, García-Verdugo JM, Alvarez-Buylla A (1999) Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 97:703–716
Engreitz JM, Pandya-Jones A, McDonel P, Shishkin A, Sirokman K, Surka C, Kadri S, Xing J, Goren A, Lander ES, Plath K, Guttman M (2013) The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 341:1237973
Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, McDonel PE, Guttman M, Lander ES (2016) Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 539:452–455
Faghihi MA, Modarresi F, Khalil AM, Wood DE, Sahagan BG, Morgan TE, Finch CE, St. Laurent G III, Kenny PJ, Wahlestedt C (2008) Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of β-secretase. Nat Med 14:723–730
Feng J, Bi C, Clark BS, Mady R, Shah P, Kohtz JD (2006) The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev 20:1470–1484
Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM, Kane M, Cleary B, Lander ES, Engreitz JM (2016) Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science 354:769–773
Geng X, Lavado A, Lagutin OV, Liu W, Oliver G (2007) Expression of Six3 opposite strand (Six3OS) during mouse embryonic development. Gene Expr Patterns 7:252–257
Gill M, Vallada H, Collier D, Sham P, Holmans P, Murray R, McGuffin P, Nanko S, Owen M, Antonarakis S, Housman D, Kazazian H, Nestadt G, Pulver AE, Straub RE, MacLean CJ, Walsh D, Kendler KS, DeLisi L, Polymeropoulos M, Coon H, Byerley W, Lofthouse R, Gershon E, Golden L, Crow T, Byerley W, Freedman R, Laurent C, Bodeau-Pean S, d’Amato T, Jay M, Campion D, Mallet J, Wildenauer DB, Lerer B, Albus M, Ackenheil M, Ebstein RP, Hallmayer J, Maier W, Gurling H, Curtis D, Kalsi G, Brynjolfsson J, Sigmundson T, Petursson H, Blackwood D, Muir W, St. Clair D, He L, Maguire S, Moises HW, Hwu H-G, Yang L, Wiese C, Tao L, Liu X, Kristbjarnason H, Levinson DF, Mowry BJ, Donis-Keller H, Hayward NK, Crowe RR, Silverman JM, Nancarrow DJ, Read CM (1996) A combined analysis of D22S278 marker alleles in affected sib-pairs: support for a susceptibility locus for schizophrenia at chromosome 22q12. Am J Med Genet 67:40–45
Gonçalves JT, Schafer ST, Gage FH (2016) Adult neurogenesis in the hippocampus: from stem cells to behavior. Cell 167:897–914
Groff AF, Sanchez-Gomez DB, Soruco MML, Gerhardinger C, Barutcu AR, Li E, Elcavage L, Plana O, Sanchez LV, Lee JC, Sauvageau M, Rinn JL (2016) In vivo characterization of Linc-p21 reveals functional cis-regulatory DNA elements. Cell Rep 16:2178–2186
Guil S, Esteller M (2012) Cis-acting noncoding RNAs: friends and foes. Nat Struct Mol Biol 19:1068–1075
Guttman M, Donaghey J, Carey BW, Garber M, Grenier JK, Munson G, Young G, Lucas AB, Ach R, Bruhn L, Yang X, Amit I, Meissner A, Regev A, Rinn JL, Root DE, Lander ES (2011) lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 477:295–300
Hart RP, Goff LA (2016) Long noncoding RNAs: central to nervous system development. Int J Dev Neurosci 55:109–116
Hasbi A, Fan T, Alijaniaram M, Nguyen T, Perreault ML, O’Dowd BF, George SR (2009) Calcium signaling cascade links dopamine D1-D2 receptor heteromer to striatal BDNF production and neuronal growth. Proc Natl Acad Sci U S A 106:21377–21382
Haupt Y, Alexander WS, Barri G, Klinken SP, Adams JM (1991) Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell 65:753–763
He D, Wang J, Lu Y, Deng Y, Zhao C, Xu L, Chen Y, Hu Y-C, Zhou W, Lu QR (2017) lncRNA functional networks in oligodendrocytes reveal stage-specific myelination control by an lncOL1/Suz12 complex in the CNS. Neuron 93:362–378
Hitoshi N, Ken-ichi Y, Jun-ichi M (1991) Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199
Horike S, Cai S, Miyano M, Cheng J-F, Kohwi-Shigematsu T (2004) Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat Genet 37:31–40
