
Abstract
Background
Anti-microtubule agents are widely used to treat ovarian cancers, but the efficacy is often compromised by drug resistance. We investigated co-targeting the actin/tropomyosin cytoskeleton and microtubules to increase treatment efficacy in ovarian cancers and potentially overcome resistance.
Methods
The presence of tropomyosin-3.1 (Tpm3.1) was examined in clinical specimens from ovarian cancer patients using immunohistochemistry. Combinatorial effects of an anti-Tpm3.1 compound, ATM-3507, with vinorelbine and paclitaxel were evaluated in ovarian cancer cells via MTS and apoptosis assays. The mechanisms of action were established using live- and fixed-cell imaging and protein analysis.
Results
Tpm3.1 is overexpressed in 97% of tumour tissues (558 of 577) representing all histotypes of epithelial ovarian cancer. ATM-3507 displayed synergy with both anti-microtubule agents to reduce cell viability. Only vinorelbine synergised with ATM-3507 in causing apoptosis. ATM-3507 significantly prolonged vinorelbine-induced mitotic arrest with elevated activity of the spindle assembly checkpoint and mitotic cell death; however, ATM-3507 showed minor impact on paclitaxel-induced mitotic defects. Both combinations substantially increased post-mitotic G1 arrest with cyclin D1 and E1 downregulation and an increase of p21Cip and p27Kip.
Conclusion
Combined targeting of Tpm3.1/actin and microtubules is a promising treatment strategy for ovarian cancer that should be further tested in clinical settings.
Similar content being viewed by others

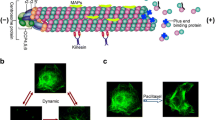

Background
As the second most common gynaecological malignancy, epithelial ovarian cancer is responsible for ~185,000 female death worldwide each year.1 The majority of patients have advanced-stage disease at initial diagnosis and have a 5-year survival of <30%.2 The chemotherapy combination of carboplatin (CBDCA) and paclitaxel (PTX) has been the standard of care for over 30 years with the most important recent progress being the introduction of maintenance therapy with poly ADP ribose polymerase (PARP) inhibitors in selected subsets of patients with high-grade serous cancer following response to first-line chemotherapy.3 Although response rates are high, the majority of patients will experience a recurrence and most will die with drug-resistant disease often after many lines of treatment, which commonly includes a taxane.3 Single agents such as weekly PTX either alone or in combination with bevacizumab is commonly used to treat patients with platinum-resistant ovarian cancer.4 The response rates with chemotherapy in platinum-resistant ovarian cancers are low and in the pivotal AURELIA trial was 12% and the progression-free survival 3.4 months.5 These numbers increased with the addition of bevacizumab to 30% and 6.4 months, respectively, although there was no difference in overall survival with the addition of bevacizumab.5 Vinorelbine (VNB) is an anti-microtubule agent that has been reported to have similar efficacy to PTX in phase 2 trials,6 but this was never extended to phase 3 trials in ovarian cancer probably due to the fact that it was out of patent and not an attractive option given there are a number of generics on the market since 2003. Thus, investigating new approaches to increase the efficacy of anti-microtubule agents is of strong rationale, particularly in patients with platinum-resistant high-grade serous cancers as well as in patients with other histotypes such as clear cell, mucinous and low-grade serous cancers where response rates to standard of care chemotherapy are even lower.4
We recently discovered a strong anti-tumour synergy that resulted from combined targeting of microtubules and tropomyosin 3.1 (Tpm3.1)-containing actin filaments.7,8 Tpm3.1 belongs to the tropomyosin family, which is a category of actin-binding proteins that form co-polymers with actin and determine the functional capability of actin filaments in an isoform-dependent manner.9 Tpm3.1 exhibits dynamic interactions with actin filaments and is able to activate myosin II ATPase, but it does not protect actin filaments from cofilin-mediated severing.10 Tpm3.1-containing actin filaments mediate the nuclear translocation of the extracellular signal-regulated kinase and thus promote cell growth and proliferation of mouse fibroblast.11 Upregulation of this isoform is associated with the process of cellular transformation and it is the major tropomyosin isoform in cancer cells.12 We have developed compounds, such as ATM-3507, that specifically target the C-terminus of Tpm3.1 and successfully inhibit tumour growth without compromising cardiac function, which is the major side effect of anti-actin agents.7,8,13,14
ATM-3507 in combination with anti-microtubule agents have shown profound synergy in a neuroblastoma xenograft mouse model.7,8 The synergy of targeting Tpm3.1 and microtubule depolymerisation was further confirmed in a lung cancer xenograft model and multiple cell lines including an ovarian cancer cell line, OVCAR-3.8 Moreover, it has long been reported that the actin cytoskeleton interacts with the microtubule network in broad cellular functions, including migration, intracellular transport and division.15,16 Synergistic effects of anti-tropomyosin compounds (ATMs) and microtubule drugs therefore represent a novel strategy of co-targeting the actin cytoskeleton and microtubules to improve the efficacy of anti-microtubule agents and could have broad application given the widespread use of these agents in multiple cancers, including ovarian cancer, breast cancer and non-small cell lung cancer among many others.17
Based on the effects on microtubules at high concentrations, anti-microtubule agents are divided into depolymerising agents (e.g. vinca alkaloids) and stabilising agents (e.g. taxanes).17 At clinically relevant concentrations, both types of anti-microtubule agents interfere with microtubule dynamics without altering the polymer mass.17 Our previous findings showed that ATM-3507 promotes vincristine (microtubule depolymeriser) induced mitotic defects,7,8 but the mechanism of synergy of anti-Tpm3.1 compounds and microtubule stabilisers has not been established. Cells exposed to anti-microtubule agents that eventually complete a prolonged mitosis are often blocked in the subsequent (or post-mitotic) interphase.18 As the actin cytoskeleton interacts with the microtubule network throughout the cell cycle,15 combined targeting of both cytoskeletons may result in a combinatorial effect on the post-mitotic interphase. Therefore, the synergy of anti-Tpm3.1 compounds and anti-microtubule agents may include enhanced interruptions in both mitosis and post-mitotic interphase.
In this study, we investigate the expression of Tpm3.1 in ovarian cancer tissues and evaluate the combined targeting of Tpm3.1 and microtubules in ovarian cancer cell lines. The combination of ATM-3507 with either the microtubule depolymeriser, VNB, or the microtubule stabiliser, PTX, synergistically reduces the viability of ovarian cancer cells. While the mitosis-dependent synergy relies on the microtubule drug in use, combinations of ATM-3507 and both microtubule drugs are sufficient to promote G1 arrest in the subsequent interphase.
Methods
Clinical samples
Tumour tissues were obtained from the Alberta Ovarian Tumour Type Study (AOV).19 and Calgary Serous Carcinoma Study (CAL).20 Five hundred and ninety-eight samples from 577 patients were received, in which 462 samples from AOV and 130 samples from 115 patients from CAL were used to generate the tumour microarrays.
Chemicals
ATM-3507 was synthesised as described previously.7 VNB (S4269) was purchased from Selleck Chemicals. PTX (T7191) and CBDCA (C2538) were purchased from Sigma Aldrich. Stock solutions (10 mM) of ATM-3507, VNB and PTX were prepared by dissolving the solid compounds in dimethyl sulfoxide (DMSO; D2650, Sigma Aldrich) and stored at −20 °C. CBDCA solutions were prepared fresh by dissolving the powder in sterilised Milli-Q water.
