
Abstract
Microtubules are intracellular components of cytoskeleton throughout the cytoplasm of all eukaryotic cells. Microtubules are dynamic polymers with continuous assembly and disassembly of tubulin dimers. This highly dynamic action of microtubule contributes to numerous cellular processes such as maintaining the structure of the cell, cell division and cell movement. For this reason, microtubule has become a notable target for chemotherapeutic achievements. Microtubule Targeting Agents (MTAs) that nowadays are used in chemotherapy, induce microtubule polymerization or depolymerization. They are categorized into two groups known as stabilizers such as texanes and destabilizers such as vincas. Either, stabilizing or destabilizing of microtubule polymer leads to spindle assembly poisoning, mitotic blockage and cell death. Yet, we are required to consider main forthcoming controversial difficulty which is resistance to MTAs. Clinical studies have documented different levels of resistance to MTAs with different durability among patients even within the same class of drugs. For instance, overexpression of P-glycoprotein (P-gp) is linked to resistance to taxanes, but not to ixabepilone, even though they have similar mechanism of action. Mutations in β-tubulin have been associated with resistance to taxanes but not to epithilones despite their mechanism of action being same. Also, there is association between poor response to taxanes and overexpression of βIII-tubulin. Either receiving benefit or harm from MTAs, depends on each individual patient considering their variable chemo-sensitivities to drugs. Therefore, it is crucial to understand the basic biology of microtubules and the molecular mechanisms by which MTAs exert their activity. This is especially important considering their current application in cancer therapy. In this chapter we discussed about MTAs in detail and illustrate their molecular mechanisms involved in various cancers.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
Cancer and Normal Cells Different Characteristics
Cancer is an ancient disease that afflicts about 40% of the global population despite improvement in the diagnosis and treatment to the disease. Rather more challenging when cancer become metastatic [1]. Surgery, chemotherapy and radiation therapy are the current strategies for cancer treatment. The conventional treatments failed achieving therapeutic selectivity to cancer cells, rather than non-specific targeting the normal cells, and this leaded to appearance of serious side effects. Therefore, the current target-based drug design and discovery requires new drugs with selectivity to cancer cells in molecular bases. Thus, differentiation of cancer cells from normal cells which includes their characteristics and behaviors, is a requirement. Based on the current knowledge of cancer cell characteristics, studies are directed to molecular–based paradigms. These paradigms are safe to say that increased drug efficacy with less side-effects [2]. In the last 25 years, countless researches have been done for understanding molecular-based strategies, which their discoveries have been improving diagnostic, prognostic and treatment of cancers [1]. In this chapter, we plan to discuss microtubule targeting and the list of microtubule targeting agents that have been reported so far (Table 1).
The hallmark characteristic attributed to cancer cells is their fast growth rate and their ability to proliferate indefinitely when compared to normal cells. To that, regulation of many key proteins involved in survival pathways and cell-cycle regulation are modified in cancer cells. In addition, apoptosis is suppressed in many of cancer cells through various pathways, and this adds to greater ability of cancer cells to survive [3]. Robust proliferative responses in normal cells, are actually sustained in cancer cells. The deregulation of Cyclin-Dependent Kinases (CDKs ) that enable cell cycle progression [4] and the abnormal expression of Myc, a strategic controller of cell proliferation, are few prominent examples [5]. It is well known that occurrence of mutation is higher in cancer cells compared to normal cells. From carcinogen exposure to clinical detection, cancer cells require to grow, divide, invade and metastases. During this period, multiple mutations in genetic stability-genes are required that initiates cascade of other mutations in other genes. Many of those second group of genes are controller of key pathways [6]. In 2013, 140 genes were reported that if are altered by mutations, can “drive” to tumorigenesis. A typical cancer cell has two to eight of these driver genes mutations. Other mutations in these cells are “passenger” mutations that do not cause any specific proliferation advantages. Overall mutations cause cell-autonomous alterations which leads to different consequences such as dividing fast and going through different cell-cycle phases faster than normal cells. “Dividing rapidly” lowers balance between supply and consumption of nutrients and oxygen. Therefore, cancer cells acquire second set of mutations to provide an appropriate condition that endures nutrient and oxygen deficiency , like mutation in EGFR (Epidermal growth factor receptor), HER2 (Human epidermal growth factor receptor 2), RAS, RAF (rapidly accelerated fibrosarcoma), PTEN (Phosphatase and tensin homolog). Many of these new set of mutations cause abnormal vascularization with well-ordered network of veins, arteries and lymphatics which bring more nutrients and oxygen for rapidly dividing cells. Beside the exterior-related help of mutations, interior-related help are also required to survive. This includes those mutations that progress the cell cycle such as CDKs, Myc, microtubule-associated protein (MAP) genes mutations [6]. There are several MAPs including motor proteins such as kinesin and dynein and several microtubule-regulatory proteins such as stathmin, survivin, MCAK (Centromere-associated kinesin), EB1(End-binding protein 1) and FHIT (Fragile histidine triad protein).The importance of MAPs is their association with microtubules in formation of mitotic spindle apparatus which is the key requirement for rapidly dividing cells. The necessity of microtubule during mitosis is by means of separating the duplicate chromosomes of mother cell into two daughter cells and finally cell division. This virtue of microtubules took a lot of attention in cancer therapy as a target. Microtubules have known to be a target of many naturally occurring toxic yet self-protective molecules, extracted from microorganisms, sea flora and plants [7].
Microtubule Dynamic as a Target for Cancer Therapy
Microtubules are made of cellular components α- and β-tubulin heterodimers by polymerizing head to tail orientation of protofilaments . Protofilaments are around 12–13 in numbers that binds together and form microtubule, a dynamic structure that are principal member of mitosis, meiosis, cell movement, maintenance of cell structure and intracellular organelles movements. Hence they have become a target in cancer therapy [8]. The way of protofilament arrangement conveys the microtubule polarity. The α-tubulins are placed in the minus end while β-tubulins are placed in plus end. In the microtubule plus end, several proteins accumulate that are known as the microtubule plus end tracking proteins [9]. This group of proteins and their function will be elaborated further in this chapter. In mitosis, the microtubule-organizing center (MTOC or centrosome) binds to minus end of microtubules and cause microtubules accumulation. From there microtubule grows toward outside of MTOC [10]. γ-Tubulin in combination with other proteins forms γ-tubulin ring complex (γ-TuRC). γ-TuRC is the α- and β-tubulin scaffold before polymerization begins in MTOC. γ-Tubulin also protects the minus end from polymerization and/or depolymerization. GTP binds to β-tubulin and its hydrolyzation is necessary for further assembly. GTP-tubulin is stable while GDP-tubulin after losing a phosphate, is susceptible to depolymerization.
An important characteristic of microtubule is the dynamic instability which is considered as nature of microtubule to switch rapidly between growth and shrinkage. Microtubule dynamicity is highly regulated via different post-translational modifications and by some MAPs that bind to tubulin dimers or at poles of microtubules. This dynamicity provides cell living-needs such as cell movement, migration and division. Prior to mitosis, the whole structure of microtubule rearranges from the interphase microtubule architecture to specialized rapidly dynamic mitotic spindle assembly. These specialized microtubule spindles direct sister chromatids toward either poles of the cell. This responsibility ultimately ensures each new daughter cell receive correct and complete genetic content [11].
In the beginning of mitosis, at prophase, microtubule grows out from the centrosome, and in case the microtubule plus end meets the chromosome, it becomes stabilized, and otherwise disassembly is initiated. These disassembled free tubulins would be available for other microtubule growths. However these events are not happening just by interaction between microtubule and chromosomes. There are different ways to achieve functional diversity of microtubule: (i) through the expression of various tubulin isotypes which each having different functions, (ii) through post-translational modifications on tubulin and (iii) through binding of numerous regulatory proteins such as MAPs . These proteins interact with soluble tubulin and/or microtubule’s surface and ends. Human tubulin isotypes include 7 forms of β-tubulin and 6 forms of α-tubulin. Their expression varies in different cell types. The post-translational modification of tubulin includes polyglutamylation, polyglycylation, acetylation, phosphorylation, tyrosination/detyrosination and removal of some residues such as the penultimate glutamic-acid from α-tubulin. Among MAPs, the motor proteins such as kinesin and dynein. Microtubule regulatory proteins include stathmin, survivin, MCAK, TOG, EB1, MAP4, dynactin 1, PAC1. Some of these proteins are associated with resistance of the cell to certain drugs [7]. Many proteins are involved in the microtubule dynamics. Stathmin is an endogenous depolimerizer/destabilizer that attaches to free tubulin dimers and avoids their bindings, therefore inhibits formation of protofilaments and microtubules. In contrary, some other proteins attach to polymerized microtubule and protect their stability or decreasing the duration and speed of depolymerization. The activity of these native stabilizers and destabilizers are regulated (phosphorylated/dephosphorylated) by cell cycle-dependent kinases.
Mutations in α- and β-tubulins affects the microtubule polymer mass and more or less effect on drug binding sites. As expected, this matter is contributing in resistance to MTAs which will be discussed broadly in this chapter. On the other hand, abnormal expression of some of β-tubulin isotypes, such as βIII-tubulin, or aberrant expressions of MAPs are also reported to be associated to resistance to MTAs . Cells survive optimally within a certain range of tubulin polymerization, about 22–58% of intracellular tubulin. Cells with lower levels of polymerized tubulin are resistant to tubulin-stabilizers such as epothilones and taxanes, but at the same time they are more sensitive to microtubule-destabilizers such as vinca alkaloids. Opposed to that, cells with increased levels of microtubules, are more resistant to microtubule-destabilizers but more sensitive to microtubule-stabilizers [12, 13].