Ip JY, Sone M, Nashiki C, Pan Q, Kitaichi K, Yanaka K, Abe T, Takao K, Miyakawa T, Blencowe BJ, Nakagawa S (2016) Gomafu lncRNA knockout mice exhibit mild hyperactivity with enhanced responsiveness to the psychostimulant methamphetamine. Sci Rep 6:27204
Ishikawa K, Tanaka M, Black JA, Waxman SG (1999) Changes in expression of voltage-gated potassium channels in dorsal root ganglion neurons following axotomy. Muscle Nerve 22:502–507
Jemc J, Rebay I (2007) The eyes absent family of phosphotyrosine phosphatases: properties and roles in developmental regulation of transcription. Annu Rev Biochem 76:513–538
Jeong J, Li X, McEvilly RJ, Rosenfeld MG, Lufkin T, Rubenstein JLR (2008) Dlx genes pattern mammalian jaw primordium by regulating both lower jaw-specific and upper jaw-specific genetic programs. Development 135:2905–2916
Johnsson P, Lipovich L, Grandér D, Morris KV (2014) Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim Biophys Acta Gen Subj 1840:1063–1071
Keppetipola N, Sharma S, Li Q, Black DL (2012) Neuronal regulation of pre-mRNA splicing by polypyrimidine tract binding proteins, PTBP1 and PTBP2. Crit Rev Biochem Mol Biol 47:360–378
Khalil AM, Guttman M, Huarte M, Garber M, Raj A, Rivea Morales D, Thomas K, Presser A, Bernstein BE, van Oudenaarden A, Regev A, Lander ES, Rinn JL (2009) Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A 106:11667–11672
Kim DS, Choi JO, Rim HD, Cho HJ (2002) Downregulation of voltage-gated potassium channel alpha gene expression in dorsal root ganglia following chronic constriction injury of the rat sciatic nerve. Brain Res Mol Brain Res 105:146–152
Kishino T, Lalande M, Wagstaff J (1997) UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet 15:70–73
Kohtz JD, Fishell G (2004) Developmental regulation of EVF-1, a novel non-coding RNA transcribed upstream of the mouse Dlx6 gene. Gene Expr Patterns 4:407–412
Kornienko AE, Guenzl PM, Barlow DP, Pauler FM (2013) Gene regulation by the act of long non-coding RNA transcription. BMC Biol 11:59
Kraus P, Sivakamasundari V, Lim SL, Xing X, Lipovich L, Lufkin T (2013) Making sense of Dlx1 antisense RNA. Dev Biol 376:224–235
Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang H-C, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee H-K, Wong PC (2005) BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. J Neurosci 25:11693–11709
Lee JT (2012) Epigenetic regulation by long noncoding RNAs. Science 338:1435–1439
Li W, Notani D, Ma Q, Tanasa B, Nunez E, Chen AY, Merkurjev D, Zhang J, Ohgi K, Song X, Oh S, Kim H-S, Glass CK, Rosenfeld MG (2013) Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature 498:516–520
Lim DA, Alvarez-Buylla A (2014) Adult neural stem cells stake their ground. Trends Neurosci 37:563–571
Lin N, Chang K-Y, Li Z, Gates K, Rana ZA, Dang J, Zhang D, Han T, Yang C-S, Cunningham TJ, Head SR, Duester G, Dong PDS, Rana TM (2014) An evolutionarily conserved long noncoding RNA TUNA controls pluripotency and neural lineage commitment. Mol Cell 53:1005–1019
Lipovich L, Dachet F, Cai J, Bagla S, Balan K, Jia H, Loeb JA (2012) Activity-dependent human brain coding/noncoding gene regulatory networks. Genetics 192:1133–1148
Liu JK, Ghattas I, Liu S, Chen S, Rubenstein JLR (1997) Dlx genes encode DNA-binding proteins that are expressed in an overlapping and sequential pattern during basal ganglia differentiation. Dev Dyn 210:498–512
Liu Q-R, Walther D, Drgon T, Polesskaya O, Lesnick TG, Strain KJ, de Andrade M, Bower JH, Maraganore DM, Uhl GR (2005) Human brain derived neurotrophic factor (BDNF) genes, splicing patterns, and assessments of associations with substance abuse and Parkinson’s disease. Am J Med Genet B 134B:93–103
