
Abstract
Background
Sunitinib has emerged as the primary treatment for advanced or metastatic clear cell renal cell carcinoma (ccRCC) due to its significant improvement in patients’ average survival time. However, drug resistance and adverse effects of sunitinib pose challenges to its clinical benefits.
Methods
The differentially expressed genes (DEGs) associated with sunitinib sensitivity and resistance in ccRCC were investigated. Cell counting kit-8, plate colony formation, flow cytometry and subcutaneous xenograft tumor model assays were employed to explore the effects of PDZK1 on ccRCC. Further research on the molecular mechanism was conducted through western blot, co-immunoprecipitation, immunofluorescence co-localization and immunohistochemical staining.
Results
We elucidated that PDZK1 is significantly downregulated in sunitinib-resistant ccRCC specimens, and PDZK1 negatively regulates the phosphorylation of PDGFR-β and the activation of its downstream pathways through interaction with PDGFR-β. The dysregulated low levels of PDZK1 contribute to inadequate inhibition of cell proliferation, tumor growth, and insensitivity to sunitinib treatment. Notably, our preclinical investigations showed that miR-15b antagomirs enhance sunitinib cytotoxic effects against ccRCC cells by upregulating PDZK1 levels, suggesting their potential in overcoming sunitinib resistance.
Conclusions
Our findings establish the miR-15b/PDZK1/PDGFR-β axis as a promising therapeutic target and a novel predictor for ccRCC patients’ response to sunitinib treatment.

Similar content being viewed by others


Introduction
Renal cell carcinoma (RCC) is the most lethal urological malignancy, ranking as the sixth most frequent malignancy in males and the ninth in females, causing more than 140,000 deaths per year worldwide [1]. Clear cell renal cell carcinoma (ccRCC) is the major subtype, accounting for 75% of all cases of RCC [2], and is characterized by frequent inactivation of the von Hippel‒Lindau (VHL) tumor suppressor gene, which causes significant accumulation of hypoxia-inducible factor (HIF), leading to overexpression of VEGF and PDGF [3, 4]. While prognosis is usually better in patients with early-stage ccRCC, advanced or metastatic ccRCC is a life-threatening disease; the five-year survival rate is only 9% after diagnosis, and the median overall survival is less than 12 months [5].
Surgery is the gold standard for localized ccRCC [6]. However, for patients with advanced or metastatic ccRCC, surgery alone is generally not considered curative or sufficient to manage the disease. ccRCC is characterized by low response rates to chemotherapy or radiotherapy, and only 20% of ccRCC patients show sensitivity toward standard cytokine treatment, a nonspecific immunotherapy for ccRCC, with an objective response rate of only 7.5% [7, 8]. Currently, molecular targeted therapy has become the primary medical treatment for advanced or metastatic ccRCC, such as tyrosine kinase inhibitors (TKIs), immune checkpoint inhibitors, and mammalian target of rapamycin (mTOR) inhibitors [9,10,11]. Sunitinib, an oral multi-targeted TKI, is extensively used in the treatment of advanced or metastatic ccRCC due to its significant improvement in the average survival time of patients, extending it from 12 to 22 months [12, 13]. Regrettably, intrinsic resistance is observed in nearly 30% of patients receiving sunitinib, and even initial responders eventually experience relapse and develop resistance after a median treatment duration of 6 to 15 months. This emergence of drug resistance significantly limits the therapeutic efficacy and provides only modest overall survival benefits [14].
The clinical application of sunitinib is also limited by its toxicity, leading to variable response rates and notable adverse effects such as fatigue, diarrhea, and hyperglycemia [15]. In the sunitinib open-access program, 8% of patients had to discontinue treatment due to serious adverse effects, and an additional 30% required dose reductions due to toxicity [16,17,18]. However, dose reduction of sunitinib may pose a risk of reducing its antitumor effects in ccRCC. Studies in metastatic ccRCC have demonstrated that patients with higher exposure to sunitinib achieved improved treatment outcomes. They had a higher probability of tumor size decrease or halting tumor growth, longer time to progression, and prolonged overall survival [19]. Combination therapies have emerged as promising treatments for advanced or metastatic ccRCC, leading to a significant improvement in overall survival. However, the occurrence of treatment-related adverse events is common among patients. The mechanisms underlying differential treatment responses and side effects are complex, but there is substantial evidence supporting the concentration-toxicity relationship of sunitinib in ccRCC [20, 21]. Therefore, enhancing the sensitivity of ccRCC to sunitinib represents a potential strategy to overcome drug resistance and reduce the risk of severe adverse events by lowering the sunitinib dosage without compromising its antitumor effects. This approach can lead to improved therapeutic efficacy against advanced or metastatic ccRCC. Understanding the molecular mechanisms of ccRCC sensitivity to sunitinib is crucial for optimizing treatment strategies and developing novel therapeutic approaches. Several mechanisms have been proposed for sunitinib resistance in ccRCC, including alterations in the tumor microenvironment, activation of alternative signaling pathways, and insufficient target inhibition [22,23,24,25]. However, further research is needed to uncover additional factors and mechanisms involved in sunitinib sensitivity and resistance in ccRCC, which may lead to the development of more effective therapies for patients with advanced or metastatic ccRCC.
In this study, we identified PDZK1 as a significantly downregulated gene in sunitinib-resistant ccRCC specimens. Through in vitro and in vivo functional assays, we demonstrated that PDZK1 interaction with PDGFR-β suppressed ccRCC cell proliferation by inhibiting the activation of the PDGFR-β pathway, thereby enhancing sunitinib sensitivity. Notably, silencing of miR-15b using inhibitors or antagomirs increased PDZK1 levels and synergistically enhanced the cytotoxic effects of sunitinib in ccRCC cells. Our findings establish the miR-15b/PDZK1/PDGFR-β axis as a potential therapeutic target and a novel predictor of response to sunitinib treatment in ccRCC patients.
Materials and methods
Data collection and ccRCC samples
Expression profiling and clinical data for ccRCC were obtained from E-MTAB-3267, GSE64052, CLINICAL PROTEOME TUMOR ANALYSIS CONSORTIUM (CPTAC), and TCGA. Paraffin embedded surgical resection specimens (n = 23, 17 males and 6 females) were collected from patients with advanced or metastatic ccRCC at the First Hospital of Shanxi Medical University between 2021 and 2023. Each of these patients had undergone nephrectomy, manifested post-operative recurrence, received sunitinib as a targeted intervention, underwent radiological assessment to determine therapeutic efficacy and the median follow-up time is 20 months. The research involving these samples was approved by the Ethics Committee of Capital Medical University (2017SY09) and the First Hospital of Shanxi Medical University (2022HLL001). Tissue microarrays containing 90 pairs of ccRCC and adjacent tissues were purchased from Shanghai Outdo Biotech. Written informed consent was obtained from all patients for the use of their tissue specimens and publication of this report.
DEG screening
The online tools NetworkAnalyst and GEO2R analyzer were used to identify differentially expressed genes (DEGs) from the E-MTAB-3267 and GSE64052 databases, comparing sunitinib-sensitive vs. sunitinib-resistant samples. DEGs were selected based on adjusted p < 0.05 and ∣Log2(fold change)∣ >0.5 as cutoff criteria. VENNY 2.1 was employed to identify the common DEGs between E-MTAB-3267 and GSE64052, and the final DEGs were defined as those appearing in both datasets.
Cell culture
Human renal cell carcinoma (RCC) cell lines ACHN and 786-O, as well as the monkey kidney cell line COS-7, were obtained from the National Infrastructure of Cell Line Resource (Beijing, China). ACHN and 786-O cells were cultured in RPMI-1640 medium, while COS-7 cells were cultured in DMEM. Both media were supplemented with 1% penicillin/streptomycin and 10% fetal bovine serum. Cells were maintained at 37 °C with 5% CO2 in a humidified environment. Prior to use, cells were authenticated using short tandem repeat (STR) DNA fingerprinting and tested negative for mycoplasma contamination. Passage number was limited to 30 continuous passages.