Antibodies
Primary antibodies: mouse anti-Tpm2.1 (CG1; 1:1000); mouse anti-Tpm3.1 (γ9/d; 1:1000); rabbit anti-Tpm4.1/4.2 (δ9/d; 1:1000); rabbit anti-cyclin E1 (sc-198, Santa Cruz; 1:1000); mouse anti-cyclin D1 (sc-8396, Santa Cruz; 1:500); mouse anti-p27 (3698S, Cell Signaling Technology; 1:1000); mouse anti-p21 (2946T, Cell Signaling Technology; 1:1000); rabbit anti-p16 (ab108349, Abcam; 1:1000); mouse anti-GAPDH (MAB374, Millipore; 1:4000), human anti-centromere antibody (15-235-0001, Antibodies Inc.; 1:200); mouse anti-BubR1 (612502, BD Biosciences; 1:500); rabbit anti-BubR1 (phospho S670; ab200062, Abcam; 1:2000); rabbit anti-phospho-Histone H3 (pSer10; Merck; 1:1000).
Secondary antibodies: rabbit anti-mouse immunoglobulin G (IgG) (horseradish peroxidase (HRP) conjugate, A9044, Sigma-Aldrich; 1:4000); goat anti-rabbit IgG (HRP conjugate, 1706515, Bio-Rad; 1:4000); chicken anti-human IgG (fluorescein isothiocyanate (FITC) conjugate, IHF-1010, Aves Labs; 1:500); goat anti-mouse IgG (AF555 conjugate, A-21422, Invitrogen; 1:500); goat anti-rabbit IgG (FITC conjugate, AB6717, Abcam; 1:1000).
Cell lines and cultures
Three human ovarian cancer cell lines, A2780 (ECACC 93112519), A2780cis (ECACC 93112517) and OVCAR4 (NCI-DTP OVCAR-4), were cultured in RPMI-1640 (11875-119, Gibco) supplemented with 10% foetal bovine serum (FBS; 10099141, Gibco). The immortalised human ovarian surface epithelium HOSE-6.3 (RRID:CVCL_7673) was cultured in 1:1 mixture of MCDB 105 medium (117–500, Sigma Aldrich) and Medium 199 (M4530, Sigma Aldrich) supplemented with 10% FBS.21 To maintain platinum resistance in A2780cis cells, medium was supplemented with 10 µM CBDCA. All cell lines were cultured in a humidified 37 °C incubator, 5% CO2. All cell lines were negative for mycoplasma using real-time PCR-based screening.
Immunohistochemistry
Immunostaining was performed at the Histopathology Facility, Garvan Institute of Medical Research (Sydney, Australia) using γ9/d anti-body (1:500 dilution in 0.1% (v/v) saponin with 2% (w/v) bovine serum albumin in phosphate-buffered saline), which has been optimised with control tissues (tonsil and uterus).22 Images were taken on a Vectra Polaris slide scanner (CLS143455, PerkinElmer) with a ×20 objective (CFI Plan Apochromat Lambda, ×20/0.75, Nikon).
Stained slides were scored by a gynaecologic pathologist (K.T.), blinded to histotype. The intensity was graded on a scale of 0 (absence), 1 (weak), 2 (moderate) and 3 (intense).
Western blot
Western blotting for proteins of interest was performed as previously described.12 To minimise the cell volume-dependent variations in the expression of housekeeping gene, a duplicate gel was incubated in Coomassie Blue R-250 staining solution (1610436, Bio-Rad) and then de-stained for total protein quantification as the loading control for the detection of tropomyosin isoforms.23
Cell viability assay
Cells were seeded into 96-well plates (5000 cells/well for A2780 and A2780cis; 2500 cells/well for OVCAR4) 24 h prior to drug treatments. The MTS-based cell viability assay was performed as previously described.8
Annexin V apoptosis assay
OVCAR4 cells were seeded into 6-well plates (2 × 105 cells/well for A2780 and A2780cis; 1 × 105 cells/well for OVCAR4) 24 h prior to drug treatments. The apoptosis assay was performed as described previously.8 Both Annexin V-positive and Annexin V/7-AAD double positive cells were scored as apoptotic.
Combination index (CI) analysis
The CI was calculated using the CompuSyn software (ComboSyn, Inc.) as described previously.8,24 The CI values based on actual experimental points were plotted as dots and the interpolated trends of CI values are presented as lines in each graph.
Live-cell imaging
OVCAR4 cells were seeded (1 × 105 cells/well) onto glass bottom 6-well plates (P06G-1.5–20-F, MatTek) 24 h prior to drug treatments. Time-lapse live-cell imaging was performed described previously using a Nikon microscope (Eclipse Ti2, Nikon) with a ×20 objective (CFI Plan Fluor DLL ×20/0.5, Nikon), phase-contrast mode.8
Immunofluorescence
OVCAR4 cells were seeded (1 × 105 cells/well) on coverslips in 6-well plates and treated as for live-cell imaging for 24 h. Immunostaining was performed as previously described.25 Images were acquired on a Zeiss LSM 900 with Airyscan 2 module using a ×63 objective (Plan-Apochromat ×63/1.4 oil, Zeiss) under the same illumination settings. ZEN software (blue edition, Zeiss) was used for image acquisition and Airyscan processing.
Image analysis
Fiji software (RRID:SCR_002285) was used for post-acquisition analysis. For live-cell imaging, time in mitosis was defined as the time from cell rounding to the exit of mitosis or mitotic cell death. BubR1 intensity was quantified as described previously.25 Threshold was set to raw images of 0.13-μm section z-stacks on each slice and all centromeres were selected under the ACA channels. Masks of the same areas were applied to the BubR1 channel. The average intensity of both channels was measured after background subtraction. All images in Fig. 4c were adjusted under the same settings.
Statistical analysis
Unpaired two-tailed Student’s t test was performed in Excel (Microsoft) for the measurement of statistical significance. p < 0.05 was considered statistically significant. All data points with error bars are presented as mean ± standard deviation (mean ± S.D.).
Results
Tpm3.1 is highly expressed in tumour tissue in all histotypes of epithelial ovarian cancer
Previous findings showed that Tpm3.1 is the dominant tropomyosin isoform consistently expressed in cancer cells;12,26 however, the expression of Tpm3.1 has not been examined in ovarian tumours. We thus investigated its expression in tumour tissues from large cohorts with a total of 577 ovarian cancer specimens using Tpm3.1 antibody.27 (Table 1 and Supplemental Tables S1 and S2). Tumour tissues collected from the AOV and CAL studies contained samples from the five main histotypes (n = 510), high-grade serous, low-grade serous, endometrioid, clear cell, mucinous carcinoma, and mucinous borderline ovarian tumours (n = 67) (Table 1). Tpm3.1 in 98% of invasive primary samples (498 out of 510 patients) was intensely stained regardless of tumour histotype, suggesting that high Tpm3.1 expression is common across ovarian tumour tissues (Table 1, Fig. 1 and Supplemental Fig. S1). Intense staining (score 3) of Tpm3.1 was also observed in 90% (60 out of 67) of mucinous borderline ovarian tumours and ubiquitous in a series of matched tumours from 8 patients from the CAL study, with consistently high expression in the ovary, omentum and recurrent tissue samples (Table 1 and Supplemental Table S3). Moreover, high levels of Tpm.3.1 were present in all sites sampled and similar at primary diagnosis and at recurrence (Supplemental Table S3). However, we could not correlate Tpm3.1 expression with FIGO staging and grade due to the lack of samples with low-to-moderate staining (Supplemental Tables S1 and S2).