Liu SJ, Nowakowski TJ, Pollen AA, Lui JH, Horlbeck MA, Attenello FJ, He D, Weissman JS, Kriegstein AR, Diaz AA, Lim DA (2016) Single-cell analysis of long non-coding RNAs in the developing human neocortex. Genome Biol 17:67
Liu SJ, Horlbeck MA, Cho SW, Birk HS, Malatesta M, He D, Attenello FJ, Villalta JE, Cho MY, Chen Y, Mandegar MA, Olvera MP, Gilbert LA, Conklin BR, Chang HY, Weissman JS, Lim DA (2017) CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 355:eaah7111
Luo X, Zhang X, Shao W, Yin Y, Zhou J (2009) Crucial roles of MZF-1 in the transcriptional regulation of apomorphine-induced modulation of FGF-2 expression in astrocytic cultures. J Neurochem 108:952–961
Maenner S, Blaud M, Fouillen L, Savoye A, Marchand V, Dubois A, Sanglier-Cianférani S, Van Dorsselaer A, Clerc P, Avner P, Visvikis A, Branlant C (2010) 2-D structure of the a region of Xist RNA and its implication for PRC2 association. PLoS Biol 8:e1000276
Matsuura T, Sutcliffe JS, Fang P, Galjaard R-J, Jiang Y, Benton CS, Rommens JM, Beaudet AL (1997) De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet 15:74–77
Mattick JS, Rinn JL (2015) Discovery and annotation of long noncoding RNAs. Nat Struct Mol Biol 22:5–7
McGuinness T, Porteus MH, Smiga S, Bulfone A, Kingsley C, Qiu M, Liu JK, Long JE, Xu D, Rubenstein JL (1996) Sequence, organization, and transcription of the Dlx-1 and Dlx-2 locus. Genomics 35:473–485
Meng S, Luo M, Sun H, Yu X, Shen M, Zhang Q, Zhou R, Ju X, Tao W, Liu D, Deng H, Lu Z (2010) Identification and characterization of Bmi-1-responding element within the human p16 promoter. J Biol Chem 285:33219–33229
Meng L, Person RE, Beaudet AL (2012) Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum Mol Genet 21:3001–3012
Meng L, Person RE, Huang W, Zhu PJ, Costa-Mattioli M, Beaudet AL (2013) Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS Genet 9:e1004039
Meng L, Ward AJ, Chun S, Bennett CF, Beaudet AL, Rigo F (2015) Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 518:409–412
Mercer TR, Mattick JS (2013) Structure and function of long noncoding RNAs in epigenetic regulation. Nat Struct Mol Biol 20:300–307
Mercer TR, Dinger ME, Sunkin SM, Mehler MF, Mattick JS (2008) Specific expression of long noncoding RNAs in the mouse brain. Proc Natl Acad Sci U S A 105:716–721
Mercer TR, Qureshi IA, Gokhan S, Dinger ME, Li G, Mattick JS, Mehler MF (2010) Long noncoding RNAs in neuronal-glial fate specification and oligodendrocyte lineage maturation. BMC Neurosci 11:14
Miyazaki K, Wakabayashi M, Chikahisa S, Sei H, Ishida N (2007) PER2 controls circadian periods through nuclear localization in the suprachiasmatic nucleus. Genes Cells 12:1225–1234
Modarresi F, Faghihi MA, Lopez-Toledano MA, Fatemi RP, Magistri M, Brothers SP, van der Brug MP, Wahlestedt C (2012) Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat Biotechnol 30:453–459
Mohammad F, Mondal T, Kanduri C (2009) Epigenetics of imprinted long non-coding RNAs. Epigenetics 4:277–286
Morikawa T, Manabe T (2010) Aberrant regulation of alternative pre-mRNA splicing in schizophrenia. Neurochem Int 57:691–704
Nagano T, Mitchell JA, Sanz LA, Pauler FM, Ferguson-Smith AC, Feil R, Fraser P (2008) The air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science 322:1717–1720
Nan X, Ng H-H, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A, Nan X, Ng H-H, Johnson CA, Laherty CD, Turner BM, Eisenman RN (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393:386–389
Nelson BR, Makarewich CA, Anderson DM, Winders BR, Troupes CD, Wu F, Reese AL, McAnally JR, Chen X, Kavalali ET, Cannon SC, Houser SR, Bassel-Duby R, Olson EN (2016) A peptide encoded by a transcript annotated as long noncoding RNA enhances SERCA activity in muscle. Science 351:271–275
Ng S-Y, Johnson R, Stanton LW (2012) Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J 31:522–533