Drugs and chemicals
Sunitinib (HY-10255A) and the PDGFR-β inhibitor CP-673451 (HY-12050) were obtained from MedChemExpress (Shanghai, China). Stock solutions were prepared in DMSO and stored at −80 °C. PDGF-BB (HY-P7055) was purchased from MedChemExpress, reconstituted to a concentration of 100 μg/ml in sterile distilled water containing 0.1% BSA, and stored at −80 °C.
The measurement of cell viability and analysis of the cell cycle
Cells were plated in 96-well plates at a density of 3000–5000 cells per well in 100 μl of cell suspension. Cell viability was assessed using an EnSpire label microplate reader (PerkinElmer, Waltham, MA) by measuring absorbance at 450 nm according to the instructions provided by the Cell Counting Kit-8 (Dojindo, Kumamoto, Japan). For cell cycle analysis, cells were fixed in 70% ethanol at 4 °C overnight, washed with PBS, stained with propidium iodide for 30 min, and analyzed using flow cytometry (EPICS@XL, Beckman Coulter, 250 S. Kraemer Boulevard Brea, CA).
Clonogenic assay
Cells were seeded in six-well plates at a density of 800–1000 cells per well and incubated at 37 °C with 5% CO2 for 10–14 days. The resulting colonies were washed with PBS, fixed with 4% paraformaldehyde for 20 min, and stained with crystal violet for 20 min. The plates were then rinsed with water, and the number of colonies (>50 cells) was counted. The assay was performed independently three times.
DNA constructs and RNA interference
pcDNA3.1-PDZK1 was obtained from Dr. Randy Hall at Emory University (Atlanta, GA) and the shPDZK1 plasmids for PDZK1 knockdown were provided by Dr. Michael R Beard at the Centre for Cancer Biology (Adelaide, Australia). Constructs encoding Flag-PDGFR-β, Flag-PDGFR-β-L1106A, and GST-PDGFR-β-CT were purchased from UNIBIO company (Chongqing, China). GST-PDGFR-β-CT consists of the C-terminal 50 amino acids of human PDGFR-β. The siRNA sequences targeting PDZK1 were previously reported [26] and obtained from Sigma (St. Louis, MO).
Stable cell line generation and cell transfection
To establish stable cell lines overexpressing or knocking down PDZK1, ACHN or 786-O cells were transfected with control vector, PDZK1 plasmid, shControl plasmid, or shPDZK1 plasmid. After 48 h, cells were selected with G418 (800 μg/ml) or puromycin (300 ng/ml) in the culture medium for 14 days.
For siRNA knockdown and miR-15b mimics or inhibitors transfection experiments, cells were seeded in a 6-well plate and transfected with siRNA (100 nmol/l) or miR-15b mimics or inhibitors (100 nmol/l) using Lipofectamine 3000. Cells were collected 48 h after transfection for protein extraction.
hsa-miR-15b control5′-UUCUCCGAACGUGUCACGUTT-3′; hsa-miR-15b mimic5′-UAGCAGCACAUCAUGGUUUACA-3′. hsa-miR-15b-5p inhibitor: 5′-UGUAAACCAUGAUGUGCUGCUA-3′; inhibitor control: 5′-UCUACUCUUUCUAGGAGGUUGUGA-3′.
Western blot analysis and reagents
Western blot assays were performed according to previously described methods [26]. Cells were harvested and lysed using lysis buffer containing Hepes, NaCl, EDTA, benzamidine, Triton X-100, protease inhibitor cocktail, and phosphatase inhibitor. The antibodies used included PDZK1 (Santa Cruz Biotechnology, sc-390932), PDGFR-β (Abcam, ab313777), p-PDGFR-β (phosphor Tyr857) (Abcam, ab62367), ERK (Cell Signaling Technology, #9102), p-ERK (Cell Signaling Technology, #4370), β-actin (Bioss Antibodies, bs-0061R), GAPDH (Bioss Antibodies, bs-8778R), His-tag (MBL, M0893), Myc-tag (MBL, 5265), Flag-tag (Bioss Antibodies, bs-0965R), anti-GST (Bioss Antibodies, bsm-52605R) and anti-mouse/rabbit secondary antibodies (Cell Signaling Technology, #7076, #7074). Immune complexes were visualized and the intensity values were quantified using NIH Image software and Photoshop software, respectively.
Coimmunoprecipitation (Co-IP)
Coimmunoprecipitation was performed as described previously [27]. Briefly, cells were lysed in lysis buffer, and the lysate was clarified. Immunoprecipitation reactions were carried out using specific antibodies and cell lysates. After incubation, protein A/G-agarose was added and incubated with rotation. The immunoprecipitants were separated by centrifugation, washed with PBS, and resolved by SDS-PAGE. Immunoblot analysis was then performed.
GST pull-down assay
GST pull-down assays were performed as previously described [28]. Briefly, GST-PDGFR-β-CT fusion protein was expressed in bacteria and purified using glutathione-Sepharose 4B beads. The purified protein was resuspended in PBS containing 0.5% Nonidet P-40 and protease inhibitors. Equal amounts of purified GST-PDGFR-β-CT or GST (conjugated to beads) were incubated with cell lysates containing His-tagged PDZ domains of PDZK1 proteins. After a 3-h incubation at 4 °C, the beads were washed with washing buffers I and II. The bound proteins were separated by SDS-PAGE and analyzed by immunoblotting using anti-GST and anti-His antibodies.
Immunohistochemistry (IHC) analysis
The IHC analysis was performed as previously described [28]. Tissue sections were incubated with anti-PDZK1 (sc-390964, Santa Cruz, Dallas, TX) and anti-p-PDGFR-β (Tyr857) (bs-3320R, Bioss) antibodies overnight at 4 °C. This was followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibody kits. Two pathologists, blinded to the clinical and pathological information, assessed the expression of PDZK1 and p-PDGFR-β on a scale of 0 to 3 for intensity (no staining, weak, moderate, and strong) and on a scale of 0 to 4 for positive tumor cell percentage (none, 1–25%, 26–50%, 51–75%, and >75%). The histochemistry score (H-score) was calculated by multiplying these two variables.
Gene set enrichment analysis (GSEA)
GSEA was conducted to evaluate the enrichment of gene sets from the Molecular Signatures Database. The analysis was performed with default settings, using 1000 permutations. A statistically significant enrichment was defined as a false discovery rate <0.25 and p < 0.05.
Animal study
Animal experiments were conducted in compliance with the National Institutes of Health guidelines for the Care and Use of Laboratory Animals and approved by the Animal Use and Care Committee of Capital Medical University (approval numbers AEEI-2020-133 and AEEI-2018-201). All animals were housed in an environment with a temperature of 22 ± 1 °C, relative humidity of 50 ± 1%, and a 12/12-h light/dark cycle.
To establish a xenograft tumor model, ACHN cells stably overexpressing PDZK1 or control were implanted subcutaneously into the flanks of male BALB/c nude mice (6 weeks old, weight 16–18 g, n = 20). Tumor volumes were monitored, and when they reached 80 to 100 mm3, mice were randomly assigned to control and treatment groups. The treatment group (n = 10) received oral sunitinib (50 mg/kg/day, 25 days), while the control group (n = 10) received the corresponding solvent.