Images represent staining scoring of 1 (weak), 2 (moderate) and 3 (intense). HGS high-grade serous carcinoma, MUC mucinous carcinoma. Note that adjacent non-tumour cells are poorly stained. Scale bar, 150 μm.
Targeting Tpm3.1 exhibits high efficacy against the viability of ovarian cancer cells
Targeting Tpm3.1 using ATM-3507 was compared with three agents (a microtubule depolymeriser, VNB, a microtubule stabiliser, PTX, and a platinum agent, CBDCA), which are commonly used in ovarian cancer treatments. The potency and efficacy of single agents on cell viability were examined in three human ovarian cancer cell lines, A2780, A2780cis and OVCAR4. A2780 is an endometrioid ovarian cancer cell line and is commonly used to test potential treatments because these cells have not been exposed to anti-cancer drugs and are highly sensitive to platinum and taxanes.28 A2780cis cells were selected due to their platinum resistance, which is the most common chemoresistance in ovarian cancer patients.29,30 Indeed, CBDCA had an approximately three times higher IC50 in A2780cis cells than that in A2780 cells (Supplemental Table S4). OVCAR4 cell line represents the most common ovarian cancer histotype, high-grade serous carcinoma.28 All three cell lines exhibited consistent high expression of Tpm3.1 while displaying variable amounts of other tropomyosin isoforms compared to an ovarian surface epithelium, HOSE-6.3 (Supplemental Fig. S2).
ATM-3507 had low-micromolar IC50s and exhibited the highest efficacy in all the tested cell lines with more than a 95% cell viability reduction at concentrations approximately 2-fold of its IC50 (Fig. 2a–c and Supplemental Table S4). Although both VNB and PTX had IC50s in the nanomolar range, they produced the maximum outcome with 80–85% viability reduction in A2780 and A2780cis cells and 60–70% in OVCAR4 cells (Fig. 2a–c and Supplemental Table S4). CDBCA is less effective than ATM-3507 with 11–53 times higher IC50s (Supplemental Table S4). While CDBCA showed similar efficacy as ATM-3507 in A2780 and A2780cis cells, it reached a dose limitation at 2.5-fold IC50 in the high-grade serous OVCAR4 cells (Fig. 2a–c). These results indicate that disrupting Tpm3.1 is highly efficient in reducing the viability of ovarian cancer cells of different histotypes, including platinum-sensitive and -resistant lines.

a–c A2780, A2780cis and OVCAR4 cells were treated with single agents as indicated for 72 h and corresponding cell viability reduction was measured by MTS assay. Concentrations of each drug were converted into fold changes to their IC50. d–f Dose–response curves of ATM, VNB and their combination. g–i Dose–response curves of ATM, PTX and their combination. j–l Chou–Talalay analysis of synergy of each drug combination in the three ovarian cancer cell lines. Lines showing the trend of CI values interpolated from actual CI values presented by each dot in the same drug combination. CI values below 0.9 indicate synergy.24 Fraction affected indicates the percentage of cell viability reduction. CBDCA carboplatin. Data were generated from at least three independent experiments and presented as mean ± S.D. if applicable.
Combined targeting of Tpm3.1 and microtubules is synergistic
In previous studies, we reported that ATM synergises with anti-microtubule agents to reduce viability in cancer cells in vitro and in vivo.7,8 While initial data showed the synergy of ATM-3507 in combination with VNB in ovarian cancer cells (OVCAR-3),8 the combinatorial effects of ATM-3507 and PTX have not yet been studied in this cancer type. Here the efficacy of ATM-3507 in combination with VNB or PTX to reduce the cell viability was evaluated in A2708, A2780cis and OVCAR4 cells, which was compared with the standard first-line combinational chemotherapy for ovarian cancer, CBDCA/PTX. The addition of ATM significantly enhanced the treatment efficacy of VNB and PTX in all three cell lines, regardless of cell types as well as platinum resistance (Fig. 2d–i). In contrast, the CBDCA/PTX combination slightly increased the viability reduction in A2780 cells, while its efficacy in A2780cis and OVCAR4 cells was lower compared to individual drugs (Supplemental Fig. S3). Quantitative analysis on the combination effects using the Chou–Talalay method showed that ATM-3507 synergises with both VNB and PTX in all tested cell lines at most of the fraction affected (percentage of reduced cell viability) where the CI values are <0.9 (Fig. 2j–l).24 However, an antagonistic effect (CI > 1.1) was observed between CBDCA and PTX (Fig. 2j–l).
Although all single agents showed a comparable activity in ovarian cancer cells versus the non-tumour HOSE-6.3 cells, the CI analysis demonstrated that ATM-3507/VNB is slightly synergistic and ATM-3507/PTX is nearly additive against HOSE-6.3 cells (Supplemental Fig. S4). Compared to the strong synergy in three ovarian cancer cell lines, data in HOSE6.3 cells indicate that combining ATM-3507 and anti-microtubule agents may have a better selectivity against cancer cells than when used as single agents.
ATM-3507 synergistically induces apoptosis with VNB but not with PTX
We previously reported that the combination of ATM-3507 and anti-microtubule agents synergistically induces apoptosis in a neuroblastoma cell line and HeLa cells.7,8 To examine whether the synergy of ATM-3507/VNB and ATM-3507/PTX is cytotoxic in ovarian cancer cells, we measured the apoptotic cell population by flow cytometry using the Annexin V/7-AAD staining. An increased population of apoptotic cells was induced by ATM-3507/VNB over that seen with ATM-3507 or VNB alone in a dose-dependent manner (Fig. 3a–c). In contrast, little difference was observed between ATM-3507/PTX and either drug alone in apoptosis induction (Fig. 3d–f). CI analysis using the Chou–Talalay method demonstrated that ATM-3507 synergised with VNB in inducing apoptosis (Fig. 3g–i), but the interaction between ATM-3507 and PTX trended towards an antagonistic or additive relationship (Fig. 3g–i). Collectively, these data show that ATM-3507 synergistically induces apoptosis with VNB but not with PTX in ovarian cancer cells.

a–f A2780, A2780cis and OVCAR4 were incubated with single agents or in combination as indicated for 48 h and the corresponding induction of apoptosis was measured by Annexin V apoptosis assay using flow cytometry. Dose–response curves of ATM, VNB and their combination are shown in a–c. Dose–response curves of ATM, PTX and their combination are shown in d–f. g–i Chou–Talalay analysis of synergy of two drug combinations in the three ovarian cancer cell lines. Lines showing the trend of CI values interpolated from actual CI values presented by each dot in the same drug combination. CI values <0.9 indicate synergy.24 Fraction affected indicates the percentage of induced apoptosis. Data were generated from at least three independent experiments and presented as mean ± S.D. if applicable.