Ng S-Y, Bogu GK, Soh BS, Stanton LW (2013) The long noncoding RNA RMST interacts with SOX2 to regulate neurogenesis. Mol Cell 51:349–359
Oltersdorf T, Ward PJ, Henriksson T, Beattie EC, Neve R, Lieberburg I, Fritz LC (1990) The Alzheimer amyloid precursor protein. Identification of a stable intermediate in the biosynthetic/degradative pathway. J Biol Chem 265:4492–4497
Ørom UA, Derrien T, Beringer M, Gumireddy K, Gardini A, Bussotti G, Lai F, Zytnicki M, Notredame C, Huang Q, Guigo R, Shiekhattar R (2010) Long noncoding RNAs with enhancer-like function in human cells. Cell 143:46–58
Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, Nagano T, Mancini-Dinardo D, Kanduri C (2008) Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell 32:232–246
Paralkar VR, Taborda CC, Huang P, Yao Y, Kossenkov AV, Prasad R, Luan J, Davies JOJ, Hughes JR, Hardison RC, Blobel GA, Weiss MJ (2016) Unlinking an lncRNA from its associated cis element. Mol Cell 62:104–110
Penny GD, Kay GF, Sheardown SA, Rastan S, Brockdorff N (1996) Requirement for Xist in X chromosome inactivation. Nature 379:131–137
Pignoni F, Hu B, Zavitz KH, Xiao J, Garrity PA, Zipursky SL (1997) The eye-specification proteins so and Eya form a complex and regulate multiple steps in drosophila eye development. Cell 91:881–891
Plath K, Mlynarczyk-Evans S, Nusinow DA, Panning B (2002) Xist RNA and the mechanism of X chromosome inactivation. Annu Rev Genet 36:233–278
Pleasure SJ, Anderson S, Hevner R, Bagri A, Marin O, Lowenstein DH, Rubenstein JL (2000) Cell migration from the ganglionic eminences is required for the development of hippocampal GABAergic interneurons. Neuron 28:727–740
Porteus MH, Bulfone A, Liu JK, Puelles L, Lo LC, Rubenstein JL (1994) DLX-2, MASH-1, and MAP-2 expression and bromodeoxyuridine incorporation define molecularly distinct cell populations in the embryonic mouse forebrain. J Neurosci 14:6370–6383
Quinn JJ, Chang HY (2015) Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet 17:47–62
Ramos AD, Diaz A, Nellore A, Delgado RN, Park K-Y, Gonzales-Roybal G, Oldham MC, Song JS, Lim DA (2013) Integration of genome-wide approaches identifies lncRNAs of adult neural stem cells and their progeny in vivo. Cell Stem Cell 12:616–628
Ramos AD, Andersen RE, Liu SJ, Nowakowski TJ, Hong SJ, Gertz CC, Salinas RD, Zarabi H, Kriegstein AR, Lim DA (2015) The long noncoding RNA Pnky regulates neuronal differentiation of embryonic and postnatal neural stem cells. Cell Stem Cell 16:439–447
Ramos AD, Attenello FJ, Lim DA (2016) Uncovering the roles of long noncoding RNAs in neural development and glioma progression. Neurosci Lett 625:70–79
Rani N, Nowakowski TJ, Zhou H, Godshalk SE, Lisi V, Kriegstein AR, Kosik KS (2016) A primate lncRNA mediates notch signaling during neuronal development by sequestering miRNA. Neuron 90:1174–1188
Rapicavoli NA, Poth EM, Blackshaw S (2010) The long noncoding RNA RNCR2 directs mouse retinal cell specification. BMC Dev Biol 10:49
Rapicavoli NA, Poth EM, Zhu H, Blackshaw S (2011) The long noncoding RNA Six3OS acts in trans to regulate retinal development by modulating Six3 activity. Neural Dev 6:32
Rinn JL, Chang HY (2012) Genome regulation by long noncoding RNAs. Annu Rev Biochem 81:145–166
Rougeulle C, Glatt H, Lalande M (1997) The Angelman syndrome candidate gene, UBE3AIE6-AP, is imprinted in brain. Nat Genet 17:14–15
Sauvageau M, Goff LA, Lodato S, Bonev B, Groff AF, Gerhardinger C, Sanchez-Gomez DB, Hacisuleyman E, Li E, Spence M, Liapis SC, Mallard W, Morse M, Swerdel MR, D’Ecclessis MF, Moore JC, Lai V, Gong G, Yancopoulos GD, Frendewey D, Kellis M, Hart RP, Valenzuela DM, Arlotta P, Rinn JL (2013) Multiple knockout mouse models reveal lincRNAs are required for life and brain development. elife 2:e01749