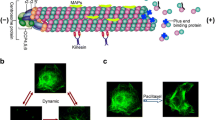
Microtubules are playing a critical role in the mitosis. In the process of mitosis, duplicated chromosomes of mother cells are divided into two identical chromosomes before division of the cell into daughter cells. Since, cancer cells divide rapidly compared to the normal cells, therefore this made microtubule a noticeable target in anticancer researches. Various MTAs through different binding sites bind to tubulin and target microtubule structure which more or less affects microtubule dynamics. Microtubule has two types of dynamic behaviors. First type is called as “dynamic instability” which is a process wherein each microtubule ends switch between two phases of growing and shrinkage (shortening). The two ends of microtubule are not equivalent. Growing and/or shortening of plus end are much faster than minus end. Dynamic instability is characterized by four key factors: by the rate of microtubule growth; by the rate of microtubule shrinkage; the frequency of transition from the growth to shrinkage which this transition is called “catastrophe ”; and the frequency of transition from shrinkage to microtubule growth which is called “rescue”. Catastrophe and rescue also are called “pause states”. The second type of dynamic behavior is “treadmilling”. Treadmilling is microtubule growth in one end and balanced shortening in the opposite end. It occurs when tubulin subunits are added to the plus (+) end rapidly, while in minus (−) end tubulins are disassembled with relatively low rate. The duration and frequency of switch between these two dynamic behaviors depends on what tubulin isotypes are involved, the degree of post-translational modifications and the type of regulatory proteins actions. For microtubule polymerization, tubulin-GTPs are added to microtubule ends and it is hydrolyzed to tubulin-GDP and Pi. Pi is eliminated from the microtubule. Those microtubules that contain tubulin-GTP or tubulin-GDP- Pi at the end of microtubule, are stable or capped. These types of microtubules do not go through depolymerization. But when the cap hydrolyses, release of Pi enhances some conformational changes in tubulin dimer and this event destabilizes the exposed microtubule polymer which causes microtubule shrinkage and final term of catastrophe [7]. Such specialized high qualified networks of mitotic spindles are remarkably sensitive to the effect of microtubule disrupting drugs. However, the action of MTAs does not confined to microtubule disruption, but also MTAs alter mitotic kinases. Further their synergism with same or different class of drugs was discovered. However, the development of resistance and side effects such as neurotoxicity, are always in the package that comes with these drugs [7] (Fig. 1).

(a) Microtubule consist of α-tubulin and β-tubulin. Those microtubules contain α-tubulin-GTP or tubulin-GDP- Pi, are capped at the end and they are stabilized. (b) When cap hydrolyses (release of Pi) microtubule destabilizes and shrinkages. Microtubule dynamic behaviors: (c) Dynamic instability where in microtubule polymer ends switch between two phases of growing and shrinkage, and (d) Treadmilling wherein microtubule grow in one end and balanced shortening in the opposite end
Microtubule-Targeting Agents as Anti-Cancer Drugs
A wide variety of small molecules, including taxoles, alkaloids, macrolides and peptides, bind to tubulin and disturb microtubule assembly, disassembly and dynamics [14]. They are known as MTAs. MTAs great contribution is to interrupt mitotic spindle formation and subsequently block the cell division. Although this class of anticancers are very successful but various sort of resistance against them have been reported [15]. The drugs that nowadays are introduced as MTAs interact with either tubulin dimers and/or microtubule polymer. MTAs are divided into two major groups: destabilizers that bind to tubulin dimers and avoid microtubule polymerization or bind to polymerized microtubules and promote depolymerization. In contrast, stabilizers are group of MTAs that bind to tubulin and/or microtubule polymer and stabilize the polymerized formation. Destabilizers include vinca alkaloids such as vinblastine, vincristine, vindesine, vinorelbine, vinflunine other destabilizers are estramustine, dolastatins, halichondrin, combret-astatins, 2-ME etc. Among stabilizers we can mention taxanes such as paclitaxel, docetaxel, epothilones such as ixabepilone (anepothilone B analogue). Also it is worth to mention other types of stabilizers with different structure but with same function such as discodernolide, sarcodictyins, eleutherobins, laulimalide, rhazinilam and some of the steroids such as polyisoprenyl benzophenones. Without a doubt MTAs are one of most active anticancer drugs.
In 1950s vincristine has been introduced as a MTA and since then the emergence of this class of drugs were recognized and introduced. Along with that, vincristine binding site on microtubule was identified and other binding sites of other MTAs were being discovered [16]. This class of MTAs comprises the agents that are microtubule destabilizers. This class includes vincristine, vinorelbine and vinblastine. Later by the discovery of paclitaxel and docetaxel (Texanes) and investigating their mode of action, the concept of stabilizers were added as second group of MTAs [17]. Unfortunately, most of cancers treated with vinorelbine and texanes, eventually exhibit resistance. Mechanisms known as being involved in resistance to MTAs, are those altering MTAs binding sites on tubulin at genetic levels (tubulin mutations) or altering the structure of tubulin dimers alone or in microtubule formation [18, 19]. Tubulin mutation may occur in α- or β-tubulin. Also alteration can be as a result of qualitative or quantitative modifications in MAPs. Altered tubulin isotype expressions and altered synthesis of tubulin are also in the list. All kinds of tubulin mutation have been connected to altered stability of microtubules [12].
Another reason that resistance is an obstacle in therapy is, there is a connection between resistance to MTAs and apoptotic pathway alteration, given to the overexpressed anti-apoptotic proteins and subsequently appearance of faulty apoptotic pathways in most cancer cells particularly metastatic cancer cells which often exhibit resistance to anticancer drugs [20]. For instance, two ovarian cancer cell-lines overexpress an anti-apoptotic protein “survivin” which is considered as one of main apoptosis inhibitors. This cell-lines also exhibit resistance to taxanes. It has been reported that taxanes induce survivin phosphorylation in Thr34 which subsequently causes high affinity of phospho-survivin for caspases and therefore inhibits caspase-dependent apoptosis [21, 22].
To know MTAs we listed out some of the natural/synthetic stabilizers and destabilizers below in detail.
Natural Stabilizers
Taxanes
Taxanes include paclitaxel and its semi-synthetized analogue docetaxel were the most significant additions to the chemotherapeutic industry in late twentieth century especially in the treatment of breast cancer [23]. Monroe Wall in 1967 extracted a compound from the bark of yew tree (Taxusbrevifolia) and named it “Taxol” which is known as Paclitaxel. Wall and his colleagues began the journey of introducing microtubule stabilizers to cancer therapy. In 1979, Peter Schiff and Susan Horwitz discovered that paclitaxel induces microtubule polymerization. By that time the development of this drug was limited due to limited storage of natural compound. Until drug discovery and developments outgrowed to introduce semi-synthetized analogues of paclitaxel. Paclitaxel discovery and development as the first known stabilizer have taken almost 30 years [24]. It was reported that HeLa cells treated with paclitaxel is blocked in metaphase but unlike vinca alkaloid- and colchicine-induced metaphase blockage, microtubules mass are not disrupted and destabilized into tubulin dimers, but microtubule polymers are organized. Nowadays, we know this reorganization of microtubule as “microtubule stabilization”. They suggested paclitaxel induces microtubule assembly and stabilizes the formation of those microtubules that already been assembled before applying the drug. The confirmation of this idea was through preparation of cold- and Ca+2-induced depolymerization condition without applying MAPs and GTP. Then paclitaxel was applied and the result was increase of microtubule assembly by increase of tubulin polymerization and continues elongation of already polymerized microtubules [25]. Taxanes bind weakly to soluble tubulin but binds tightly to tubulins that are embedded along the length of microtubule. Taxane-binding site is on β-subunit located in inner side of microtubule lumen. The exact binding site of this drug was found by electron crystallography of tubulin bound to paclitaxel [26]. It is suggested that the way paclitaxel accesses its binding site inside the microtubule lumen is by diffusion mechanism or by fluctuations of the microtubule lattice. The exact mechanism of action is after binding to its binding site, paclitaxel changes the conformation of tubulin by an unknown mechanism which increases tubulin’s affinity for neighbor tubulins therefore stabilizes the microtubule and causes increase of polymerization [27]. Exact binding site of paclitaxel is on β-tubulin [28]. Development of various photoactive group conjugated-paclitaxel analogues, revealed more details of paclitaxel binding site on β-tubulin until eventually by electron crystallography they determined the exact binding site [29]. Each tubulin has one paclitaxel binding site. Paclitaxel at high dosage (>10 nM) stabilizes microtubule dynamics with few paclitaxel molecules without increasing microtubule polymerization. In this scenario, just one paclitaxel molecule is required to bound to microtubule per hundreds of tubulin molecules and this paclitaxel molecule can reduce extend and rate of microtubule shrinkage around 50% [30]. Paclitaxel was approved for clinical use by 1995, and nowadays it is used to treat ovarian, breast, non-small-cell lung cancers and kaposi’s sarcoma. The side effects of paclitaxel are neurotoxicity and myelosuppression [31].
Another interesting fact about paclitaxel’s efficacy is paclitaxel-induced reduction of protofilaments that form the microtubule from 13 to 12 [32]. However exposure of microtubule to high dosage of paclitaxel, increases microtubule polymer mass but researches showed at the dosage of below 10 nM, there is no significant effect on microtubule polymerization and stabilization [30, 33]. There are other class of stabilizers such as epothilones, sarcodictyins, discodermolide and eleutherobin that compete with paclitaxel to bind to taxane-binding site or nearby [34]. Another advantage of stabilizers in regard of avoid cancer cell proliferation is causing multipolar spindle assembly formation [35].
Docetaxel (Taxotere)
Docetaxel was discovered after paclitaxel. Docetaxel is actually semi-synthesis from 10-deacetyl baccatin III which is an inactive taxoid precursor isolated from needles of European yew Taxusbaccata. Docetaxel promotes tubulin assembly in microtubule polymer and suppresses their depolymerization [36]. This discovery prompted scientists to discover more stabilizers. Years of research resulted in approaching several other natural stabilizers which were not related to taxanes [37].