In the miR-15b-5p antagomir treatment experiment, ACHN cells were injected into male BALB/c nude mice (n = 14), and when the tumors reached 80 to 100 mm3, the mice were randomly assigned to treatment and control groups and treated with sunitinib (oral, 50 mg/kg/day) combined with miR-15b antagomirs (n = 7, local injection, 5 mg/kg, volume 40 ul, 4 injection sites, once every 3 days, 21 days) or antagomir control (n = 7). Tumor growth was monitored, and tumor volumes were calculated. When the tumor volume reached ~1000 mm3, mice were sacrificed and tumors were dissected and weighed. The tumor was fixed in 4% paraformaldehyde, then paraffin embedded and IHC staining was performed.
Mice were randomly allocated to either the control or treatment groups using a random number method. Grouping was conducted without utilizing a blind method, and sample sizes were determined based on previous experimental experience to ensure that statistical differences could be achieved.
The hsa-miR-15b-5p antagomir and antagomir control were synthesized by ChemShine Biotechnology Inc. (Shanghai, China). hsa-miR-15b-5p antagomir: 5′-UGUAAACCAUGAUGUGCUGCUA-3′; antagomir control: 5′-UCUACUCUUUCUAGGAGGUUGUGA-3′.
The Pdzk1 knockout (Pdzk1−/−) and wild-type (Pdzk1+/+) mice on the C57BL/6J background were obtained from GemPharmatech Co., Ltd. (Nanjing, China). Tissue samples from the kidneys of Pdzk1−/− (n = 3) and wild-type mice (n = 3) were used for IHC analysis.
Statistical analysis
Statistical analyses were performed using GraphPad Prism software. Data are presented as the mean with standard error of the mean (SEM) for at least three independent experiments. Measurement data were assessed for normal distribution and homogeneity of variance using appropriate tests (homogeneity of variance was considered present when p > 0.1). Measurement data were analyzed using analysis of variance or t-tests, and nonparametric tests were used for non-normal distributed data. Pearson correlation was used for variables assuming a Gaussian distribution, while Spearman correlation was used for non-Gaussian distributed data. The log-rank test was used to compare survival rates between groups based on Kaplan-Meier survival curves. Two-way ANOVA with Bonferroni posttests was used for comparing cell viability or tumor size at multiple cutoff times. The determination of sample size was based on statistical power analysis to ensure sufficient ability to detect a pre-specified effect size. We assumed a moderate effect size, with an alpha level set at 0.05, and a power (1-β) set at 0.80. A significance level of p < 0.05 (two-tailed) was considered statistically significant.
Results
The downregulation of PDZK1 is correlated with decreased sensitivity to sunitinib in ccRCC
Differentially expressed genes (DEGs) were identified by analyzing the E-MTAB-3267 and GSE64052 datasets, resulting in the discovery of 9 common DEGs (Fig. 1a, b). Among these DEGs, PDZK1 showed the most significant correlation with patient prognosis (Fig. 1c and Supplementary Fig. 1) and was higher in patients who responded favorably to sunitinib treatment (Fig. 1d). Xenograft ccRCC tumors with high PDZK1 levels were also more sensitive to sunitinib (Fig. 1e). Furthermore, the PDZK1 mRNA value showed predictive potential for sunitinib response, as indicated by the area under the receiver operating characteristic (ROC) curve (AUC) of 0.70 ± 0.08 (Fig. 1f).

a Volcano plot of DEGs between sunitinib-resistant and sunitinib-sensitive RCC samples from E-MTAB-3267 and GSE64052. X-coordinate: log2 (fold change); Y-coordinate: −log10 (p value). b Venn diagram showing 9 common DEGs identified from E-MTAB-3267 and GSE64052, including PDZK1. c KM survival plots for progression-free survival (PFS) based on PDZK1 mRNA expression in sunitinib-treated ccRCC patients from E-MTAB-3267 (p < 0.01, log-rank test). d Higher PDZK1 mRNA levels in good responders to sunitinib compared to poor responders. Mean ± SEM. Mann–Whitney test. *p < 0.05. e Significant downregulation of PDZK1 mRNA in sunitinib-resistant ccRCC xenografts (GSE64052). Mean ± SEM. Two-tailed unpaired t-test. ***p < 0.001. f ROC curve with PDZK1 mRNA value in ccRCC specimens from E-MTAB-3267 dataset to predict sunitinib response. g Enrichment plots of GSEA showing significant enrichment of PDGF/PDGFR-β and ERK pathways in ccRCC specimens with low PDZK1 expression from E-MTAB-3267 dataset.
To investigate the molecular mechanisms underlying the correlation between PDZK1 and sunitinib sensitivity, gene set enrichment analysis (GSEA) was performed between PDZK1 level and the well-known sunitinib targeting pathways, including EGFR, VEGFR, KIT, RET and PDGFR. The analysis revealed that patients with lower PDZK1 levels exhibited significant enrichment of gene signatures associated with the PDGFR-β pathway and its downstream ERK pathway (Fig. 1g and Supplementary Fig. 2), highlighting their potential involvement in PDZK1-mediated sunitinib sensitivity.
PDZK1 negatively regulates PDGFR-β phosphorylation (Tyr 857) and its downstream signaling in ccRCC cells by directly interacting with PDGFR-β
To investigate the relationship between PDZK1 and the PDGFR-β pathway, we examined their expression correlation at both the mRNA and protein levels. While no correlation was found at the mRNA level in clinical specimens from TCGA (Fig. 2a), there was a negative correlation observed between PDZK1 expression and phosphorylated PDGFR-β at the protein level (Fig. 2b), suggesting a potential role for PDZK1 in regulating PDGFR-β phosphorylation. PDZK1 is a scaffold protein containing PDZ domains, and PDGFR-β possesses a PDZ domain binding motif (PBM) at its C-terminus (D-S-F-L). Therefore, there is a molecular structural basis for their interaction. Co-immunoprecipitation experiments confirmed the association between endogenous PDGFR-β and PDZK1 in ACHN cells (Fig. 2c). To further understand the interaction between PDGFR-β and PDZK1 at a structural level, we performed transient co-transfection experiments in COS-7 cells. Overexpression of Myc-PDZK1 along with either wild-type PDGFR-β (Flag-PDGFR-β-WT) or a mutant form with a C-terminus L1106A mutation (Flag-PDGFR-β-MT) revealed that the L1106A mutation disrupted the association between PDGFR-β and PDZK1, indicating a direct binding between PDZK1 and the carboxyl terminus of PDGFR-β (Fig. 2d). GST-pulldown assays further demonstrated that PDGFR-β-CT strongly interacted with PDZ2 and PDZ3 domains, while exhibiting weaker interaction with PDZ1 and PDZ4 domains (Fig. 2e). Immunofluorescence colocalization studies supported the interaction between PDZK1 and wild-type PDGFR-β, while no significant colocalization was observed with the PDGFR-β L1106A mutant (Fig. 2f, g).

a Correlation analysis (Spearman) between PDZK1 and PDGFR-β mRNA levels in ccRCC specimens from the TCGA dataset. b Correlation analysis (Spearman) between PDZK1 mRNA level and phosphorylated PDGFR-β level in ccRCC specimens from the CPTAC dataset. c Coimmunoprecipitation of endogenous PDGFR-β and PDZK1 proteins in ACHN cells. d Impaired interaction between PDGFR-β and PDZK1 due to the PDGFR-β L1106A mutation. Immunoprecipitation and Western blot analysis in COS-7 cells overexpressing Myc-PDZK1 and either Flag-PDGFR-β-WT or Flag-PDGFR-β-L1106A mutant. e Specific binding of PDGFR-β-CT to the PDZ2 and PDZ3 domains of PDZK1. Pull-down assay using purified proteins and immunoblotting. f, g Colocalization of PDZK1 and PDGFR-β in renal cells. Confocal microscopy images of COS-7 cells expressing PDGFR-β-WT or PDGFR-β-MT (L1106A mutant) (f, a and b) and PDZK1 (f, c). Colocalization of PDGFR-β-WT and PDZK1 is shown in yellow at the plasma membrane (g, a–d). The PDGFR-β L1106A mutation impairs their colocalization (g, e–h).