ATM-3507 enhances VNB-induced mitotic arrest with elevated activity of the spindle assembly checkpoint (SAC)
Our previous studies demonstrated a mitosis-dependent synergy between ATM-3507 and microtubule depolymerisers.7,8 To further investigate the difference in the mechanism of action between ATM-3507/VNB and ATM-3507/PTX in ovarian cancer cells, we monitored the response of single cells and determined their fates upon drug treatments using live-cell imaging (Supplemental Fig S5). OVCAR4 cells were chosen because they are the most suitable model for imaging among the three studied cell lines. Moreover, OVCAR4 line is from the high-grade histotype, which is the most common type of ovarian cancer. ATM-3507 did not perturb mitotic progression with an average of 1.1 h to complete the first mitosis, as compared with the control (Fig. 4a). VNB and PTX alone significantly arrested cells in mitosis to an average of 4.3 h (Fig. 4a). However, the VNB-induced mitotic arrest was prolonged by ATM-3507 with an average of 13 h, whereas ATM-3507 only slightly increased the mitotic duration in PTX-treated cells by about 1 h (Fig. 4a).

a Time that cells spent in the first mitosis. OVCAR4 cells were treated with DMSO, ATM (5 μM), VNB (8 nM), A + V (5 μM ATM + 8 nM VNB), paclitaxel (PTX, 5 nM) and A + P (5 μM ATM + 5 nM PTX), respectively, and then subjected to time-lapse microscopy. Images were recorded every 2 min for 72 h. For each treatment, 100 cells from each of at least three independent experiments were analysed. b The percentage of OVCAR4 cells in a that underwent different cell fates before entering the next interphase. c Representative images showing different BubR1 immunostaining in cells treated as in a for 24 h before fixation and antibody staining. Scale bar, 2 μm. d The ratio of fluorescent intensity of BubR1 relative to centromere (ACA). The average ratio of a cell is presented as an individual dot. In each treatment, 30 randomly imaged late prometa/metaphase cells were analysed. e The expression of p-BubR1 (S670) in cells treated with VNB (8 nM), A + V (5 μM ATM + 8 nM VNB), PTX (5 nM) and A + P (5 μM ATM + 5 nM PTX), respectively, for 24 h. Treated cells collected via mitotic shake-off was lysed for western blotting analysis. The proportion of mitotic cells in each shake-off cell population was measured using propidium iodide (PI) and anti-p-Histone H3 (pSer10) antibody via flow cytometry.8 GAPDH was detected as the loading control. Error bars are mean ± S.D.; n.s. not significant; *p < 0.05; **p < 0.001.
Moreover, 21% of VNB-treated cells exited mitosis without division, a phenomenon known as mitotic slippage,31 while 75% of them underwent division, 1% dying in mitosis and 3% remaining in interphase without entering into mitosis (Fig. 4b and Supplemental Fig. S6). However, the addition of ATM-3507 markedly increased the slipped cell population (42%) or death in the first mitosis (28%; Fig. 4B and Supplemental Fig. S6). In contrast, cells treated with PTX or ATM-3507/PTX had similar cell fate during the first cell cycle, with 42 versus 45% cells slipped (Fig. 4B and Supplemental Fig. S6). These data suggest that the anti-mitotic synergy is unique to the combination of ATM-3507 and microtubule depolymerisers in ovarian cancer cells.
During mitosis, the SAC plays an essential role in maintaining the accuracy of chromosome segregation by delaying the anaphase onset.32 Kinetochores unattached and improperly attached to microtubules lead to SAC activation by generating a protein complex consisting of Mad-2, Bub1, BubR1 and Cdc20, which inhibits the anaphase-promoting complex/cyclosome.32 Moreover, phosphorylation on BubR1 at S670 is found significantly enhanced at unattached kinetochores but not at those lacking tension.33,34 Anti-microtubule drugs interfere with the dynamics of microtubules, leading to aberrant spindle formation and activation of the SAC (Fig. 4c).35 Since a significantly prolonged mitotic arrest resulted from ATM-3507 with VNB, but not with PTX, we reasoned that combining ATM-3507 with VNB may reinforce the activity of the SAC.
The fluorescence intensity of BubR1 on kinetochores was measured to indicate the activity of the SAC.36,37 We found that the activity of the SAC was significantly stronger in ATM-3507 and VNB co-treated cells than VNB alone as indicated by the increased BubR1 intensity on kinetochores (Fig. 4c, d). In contrast, ATM-3507/PTX caused no change in SAC activity as compared to PTX alone (Fig. 4c, d). To confirm the drug impacts on SAC activation, p-BubR1 (S670) expression in treated cells was evaluated. After drug treatments, mitotic shake-off was performed to increase the proportion of mitotic cells for western blot analysis. We observed that the level of p-BubR1 was increased in VNB-treated cells containing 60% of cells in mitosis, as compared to the control (without mitotic shake-off) with 2.6% mitotic population (Fig. 4e). The upregulation of p-BubR1 was strikingly enhanced in cells co-treated with ATM-3507/VNB, 70% of which were mitotic cells (Fig. 4e). This enhancement is more likely to be a result of a strengthened activation of the SAC, rather than the slightly increased mitotic population (i.e. 10%). In contrast, PTX (28% mitotic cells) and its combination with ATM-3507 (30% mitotic cells) showed similar impact on p-BubR1 expression (Fig. 4e). Taken together, these data suggest that ATM-3507 enhances VNB-induced mitotic arrest with elevated activity of the SAC, resulting in a portion of cells dying in mitosis, while ATM-3507 has marginal impact on PTX-induced mitotic defects.
Co-targeting Tpm3.1 and microtubules promotes post-mitotic arrest in the subsequent G1 phase
Since a substantial cell population survived the first mitosis in the live-cell imaging experiment, we questioned whether the combinations lead to a different effect on the subsequent interphase, which may in turn account for the synergy between ATM-3507 and PTX. We observed that both ATM-3507/PTX and ATM-3507/VNB resulted in more daughter cells remaining in the subsequent interphase (50 and 65%, respectively) at the end of imaging without further division, compared to ATM-3507 (23%), PTX (30%) and VNB (15%) alone (Fig. 5a). Cells treated with single agents or their combinations spent significantly longer time in the next interphase than with DMSO, indicating a post-mitotic arrest (Fig. 5b). The expression of two essential interphase cyclins that regulate the G1–S transition, cyclin D1 and E1,38,39 were examined to confirm the state of subsequent interphase arrest. Immunoblots revealed that single drug treatments decreased cyclin D1 and E1 expression. However, a much more substantial decrease was observed in cells co-treated with ATM-3507/VNB or ATM-3507/PTX (Fig. 5c), suggesting that both drug combinations induce an enhanced G1 arrest in daughter cells.

a The percentage of OVCAR4 cells that underwent different cell fates after completing their first mitosis. At least 85 cells from three independent experiments were analysed for each treatment. b Time cells spent in the next interphase. c Immunoblots and quantification of cyclin D1 and E1 in cell lysates from OVCAR4 cells treated with DMSO, ATM (5 μM), VNB (8 nM), A + V (5 μM ATM + 8 nM VNB), PTX (5 nM) and A + P (5 μM ATM + 5 nM PTX), respectively, for 36 h. Dead and mitotic cells were removed by shake-off. d Immunoblots and quantification of p16, p21 and p27 in cell lysates as in c. Data were collected from at least three independent experiments. Error bars are mean ± S.D.; *p < 0.05; **p < 0.001.