Epothilones
The Epothilones are natural macrolides, considered as non-taxane microtubule stabilizers including epothilone A and epothilone B. This family of stabilizers are secondary metabolites isolated from myxobacterium Sorangiumcellulpsum in the 1990s [38]. The myxobacterial source of epothilones causes easy culture and isolation of this compound. Epothilones are classified into two groups, epoxides and olefins based on the absence or presence of epoxide group in their C-12 to C-13 position on macrolide. Examples include epoxides such as epothilones A, B, E and F and olefins such as epothilones C and D [39]. Epothiones may target taxanes binding site or a region near that because studies showed, epothilone competes with paclitaxel in binding to tubulin. Epotilone causes tubulin polymerization, induces microtubule elongation and stabilizes microtubule polymer [40]. Two main advantages of epothilones are: unlike paclitaxel it can overcome resistance caused by P-gp, therefore compared to taxanes, epothilones are improved MTA. Another advantage of epothilone is chemically offers a unique chemotype that makes it available for fermentation-based semi-synthetic approach to synthesiszing its analogues [41]. Currently several members of epothilone family are under clinical trials, such as patupilone a natural epothilone B (EPO906), a second generation epothilone B (BMS-310705), a third generation epothilone B (ZK-EPO), an epothilone D (KOS-862) and a second generation epothilone D (KOS-1584) [42].
Peloruside A
Peloruside A is a macrolide MTA that was isolated from a New Zealand marine sponge called Mycale hentscheli by West and Northcote. Peloruside A shows microtubule stabilizing activity and mitotic blockage at dosage of 10 nM and it has shown a synergistic effect with paclitaxel, even though its binding site is not taxane-binding site. Peloruside is a rare natural product and contains an intriguing structural feature, therefore became attractive for synthetic studies [43] .
Laulimalides
Laulimalides are extracted from marine sponge Cacospongiamycofijiensis [44] and this compound also displays microtubule stabilizing activity, binding to microtubule in a non-texane site [45]. The compound actually binds to α-tubulin [46]. This compound is reported to exhibit a synergistic stabilizing activity in combination with paclitaxel or epothilones [47]. These properties make laulimalides considered for next generation of stabilizers in combination therapy. But the mechanistic action of this compound and how they have synergism with taxane-site binding drugs is not completely understood.
Prota et al. [48] have showed laulimalides and peloruside A stabilize β-tubulin M-loop without forming any secondary structure plus causing formation of a bridging between two tubulin dimers across protofilaments in structure of microtubule. Also in same study they reported that there is an allosteric crosstalk activity between laulimalide/peloruside and taxane-binding site in either assembled or unassembled tubulins [48]. Ligand that binds to laulimalide/peloruside-binding site, stabilizes the conformation of the taxane site, including the M-loop [49]. On the contrary, ligands which binding to taxane-binding site, stabilize the elements that form laulimalide/peloruside-binding site in microtubule [49]. This study is the first study that showed the structural framework for this crosstalk and therefore they proposed the synergistic effect of laulimalide/pelorusideand taxane-binding site ligands on tubulin [48].
Taccalonolide
Taccalonolides are isolated from roots and rhizomes of Taccachantrieri species. They are another class of microtubule stabilizers that do not bind to taxane-binding site. The uniqueness of taccalonolides is that they do not bind directly to tubulin or microtubule and also do not induce polymerization of purified tubulin. They are able to bind to β-tubulin covalently and this is how they contribute considerable efficacy at low concentrations [50]. Taccalonolide causes bundling of microtubules in interphase. There is a group of this compound include at least 25 members. Among them taccalonolide A and E are the most rare naturally occurring secondary metabolite. The greatly acetylated pentacyclic skeleton in taccalonolides structure makes them distinct from other stabilizers. Taccalonolides A and E specifically cause bundling of microtubules in interphase and mitotic arrest of cancer cells which includes multiple aberrant spindles that is able to initiate apoptosis in same manner as paclitaxel. However, taccalonolides keep their efficacy in cells that are mutated in paclitaxel-binding site [51] as well as those cells that overexpress P-gp [52], or cells that show resistance due to expression of βIII-isotype of tubulin. Taccalonolides A and E showed excellent efficacy in paclitaxel and doxorubicin resistant mammary tumor model in vivo [53].
Cyclostreptin
Cyclostreptin is a bacterial product that was reported to have a weak paclitaxel-like activity on tubulin and exhibited anticancer activity in vivo [54]. Cyclostreptin is noteworthy for its characteristic feature of not being a taxane-binding compound but displaces taxane-binding ligands from their binding sites. Cyclostreptin poorly induces microtubule polymerization [55]. Buey et al. [56] reported that cyclostreptin binds to microtubule covalently. Mass spectrometry (MS) results showed cyclostreptin covalently binds to microtubule with either Thr220 or Asn228. With the preference for Thr220 in case of binding to the free tubulin dimer. This was the first MTA agent that has been shown to bind to microtubule in such particular way. This covalent type of binding explains unusual properties of cyclostreptin including requirement for higher temperature for polymerization induction. Cyclostreptin binds to microtubules irreversibly and is thought to be much more stable in complex compared to unbound microtubules and even more stable than paclitaxel-induced microtubule. Cyclostreptin’s activity retains in paclitaxel-resistant cancer cells. The cells that overexpression of P-gp or the expression of mutant β-tubulin is thought to be responsible for resistance to paclitaxel. It was suggested the covalent binding of cyclostreptin to microtubules could be one of the reasons for overcoming resistance as observed with paclitaxel. It is interesting that cyclostreptin is 40-fold less toxic than paclitaxel [56]. However, we must consider the fact that drugs that react covalently with any specific target, could be extremely toxic for humans [57].
Discodermolide
Discodermolide, is a polyketide natural product extracted from Caribbean marine sponge Discodermia dissolute, that was initially reported to be an immunosuppressive and an antifungal agent [58]. Later discodermolide was reported to be a potential microtubule stabilizer. Recent studies reported this stabilizer or its analogues may have advantages compared to other class of microtubule stabilizers [59]. Discodermolide has demonstrated its potent effects against those cancer cells that express P-gp and the cells that exhibit resistance to taxanes via incorporating mutated tubulins. Discodermolide also has been shown to have a synergistic effect with paclitaxel [60].
Dictyostatin
Macrolactone (-)-dictyostatin first was isolated from a Maldives marine sponge Sponqia sp.[61]. Dictyostatin stabilizes microtubule polymer by preventing depolymerization of tubulin dimers in the same way as discodermolide does. Dictyostatin proved to be effective against multidrug-resistant (MDR) cancer cells. In spite of this interesting result, further study of dictyostatin was not undertaken due to rarity of this natural agent and its unknown structure. Eventually, researchers elucidated its structure to be related to discodermolide [62]. In 2002, Shin and his colleagues reported the synthesis of first discodermolide-dictyostatin hybrid [63].
Eleutheside
Sarcodictyin and eleutherobin are two members of diterpene glycosides that belong to eleutheside family. They are natural microtubule stabilizers with the activity profiles different from paclitaxel [64]. Sarcodictyin was first extracted from Mediterranean stolonifer Sarcodictyonroseum. Later, it was isolated from the South African soft coral Eleutherobiaaurea along with isolation of two glycosidated congeners, eleuthosides A and B. Sarcodictyin was reported to stabilize microtubule and compete with paclitaxel in binding to taxane-binding sites [65]. Sarcodictyin and eleutherobin are active against paclitaxel-resistant human cancer cells therefore are classified as second generation of natural microtubule stabilizers. Eleuthesides are marine products therefore their isolation is difficult and obtained compound is in a very limited quantities. Therefore, once again, the synthesis of these natural products analogues are required for further studies [66].
Synthetized Stabilizers
Discodermolide Analogues
Minguez et al. [59] produced analogues of discodermolide in simplest synthetic steps wherein they described it as simplified discodermolide analogues. By that time synthesize of analogues have not been reported in simpler production steps (30 or more steps starting from commercial material to the final product). All of the analogues kept the C8-C14 core of discodermolide because three stereocenters and two alkenes of this core form a characteristic shape for the molecule. These analogues have been tested beside discodermolide and paclitaxel for their cytotoxic effects. The result showed even drastic structural simplification has microtubule targeting activity. Six novel derivatives of discodermolide were produced. Here we mention the cytotoxic activity of few of these analogues.
Analogue 1 was tested on microtubule assembly assay in isolated bovine brain tubulin and the results showed that this agent caused very low tubulin nucleation in vitro. Moreover, this agent caused displacement of (3H) paclitaxel bound to microtubules. High content multi-parameter fluorescent cell profile displayed disruption of microtubule assembly by this discodermolide analogue and blocked the cell in G2-M cell cycle phase. Analogue 6 showed targeted multiple cellular survival patterns in cells that are resistance to paclitaxel and its own parental agent, discodermolide [59].
Discodermolide-Dictyostatin Hybrid
Shin et al. [63] reported the first series of discodermolide-dictyostatin hybrid agents by inverting the absolute configuration of dictyostatin, therefore this way it resembles discodermolide. Some of these new hybrids showed 50% anti-proliferation activity against human breast cancer cell-line MDA-MB-231 and human ovarian cancer cell-line 2008 at 1 μM. Further study showed this hybrid displaced (3H) paclitaxel bound in microtubules [63].