To investigate the impact of PDZK1 expression on PDGFR-β signaling in ccRCC, we evaluated the phosphorylation of PDGFR-β (Tyr 857) and its downstream target ERK1/2 in ccRCC cells. Overexpression of PDZK1 led to decreased phosphorylation of PDGFR-β and ERK1/2, while knockdown of PDZK1 increased their phosphorylation levels (Fig. 3a–c). Moreover, in COS-7 cells expressing wild-type PDGFR-β, PDZK1 overexpression significantly reduced the phosphorylation of ERK1/2, whereas no such effect was observed in cells expressing the L1106A mutant (Fig. 3d). These findings indicate that the interaction between PDZK1 and PDGFR-β is necessary for PDZK1 to inhibit PDGFR-β signaling.

a PDZK1 overexpression inhibits PDGFR-β pathway activation in ccRCC cells. Western blot analysis of the indicated proteins in cells overexpressing PDZK1 or vector, stimulated with PDGF-BB. b, c Knockdown of PDZK1 enhances PDGFR-β pathway activation in ccRCC cells. Western blot analysis of the indicated proteins in cells transfected with siPDZK1 or siCtrl. d The interaction between PDZK1 and PDGFR-β reduced PDGFR-β pathway activation. Western blot analysis in COS-7 cells transfected with Myc-PDZK1 and PDGFR-β-WT or PDGFR-β-L1106A mutant constructs, stimulated with PDGF-BB after serum starvation for 24 h. e The protein structure prediction of PDGFR-β (a), PDZK1 (b) and the molecular dynamics simulation of the carboxyl terminus of PDGFR-β bound with PDZK1 (c) to block the lysine at positions 579 and 581. Right panels (d) show magnifications of the wireframe area on the left (c). The kinase domains of PDGFR-β (faint yellow); Tyr579, Tyr581, Tyr857 (red); PDZK1 (green). f Schematic diagram of PDZK1 bound to the carboxyl terminus of PDGFR-β, blocking access to the tyrosine residues at positions 579 and 581.
To understand the molecular mechanism underlying the inhibition of PDGFR-β phosphorylation by PDZK1, we employed protein structure prediction and modeling. The results revealed that PDZK1 interacted with PDGFR-β and blocked access to the tyrosine residues at positions 579 and 581, and may lead to a reduction in phosphorylation of tyrosine 857 (Fig. 3e, f).
PDZK1 plays a role in suppressing ccRCC cell proliferation by inhibiting PDGFR-β signaling
Activation of the PDGFR-β pathway is closely associated with cell proliferation [29, 30]. Given that PDZK1 has been demonstrated to effectively inhibit PDGF-induced PDGFR-β phosphorylation at Tyr857, we investigated its role in cell proliferation. CCK8 experiments were conducted following knockdown and overexpression of PDZK1 in 786-O cells. The results showed that knocking down PDZK1 promoted the proliferation of 786-O cells (Fig. 4a), whereas overexpression of PDZK1 significantly inhibited proliferation in both 786-O (Fig. 4b) and ACHN cells (Supplementary Fig. 3A). Consistent with these results, the clone formation assay also showed decreased colony formation in ACHN cells with PDZK1 overexpression and increased colony formation in 786-O cells with PDZK1 knockdown (Fig. 4c, d). To investigate the impact of the interaction between PDGFR-β and PDZK1 on cell proliferation, COS-7 cells were engineered to stably overexpress wild-type or L1106A mutant PDGFR-β with or without coexpressed PDZK1. The results demonstrated that cells overexpressing wild-type or mutant PDGFR-β exhibited enhanced proliferation compared to the control group. However, coexpression of PDZK1 significantly inhibited the growth of COS-7 cells overexpressing wild-type PDGFR-β but not the L1106A mutant (Fig. 4e). Furthermore, PDZK1 was found to regulate cell cycle progression. Knockdown of PDZK1 resulted in increased accumulation of ccRCC cells in the S phase (shCtrl, 25.3% vs. shPDZK1, 40.6%), indicating disrupted cell cycle progression. However, when cells were pretreated with CP-673451, an inhibitor of PDGFR-β, PDZK1 knockdown no longer promoted cell accumulation in the S phase (shCtrl, 25.3% vs. shPDZK1+ CP-673451, 27.0%), suggesting that the effects of PDZK1 on cell cycle progression were dependent on PDGFR-β signaling (Fig. 4f, g).

a PDZK1 knockdown enhances 786-O cell proliferation. b PDZK1 overexpression inhibits 786-O cell proliferation. c, d Clone formation assays in ACHN and 786-O cells demonstrate the impact of PDZK1 on cell proliferation. The cells were seeded in 6-well plates and cultured in 5% FBS medium for 14 days. Clones with more than 50 cells were counted. e PDZK1 inhibits the proliferation of COS-7 cells expressing PDGFR-β-WT, but not the L1106A mutant. The cells were stimulated with PDGF-BB (10 ng/ml), and cell proliferation was assessed as described above. **p < 0.01, PDZK1 + PDGFR-β-WT vs. PDGFR-β-WT; ns not significant, PDZK1 + PDGFR-β-MT vs. PDGFR-β-MT. f, g Inhibition of PDGFR-β activation reverses the PDZK1 knockdown-induced G1-S cell cycle transition, as shown by flow cytometry analysis (f) and Western blot analysis (g). CP-673451 (4 μM). Statistical significance is indicated as *, **, *** (p < 0.05, p < 0.01, p < 0.001).
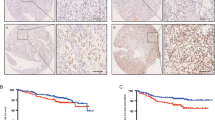
To validate the in vitro findings, Pdzk1 knockout mice (Pdzk1−/−) were generated and examined. Immunohistochemical staining of kidney tissues from Pdzk1−/− mice showed higher levels of PDGFR-β phosphorylation (Tyr857) compared to wild-type controls (Fig. 5a). Additionally, analyzing 90 ccRCC specimens from a tissue microarray, it was found that PDZK1 levels were significantly reduced in tumor tissues from late-stage patients compared to early-stage patients (Fig. 5b, c). Kaplan-Meier survival analysis revealed that ccRCC patients with high PDZK1 levels had better overall survival (Fig. 5d), while high levels of phosphorylated PDGFR-β (Tyr 857) were associated with poor prognosis (Fig. 5e–g). Importantly, there was a negative correlation between PDZK1 protein levels and phosphorylated PDGFR-β levels (Fig. 5h). Moreover, patients with low PDZK1 and high phosphorylated PDGFR-β levels had the worst prognosis, while those with high PDZK1 and low phosphorylated PDGFR-β levels had significantly better survival (Fig. 5i). These findings demonstrate the critical role of PDZK1 in PDGFR-β signaling and its impact on ccRCC progression and prognosis.

a Representative IHC staining of PDZK1 and phosphorylated PDGFR-β (Tyr 857) in kidney tissues from Pdzk1−/− or Pdzk1+/+ mice. Scale bars: 200 μm. Right panels show magnifications of the dashed areas on the left. Scale bars: 50 μm. b, c IHC analysis of PDZK1 protein in ccRCC specimens. Representative staining images of PDZK1 in stage T1 or T3 ccRCC specimens. Scale bars: 200 μm. Right panels show magnifications of the dashed areas on the left. Scale bars: 50 μm. Dot plot showing the corresponding quantification of the PDZK1 histochemistry score (H-score). Data presented as mean ± SEM. Statistical significance calculated by the Mann‒Whitney test. **p < 0.01. d KM survival plots for overall survival (OS) according to PDZK1 H-score in ccRCC specimens (log-rank test). e, f IHC analysis of phosphorylated PDGFR-β (Tyr 857) protein in ccRCC specimens. Representative staining images of p-PDGFR-β in stage T1 or T3 ccRCC specimens. Scale bars: 200 μm. Right panels show magnifications of the dashed areas on the left. Scale bars: 50 μm. Dot plot showing the corresponding quantification of the p-PDGFR-β H-score. Data presented as mean ± SEM. Statistical significance calculated by the Mann–Whitney test. ***p < 0.001. g KM survival plots for OS according to the p-PDGFR-β H-score in ccRCC specimens (log-rank test). h Correlation analysis between PDZK1 H-score and p-PDGFR-β H-score in ccRCC specimens (Spearman). i KM survival plots for OS according to PDZK1 H-score and p-PDGFR-β H-score in ccRCC specimens (log-rank test).