The cyclin-dependent kinase (CDK) inhibitors (CKIs) are negative regulators of interphase progression, including proteins from two gene families, CIP/KIP (CDK interacting protein/Kinase inhibitory protein) and INK4 (inhibitors of CDK4).40,41 We found that the expression of p21Cip and p27Kip from the CIP/KIP family were upregulated in ATM-3507/VNB or ATM-3507/PTX co-treated cells by approximately 2-fold compared to single drug-treated cells (Fig. 5d).40 However, while slightly increased in VNB- or PTX-treated cells, the expression of p16INK4a from the INK4 family displayed a significant downregulation in cells when ATM-3507 was present,40 irrespective of its presence as a single agent or in combination with microtubule drugs, suggesting that Tpm3.1 may locate upstream of microtubule-regulated p16INK4a expression (Fig. 5d). Therefore, the synergy of ATM-3507 and both anti-microtubule drugs involves an enhanced post-mitotic G1 arrest with upregulation of p21Cip and p27Kip.
Discussion
In this study, we demonstrate for the first time that Tpm3.1 is abundant in 97% of 577 ovarian cancer tumour samples representing all histotypes of epithelial ovarian cancer. Tpm3.1 expression was homogeneous and observed in primary and metastatic sites as well as at initial diagnosis and at recurrence. Upregulation of TPM3, the gene encoding Tpm3.1 and a dozen of other Tpm isoforms, has been implicated in haematopoietic malignancy,42 gliomas,43 and oesophageal squamous cell carcinoma.44 Using isoform-specific reagents, it has been well documented that, among all isoforms encoded by TPM3, Tpm3.1 is consistently expressed in a variety of cancer cell lines,12,13,26 resulting in the development of anti-Tpm3.1 as a cancer therapy. However, Tpm3.1 expression in tumour samples of a particular cancer type has not been analysed previously.
Although there have been advances in the treatment of a subset of patients with recurrent ovarian cancer including targeting angiogenesis and inhibiting PARP, the majority of patients progress and most will ultimately die due to platinum-resistant ovarian cancer.45,46,47 Despite an enormous effort and multiple clinical trials in patients with platinum-resistant ovarian cancer, the response rates are low and median survival is around 12 months.4 Improving treatment for these patients remains an important unmet need. The abundance of Tpm3.1 in almost all tumour samples make anti-Tpm3.1 a potential and generic strategy to treat epithelial ovarian cancer of all histotypes including clear cell and mucinous cancers as well as low-grade serous cancers, which all have a low response to platinum- and taxane-based chemotherapies.4 This is supported by the high efficacy of ATM-3507 in the three tested ovarian cancer cell lines that represent high-grade serous (OVCAR4), endometrioid (A2780) and platinum-resistant (A2780cis) ovarian cancers, which validates the feasibility of inhibiting Tpm3.1 for ovarian cancer treatment. Moreover, ATM-3507 was well tolerated in animal models without compromising muscle structure and functions, which is the major side effect of anti-actin agents, suggesting that ATM-3507 is likely to specifically target the cytoskeleton of tumour cells while sparing those in muscle tissues.7
Simultaneously targeting Tpm3.1 and microtubules has shown synergy in multiple cancer cell types.7,8 This study comprehensively demonstrated a similar synergy in ovarian cancer cell lines. The combination of CBDCA and PTX has been the first-line chemotherapy for ovarian cancer since the late 1990s.48 Although response rates are high particularly in patients with high-grade serous cancers, the majority of patients with advanced-stage disease will relapse and either have platinum-resistant disease at first recurrence or after multiple lines of further chemotherapy.49 Moreover, the CBDCA/PTX combination is not synergistic;50 some studies have even reported an antagonistic interaction between these two drugs,51 which is also seen in our study using ovarian cancer cell lines. Therefore, the combination of ATM-3507 and anti-microtubule drugs is promising as a supplemental approach to CBDCA/PTX. Our findings support the further establishment of the activity and tolerability of our drug combinations in in vivo models of platinum-resistant ovarian cancer.
The drug synergy in reducing the viability of ovarian cancer cells is observed with ATM-3507 in combination with either VNB or PTX. However, only VNB but not PTX synergises with ATM-3507 to induce apoptotic cell death in the studied cell lines. This discrepancy is also seen in mitotic defects, where the mitotic arrest caused by VNB but not PTX is significantly prolonged by ATM-3507. It has been demonstrated that during a prolonged mitotic arrest certain anti-apoptotic proteins (e.g. IAP and Mcl-1) are depleted due to the absence of transcription, which can trigger apoptotic cell death.52 This might partially explain why ATM-3507/VNB shows synergy in inducing apoptosis, whereas AMT-3507/PTX does not. On the other hand, cells that exit mitosis upon the co-treatment with ATM-3507/PTX are synergistically blocked in the subsequent G1 phase, which likely underlies the synergy of this combination in reducing cell viability.
In contrast to the synergy with VNB, ATM-3507 has marginal impacts on PTX-induced mitotic defects suggesting that the mitosis-dependent synergy relies on the anti-microtubule drug used in combination. The distinct impacts of VNB and PTX on microtubule dynamics (i.e. depolymerising versus stabilising) during mitosis may subsequentially cause different consequences in disrupting the interactions between the microtubule network and the actin cytoskeleton when AMT-3507 is used simultaneously. There are numerous studies reporting the crosstalk between the actin cytoskeleton and microtubules in spindle positioning and cortical force generation.53,54,55 Given that Tpm3.1 or Tpm3.1-containing actin filaments are enriched at the cell cortex during mitosis,8 where astral microtubules interact with cortical actin via protein complexes (e.g. NuMA-dynein-dynactin and LGN-Gαi) required for mitotic spindle assembly and positioning,56 it is not surprising that anti-Tpm3.1 by ATM-3507 produces a better combinational impact with the microtubule depolymeriser VNB than the stabiliser PTX on mitosis. Moreover, the fact that ATM-3507 reinforces VNB- but not PTX-induced activation of the SAC also supports this statement. Therefore, our data suggest the possibility that disruption of the interaction between Tpm3.1-containing actin filaments and microtubules during mitosis is exclusively triggered by the consequences of the depolymerisation of microtubules.
The increased arrest in the post-mitotic G1 phase with cyclin D1 and E downregulation underlies the synergy of ATM-3507/PTX and also contributes to the combinatorial effects of ATM-3507/VNB. Given that disruption of actin filaments downregulates the expression of cyclin D1 and E57,58 and prolonged mitosis leads to the blockage of the subsequent interphase,18 combined targeting of both systems may be expected to enhance a post-mitotic interphase block. A similar explanation can be applied to the upregulation of p21Cip and p27Kip in daughter cells exposed to the drug combinations because the expression of both CKIs is either directly or indirectly regulated by actin and microtubules.59,60,61 Interestingly, p16INK4a is downregulated when ATM-3507 is used in the combinations. Although p16INK4a has long been regarded as a tumour suppressor, recent studies suggest that this CKI is not a favourable prognostic marker for ovarian cancer, particularly high-grade serous carcinoma;62 rather downregulation of p16INK4a inhibits cell proliferation and induces G1 arrest in cervical cancer cells.63 Thus, decreased p16INK4a may potentially contribute to the efficacy of both studied combinations in the treatment of ovarian cancer.