Ixabepilone
The Ixabepilone (aza-epothilone B, BMS-247550, trade name: Ixempra®) is a microtubule stabilizer and it is semi-synthetized analogue of epothilone B by exchange of an azide group with oxygen at position 16 on the macrolide ring. Ixabepilone is one of latest epothilones being approved by FDA for clinical treatment in 2007. Ixabepilone binds to taxane-binding site but its interaction with tubulin is different than that by paclitaxel. Once it binds to β-tubulin subunit, it suppresses the dynamic instability of microtubules. Ixabepilone exhibited activity against cells that show multiple drug-resistance such as that induced by P-gp [67]. A phase III randomized study compared ixabepilone plus capecitabine v against capecitabine alone in advanced or metastatic breast cancer patients who showed resistant to taxanes and anthracyclins. Ixabepilone in combination with capecitabine resulted in a 25% reduction in the disease progression compared with capecitabine alone [68]. It is also recommended for treatment in the cancers of bladder, colon, pancreatic, breast, kidney etc. It is noteworthy to mention that Ixabepilone side effects are low and manageable [67].
Natural Destabilizers
Vinca Alkaloids
The Vinca alkaloids
are successful MTAs from their introduction to clinical trials. Vinblastine and vincristine are two well-known members of vinca alkaloid family. They are extracted from leaves of the periwinkle plant Catharanthusroseus (L) G. Don. Periwinkle plant leaves were used for their traditional medicinal properties from the seventieth century. But in late 1950s, their anti-mitotic property was discovered by Eli Lily Research Laboratories and at the University of Western Ontario [69, 70]. Initially, vinca alkaloids were used for treatment of haematological malignancies. They were called “wonder drugs” for their successful outcome, and their efficacy in several combination therapies was also remarkable. The success of vinca alkaloids persuaded development of numerous semi-synthetic analogues such as vinflunine, vindesine and vinorelbine. Myelosuppression and peripheral neuropathy are the major side effects of vinca alkaloids [71]. Especially, myelosuppression occurs due to the mitotic blockage of rapidly dividing bone-marrow cells. Vinca alkaloids depolymerize microtubule polymers and disrupt mitotic spindle at high dosage (10–100 nM in HeLa cell-line) [72]. But at low clinically relevant dosage (0.8 nM in HeLa cell-line), vinblastine blocks mitosis without depolymerizing microtubules involved in spindle assembly and cells finally die by apoptosis [72]. This mitotic blockage happens due to inhibition of microtubule dynamics rather than depolymerization. Vinblastine is a dimeric alkaloid extracted from the genus Vinca [73]. In 2005, the exact mechanisms by which vinblastine exerts its action structurally have been characterized. Its major effect is the ability to induce formation of spiral-like tubulin accumulation by interacting with both α-tubulin and β-tubulin. This type of contacts would form and stabilize a curved proto-filament. Vinblastine also induces accumulation of ternary complex of two tubulin dimers which are helical assemblies of complexes in which vinblastine act like a bridge between them. This bridge is formed by the interaction of vinblastine with α-tubulin of the first dimer and β-tubulin of the second dimer. Electron micrographs revealed long proto-filament curls upon vinblastine treatment. These curls are shorter and more flexible than those formed under colchicine treatment under the same condition. Vinblastine at its low dosage suppresses microtubules plus (+) end’s dynamic instability. Therefore, cells that are under clinically relevant dosage of vinblastine, encounter mitotic blockage. But by increasing its dosage, vinblastine depolymerizes the microtubule by increasing the proto-filaments spiral-like structures and curls. Gigant et al. [74] was the first study that introduced curvature into tubulin and tubulin assemblies. Compared to colchicne, another natural destabilizer, vinblastine binds in different binding sites. Vinblastine acts at the inter-dimer interface of tubulin while colchicine acts at the intra-dimer interface of tubulin. Compare to other MTAs such as paclitaxel and colchicine, vinca-binding site is equally shared between two tubulin dimers [74]. Variety of other drugs such as vincristine, vinorelbine, viinfluine, cryptophysin 52, halichondrins, doolastatins and hemiasterline also bind at vinca-binding site [7]. Vinblastine binds to soluble tubulin rapidly and it is reversible [75]. Binding of vinblastine to tubulin enhances a conformational change in connection with tubulin self-association [76], meaning vinblastine increases the affinity of tubulin for itself. In already polymerized microtubules, vinblastine binds to last tubulin at the far ends of microtubule with high affinity but it binds with very low affinity to those tubulins which are localized deep in the microtubule lattice [77]. One or two vinblastine molecules are enough for each microtubule to lose both dynamic instability and treadmilling without going through any microtubule depolymerization
. To be more specific, vinblastine causes reduction of extent and rate of microtubule growth and shrinkage but causes increase of microtubule pause state with neither grow nor shrinkage. Reduction of dynamic instability and treadmilling causes mitotic blockage by reducing normal mitotic spindle assembly and reducing the tension at the kinetochore of chromosomes. Cells in this step are stuck in metaphase-like state, chromosomes are stuck at the spindle poles, unable to move to spindle equator. Also in this step the signal to anaphase-promoting complex/cyclosome (APC/C) is blocked therefore cell is unable to transit from metaphase to anaphase. These cells eventually will die through apoptosis [7]. One significant property of vincristine and vinblastine is they bind to tubulin in lower than 5 min, while colchicine binding takes over 4 h
[78].
Estramustine
Estramustine is a nitrogen mustard derivative of estradiol-17β-phosphate that is currently used alone or in combination with other anticancer drugs for prostate cancer treatment. Estramustine inhibits the microtubule dynamics and arrests the cells in G2-M phase. The Considerable side effects of estramustine exist and include the development of anaemia [79].
Dolastatin
Dolastatin is a natural peptide and it was originally found in Indian Ocean sea hare, Dolabellaauricu-laria. A variety of dolastatin analogues have been reported to disrupt microtubules and therefore induce mitotic arrest [80]. Some of dolastatin analogues such as dolastatin 15 (DL15) have been reported to cause significant regression of tumors in clinical trials [81]. Mitra et al. [82] found that another analogue, dolastatin 10 binds to β-tubulin in a domain near the exchangeable GTP site [82]. But Cruz-Monserrate et al. [83] found that DL15 binds to tubulin at vinca-binding site [83]. Dolastatin 10 and DL15 both inhibit microtubule assembly dynamics, microtubule polymerization and cause the formation of non-microtubule structure assemblies of tubulin. Disruption of microtubule assembly dynamics caused inter-polar distance reduction in HeLa cervical cancer cells. Also, dolastatin causes loss of tension across the chromosomes kinetochores. Whenever tension is lost, tension-sensing checkpoint proteins such as BubR1 accumulate at the kinetochores during mitosis. Higher dosage of DL15 causes multi-polarity in the cells. Besides DL15 suppressed naturally decay of tubulin which is time- and temperature-dependent in vitro. This fact suggested DL15 causes conformational alterations in tubulin structure [84].
Colchicine
Colchicine was first isolated from the meadow saffron, Colchicum autumnale. In fact tubulin was referred to as “colchicine binding protein”. First it was reported to bind unpolymerized tubulin dimer and ultimately avoid polymerization [85]. Colchicine is known as a microtubule destabilizer. Its efficacy is in the dosage of 0.015 mg/kg. And it is toxic in the dosage greater than 0.1 mg/kg and it is lethal in dosage of 0.8 mg/kg [86]. Ravelli et al. (2004) reported that colchicine-binding site on tubulin is at a location of where it prevents curved tubulin from reforming a straight formation therefore it inhibits tubulin assembly. Colchicine binds to β-tubulin at the interface. At low dosage of colchicine, microtubule growth is delayed and microtubule dynamic is suppressed, but at high dosage, microtubule depolymerizes. To elaborate mechanistic action of colchicine, we must know that microtubules in order to gain stability, require lateral and longitudinal interactions between tubulin dimers. The M loops of straight tubulins are main area for lateral interaction. When colchicine is added to microtubule, M loop will be displaced therefore the lateral interaction of newly formed protofilaments are not conducted. This is because the straight tubulin formation is not selected. Until missing lateral interaction portion is small, the microtubule mass will stay same. This scenario case occurs when colchicine is applied at low dosage. When a higher dosage is applied, the proportion of missing lateral interaction will increase which leads to destabilization of microtubule ends and causes disassembly [87].
Nocodazole
Nocodazole is a natural destabilizer with reversible and rapid activity. It interferes with microtubules polymerization and the therapeutic efficacy of nocodazole is limited due to occurrence of different side effects such as neutropenia, leukopenia, bone marrow suppression and anaemia. This agent currently is used as a lead agent for design and discovery of novel analogues and also used as a reference compound in cancer studies [88].
Cryptophysin
The Cryptophycins are the macrocyclic depsipeptides and they were isolated initially from cultivated cyanobacteria Nostoc sp. in 1990. Investigation on different member of this group of compounds leaded to discovery of cryptophycin-1 as the major toxin among this species. Almost in same period of time another group of scientists isolated nastatin A from marine sponge Dysideaarenaria which later was known as cryptophysin-24. Cryptophycin-1 and the highly bioactive synthetic cryptophycin-52 display remarkable anti-mitotic activity even against those cells with MDR. Cryptophysin binds to β-tubulin and suppress tubulin polymerization and also depolymerization of microtubule has been observed in vitro. The cryptophycin binding site on tubulin is close to vinca-binding site. This location is known as “peptide-site”. But until today no structural information have been proposed to validate tubulin-cryptophysin complex [89].
Halichondrin B
The Halichondrin B was first isolated from the marine sponge Halichondriaokadai. It is also found in Axinellasp. Phakelliacarterisp. and Lissondendryxsp. Halichondrin B has been reported to inhibit tubulin polymerization and microtubule assembly in vitro and in vivo. It binds to the GTP and vinca-binding site in tubulin and inhibits tubulin-dependent GTP hydrolysis [90].
Combretastatins
Combretastatin is a stilbenoid phenols from root bark of a South African tree Combretumcaffrum which were found to be effective for primitive cancer treatment [91]. Combretastatin resembles colchicine and binds to the colchicine-binding site. Since 1990, this compound has been under extreme development as a vascular-disrupting agent (VDA). When combretastatin is applied to endothelial cells, cellular microtubules begin to depolymerize rapidly [92]. When combretastatin was treated to rodents their blood flow drops up to 95% less than 1 h [93]. A most potent natural combretastatin is combretastin A-4 (CA-4,3) which is unstable in vivo because of the transformation from the active cis-configuration to the more stable but inactive trans-configuration [88].