PDZK1 enhances sunitinib sensitivity in ccRCC by suppressing PDGFR-β signaling
Over activation of the PDGFR-β/ERK signaling pathway is associated with sunitinib resistance in RCC cells [31, 32], suggesting that dysregulated low levels of PDZK1 in ccRCC may reduce sunitinib sensitivity. To investigate the role of PDZK1 in sunitinib sensitivity, ccRCC cells (786-O and ACHN) with PDZK1 overexpression were treated with different doses of sunitinib. PDZK1 overexpression significantly reduced the IC50 of sunitinib, indicating improved sunitinib sensitivity in ccRCC cells (Fig. 6a, b). Colony formation assays also revealed that PDZK1 overexpression notably augmented the responsiveness of both 786-O (Fig. 6c) and ACHN cell lines (Supplementary Fig. 3B) to sunitinib. Conversely, depletion of PDZK1 resulted in decreased sensitivity of 786-O cells to sunitinib (Fig. 6d). In subcutaneous xenograft tumor models, PDZK1 overexpression inhibited the growth of ACHN xenograft tumors, and the tumor volume and weight decreased to a greater extent after treatment with sunitinib (Fig. 6e–g). IHC staining of xenograft tissues showed decreased phosphorylated PDGFR-β (Tyr 857) levels in PDZK1-overexpressing tumors (Fig. 6h). These results indicated that PDZK1 overexpression synergistically enhanced the antitumor effects of sunitinib by suppressing PDGFR-β signaling.

a, b PDZK1 overexpression reduces sunitinib IC50 in ACHN and 786-O cells. Dose-response survival curves of PDZK1-overexpressing ACHN (a) or 786-O (b) cells exposed to increasing concentrations of sunitinib for 48 h. Mean ± SEM; n = 3; two-way ANOVA. c, d PDZK1 overexpression inhibits while knockdown promotes clonogenic formation in sunitinib-treated 786-O cells. Quantification analysis of clone formation assays. e–h PDZK1 enhances sunitinib sensitivity in renal cell carcinoma xenografts. Nude mice transplanted with PDZK1-overexpressing ACHN cells or control cells were treated with or without sunitinib. Representative images of xenograft tumors (e), tumor masses (f), and tumor growth curves (g) are shown. Representative IHC staining of PDZK1 and p-PDGFR-β in xenografted tumors are shown in (h). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, sunitinib-treated PDZK1-overexpressed vs. sunitinib-treated control; #p < 0.05, ##p < 0.01, ###p < 0.001, PDZK1-overexpressed vs. control.
To further elucidate the correlation between PDZK1 protein expression and sunitinib responsiveness in a clinical setting, we conducted immunohistochemical analysis of PDZK1 in the primary lesions of 23 advanced or metastatic ccRCC patients who had experienced disease recurrence following nephrectomy and had undergone sunitinib treatment. Our findings indicated that patients who responded favorably to sunitinib demonstrated high PDZK1 expression and low phosphorylated PDGFR-β (Tyr857) expression. Conversely, those with a poorer response to sunitinib exhibited low PDZK1 expression and elevated phosphorylated PDGFR-β (Tyr857) expression (Fig. 7a–c). The PDZK1 protein level was negatively correlated with phosphorylated PDGFR-β (Fig. 7d), and patients with higher PDZK1 levels had better progression-free survival (Fig. 7e). Moreover, based on the H-score of PDZK1, we plotted a ROC curve to assess patient sensitivity to sunitinib treatment. The area under the curve (AUC) registered at 0.89 ± 0.07, highlighting the significant predictive value of PDZK1 (Fig. 7f). Consistently, analysis of TCGA data also showed that patients who received sunitinib treatment with high PDZK1 mRNA levels had better overall survival and progression-free survival (Fig. 7g). These findings indicate that PDZK1 can enhance sunitinib sensitivity in ccRCC by suppressing PDGFR-β signaling.

a–c IHC analysis of PDZK1 and p-PDGFR-β (Tyr 857) in ccRCC specimens from sunitinib-treated patients. Representative staining images of PDZK1 and p-PDGFR-β in responders (R) and non-responders (NR) to sunitinib. Scale bars: 200 μm. Right panels show magnifications of the dashed areas on the left. Scale bars: 50 μm (a). Dot plots showing the quantification of PDZK1 H-score (b) and p-PDGFR-β H-score (c) in 23 ccRCC specimens. Data presented as mean ± SEM. Statistical significance determined using the Mann–Whitney test. *p < 0.05, **p < 0.01. d Correlation analysis (Spearman) between PDZK1 H-score and p-PDGFR-β H-score in 23 ccRCC specimens. e Kaplan-Meier (KM) survival plots for progression-free survival (PFS) based on PDZK1 H-score in 23 ccRCC specimens (log-rank test). f ROC curve with PDZK1 immunohistochemical scores (H-score) in ccRCC specimens from sunitinib-treated patients to predict sunitinib response. g KM survival plots for overall survival (OS) and PFS based on PDZK1 mRNA expression in ccRCC specimens from TCGA patients treated with sunitinib (log-rank test).
To investigate potential therapeutic strategies targeting the PDZK1/PDGFR-β axis, we examined the impact of miR-15b inhibition. Previous studies have identified PDZK1 as a target gene of miR-15b [33], and inhibiting miR-15b has been shown to enhance sunitinib sensitivity in ccRCC cells [34]. In our research, we conducted additional experiments using both miR-15b mimics and inhibitors. Our results demonstrated that miR-15b mimics suppressed PDZK1 expression and increased activity in the PDGFR-β and ERK pathways, while miR-15b inhibitors produced the opposite effects (Supplementary Fig. 4). Combination treatment with miR-15b inhibitor and sunitinib synergistically inhibited ccRCC cell proliferation (Fig. 8a–d). In subcutaneous xenograft experiments, miR-15b antagomirs combined with sunitinib resulted in decreased tumor growth rate and size. IHC staining showed upregulated PDZK1 levels and downregulated phosphorylated PDGFR-β (Tyr 857) in the tumors, indicating the suppression of ccRCC cell growth by upregulating PDZK1 and blocking PDGFR-β signaling (Fig. 8e–h).

a, b hsa-miR-15b-5p inhibitors reduce the IC50 of sunitinib in ACHN and 786-O cells. Dose-response survival curves of ACHN (a) and 786-O (b) cells transfected with hsa-miR-15b-5p inhibitors and treated with increasing concentrations of sunitinib for 48 h. Mean ± SEM; n = 3; two-way ANOVA. c, d hsa-miR-15b-5p inhibitors enhance the ability of sunitinib to inhibit clonogenic formation in ccRCC cells. Clone formation assays of ACHN (c) and 786-O (d) cells transfected with hsa-miR-15b-5p inhibitors or inhibitor control and treated with increasing concentrations of sunitinib for 14 days. Quantification analysis of clones with more than 50 cells is shown (n = 3). e–h hsa-miR-15b-5p antagomir enhances sunitinib sensitivity in renal cell carcinoma xenografts. Nude mice injected with ACHN cells were treated with sunitinib (oral, 50 mg/kg/day) or sunitinib combined with antagomir (local injection, 5 mg/kg, volume 40 ul, 4 injection sites, once every 3 days, 21 days). Representative images of xenograft tumors (e), tumor masses (f), and tumor growth curves (g) are shown. Immunohistochemical staining demonstrates PDZK1 and p-PDGFR-β expression in xenografted tumors (h). Scale bars: 50 μm. Data are presented as mean ± SEM. Significance levels are indicated as *, **, *** (p < 0.05, p < 0.01, p < 0.001).