Furthermore, it is noted that cells surviving mitosis via slippage often become tetraploid, especially upon PTX treatment, which is a state that some studies argue may promote chromosome instability and thus cancer progression.64,65 However, it has also been shown by many others that tetraploid cells would be arrested in the next interphase (i.e. post-mitotic G1 phase) and die through apoptosis.66,67 Indeed, the PTX-induced post-slippage multinucleation in cancer cells triggers extensive DNA damage and subsequent apoptosis in the following cell generations.68 The dual cytotoxicity (mitotic and post-slippage) of PTX by two distinct mechanisms makes it much more potent in triggering cell death than anti-mitotic agents, especially in cancer cells resistant to apoptosis during mitotic arrest.68 The enhanced post-slippage G1 arrest caused by the combination of PTX/VNB and ATM-3507 indicates a potentially increased treatment efficacy as compared to PTX or VNB alone, which is supported by the synergy of these drug combinations in reducing the viability of ovarian cancer cells.
It also needs to be noted that our study is limited by a lack of tissues from normal ovary or other organs, which may in turn result in concerns such as targeting Tpm3.1 may not have a sufficient therapeutic window. Nevertheless, data from public database provide important insights. For example, Ensembl records that the expression of TPM3 gene is at a level from low to medium in most organs.69 The Human Protein Atlas shows that TPM3 is low at RNA level while moderate at protein level throughout the body.70 These data together with our findings support the rationale of introducing anti-Tpm3.1-based therapy for the treatment of ovarian cancer, even though further comparisons of the expression of Tpm3.1 between cancerous and normal tissues is needed in future studies. We conclude that synergy resulting from combined targeting of Tpm3.1 and microtubules in ovarian cancer cell lines demonstrates that this is a promising strategy that deserves to be explored in in vivo models and subsequently the clinic. The abundance of Tpm3.1 in almost all epithelial ovarian tumour histotypes suggests the potential broad applicability of this combinatorial approach.
References
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A. & Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018).
Howlader, N., Noone, A. M., Krapcho, M., Miller, D., Brest, A., Yu, M. et al. SEER Cancer Statistics Review, 1975-2016 (National Cancer Institute, 2019).
Jelovac, D. & Armstrong, D. K. Recent progress in the diagnosis and treatment of ovarian cancer. CA Cancer J. Clin. 61, 183–203 (2011).
Lheureux, S., Gourley, C., Vergote, I. & Oza, A. M. Epithelial ovarian cancer. Lancet 393, 1240–1253 (2019).
Pujade-Lauraine, E., Hilpert, F., Weber, B., Reuss, A., Poveda, A., Kristensen, G. et al. AURELIA: a randomized phase III trial evaluating bevacizumab (BEV) plus chemotherapy (CT) for platinum (PT)-resistant recurrent ovarian cancer (OC). J. Clin. Oncol. https://doi.org/10.1200/jco.2012.30.18_suppl.lba5002 (2012).
Burger, R. A., DiSaia, P. J., Roberts, J. A., O’Rourke, M., Gershenson, D. M., Homesley, H. D. et al. Phase II trial of vinorelbine in recurrent and progressive epithelial ovarian cancer. Gynecol. Oncol. 72, 148–153 (1999).
Currier, M. A., Stehn, J. R., Swain, A., Chen, D., Hook, J., Eiffe, E. et al. Identification of cancer-targeted tropomyosin inhibitors and their synergy with microtubule drugs. Mol. Cancer Ther. 16, 1555–1565 (2017).
Wang, Y., Stear, J. H., Swain, A., Xu, X., Bryce, N. S., Carnell, M. et al. Drug targeting the actin cytoskeleton potentiates the cytotoxicity of low dose vincristine by abrogating actin-mediated repair of spindle defects. Mol. Cancer Res. 18, 1074–1087 (2020).
Gunning, P. W., Hardeman, E. C., Lappalainen, P. & Mulvihill, D. P. Tropomyosin - master regulator of actin filament function in the cytoskeleton. J. Cell Sci. 128, 2965–2974 (2015).
Gateva, G., Kremneva, E., Reindl, T., Kotila, T., Kogan, K., Gressin, L. et al. Tropomyosin isoforms specify functionally distinct actin filament populations in vitro. Curr. Biol. 27, 705–713 (2017).
Schevzov, G., Kee, A. J., Wang, B., Sequeira, V. B., Hook, J., Coombes, J. D. et al. Regulation of cell proliferation by ERK and signal-dependent nuclear translocation of ERK is dependent on Tm5NM1-containing actin filaments. Mol. Biol. Cell 26, 2475–2490 (2015).
Meiring, J. C. M., Bryce, N. S., Wang, Y., Taft, M. H., Manstein, D. J., Lau, S. L. et al. Co-polymers of actin and tropomyosin account for a major fraction of the human actin cytoskeleton. Curr. Biol. 28, 2331–2337 (2018).
Stehn, J. R., Haass, N. K., Bonello, T., Desouza, M., Kottyan, G., Treutlein, H. et al. A novel class of anticancer compounds targets the actin cytoskeleton in tumor cells. Cancer Res. 73, 5169–5182 (2013).
Janco, M., Rynkiewicz, M. J., Li, L., Hook, J., Eiffe, E., Ghosh, A. et al. Molecular integration of the anti-tropomyosin compound ATM-3507 into the coiled coil overlap region of the cancer-associated Tpm3.1. Sci. Rep. 9, 11262 (2019).
Goode, B. L., Drubin, D. G. & Barnes, G. Functional cooperation between the microtubule and actin cytoskeletons. Curr. Opin. Cell Biol. 12, 63–71 (2000).
Rodriguez, O. C., Schaefer, A. W., Mandato, C. A., Forscher, P., Bement, W. M. & Waterman-Storer, C. M. Conserved microtubule-actin interactions in cell movement and morphogenesis. Nat. Cell Biol. 5, 599–609 (2003).
Mukhtar, E., Adhami, V. M. & Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 13, 275–284 (2014).
Uetake, Y. & Sluder, G. Prolonged prometaphase blocks daughter cell proliferation despite normal completion of mitosis. Curr. Biol. 20, 1666–1671 (2010).
Kelemen, L. E., Kobel, M., Chan, A., Taghaddos, S. & Dinu, I. Differentially methylated loci distinguish ovarian carcinoma histological types: evaluation of a DNA methylation assay in FFPE tissue. Biomed. Res. Int. 2013, 815894 (2013).
Bromley, A. B., Altman, A. D., Chu, P., Nation, J. G., Nelson, G. S., Ghatage, P. et al. Architectural patterns of ovarian/pelvic high-grade serous carcinoma. Int. J. Gynecol. Pathol. 31, 397–404 (2012).
Tsao, S. W., Mok, S. C., Fey, E. G., Fletcher, J. A., Wan, T. S. K., Chew, E. C. et al. Characterization of human ovarian surface epithelial-cells immortalized by human papilloma viral oncogenes (Hpv-E6e7 Orfs). Exp. Cell Res. 218, 499–507 (1995).