Hemiasterlins
Hemiasterlin was isolated from sponge Hemiasterella minor in Sodwana Bay, South Africa. It displays anticancer activity against human colon carcinoma, lung carcinoma and melanoma in vivo [94]. This compound inhibits depolymerization of existing microtubules and inhibits new microtubule assembly. Hemiasterlins are poor substrates for P-gp, therefore they are interesting candidates for cancer therapy and they are under clinical trials [12]. Hemiasterlin efficacy is banned by overexpressed P-gp mouse tumor xenograft models. But synthesis of several analogues of hemiasterlin, their in vitro and in vivo anticancer activities and their indifference reaction to P-gp-mediated drug efflux, could overcome the flaw [95].
Podophyllotoxin
Podophyllotoxinalso known as podofilox and it is a toxic lignin isolated from rhizomes and root of Podophyllumpeltatum. This agent compete with colchicine in colchicine-binding site and it binds to tubulin faster than colchicine [88]. Podophyllotoxin semi-synthetic derivatives teniposide, etoposide andetopophos (etoposide phosphate) are used for therapy of numerous malignant conditions [96]. Podophyllotoxin mechanism of action is through the inhibition of tubulin polymerization and inhibition of microtubule assembly in the mitotic spindle apparatus. However, its two derivatives, etoposide and teniposide were reported not to follow same mechanism of action as parent compound but their efficacy is through interaction with DNA and suppressing DNA topoisomerase II [97].
Curacin A
Curacin A is a lipid component isolated from a strain of the cyano bacterium Lyngbyamajuscule. Curacin A binds tightly and rapidly to colchicine-binding site on tubulin. It is not developed in clinical trials owing to its poor water-solubility and lack of stability, however development of its synthetic analogues with improved water-solubility and bioavailability may provide new hopes [88].
2-Methoxyestradiol (2-ME)
2-Methoxyestradiol is an endogenous estrogen metabolite, formed by hepatic cytochrome P450 2-hydroxylation of β-estradiol and 2-O-methylation. 2-ME targets tubulin polymerization by binding to colchicine-binding site and suppresses tumor vascularization in vivo. Its main side effects are nausea, fatigue, edema, diarrhea, neuropathy and dyspnea. Therefore, the development of metabolically stable analogues of 2-ME has started with the aim of improved properties. In this regard, ENMD-1198 was generated through the chemical modifications at 3 and 17 position of 2-ME [88].
Synthetized Destabilizers
Eribulin (E7389)
Eribulin is a synthetized analogue of halichondrin B already it is in phase III clinical trial for breast cancer treatment. At low dose, it binds to tubulin dimer and suppresses the polymerization of microtubule and microtubule dynamic instability in interphase, arrests mitosis and causes apoptosis. In one study they measured the effects of eribulin on centromere dynamics and the microtubules that attached to centromere and kinetochore by time-lapse confocal microscopy in U-2 OS human osteosarcoma cell-line. Eribulin suppressed centromere dynamics at dosage that arrest mitosis (60 nmol/L). The result showed that dynamicity decreased 35% without centromere separation. This indicated that eribulin decreased microtubule-dependent spindle tension at the kinetochores, preventing the signal for mitotic checkpoint passage [14]. A study in 2009, in order to understand the exact molecular interaction between halichondrin B and tubulin, investigated the binding of two halichondrin B analogues, eribulin and ER-076349 to tubulin by quantitative analytical ultracentrifugation . Under critical dosage of tubulin for microtubule assembly and in the presence of GDP, tubulin undergoes weak self-association into short curved oligomers. In the presence of eribulin, this oligomer formation is suppressed 4–6-fold, while ER-076349 slightly induces oligomer formation by 2-fold. This is exactly opposite of the vinblastine effect. Vinblastine strongly induces large spiral polymers around 1000-fold under same condition. Vinblastine-induced spiral formation is suppressed by both eribulin and ER-076349. Colchicine does not have any significant effect on small oligomer formation or does not undergo inhibitory effect of eribulin. These results suggest that halichondrin B analogues bind to the interdimer interface of tubulin or to the β-tubulin alone, disrupt polymer stability, and compete with vinblastine-induced spiral formation. Eribulin is a comprehensive inhibitor of tubulin polymer formation while ER-076349 also perturbs tubulin-tubulin contacts, but in more polymer formation. These results suggested that halichondrin B analogues show unique tubulin-based activities [98].
SMART
Among synthetized destabilizers, a class of 4-substituted methoxybenzoylaryl-thiazoles (SMART ) including 3 compounds known as SMART-H (H), SMART-F (F) and SMART-OH (OH) were recently reported. These compounds are with varying substituents at the 4-position of aryl ring, showed their potency to colchicine-binding site on tubulin, which causes suppression of tubulin polymerization, arrest cancer cells in G2-M phase and induce apoptosis. A remarkable characteristic of SMART compounds is that they can inhibit the growth of MDR overexpressing cells in vitro. This potency indicates that they can overcome MDR. SMART compounds suppressed the growth of 4 human prostate cancer cell-lines, and 2 melanoma cell-lines at a nanomolar range. In human prostate (PC-3) and melanoma (A375) cancer xenograft models, SMART-H and SMART-F treatments resulted in G2-M arrest and induced apoptosis. Incubating SMART compounds with bovine brain tubulin (>97% pure) showed effect of SMART compounds on tubulin polymerization. SMART-H and SMART-F suppressed tubulin polymerization by 90%, SMART-OH inhibited the polymerization by only 55%. In vivo treatment of SMART-H for 21 days at the higher dosage (15 mg/kg) failed to produce any apparent neurotoxicity. In the same study under same experimental conditions, the IC50 for SMART-H (4.23 mmol/L) was close to colchicine’s IC50 (4.91 mmol/L). Also by using novel MS competitive binding assay which corresponding to the 3 binding sites on tubulin, colchicine, vinca alkaloid and paclitaxel, they found out that SMART-H specifically competed for colchicine-binding site on tubulin, but it did not compete for either vinca alkaloid-or paclitaxel-binding sites. Among them, SMART-OH had the least potent anti-proliferative effects [99].
Methyl 2-(5-Fluoro-2-Hydroxyphenyl)-1H-Benzo (d) Imidazole-5-Carboxylate (MBIC )
A recent study in 2016, a benzimidazole-derivative, Methyl 2-(5-fluoro-2-hydroxyphenyl)-1H-benzo (d) imidazole-5-carboxylate known as MBIC was introduced as a potential MTA more specifically a tubulin destabilizer. Tubulin polymerization assay demonstrated MBIC disrupted tubulin nucleation and polymerization at similar dosage as nocodazole, colchicine and paclitaxel. The maximal velocity (Vmax) for MBIC was 2.45mOD/min which was more close to colchicine (Vmax: 2.25 mOD/min) rather than nocodazole (Vmax: 3 mOD/min), in contrast with paclitaxel (Vmax: 33 mOD/min) and untreated cells (Vmax: 12 mOD/min). This result indicated the resemblance of MBIC to colchicine. Also in this study, live-cell imaging result showed untreated HeLa cells formed bipolar spindle assembly but MBIC-treated cells did not form proper mitotic spindle and stayed in mitotic arrest for long time until cells undergo apoptosis. A remarkable characteristic of MBIC is the cytotoxic effect of this novel drug against HeLa cancer cell-lines is about 0.02 μM while its toxicity against normal cell WRL-68 is around 10.09 μM. Another considerable characteristics of MBIC is its synergistic effect with conventional drugs such as colchicine, nocodazole, doxorubicin and even paclitaxel (due to overall mutual interruption of microtubule dynamics) [100].
N-Acetylcolchinol O-Methyl Ether and Thiocolchicine
N-Acetylcolchinol O-Methyl Ether (NCME ) and Thiocolchicine are two colchicine analogues with modification only in C ring that are reported to be a better destabilizer than colchicine. Radio-labelled thio colchicine (with a thiomethyl instead of a methoxy group at position C-10) and NCME (with amethoxy-substituted benzenoid instead of the methoxy-substituted tropone C ring) were produced to be compared with colchicine. The result showed that, NCME and thiocolchicine bind to tubulin much faster than colchicine even though there is differences between these two analogues and colchicine. The binding of thiocolchicine to tubulin is temperature-dependent but thiocolchicine has similar rate of binding to tubulin with colchicine so as a function of temperature, almost there are no differences in activation energy of thiocolchicine and colchicine binding reaction. In a contrary, NCME binds to tubulin at low temperatures and reaction is done at low tubulin and drug dosage. The binding rate of NCME to tubulin is 16 times faster than colchicine binding and it is constant from 10C to 37C. The high binding rate is constant therefore the reaction eventually increases relatively while temperature rises. On the other hand the activation energy is only 40% of colchicine activation energy [101].
ZD6126
ZD6126 is a synthetic form of a water-soluble phosphate prodrug N-acetylcolchinol. In vitro studies have shown appearance of pronounced and reversible changes in the morphology of endothelial cells that being treated with ZD6126 compared to those being treated with N-acetylcolchinol, at sub-cytotoxic dosage. None of ZD6126 nor N-acetylcolchinol have induced changes in growth of human umbilical vein endothelial cells at dosages below 100 μM. But, changes in endothelial cell morphology were detectable at 0.1 μM of ZD6126. In vivo studies using a murine tumor model (CaNT) with dosage below the maximum tolerated dosage have showed a significant reduction in vascularization volume and induction of extreme necrosis in the tumor. Another in vivo study in the human xenograft FaDu, paclitaxel stabilizing activity was enhanced by adding a single dose of ZD6126 in the combination. This overall pronounced growth delay given by paclitaxel and ZD6126 combination was much higher than the effect of each individual drug alone. These finding offers ZD6126 as a promising anti-vascular agent for the treatment of solid tumors .