Overall, these findings demonstrate that PDZK1 enhances sunitinib sensitivity in ccRCC by retarding PDGFR-β signaling. Additionally, miR-15b inhibitors have synergistic effects with sunitinib in suppressing ccRCC cell growth by upregulating PDZK1 and blocking PDGFR-β signaling.
Discussion
Advanced or metastatic ccRCC is a life-threatening disease. Sunitinib has been widely used for the treatment of advanced or metastatic ccRCC and has greatly benefited patients in recent years. However, drug insensitivity and dose-limiting toxicity have limited its effectiveness in improving patient survival [35,36,37]. Therefore, it is urgent to understand the molecular events involved in ccRCC progression and the mechanisms underlying sunitinib sensitivity in order to develop novel approaches for effective therapies.
In this study, we observed significantly reduced expression levels of PDZK1 in sunitinib-resistant ccRCC tissues or cells through screening and validating data from multicenter cohorts (Fig. 1). Functional experiments in ccRCC cells demonstrated that PDZK1 expression conferred sunitinib sensitivity by suppressing cell viability (Fig. 6a–d). These findings were further confirmed in in vivo xenograft tumor models (Fig. 6e–g) and a clinical study (Fig. 7). Collectively, these results highlight the crucial role of PDZK1 in conferring sunitinib sensitivity to ccRCC.
While most studies on ccRCC have focused on VEGF/VEGFR signaling [38,39,40], the significance of PDGF/PDGFR-β signaling in ccRCC development and progression has been underestimated. PDGFR-β is significantly expressed in ccRCC [32, 40], and its activation is associated with poor prognosis [31]. In our study, we analyzed PDGFR-β gene alterations in ccRCC specimens and observed no consistent dysregulation at the DNA or mRNA level (Supplementary Fig. 5A–C). However, we found a positive correlation between the level of phosphorylated PDGFR-β and tumor size, as well as poor prognosis in ccRCC (Fig. 5e–g and Supplementary Fig. 5D). Additionally, our findings revealed that PDZK1 physically interacts with PDGFR-β through the PDZ binding motif, thereby negatively regulating PDGFR-β phosphorylation at Tyr857 and downstream signaling activation, resulting in the inhibition of ccRCC cell proliferation (Figs. 3 and 4). Prior research has established the importance of phosphorylation at tyrosine 857 in PDGFR-β for its full activation and propagation of the PDGF-mediated signal [41,42,43]. Upon PDGF binding to PDGFR-β, specific tyrosine residues, including tyrosines 579 and 581, undergo autophosphorylation. Subsequently, the autophosphorylation of tyrosines 579 and 581 recruits and activates Src-family kinases, which in turn phosphorylate tyrosine 857 in PDGFR-β [41]. Remarkably, our molecular dynamics simulation revealed that PDZK1 interacted with PDGFR-β and blocked access to the tyrosine residues at positions 579 and 581, and may lead to a reduction in phosphorylation of tyrosine 857 (Fig. 3e). This find sheds light on how PDZK1 suppresses PDGFR-β activation, thereby conferring sunitinib sensitivity to ccRCC cells. This was further validated in PDZK1 deficient mouse models (Fig. 5a). Moreover, low PDZK1 expression in ccRCC specimens was associated with PDGFR-β signaling activation and poor prognosis (Figs. 1 and 5b–i).
Given sunitinib’s ability to target and inhibit PDGFR and VEGFR in endothelial cells, an intriguing question arises: does PDZK1 inhibition of PDGFR and VEGFR signaling in endothelial cells impact tumor progression? Several studies have indicated that the loss of PDZK1 expression in endothelial cells has no effect on cell migration or angiogenesis induced by VEGF [44, 45]. Additionally, our GSEA of transcriptome data from Human Umbilical Vein Endothelial Cells (HUVEC) revealed no enrichment of the PDGFR or VEGFR pathways associated with PDZK1 expression. These findings suggest that PDZK1 may not modulate these interactions in endothelial cells (Supplementary Fig. S6). Overall, our study confirms that the loss of PDZK1 expression in ccRCC cells contributes to sustained PDGFR-β signaling activation, thereby promoting ccRCC carcinogenesis and progression.
Systemic treatment with tyrosine kinase inhibitors (TKIs), such as sunitinib, is the standard first-line therapy for advanced or metastatic ccRCC. However, drug insensitivity remains a significant challenge in achieving effective treatment outcomes [46,47,48]. Several factors contribute to drug insensitivity, including the tumor microenvironment, invasiveness, alternative signaling pathways, noncoding RNAs, and insufficient target inhibition [22,23,24,25]. Insufficient target inhibition refers to the situation where the intended target of a TKI is not effectively inhibited or suppressed, leading to reduced therapeutic effectiveness and limited response to treatment. Examples include mutations, such as EGFR T790M, that reduce the binding affinity of TKIs, resulting in inadequate target inhibition. In such cases, higher doses of TKIs or alternative treatment options may be necessary to overcome resistance and achieve effective target inhibition.
In our study, we observed that cells with low levels of PDZK1 exhibited insensitivity to sunitinib treatment, leading to insufficient inhibition of cell proliferation and tumor growth compared to cells with high PDZK1 expression (Figs. 6 and 7). This suggests that consistent activation of PDGFR-β in PDZK1-low-expressing ccRCC cells may require higher doses of sunitinib to achieve therapeutic efficacy, which can lead to dose-dependent toxicity. Therefore, it is crucial to develop strategies that maintain or enhance sunitinib sensitivity in ccRCC cells. These approaches can potentially improve treatment outcomes and minimize the need for high-dose sunitinib, reducing the risk of toxicity.
RNA-based therapies, including RNA-targeted small-molecule drugs, have emerged as promising strategies for cancer therapy. We previously identified miR-15b as a suppressor of PDZK1 expression in ccRCC, and its high level has been associated with sunitinib resistance [34]. In our study, we demonstrated that miR-15b inhibitor or antagomir upregulated PDZK1 expression, resulting in increased sunitinib sensitivity in ccRCC cells (Fig. 8). These findings support the development of miR-15b antagomirs as a potential therapeutic intervention to overcome sunitinib resistance in advanced ccRCC.
Taken together, our study revealed the association of the miR-15b/PDZK1/PDGFR-β axis with ccRCC progression and sunitinib sensitivity. Preclinical investigations indicate that miR-15b antagomirs could be promising drugs to overcome sunitinib resistance by upregulating PDZK1 levels and attenuating PDGFR-β signaling. This provides a theoretical basis for further development of this combination therapy in a clinical setting to enhance the effectiveness of sunitinib treatment.
Data availability
The previously published data underlying Figs. 1, 2, 7 and Supplementary Figs. 1–6 are openly available in [E-MTAB-3267] at [https://www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-3267]; [GSE64052] at [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE64052]; [TCGA, Kidney Renal Clear Cell Carcinoma, PanCancer Atlas] at [https://www.cbioportal.org/study/summary?id=kirc_tcga_pan_can_atlas_2018]; [CPTAC, Clear Cell Renal Cell Carcinoma Discovery Cohort] at [https://pdc.cancer.gov/pdc/study/PDC000128]; [GSE210189] at [https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE210189]. The data underlying Fig. 5 (tissue microarrays containing 90 pairs of ccRCC and adjacent tissues) and Fig. 7 (23 human ccRCC samples from the First Hospital of Shanxi Medical University) in this study are not publicly available due to patient privacy issues. However, the data are available from the corresponding author upon reasonable request (for research only).