Schevzov, G., Vrhovski, B., Bryce, N. S., Elmir, S., Qiu, M. R., O’Neill, G. M. et al. Tissue-specific tropomyosin isoform composition. J. Histochem. Cytochem. 53, 557–570 (2005).
Aldridge, G. M., Podrebarac, D. M., Greenough, W. T. & Weiler, I. J. The use of total protein stains as loading controls: an alternative to high-abundance single-protein controls in semi-quantitative immunoblotting. J. Neurosci. Methods 172, 250–254 (2008).
Chou, T. C. Preclinical versus clinical drug combination studies. Leuk. Lymphoma 49, 2059–2080 (2008).
Saurin, A. T., van der Waal, M. S., Medema, R. H., Lens, S. M. A.,Kops, G. J. P. L. Aurora B potentiates Mps1 activation to ensure rapid checkpoint establishment at the onset of mitosis. Nat. Commun. 2, 316 (2011).
Stehn, J. R., Schevzov, G., O’Neill, G. M. & Gunning, P. W. Specialisation of the tropomyosin composition of actin filaments provides new potential targets for chemotherapy. Curr. Cancer Drug Targets 6, 245–256 (2006).
Schevzov, G., Whittaker, S. P., Fath, T., Lin, J. J. & Gunning, P. W. Tropomyosin isoforms and reagents. Bioarchitecture 1, 135–164 (2011).
Domcke, S., Sinha, R., Levine, D. A., Sander, C., Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 4, 2126 (2013).
Behrens, B. C., Hamilton, T. C., Masuda, H., Grotzinger, K. R., Whang-Peng, J., Louie, K. G. et al. Characterization of a cis-diamminedichloroplatinum(II)-resistant human ovarian cancer cell line and its use in evaluation of platinum analogues. Cancer Res. 47, 414–418 (1987).
Matulonis, U. A., Sood, A. K., Fallowfield, L., Howitt, B. E., Sehouli, J. & Karlan, B. Y. Ovarian cancer. Nat. Rev. Dis. Prim. 2, 16061 (2016).
Brito, D. A. & Rieder, C. L. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr. Biol. 16, 1194–1200 (2006).
Lara-Gonzalez, P., Westhorpe, F. G. & Taylor, S. S. The spindle assembly checkpoint. Curr. Biol. 22, R966–R980 (2012).
Elowe, S., Dulla, K., Uldschmid, A., Li, X., Dou, Z. & Nigg, E. A. Uncoupling of the spindle-checkpoint and chromosome-congression functions of BubR1. J. Cell Sci. 123, 84–94 (2010).
Huang, H., Hittle, J., Zappacosta, F., Annan, R. S., Hershko, A. & Yen, T. J. Phosphorylation sites in BubR1 that regulate kinetochore attachment, tension, and mitotic exit. J. Cell Biol. 183, 667–680 (2008).
Yamada, H. Y. & Gorbsky, G. J. Spindle checkpoint function and cellular sensitivity to antimitotic drugs. Mol. Cancer Ther. 5, 2963–2969 (2006).
Howell, B. J., Moree, B., Farrar, E. M., Stewart, S., Fang, G. & Salmon, E. D. Spindle checkpoint protein dynamics at kinetochores in living cells. Curr. Biol. 14, 953–964 (2004).
Zhang, G., Mendez, B. L., Sedgwick, G. G. & Nilsson, J. Two functionally distinct kinetochore pools of BubR1 ensure accurate chromosome segregation. Nat. Commun. 7, 12256 (2016).
Sherr, C. J. Mammalian G1 cyclins. Cell 73, 1059–1065 (1993).
Resnitzky, D. & Reed, S. I. Different roles for cyclins D1 and E in regulation of the G1-to-S transition. Mol. Cell Biol. 15, 3463–3469 (1995).
Sherr, C. J. & Roberts, J. M. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 13, 1501–1512 (1999).
Besson, A., Dowdy, S. F. & Roberts, J. M. CDK inhibitors: cell cycle regulators and beyond. Dev. Cell 14, 159–169 (2008).
Kim, T. M., Yim, S. H., Shin, S. H., Xu, H. D., Jung, Y. C., Park, C. K. et al. Clinical implication of recurrent copy number alterations in hepatocellular carcinoma and putative oncogenes in recurrent gains on 1q. Int. J. Cancer 123, 2808–2815 (2008).
Tao, T., Shi, Y., Han, D., Luan, W., Qian, J., Zhang, J. et al. TPM3, a strong prognosis predictor, is involved in malignant progression through MMP family members and EMT-like activators in gliomas. Tumour Biol. 35, 9053–9059 (2014).
Lin, W. W., Lin, J. H., Chen, B. Y., Tang, W. F., Yu, S. B., Chen, S. C. et al. Tropomyosin3 is associated with invasion, migration, and prognosis in esophageal squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 9, 11313–11323 (2016).
Vaughan, S., Coward, J. I., Bast, R. C. Jr., Berchuck, A., Berek, J. S., Brenton, J. D. et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat. Rev. Cancer 11, 719–725 (2011).
Jayson, G. C., Kerbel, R., Ellis, L. M. & Harris, A. L. Antiangiogenic therapy in oncology: current status and future directions. Lancet 388, 518–529 (2016).
Franzese, E., Centonze, S., Diana, A., Carlino, F., Guerrera, L. P., Di Napoli, M. et al. PARP inhibitors in ovarian cancer. Cancer Treat. Rev. 73, 1–9 (2019).
du Bois, A., Neijt, J. P. & Thigpen, J. T. First line chemotherapy with carboplatin plus paclitaxel in advanced ovarian cancer-a new standard of care?. Ann. Oncol. 10(Suppl. 1), 35–41 (1999).
Luvero, D., Milani, A. & Ledermann, J. A. Treatment options in recurrent ovarian cancer: latest evidence and clinical potential. Ther. Adv. Med. Oncol. 6, 229–239 (2014).
Engblom, P., Rantanen, V., Kulmala, J. & Grenman, S. Carboplatin-paclitaxel- and carboplatin-docetaxel-induced cytotoxic effect in epithelial ovarian carcinoma in vitro. Cancer 86, 2066–2073 (1999).
Xiong, X., Sui, M., Fan, W. & Kraft, A. S. Cell cycle dependent antagonistic interactions between paclitaxel and carboplatin in combination therapy. Cancer Biol. Ther. 6, 1067–1073 (2007).
Blagosklonny, M. V. Mitotic arrest and cell fate: why and how mitotic inhibition of transcription drives mutually exclusive events. Cell Cycle 6, 70–74 (2007).
Woolner, S., O’Brien, L. L., Wiese, C. & Bement, W. M. Myosin-10 and actin filaments are essential for mitotic spindle function. J. Cell Biol. 182, 77–88 (2008).
Luxenburg, C., Pasolli, H. A., Williams, S. E. & Fuchs, E. Developmental roles for Srf, cortical cytoskeleton and cell shape in epidermal spindle orientation. Nat. Cell Biol. 13, 203–214 (2011).