E7974
E7974 is a synthetic analogue of hemiasterlin and the benefit of E7974 over hemiasterlin is exhibition of same efficacy against numerous human cancer cell-lines at nanomolar dosage. A significant in vivo anticancer activity of E7974 in numerous human tumor xenograft models was observed. E7974 displayed a very low cytotoxicity against non-dividing human fibroblasts and quiescent. E7974 retains significant potency in cells with overexpressed P-gp. E7974 also exhibited a strong potency in paclitaxel-resistant ovarian cancer cell-line that their resistance is based on mutations in β-tubulin gene. Among xenograft models, those that are resistant to taxanes show high sensitivity to E7974 [102].
HTI-286
HTI-286 is another synthetic analogue of hemiasterlin and like other members of this family, as a destabilizer, hinders tubulin polymerization and induces microtubule’s dissolution by binding to tubulin dimer [103]. This compound also causes mitotic arrest and cell death in cancer cells [104]. Nunes et al. [105] radiolabeled a photo-affinity analogue of HTI-286 and they reported HTI-286 binds to α-tubulin subunit [105]. HTI-286 suppresses growth of human tumor xenografts models which are resistant to paclitaxel and vincristine through MDR1 expression [106]. HTI-266 is also effective against those cell-lines that are resistant to paclitaxel, epothilone and those that contain point mutations in β-tubulin at the taxane binding site, therefore this drug is able to bypass different resistance mechanisms [107].
CA-4P , Oxi4503 , AVE8062 , Plenstatin and CC-5079
As it is mentioned above combretastatin has anti-tubulin activity by binding to colchicine-binding site but faced some limitations in vivo. A prodrug of CA-4 is CA-4P (zybrestat) currently is in phase II trials for various types of cancers such as relapsed ovarian cancer, non-small cell lung cancer and anaplastic thyroid cancer. Oxi4503 is another analogue of combretastatin (combretastatin A-1 diphosphate, CA-1P) which targets tumor vasculature. AVE8062 is a CA-4 analogue which disrupts the formation of blood vessels in the tumors. It has the better water solubility when compared to CA-4. This analogue recently started its phase III trials. Plenstatin is another analogue of CA-4 and it is more stable than CA-4 while has exact same anti-tubulin activity as CA-4. CC-5079 is another analogue of CA-4 also known as isocombretastamins A . This analogue acts as a dual inhibitor which inhibits polymerization of tubulin and inhibits activity of phosphodiesterase (PDE4), therefore contains not only anticancer but also anti-angiogenic properties [88].
ABT-751 (E7010)
ABT-751 is a synthetized destabilizer and a novel sulfonamide which is currently in phase II clinical trials. ABT-751 binds to colchicine-binding site on β-tubulin. It is reported to have side effects included fatigue, abdominal pain and constipation [88].
T138067
T138067 is another synthetized destabilizer which binds to β-tubulin covalently which causes specific modification that prevents α- and β-tubulin dimers polymerization. T138067 showed efficacy against those cancer cells that express MDR phenotype. A phase II clinical trial reported that T138067 application was tolerable with moderate hematologic and gastrointestinal toxicity. Other expected side effects such as neurotoxicity was minimal [88].
Indibulin (D-24851, ZIO-301)
Indibulin is a promising candidate because this agent exhibited a great property which is lack of neurotoxicity in its curative dosages while this side effect is largely associated with other MTAs such as paclitaxel and vincristine. Indibulin has efficacy against taxane-resistant cancer cells in vitro and xenograft model. In addition, it has efficacy against MDR expressing cancer cells. Also, indibulin retains its efficacy in the cells that show resistance to cisplatin, thymidylate synthase inhibitor 5-Flourouracil and topoisomerase-I-inhibitor. This agent orally is available. Another great property of indibulin is partially competes with colchicine-binding site binders without overlapping in colchicine-binding site [88].
Cryptophysin 52 (LY355703)
Cryptophycin-52 (LY355703) is a synthetic form of cryptophysin family. It acts as a destabilizer, and it depolymerizes microtubules that are involved in spindle apparatus and suppressing their dynamics. Under effect of this drug, inhibition of cell proliferation was observed even in absence of noticeable spindle microtubule depolymerization [108]. Cryptophysin 52 has passed clinical phase I trials but clinical phase II trials were left uncontinued due to increase of dose limiting toxicity in vivo. Cryptophysin 52 manifested a remarkable experience that of those agents are highly effective in vitro do not necessarily exhibit same efficacy in vivo. From here researches are undertaken for synthetize of second-generation structure of this drug, most likely with more water solubility and greater selectivity for cancer cells [89]. Some of modified analogues of cryptophycins that contain polar residues display significant lower cytotoxicity against cancer cells with MDR because cryptophycins are good substrates for the P-gp efflux pump. Recently, a cryptophycin 52 analogue named cryptophycin-fluorescein-RGD-peptide conjugate was synthesized. Cyclic RGD-peptides are known for their affinity for αvβ3integrins which are highly expressed on some of cancer cells. This compound was found in the lysosomes of WM-115 human epithelial cancer cells. This property of cryptophycin-RGD-peptide conjugates make them potential to have a selectivity for cancer cells [89] (Fig. 2).

Microtubule stabilizers and destabilizers binding sites in microtubule
End-Binding Proteins; MTAs Assistants
Despite the fact, MTAs are widely used , still researches continue to find more effective and less toxic agents to substitute and/or combine with conventional MTAs. Some MTAs at low concentrations interact with microtubule ends [7]. As we explained earlier in this chapter, the plus (+) end of growing microtubule has a stabilizing GTP-bound tubulin cap which if is lost, microtubule depolymerizes (catastrophe). In fact catastrophe depends on microtubule age and happens after several steps [109]. There are several endogenous microtubule destabilizing proteins that increase the speed of microtubule ageing or decrease the number of steps leading to catastrophe [110]. At plus end of microtubule a group of proteins known as microtubule plus end-tracking proteins (+TIPs ) are accumulated which regulate dynamics and function of microtubule [111]. +TIP includes end-binding proteins (EB) in the core of +TIP. EB proteins recognize a stabilizing cap at microtubule growing end [112]. EBs affect microtubule dynamics therefore it has been suggested that EBs might affect MTAs efficacies. Maurer et al. [113] reported that EBs effecting the interfilaments contacts and/or alter the rate of GTP hydrolysis in the microtubule stabilizing cap [113]. In 2013, a group of researchers investigated the EB proteins effect on various types of MTAs and its function on microtubule dynamics. They found out in the absence of EBs, destabilizer agents suppress elongation of microtubules by delaying the polymerization. In contrast, in the presence of EBs, the elongation was inhibited due to highly increased rate of catastrophe at a dosage of destabilizer which is insufficient to effect the microtubule growth. On the other hand, in the EBs supporting stabilizer side, to inhibit the catastrophe stabilizer agents do not require any EBs’ existence but they mildly increase the catastrophe rate when EBs are added. Regardless MTAs mechanism of action and their binding site, they increase the frequency of catastrophe if EBs are bound to microtubules. In the presence of EBs, stabilizers and destabilizers could be applied in lower dosage, yet exhibit the promoted catastrophe [9]. Analyzing microtubule growth times proved that destabilizer agents increased the frequency of catastrophe in a dose-dependent manner by increasing the occurrence rate, not by number of intermediate-catastrophe-promoting steps. This result suggests that these destabilizer agents do not alter the microtubule aging mechanism but in interaction with EBs, destabilizer agents accelerate microtubule aging progression. Moreover stabilizing agents increase rate of both catastrophe and rescue in EBs presence [9]. In the presence of EBs, both stabilizers and destabilizers induce the formation or increase of microtubule lattice defects for example by altering the protofilaments structure [114]. The EB proteins are extensive and ubiquitous in the cells, the MTAs and EBs synergism in induction of catastrophe has important consequences for the final MTAs effect in the treated cells. For example in the interphase, increased catastrophe causes decrease of effective microtubule penetration into lamellae or into actin-rich cortex of cell-cell junctions, which defect cell migration and adhesion (final defect on cancer cell metastasis) [115]. At mitosis, highly increased catastrophe effects the mitotic spindle organization, microtubule attachment to kinetochore and manipulation of spindle assembly checkpoint [116].
MTAs Extra Tasks in Interphase
Since mitotic kinases have fundamental roles in mitosis, they became noticeable as targets in anticancer studies. There was idea of developing MTAs that co-target mitotic kinases as well, to achieve greater result. However, until today, many non-tubulin targeting mitotic inhibitors exhibited unsuccessful results in clinical trials [117]. Since microtubules are always present in the cells, both in interphase and mitosis, but mitotic kinases are only present in M-phase of the cell cycle, therefore they can be suppressed by desired drug about 10% of the whole cell cycling [118]. On the other hand cancer cell-lines have flaw of overexpression of some certain proteins which may involve some of mitotic kinases. If a protein is overexpressed, the chance of being more effected by a certain potential drug is much more. Aurora kinases and polo-like kinases are part of mitosis regulatory system, at the same time they are overexpressed in numerous human cancer cell-lines. This fact validating them as anticancer drug target [119, 120]. The Aurora kinase and Polo-like kinase inhibitors have been developed and introduced; nonetheless they have shown infrequent clinical results with limited efficacies [118].