References
Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17–48. https://doi.org/10.3322/caac.21763.
Capitanio U, Bensalah K, Bex A, Boorjian SA, Bray F, Coleman J, et al. Epidemiology of renal cell carcinoma. Eur Urol. 2019;75:74–84. https://doi.org/10.1016/j.eururo.2018.08.036.
Jonasch E, Walker CL, Rathmell WK. Clear cell renal cell carcinoma ontogeny and mechanisms of lethality. Nat Rev Nephrol. 2021;17:245–61. https://doi.org/10.1038/s41581-020-00359-2.
Choueiri TK, Kaelin WG. Targeting the HIF2–VEGF axis in renal cell carcinoma. Nat Med. 2020;26:1519–30. https://doi.org/10.1038/s41591-020-1093-z.
Gupta K, Miller JD, Li JZ, Russell MW, Charbonneau C. Epidemiologic and socioeconomic burden of metastatic renal cell carcinoma (mRCC): a literature review. Cancer Treat Rev. 2008;34:193–205. https://doi.org/10.1016/j.ctrv.2007.12.001.
Kotecha RR, Motzer RJ, Voss MH. Towards individualized therapy for metastatic renal cell carcinoma. Nat Rev Clin Oncol. 2019;16:621–33.
Najjar YG, Rini BI. Novel agents in renal carcinoma: a reality check. Ther Adv Med Oncol. 2012;4:183–94. https://doi.org/10.1177/1758834012443725.
Oudard S, George D, Medioni J, Motzer R. Treatment options in renal cell carcinoma: past, present and future. Ann Oncol. 2007;18:x25–31. https://doi.org/10.1093/annonc/mdm411.
Rathmell WK, Rumble RB, Van Veldhuizen PJ, Al-Ahmadie H, Emamekhoo H, Hauke RJ, et al. Management of metastatic clear cell renal cell carcinoma: ASCO guideline. J Clin Oncol. 2022;40:2957–95. https://doi.org/10.1200/JCO.22.00868.
Hsieh JJ, Purdue MP, Signoretti S, Swanton C, Albiges L, Schmidinger M, et al. Renal cell carcinoma. Nat Rev Dis Primers. 2017;3. https://doi.org/10.1038/nrdp.2017.9.
Posadas EM, Limvorasak S, Figlin RA. Targeted therapies for renal cell carcinoma. Nat Rev Nephrol. 2017;13:496–511. https://doi.org/10.1038/nrneph.2017.82.
Motzer RJ, Rini BI, McDermott DF, Arén Frontera O, Hammers HJ, Carducci MA, et al. Nivolumab plus ipilimumab versus sunitinib in first-line treatment for advanced renal cell carcinoma: extended follow-up of efficacy and safety results from a randomised, controlled, phase 3 trial. Lancet Oncol. 2019;20:1370–85. https://doi.org/10.1016/S1470-2045(19)30413-9.
Méjean A, Ravaud A, Thezenas S, Chevreau C, Bensalah K, Geoffrois L, et al. Sunitinib alone or after nephrectomy for patients with metastatic renal cell carcinoma: is there still a role for cytoreductive nephrectomy? Eur Urol. 2021;80:417–24. https://doi.org/10.1016/j.eururo.2021.06.009.
Morais C. Sunitinib resistance in renal cell carcinoma. J Kidney Cancer Vhl. 2014;1:1–11.
Dranitsaris G, Schmitz S, Broom RJ. Small molecule targeted therapies for the second-line treatment for metastatic renal cell carcinoma: a systematic review and indirect comparison of safety and efficacy. J Cancer Res Clin Oncol. 2013;139:1917–26. https://doi.org/10.1007/s00432-013-1510-5.
Ravaud A. Treatment-associated adverse event management in the advanced renal cell carcinoma patient treated with targeted therapies. The Oncologist. 2011;16:32–44. https://doi.org/10.1634/theoncologist.2011-S2-32.
Bracarda S, Iacovelli R, Boni L, Rizzo M, Derosa L, Rossi M, et al. Sunitinib administered on 2/1 schedule in patients with metastatic renal cell carcinoma: the RAINBOW analysis. Ann Oncol. 2015;26:2107–13. https://doi.org/10.1093/annonc/mdv315.
Bracarda S, Sisani M, Marrocolo F, Hamzaj A, Del Buono S, De Simone V. GOAL: an inverse toxicity-related algorithm for daily clinical practice decision making in advanced kidney cancer. Crit Rev Oncol Hematol. 2014;89:386–93. https://doi.org/10.1016/j.critrevonc.2013.09.002.
Knox JJ, Barrios CH, Kim TM, Cosgriff T, Srimuninnimit V, Pittman K, et al. Final overall survival analysis for the phase II RECORD-3 study of first-line everolimus followed by sunitinib versus first-line sunitinib followed by everolimus in metastatic RCC. Ann Oncol. 2017;28:1339–45. https://doi.org/10.1093/annonc/mdx075.
Yamada Y, Ohno Y, Kato Y, Kobayashi R, Hayashi H, Miyahara S, et al. Optimal dose of sunitinib for long-term treatment in Japanese patients with renal cell carcinoma. Cancer Chemother Pharmacol. 2019;84:987–92. https://doi.org/10.1007/s00280-019-03935-x.
Demlova R, Turjap M, Pes O, Kostolanska K, Jurica J. Therapeutic drug monitoring of sunitinib in gastrointestinal stromal tumors and metastatic renal cell carcinoma in adults—a review. Ther Drug Monit. 2020;42:20–32. https://doi.org/10.1097/FTD.0000000000000663.
Jiao Q, Bi L, Ren Y, Song S, Wang Q, Wang Y. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol Cancer. 2018;17. https://doi.org/10.1186/s12943-018-0801-5.
Neul C, Schaeffeler E, Sparreboom A, Laufer S, Schwab M, Nies AT. Impact of membrane drug transporters on resistance to small-molecule tyrosine kinase inhibitors. Trends Pharmacol Sci. 2016;37:904–32. https://doi.org/10.1016/j.tips.2016.08.003.
Westover D, Zugazagoitia J, Cho BC, Lovly CM, Paz-Ares L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol. 2018;29:i10–9. https://doi.org/10.1093/annonc/mdx703.
Sekino Y, Teishima J, Liang G, Hinata N. Molecular mechanisms of resistance to tyrosine kinase inhibitor in clear cell renal cell carcinoma. Int J Urol. 2022. https://doi.org/10.1111/iju.15042.
Tao T, Yang X, Zheng J, Feng D, Qin Q, Shi X, et al. PDZK1 inhibits the development and progression of renal cell carcinoma by suppression of SHP-1 phosphorylation. Oncogene. 2017;36:6119–31. https://doi.org/10.1038/onc.2017.199.
Zhao C, Tao T, Yang L, Qin Q, Wang Y, Liu H, et al. Loss of PDZK1 expression activates PI3K/AKT signaling via PTEN phosphorylation in gastric cancer. Cancer Lett. 2019;453:107–21. https://doi.org/10.1016/j.canlet.2019.03.043.
Wang H, Yang W, Qin Q, Yang X, Yang Y, Liu H, et al. E3 ubiquitin ligase MAGI3 degrades c-Myc and acts as a predictor for chemotherapy response in colorectal cancer. Mol Cancer. 2022;21. https://doi.org/10.1186/s12943-022-01622-9.
Dong LH, Wen JK, Miao SB, Jia Z, Hu HJ, Sun RH, et al. Baicalin inhibits PDGF-BB-stimulated vascular smooth muscle cell proliferation through suppressing PDGFRbeta-ERK signaling and increase in p27 accumulation and prevents injury-induced neointimal hyperplasia. Cell Res. 2010;20:1252–62. https://doi.org/10.1038/cr.2010.111.