Dugina, V., Alieva, I., Khromova, N., Kireev, I., Gunning, P. W. & Kopnin, P. Interaction of microtubules with the actin cytoskeleton via cross-talk of EB1-containing +TIPs and γ-actin in epithelial cells. Oncotarget 7, 72699–72715 (2016).
di Pietro, F., Echard, A. & Morin, X. Regulation of mitotic spindle orientation: an integrated view. EMBO Rep. 17, 1106–1130 (2016).
Reshetnikova, G., Barkan, R., Popov, B., Nikolsky, N. & Chang, L. S. Disruption of the actin cytoskeleton leads to inhibition of mitogen-induced cyclin E expression, Cdk2 phosphorylation, and nuclear accumulation of the retinoblastoma protein-related p107 protein. Exp. Cell Res. 259, 35–53 (2000).
Welsh, C. F., Roovers, K., Villanueva, J., Liu, Y. Q., Schwartz, M. A. & Assoian, R. K. Timing of cyclin D1 expression within G1 phase is controlled by Rho. Nat. Cell Biol. 3, 950–957 (2001).
Liu, X. M., Jiang, J. D., Ferrari, A. C., Budman, D. R. & Wang, L. G. Unique induction of p21(WAF1/CIP1)expression by vinorelbine in androgen-independent prostate cancer cells. Br. J. Cancer 89, 1566–1573 (2003).
Coleman, M. L., Densham, R. M., Croft, D. R. & Olson, M. F. Stability of p21Waf1/Cip1 CDK inhibitor protein is responsive to RhoA-mediated regulation of the actin cytoskeleton. Oncogene 25, 2708–2716 (2006).
Panno, M. L., Giordano, F., Mastroianni, F., Morelli, C., Brunelli, E., Palma, M. G. et al. Evidence that low doses of Taxol enhance the functional transactivatory properties of p53 on p21 waf promoter in MCF-7 breast cancer cells. FEBS Lett. 580, 2371–2380 (2006).
Shandiz, F. H., Kadkhodayan, S., Ghaffarzadegan, K., Esmaeily, H., Torabi, S. & Khales, S. A. The impact of p16 and HER2 expression on survival in patients with ovarian carcinoma. Neoplasma 63, 816–821 (2016).
Zhang, C. Y., Bao, W. & Wang, L. H. Downregulation of p16(ink4a) inhibits cell proliferation and induces G1 cell cycle arrest in cervical cancer cells. Int. J. Mol. Med. 33, 1577–1585 (2014).
Lv, L., Zhang, T., Yi, Q., Huang, Y., Wang, Z., Hou, H. et al. Tetraploid cells from cytokinesis failure induce aneuploidy and spontaneous transformation of mouse ovarian surface epithelial cells. Cell Cycle 11, 2864–2875 (2012).
Fujiwara, T., Bandi, M., Nitta, M., Ivanova, E. V., Bronson, R. T. & Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 437, 1043–1047 (2005).
Castedo, M., Coquelle, A., Vivet, S., Vitale, I., Kauffmann, A., Dessen, P. et al. Apoptosis regulation in tetraploid cancer cells. EMBO J. 25, 2584–2595 (2006).
Fuhrken, P. G., Apostolidis, P. A., Lindsey, S., Miller, W. M. & Papoutsakis, E. T. Tumor suppressor protein p53 regulates megakaryocytic polyploidization and apoptosis. J. Biol. Chem. 283, 15589–15600 (2008).
Zhu, Y., Zhou, Y. & Shi, J. Post-slippage multinucleation renders cytotoxic variation in anti-mitotic drugs that target the microtubules or mitotic spindle. Cell Cycle 13, 1756–1764 (2014).
Yates, A. D., Achuthan, P., Akanni, W., Allen, J., Allen, J., Alvarez-Jarreta, J. et al. Ensembl 2020. Nucleic Acids Res. 48, D682–D688 (2020).
Uhlén, M., Fagerberg, L., Hallström, B. M., Lindskog, C., Oksvold, P., Mardinoglu, A. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Acknowledgements
We thank the Health Science Alliance Biobank from the Translational Cancer Research Network (UNSW Sydney, South Eastern Sydney Local Health District, NSW Health Pathology, Sydney, Australia) for access to tumour tissues that were used in a preliminary study to identify the extent of expression of Tpm3.1 in ovarian cancer. We thank Mr. Christopher Brownlee and Dr. Emma J. Beves (The Flow Cytometry Facility, UNSW Sydney, Sydney, Australia) for flow cytometer training and support; Dr. Michael Cornell, Dr. Elvis Pandzic, Dr. Alex Macmillan, Mrs. Iveta Slapetova and Ms. Florence C. J. Tomasetig (Biomedical Imaging Facility, UNSW Sydney, Sydney, Australia) for microscope training and support; and Dr. Michael Cornell (Biomedical Imaging Facility, UNSW Sydney, Sydney, Australia) for consulting on image data analysis.
Author information
Authors and Affiliations
Contributions
X.X. designed and performed experiments, analysed the data and drafted the manuscript. Y.W. supervised the project, designed and performed experiments and drafted the manuscript. N.S.B. supervised the project and interpreted the results. K.T. analysed the data and interpreted the results. N.S.M., E.Y.K., L.E.K., M.K. and S.J.R. designed experiments and interpreted the results. M.F. initiated the research concept, designed the experimental strategy and drafted the manuscript. C.E.F. initiated the research concept and designed the experimental strategy. E.C.H. and P.W.G. initiated the research concept, designed the experimental strategy and supervised the project. All authors revised the manuscript and approved the submission of this work.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was performed with approval from the UNSW Human Research Ethics Committee (Approval # HC15771 and # HC16299). AOVT and CAL study protocols were approved by Alberta Health Services, Research Ethics and the University of Calgary, Faculty of Medicine, Office of Medical Bioethics, respectively. These study protocols used retrospective data collection from pathology reports and medical charts and were exempt by the ethics boards from requiring informed patient consent. We also declare that the study was performed in accordance with the Declaration of Helsinki.
Consent to publish
Not applicable.
Data availability
Digital data are kept on the server of School of Medical Sciences, UNSW Sydney and available from the corresponding author upon request.
Competing interests
P.W.G. and E.C.H. receive funding from TroBio Therapeutics, a company commercialising anti-tropomyosin drugs. P.W.G. and E.C.H. are directors and shareholders of TroBio. Other authors declare no competing interests.
Funding information
This work was supported by an Australian Department of Industry, Innovation and Science Cooperative Research Centre Project (CRC-P) grant to P.W.G. and E.C.H. and grants from the Australian Research Council (ARC grant DP160101623), the Australian National Health and Medical Research Council (NHMRC grant APP1100202, APP1079866) and The Kid’s Cancer Project to P.W.G. and E.C.H.; N.S.M. is supported by the NSW Ministry of Health and UNSW Sydney under the NSW Health PhD Scholarship Program and the Translational Cancer Research Network, a translational cancer research centre program funded by the Cancer Institute NSW (RG171797 and 15/TRC/1-03).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Xu, X., Wang, Y., Bryce, N.S. et al. Targeting the actin/tropomyosin cytoskeleton in epithelial ovarian cancer reveals multiple mechanisms of synergy with anti-microtubule agents. Br J Cancer 125, 265–276 (2021). https://doi.org/10.1038/s41416-021-01420-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-021-01420-y
- Springer Nature Limited