The main problem may be the fact that mitotic kinases are involved in different cellular pathways therefore, even if they totally are inhibited by a certain drug, their inhibition may remain only for a period of time. While an alternative pathway may carry out the rest of cell cycle regulation and tumor is likely to return. To explain it better at molecular base, drug that causes defect in one signal-transduction pathway, may up-regulate other signaling pathways that could lead to raise of resistance against same drug [121]. Therefore, worth to mention besides MTAs there is no other cellular structure that can compensate mitotic blockage and cell division interruption therefore, there is no back-up signaling pathway to retreat MTA induced-defects of cell division. Thus, safe to say that effects of MTAs are potential to cover both microtubules disruption and mitotic regulatory system alteration. The MTAs success does not stop here. Since there are microtubules in interphase, these groups of drugs are also alters interphase events. Paclitaxel has been reported to correlate with apoptotic events more than mitotic blockage [122]. Several studies reported MTAs alter mitotic signaling pathway by manipulating mitotic proteins in expression levels and/or in post-translational modification levels [123]. MTAs were believed for long time, to prolong spindle assembly checkpoint activation which resulted in incomplete mitosis [124].
MTAs even impair non-mitotic proteins . There are studies that reported paclitaxel effects on bcl-2 protein level and subsequently causes apoptosis [125]. Microtubule network provided an extreme delicate surface area for protein-protein contact and with polarity of microtubule it allows various proteins and organelles movement throughout the cell by the help of motor proteins such as dynein and kinesin. Moreover, microtubule is required for cell migration, cytokines secretions and vascularization.
MTAs neurotoxicity characteristics proved the efficacy of MTAs in interphase. Because neurons are not dividing rapidly unlike cancer cells, therefore they enter mitosis so rarely, but yet the effect of MTAs were detected [124]. The evidence of MTAs on cellular secretion came from studies that have been conducted on impairment of T-cells with disrupted microtubules. This disruption in T-cells effected on antigen presenting cells during immune response [126]. In addition, in interphase microtubules track transportation of numerous proteins and transcription factors such as p53, androgen receptor and hypoxia inducible factor-1 (HIF-1) [127–129]. Pathways that are controlled by microtubules in interphase are complex and poorly understood. Many studies reported the transportation of oncogenic proteins by interphase microtubules. This emphasizes the role of interphase microtubule in cancer cell survival, therefore efficacy of MTAs on interphase microtubules are undeniable. The tumor suppressor p53 is transported into nucleus by microtubules when DNA is damaged. Even low dosage of MTAs changes nuclear accumulation of p53 and consequently activates target genes that cause apoptosis [127]. Another example is the effect of taxane on interphase microtubule dynamics which prevents dynein-mediated nuclear accumulation of androgen receptor, therefore suppresses transcriptional activation of genes that are involved in prostate cancer [128].
MTAs target cancer cells vascularization and this is a remarkable approach given to cancer cells accessibility to blood, the source of oxygen and nutrients, while they typically go through lack of these vital materials due to their fast division. Hence, the development of blood vessels is crucial for cancer growth and metastasis. MTAs are reported to suppress endothelial cell angiogenic function including cell proliferation, migration and vascular tubes formation [130]. Disruption of cytoskeletal tubulin in vascular cells causes vascular permeability and stops blood supply for tumors [131]. The importance of MTAs effect against tumor vascularization is clearer when we understand blood vessels in tumors are much more than in normal tissues due to endothelial cells high proliferation and their elevated vascular permeability. To be more sensitive to MTAs is associated to increase of vascular permeability [131]. For instance, opaxio™ (paclitaxel bound to biodegradable polyglutamate polymer) has anticancer advantages for highly permeable (leaky) vessels. When paclitaxel is bound to this polymer, it is inactive, thus it does not affect normal tissues. Meanwhile tumor blood vessels are highly penetrable for macromolecules, allowing opaxio™ to pass through tumor tissues. When opaxio™ penetrated into tumor, it is cleaved by lysosomal enzymes, therefore it lets paclitaxel to stabilize the microtubules. Opaxio™ is currently in phase II of clinical trial of glioblastoma and in phase III of clinical trial for ovarian cancer [132].
It is well-known that cancer cells are potential to be metastatic and this requires newly formed blood vessels which are made by angiogenic proliferation and migration of endothelial cells. During cell migration, there is cell to cell connection and cell to extracellular matrix connection. These processes have been reported to be suppressed by MTAs [133]. For example effect of taxanes on HUVEC cells migration has been reported much more than effect of this drug on HUVEC mitosis [134]. Focal adhesion of cell is necessary for migration. Focal adhesion to be formed requires Rho GTPase activity which microtubule disruption can affect them negatively [130].
HIF-1α activates tumor angiogenesis therefore it causes cancer cell survival. HIF-1α is overexpressed in most of the solid tumors. MTAs inhibits HIF-1α activity by disrupting HIF-1α mRNA trafficking on altered microtubules [129]. Also in protein level, those HIF-1α proteins that already have been expressed and exist in the cells, are supposed to be transferred into nucleus via microtubule by the aid of dynein. Therefore, MTAs inhibit HIF-1α transcriptional activity as well [135]. Hence, MTAs are involved in anti-angiogenic activity. There are other evidences that prove “MTAs effect on interphase microtubules” are important as well as their effect on mitosis. For example renal cell carcinomas shows resistance to MTA in interphase and it might be due to this cell-lines microtubule-independent trafficking [45].
Compretatins are vascular disrupting agents (VDAs) that disrupt cytoskeletal tubulin. Several of this class of agents are in clinical trials now to serve as an anticancer drugs [131].
MTAs Against Actin and Intermediate Filaments
For successful spindle positioning and suppression of cytokinesis, tubulin and actin interaction is important. In addition, there are numerous non-mitotic functions that require microtubule and microfilaments interactions such as cell migration [136], or microtubule and actin interactions, such as wound healing [137]. MTAs have been reported to disrupt intermediate filaments and actin in addition to their effect on microtubules [138]. However, actin and microtubule interaction are needed for neuronal growth cones which are highly mobile [139], therefore, disruption of microtubule impairs axon path-finding [140].
MTAs Against Mitotic Kinases
It has been reported that there are variety of cell responses against anti-mitotic drugs even among cells from same cell-line [141]. To know this fact, preserve us to expect that each individual tumor may response differently to different MTAs. This understanding is helpful in the design and development of the future new MTAs. As mitosis has been identified further, mitotic mediators have being known and classified. Targeting these proteins in virtue of their significant responsibility in controlling, guarding and processing mitosis is a rational extension to successful attempts of targeting microtubules. For instance, Polo-like kinase 1 is part of centrosome maturation and construction of mitotic spindle apparatus. This protein is also required for pass the mitosis and for separation of sister chromatids [142]. Aurora family members are required for occurrence of multiple events in mitosis. Aurora A is required for spindle assembly, Aurora B is required to phosphorylate histone H3, chromosome segregation and cytokinesis [119]. Kinesin spindles are motor proteins which are required for the formation of spindle apparatus in the beginning of mitosis [143]. Focusing on these mediators and their roles broadened the efforts of targeting cancer cell division in other ways besides microtubule disruption.
Some of agents that target these proteins happened to have synergistic effects with MTAs. For instance, AZD1152 is an Aurora B inhibitor and synergically enhances the anti-proliferative activity of vincristine in human acute leukemia cells in vitro and in vivo [144].
As we mentioned earlier, BubR1 is a tension-sensing protein. If kinetochores and microtubules are not attached together correctly, the lack of sufficient tension causes BubR1 accumulation in kinetochore and causes mitotic arrest until all kinetochore-microtubule attachments are correct. A study was done in absence and presence of DL15 in two groups of cells and they were incubated for one cell cycle. Later cells were stained by anti-BubR1 antibody. The result showed DL15 promoted BubR1 accumulation, caused loss of tension all across the kinetochores and subsequently caused mitotic arrest [84].
MBIC a synthetic destabilizer class of MTAs is also reported to alter some of mitotic kinases such as Aurora-B, BubR1, Cyclin B1 and CDK1 in human cervical cancer cell-line (HeLa) which along with disruption of tubulin polymerization, caused mitotic arrest and finally apoptosis [100].
Resemblance Between Stabilizers and Destabilizers
MTAs are divided into two classes and characteristics of these agents as stabilizers and destabilizers refer to their efficacy when drugs dosages are high and tubulin concentration is high. Nevertheless the main resemblance between these two classes of drugs is that they suppress microtubule dynamic (grow and shrinkage) instability. This resemblance is central in all MTAs [145]. That is why in some drug combination among combined stabilizer and destabilizer we can see great synergism. Each individual MTA may regulate different mechanism or mode of action but together in combination they increase the efficacy of treatment. For example the roots and rhizomes of Tacca species have a stabilizer compound named Taccalonolide and a destabilizer compound named Taccabulin A. These two compounds with their two opposite actions on microtubule polymerization, together they interfere with microtubule dynamics and overall effect is interrupting the proliferation of cancer cells. During mitosis separation of two sister chromatids are necessary and their separation requires mitotic spindles grow and shrink fast (3.6-fold more rapid than interphase). Therefore combination of stabilizer and destabilizer interferes with microtubule overall dynamics. This ability is considerable in cancer cells because of their rapid cell division compared to normal cells [146]. Moreover, in a recent study synergism between paclitaxel (stabilizer) and a newly introduced MTA , MBIC which acts as a destabilizer, was reported in treatment of cervical cancer cells in vitro [100].
Abnormal Genetic Expression and Genetic Mutations: Basic Crisis in Resistance to MTAs
α-Tubulin Mutation
There are two paclitaxel-resistant A549 cell-lines namely: A549-T12 and A549-T24. For normal growth these two cell-lines require low dose of paclitaxel [60]. In lack of paclitaxel, A549-T24 showed a most dramatic increase of microtubule dynamic and A549-T12 showed increase of dynamics in lesser degree both compared with their parental A549 cell-line. Therefore, these cell-lines are paclitaxel resistant/dependent [147]. The studies revealed that mutations on α-tubulin gene can modulate the sensitivity to agents that interact with β-tubulin. The mutation that was identified at Ser379 in A549-T12 cells corresponds to α-tubulin Ser 380 in the yeast which is located between resistant and sensitive loci in yeast genes [148]. Hence, mutation of α-tubulin in the yeast alters the binding site. In the α-tubulin gene region, mutation at α 379 in the paclitaxel-resistant cells is located between helix 10 (that contains stathmin gene [149]) and helix 11 (that is inside a domain containing MAPs genes). Therefore a mutation at α 379 cause changes in stathmin and MAPs genes leading to alter the microtubule stability and dynamics [150]. Curmi et al. [151] evaluated the expression levels of stathmin and MAP4 and they found stathmin is up-regulated in aggressive breast cancer patients [151]. It was same increase as in paclitaxel-resistant cell-lines. Thus, one suggestion is α-tubulin mutation may cause altered binding position of stathmin to β-tubulin combined with elevated protein expression level of stathmin [150].