Shearwin KE, Callen BP, Egan JB (2005) Transcriptional interference — a crash course. Trends Genet 21:339–345
Shibasaki T, Tokunaga A, Sakamoto R, Sagara H, Noguchi S, Sasaoka T, Yoshida N (2013) PTB deficiency causes the loss of adherens junctions in the dorsal telencephalon and leads to lethal hydrocephalus. Cereb Cortex 23:1824–1835
Sone M, Hayashi T, Tarui H, Agata K, Takeichi M, Nakagawa S (2007) The mRNA-like noncoding RNA Gomafu constitutes a novel nuclear domain in a subset of neurons. J Cell Sci 120:2498–2506
Spadaro PA, Flavell CR, Widagdo J, Ratnu VS, Troup M, Ragan C, Mattick JS, Bredy TW (2015) Long noncoding RNA-directed epigenetic regulation of gene expression is associated with anxiety-like behavior in mice. Biol Psychiatry 78:848–859
Su Y, Ryder J, Ni B (2003) Inhibition of Abeta production and APP maturation by a specific PKA inhibitor. FEBS Lett 546:407–410
Sugitani Y, Nakai S, Minowa O, Nishi M, Jishage K-I, Kawano H, Mori K, Ogawa M, Noda T (2002) Brn-1 and Brn-2 share crucial roles in the production and positioning of mouse neocortical neurons. Genes Dev 16:1760–1765
Takahashi S, Ohtsuki T, Yu S-Y, Tanabe E, Yara K, Kamioka M, Matsushima E, Matsuura M, Ishikawa K, Minowa Y, Noguchi E, Nakayama J, Yamakawa-Kobayashi K, Arinami T, Kojima T (2003) Significant linkage to chromosome 22q for exploratory eye movement dysfunction in schizophrenia. Am J Med Genet 123B:27–32
Talkowski ME, Maussion G, Crapper L, Rosenfeld JA, Blumenthal I, Hanscom C, Chiang C, Lindgren A, Pereira S, Ruderfer D, Diallo AB, Lopez JP, Turecki G, Chen ES, Gigek C, Harris DJ, Lip V, An Y, Biagioli M, MacDonald ME, Lin M, Haggarty SJ, Sklar P, Purcell S, Kellis M, Schwartz S, Shaffer LG, Natowicz MR, Shen Y, Morton CC, Gusella JF, Ernst C (2012) Disruption of a large intergenic noncoding RNA in subjects with neurodevelopmental disabilities. Am J Hum Genet 91:1128–1134
Tamagno E, Bardini P, Guglielmotto M, Danni O, Tabaton M (2006) The various aggregation states of beta-amyloid 1-42 mediate different effects on oxidative stress, neurodegeneration, and BACE-1 expression. Free Radic Biol Med 41:202–212
Terranova R, Yokobayashi S, Stadler MB, Otte AP, van Lohuizen M, Orkin SH, Peters AHFM, Bickmore WA, Feil R, Segal E, Chang HY (2008) Polycomb group proteins Ezh2 and Rnf2 direct genomic contraction and imprinted repression in early mouse embryos. Dev Cell 15:668–679
Tsuiji H, Yoshimoto R, Hasegawa Y, Furuno M, Yoshida M, Nakagawa S (2011) Competition between a noncoding exon and introns: Gomafu contains tandem UACUAAC repeats and associates with splicing factor-1. Genes Cells 16:479–490
Ulitsky I (2016) Evolution to the rescue: using comparative genomics to understand long non-coding RNAs. Nat Rev Genet 17:601–614
Ulitsky I, Shkumatava A, Jan CH, Sive H, Bartel DP (2011) Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 147:1537–1550
Vassar R (2001) The beta-secretase, BACE: a prime drug target for Alzheimer’s disease. J Mol Neurosci 17:157–170
Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M (1999) Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286:735–741
Venkatesh S, Workman JL (2015) Histone exchange, chromatin structure and the regulation of transcription. Nat Rev Mol Cell Biol 16:178–189
Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Südhof TC, Wernig M (2010) Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463:1035–1041
Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden J-M, Bollen M, Esteller M, Di Croce L, de Launoit Y, Fuks F (2005) The Polycomb group protein EZH2 directly controls DNA methylation. Nature 439:871–874
Vu TH, Hoffman AR (1997) Imprinting of the Angelman syndrome gene, UBE3A, is restricted to brain. Nat Genet 17:12–13