Xue Y, Lim S, Yang Y, Wang Z, Jensen LDE, Hedlund E, et al. PDGF-BB modulates hematopoiesis and tumor angiogenesis by inducing erythropoietin production in stromal cells. Nat Med. 2012;18:100–10. https://doi.org/10.1038/nm.2575.
Shim M, Song C, Park S, Choi S, Cho YM, Kim C, et al. Prognostic significance of platelet-derived growth factor receptor-β expression in localized clear cell renal cell carcinoma. J Cancer Res Clin Oncol. 2015;141:2213–20. https://doi.org/10.1007/s00432-015-2019-x.
Carrato Mena A, Grande Pulido E, Guillén-Ponce C. Understanding the molecular-based mechanism of action of the tyrosine kinase inhibitor: sunitinib. Anticancer Drugs. 2010;21:S3–11. https://doi.org/10.1097/01.cad.0000361534.44052.c5.
Qi Y, Ma Y, Peng Z, Wang L, Li L, Tang Y, et al. Long noncoding RNA PENG upregulates PDZK1 expression by sponging miR-15b to suppress clear cell renal cell carcinoma cell proliferation. Oncogene. 2020;39:4404–20. https://doi.org/10.1038/s41388-020-1297-1.
Lu L, Li Y, Wen H, Feng C. Overexpression of miR-15b promotes resistance to sunitinib in renal cell carcinoma. J Cancer. 2019;10:3389–96. https://doi.org/10.7150/jca.31676.
Lee WJ, Lee JL, Chang SE, Lee MW, Kang YK, Choi JH, et al. Cutaneous adverse effects in patients treated with the multitargeted kinase inhibitors sorafenib and sunitinib. Br J Dermatol. 2009;161:1045–51. https://doi.org/10.1111/j.1365-2133.2009.09290.x.
Lankhorst S, Baelde HJ, Kappers MHW, Smedts FMM, Hansen A, Clahsen-van Groningen MC, et al. Greater sensitivity of blood pressure than renal toxicity to tyrosine kinase receptor inhibition with sunitinib. Hypertension. 2015;66:543–9. https://doi.org/10.1161/HYPERTENSIONAHA.115.05435.
Narayan V, Keefe S, Haas N, Wang L, Puzanov I, Putt M, et al. Prospective evaluation of sunitinib-induced cardiotoxicity in patients with metastatic renal cell carcinoma. Clin Cancer Res. 2017;23:3601–9. https://doi.org/10.1158/1078-0432.CCR-16-2869.
Stitzlein L, Rao P, Dudley R. Emerging oral VEGF inhibitors for the treatment of renal cell carcinoma. Expert Opin Investig Drugs. 2019;28:121–30. https://doi.org/10.1080/13543784.2019.1559296.
Hirsch L, Flippot R, Escudier B, Albiges L. Immunomodulatory roles of VEGF pathway inhibitors in renal cell carcinoma. Drugs. 2020;80:1169–81. https://doi.org/10.1007/s40265-020-01327-7.
Song SH, Jeong IG, You D, Hong JH, Hong B, Song C, et al. VEGF/VEGFR2 and PDGF-B/PDGFR-beta expression in non-metastatic renal cell carcinoma: a retrospective study in 1,091 consecutive patients. Int J Clin Exp Pathol. 2014;7:7681–9.
Baxter RM, Secrist JP, Vaillancourt RR, Kazlauskas A. Full activation of the platelet-derived growth factor beta-receptor kinase involves multiple events. J Biol Chem. 1998;273:17050–5. https://doi.org/10.1074/jbc.273.27.17050.
Kazlauskas A, Durden DL, Cooper JA. Functions of the major tyrosine phosphorylation site of the PDGF receptor β subunit. Cell Regul. 1991;2:413–25.
Nickas ME, Bernard A, Kazlauskas A. The requirement of tyrosines 579 and 581 for maximal ligand-dependent activation of the βPDGFR is influenced by noncytoplasmic regions of the receptor. Exp Cell Res. 2001;265:80–9. https://doi.org/10.1006/excr.2001.5169.
Turner EC, Mulvaney EP, Reid HM, Kinsella BT. Interaction of the human prostacyclin receptor with the PDZ adapter protein PDZK1: role in endothelial cell migration and angiogenesis. Mol Biol Cell. 2011;22:2664–79. https://doi.org/10.1091/mbc.E11-04-0374.
Zhu W, Saddar S, Seetharam D, Chambliss KL, Longoria C, Silver DL, et al. The scavenger receptor class B type I adaptor protein PDZK1 maintains endothelial monolayer integrity. Circ Res. 2008;102:480–7. https://doi.org/10.1161/CIRCRESAHA.107.159079.
Dai C, Liang Y, Wang Y, Tiwari AK, Yan Y, Wang F, et al. Sensitization of ABCG2-overexpressing cells to conventional chemotherapeutic agent by sunitinib was associated with inhibiting the function of ABCG2. Cancer Lett. 2009;279:74–83. https://doi.org/10.1016/j.canlet.2009.01.027.
Shukla S, Robey RW, Bates SE, Ambudkar SV. Sunitinib (Sutent, SU11248), a small-molecule receptor tyrosine kinase inhibitor, blocks function of the ATP-binding cassette (ABC) transporters P-glycoprotein (ABCB1) and ABCG2. Drug Metab Dispos. 2009;37:359–65. https://doi.org/10.1124/dmd.108.024612.
Jin J, Xie Y, Zhang JS, Wang JQ, Dai SJ, He WF, et al. Sunitinib resistance in renal cell carcinoma: from molecular mechanisms to predictive biomarkers. Drug Resist Updat. 2023;67:100929. https://doi.org/10.1016/j.drup.2023.100929.
Acknowledgements
We would like to thank Doctor Tongmei Zhang (Beijing Chest Hospital, Capital Medical University) and Doctor Jinwei Miao (Beijing Obstetrics and Gynecology Hospital, Capital Medical University) for kindly providing some clinical samples. We thank Dr. Randy Hall at Emory University (Atlanta, GA) and Dr. Michael R Beard at the Centre for Cancer Biology (Adelaide, Australia) for plasmids materials.
Funding
This study was supported by the National Natural Science Foundation of the People’s Republic of China (Grants Nos. 81772707, 81972732, 82273965); China Postdoctoral Foundation (Grants No. 2023M732409). The funding agency did not participate in the study design, data collection, analysis and interpretation, or the writing of this manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization: JH and HW; data curation: LZ, HL and HW; methodology: HW and LZ; investigation: RS and QQ; validation: XY, DF and SW; visualization: HW and WL; formal analysis: XC and YY; project administration: SW and JH; resources: TB; supervision: JH and TB; funding acquisition: JH and HW; writing—original draft: HL and HW; writing—review & editing: JH and TB.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Animal experiments were conducted in compliance with the National Institutes of Health guidelines for the Care and Use of Laboratory Animals and approved by the Animal Use and Care Committee of Capital Medical University (approval numbers AEEI-2020-133 and AEEI-2018-201). The research involving ccRCC samples was approved by the Ethics Committee of Capital Medical University (2017SY09) and the First Hospital of Shanxi Medical University (2022HLL001).
Consent for publication
Written informed consent was obtained from all patients for the use of their tissue specimens and publication of this report.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, H., Zhang, L., Liu, H. et al. PDZK1 confers sensitivity to sunitinib in clear cell renal cell carcinoma by suppressing the PDGFR-β pathway. Br J Cancer 131, 347–360 (2024). https://doi.org/10.1038/s41416-024-02725-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-024-02725-4
- Springer Nature Limited