Wang W, Côté J, Xue Y, Zhou S, Khavari PA, Biggar SR, Muchardt C, Kalpana GV, Goff SP, Yaniv M, Workman JL, Crabtree GR (1996) Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J 15:5370–5382
Willem M, Garratt AN, Novak B, Citron M, Kaufmann S, Rittger A, DeStrooper B, Saftig P, Birchmeier C, Haass C (2006) Control of peripheral nerve myelination by the beta-secretase BACE1. Science 314:664–666
Xue Y, Ouyang K, Huang J, Zhou Y, Ouyang H, Li H, Wang G, Wu Q, Wei C, Bi Y, Jiang L, Cai Z, Sun H, Zhang K, Zhang Y, Chen J, Fu X-D (2013) Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell 152:82–96
Yamasaki K, Joh K, Ohta T, Masuzaki H, Ishimaru T, Mukai T, Niikawa N, Ogawa M, Wagstaff J, Kishino T (2003) Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum Mol Genet 12:837–847
Yang Y, Wen L, Zhu H (2015) Unveiling the hidden function of long non-coding RNA by identifying its major partner-protein. Cell Biosci 5:59
Yin Y, Yan P, Lu J, Song G, Zhu Y, Li Z, Zhao Y, Shen B, Huang X, Zhu H, Orkin SH, Shen X (2015) Opposing roles for the lncRNA haunt and its genomic locus in regulating HOXA gene activation during embryonic stem cell differentiation. Cell Stem Cell 16:504–516
Yoshimura R, Ito K, Endo Y (2009) Differentiation/maturation of neuropeptide Y neurons in the corpus callosum is promoted by brain-derived neurotrophic factor in mouse brain slice cultures. Neurosci Lett 450:262–265
Young TL, Matsuda T, Cepko CL (2005) The noncoding RNA Taurine Upregulated gene 1 is required for differentiation of the murine retina. Curr Biol 15:501–512
Zerucha T, Stühmer T, Hatch G, Park BK, Long Q, Yu G, Gambarotta A, Schultz JR, Rubenstein JL, Ekker M (2000) A highly conserved enhancer in the Dlx5/Dlx6 intergenic region is the site of cross-regulatory interactions between Dlx genes in the embryonic forebrain. J Neurosci 20:709–721
Zhang J-W, Tang Q-Q, Vinson C, Lane MD (2004) Dominant-negative C/EBP disrupts mitotic clonal expansion and differentiation of 3T3-L1 preadipocytes. Proc Natl Acad Sci U S A 101:43–47
Zhang L, Yang Z, Trottier J, Barbier O, Wang L (2017) Long noncoding RNA MEG3 induces cholestatic liver injury by interaction with PTBP1 to facilitate shp mRNA decay. Hepatology 65:604–615
Zhao X, Tang Z, Zhang H, Atianjoh FE, Zhao J-Y, Liang L, Wang W, Guan X, Kao S-C, Tiwari V, Gao Y-J, Hoffman PN, Cui H, Li M, Dong X, Tao Y-X (2013) A long noncoding RNA contributes to neuropathic pain by silencing Kcna2 in primary afferent neurons. Nat Neurosci 16:1024–1031
Zhou Q-P, Le TN, Qiu X, Spencer V, de Melo J, Du G, Plews M, Fonseca M, Sun JM, Davie JR, Eisenstat DD (2004) Identification of a direct Dlx homeodomain target in the developing mouse forebrain and retina by optimization of chromatin immunoprecipitation. Nucleic Acids Res 32:884–892
Zhu CC, Dyer MA, Uchikawa M, Kondoh H, Lagutin OV, Oliver G (2002) Six3-mediated auto repression and eye development requires its interaction with members of the Groucho-related family of co-repressors. Development 129:2835–2849
Zhu S, Li W, Liu J, Chen C-H, Liao Q, Xu P, Xu H, Xiao T, Cao Z, Peng J, Yuan P, Brown M, Liu XS, Wei W (2016) Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR–Cas9 library. Nat Biotechnol 34:1279–1286
Ziats MN, Rennert OM (2013) Aberrant expression of long noncoding RNAs in autistic brain. J Mol Neurosci 49:589–593
Funding
This work was supported by National Institutes of Health (NIH) 1F31NS098562–01 to R.E.A. and NIH 5R01NS091544–02, NIH 1R21NS101395–01, and Veterans Affairs 5I01 BX000252–07 to D.A.L.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Andersen, R.E., Lim, D.A. Forging our understanding of lncRNAs in the brain. Cell Tissue Res 371, 55–71 (2018). https://doi.org/10.1007/s00441-017-2711-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-017-2711-z