β-Tubulin Mutations
Since β-tubulin is a target of many MTAs, several studies have investigated the DNA sequences of this protein. Reports indicated β-tubulin mutations and mutation-related clinical observations resulted from non-functional β-tubulin pseudo genes. All these known pseudo-genes, share substantial sequence homology with β-tubulin functional gene. β-Tubulin includes six isotypes. It is very well-conserved between species with similar amino-acid sequences. β-Tubulin isotypes differences are in regions of sequences that can be used for classifications between different species. In human cancer cells, class I β-tubulin is most commonly expressed β-tubulin isotype [152]. Each six isotypes has their own gene plus β-tubulin family includes pseudo-genes. Pseudo-gene sequences are not functional and probably are generated from mutations on duplicated functional β-tubulin genes [153]. Many studies have been conducted to investigate association between resistance to MTAs and changes in β-tubulin isotypes expressions and mutations [18].
Giannakakou et al. [154] performed in vitro tubulin polymerization assay and they have shown paclitaxel caused microtubule polymerization of wild-type tubulin but it did not show same efficacy in mutant tubulin purified from paclitaxel-resistant cells [154]. A study was done in 1A9 cell-line which is a paclitaxel-resistant ovarian cancer cell-line, and this cell-line carries functionally inactive mutant p53. Cells that carry p53 mutations are more likely to have additional mutations due to increase of genome instability. Among various studies that investigated this theory, one study identified presence of p53 response element in promoter region of human MSH2 gene in human ovarian cancer cell-line (A2780) which carries mutated p53. Apparently, this element is required for expression of hMSH2, Therefore non-functional p53 prevents expression of hMSH2 [155]. MSH2 is member of human mismatch repair (MMR) gene, therefore hMSH2 (human mutS homolog-2) is a DNA repair gene [156]. This study proved the role of P53 in genomic stability. Therefore, p53 mutation itself accelerates acquisition of more mutations within the cells. Giannakakou et al. [127, 157] selected several clones which were isolated from 1A9 human ovarian cell-line (PTX10 and PTA22). DNA sequencing experiment revealed that PTX22 included both mutated and wild-type p53. Silencing of P53 caused accumulation of mutated β-tubulin. Once mutation in β-tubulin genes occurred, even restoring wild-type p53 could not return the cells back to paclitaxel-sensitive state. Therefore p53 silenced cells had better chance to confer paclitaxel-resistance [157]. Although, another study in 2000 reported patients with ovarian cancer who are carriers of mut-P53 gene, had better sensitivity to paclitaxel (86%) than those patients with wt-p53 gene (47%) [158].
Investigation of connection between β-tubulin gene mutation and resistant to MTA also is done for vinca alkaloids. Kavallaris et al. [159] found point mutation of β-tubulin gene in leukemia cell-lines resistant to vinblastine (CEM/VLB 100) and to vincristine (CEM/VCR R). However both cell-lines contained elevated level of drug efflux pump P-gp but overexpression of this protein did not match the whole drug resistant phenotype [159], therefore, we can conclude β-tubulin mutation can confer resistance to MTAs.
Alteration in Class III β-Tubulin Isotype Expression
The class III β-tubulin overexpression is correlated to resistant to MTAs such as paclitaxel and vinorelbine in vitro and in vivo for various cancers especially for breast cancer. Alteration in tubulin isoform expression is associated to resistance to taxanes. Although identifying the connection of class III β-tubulin and drug resistance is complicated due to complexity of tubulin auto-regulation systems, but there are studies that investigated effects of changes in tubulin isoform expressions on efficacy of several new tubulin-binding agents. For instance, Suzuki et al. [160] reported that STX140 is able to suppress proliferation of resistant MCF-7 dox cell-line with overexpressed P-gp [160]. When the level of soluble tubulin increases, β-tubulin mRNA degrades through a co-translational degradation that also regulates the expression of β-tubulin isotypes [161]. In a study, Hari et al. [162] overexpressed the class III β-tubulin in CHO cell-line. This experiment increased resistance to paclitaxel by decrease of paclitaxel suppression on microtubule dynamics [162]. Data showed breast cancer patients with resistance to docetaxel, had five-fold increase of class III β-tubulin [163]. Joe et al. [164] transfected class III β-tubulin in cervical carcinoma HeLa cell-line and tested taccalonolides efficacy. It was reported there is no evidence of resistance against this drug in cells with overexpressed class III β-tubulin [53], however, overexpression of class III β-tubulin caused resistance to paclitaxel [164]. Gan et al. [165] investigated the effect of class III β-tubulin expression in different pattern. They silenced class III β-tubulin and observed that this experiment induced resistance to vincristine, paclitaxel and DNA-damaging agents in non-small cell lung carcinoma [165]. Stengel et al. [166] have shown that alteration in class III β-tubulin expression (both silencing and overexpression) in two breast cancer cell-lines, MCF-7 and MDA-MB-231 did not affect the efficacies of STX140, STX243, ENMD1198, 2-MeOE2 and colchicine. While efficacies of paclitaxel and vinorelbine have changed [166]. Small difference between class I and III of β-tubulin is one amino-acid which causes different final three dimensional formations. Class III β-tubulin carries Arg 277 instead of Ser 277 which exist in class I. Ser277 and Arg278 are key amino-acid for stable binding of tubulin to paclitaxel in its binding site at class I β-tubulin [167], but in class III β-tubulin structure changed and lost its stable taxane-binding site [168]. Class III β-tubulin is correlated with increased microtubule dynamics which is opposite of assembling activity of paclitaxel at microtubules plus (+) end [169]. The reason that high contained class III β-tubulin cancer cells are not resistant to STX140, STX243, ENMD1198, 2-MeOE2 and colchicine, but are resistant to paclitaxel, is these agents bind to colchicine-binding site which includes a different conformation and leads to formation of a stable complex containing class III β-tubulin. Besides, these agents are destabilizers, therefore overexpression of class III β-tubulin is even an advantage for this class of drugs due to class III β-tubulin increases microtubule dynamics. Although, vinorelbine which binds to vinca-binding site appeared to have less efficacy in cancer cells with class III β-tubulin overexpression in small lung cancer cell-line [165], breast cancer cell-line [166] and in vivo[170]. These observations suggest that resistant cancer cells with overexpressed class III β-tubulin is related to drug binding-site. This idea is supported by study of Stengel et al. [166], wherein they showed those MTAs that binds to colchicine-binding site in cells which contain altered class III β-tubulin, do not exhibit resistance to these drugs, but those MTAs binding to taxane- and vinca-binding sites , alteration in class III β-tubulin, affected their efficacies. A satisfactory conclusion here could be the importance of design and development of new agents that bind to colchicine-binding site as taxanes alternatives for resistant cancers [166].
Alteration in Class VI β-Tubulin Isotype Expression
Lack of β-tubulin VI in non-hematopoietic tissues confirmed that β-tubulin VI is a hematology-specific isotype and consequently it is a marker that mediates hematologic toxicity of β-tubulin binding drugs . These cells express a specific variant of β-tubulin VI named β-tubulin VI 274 M variant. This variant is less sensitive to paclitaxel compared to other variants and compared to β-tubulin VI wild-type [171]. Within paclitaxel-binding site of all human β-tubulin isotypes, there is a conserved residue (residue 274) [172]. This residue is necessary for binding of paclitaxel to tubulin. Mutation in this residue (T274 M) leads to resistance to paclitaxel [173], therefore patients who carrying this mutation could be resistant to myelosuppressive effects of MTAs. Leandro-Garcia et al. [171] for first time showed β-tubulin class VI is a hematology-specific isotype which differentiates from other β-tubulin genes by few genetic and expression variability. Also they have shown patients who are carriers of TUBB1 T274 M in their β-tubulin class VI gene, had protection against paclitaxel effect [171].
P-Glycoprotein Overexpression
One of the major current problem in chemotherapy is efflux of drugs into extracellular matrix. This occurs by activity of ATP-binding cassette protein known as P-gp product of MDR1 gene and it is part of MDR mechanism [174]. The main natural positive role of P-gp is pumping the toxic substances out of cells in brain and gastrointestinal tracts [175]. In many cancer types, overexpression of P-gp seemed to be related to resistance to taxanes [176], but in same cancers ixabepilone a class of epothilone, showed cytotoxicity [40]. A meta-analysis of 31 breast cancer trials showed that the P-gp was overexpressed in 41% of patients after treatment and interestingly, there was 3-fold reduction of response to paclitaxel in same patients. This result suggested chemotherapy induces P-gp expression [177]. Investigation on connection between other taxanes such as docetaxel and expression of P-gp in breast cancer were done and no significant correlation has been reported [178]. Epothilones and taxanes bind to same tubulin binding site but they are functioning differently. The main advance of epothilones is that unlike taxanes they are not P-gp substrate [179]. In this regard, epothilones are active in models that are taxane resistance because epothilones tolerance is provided. As an example, epothilone B causes much higher levels of tubulin polymerization when it is compared with equimolar dosages of paclitaxel in vitro [180]. Other experiments showed almost all models that are resistant to paclitaxel display sensitivity to epothilone A and epothilone B. Recent studies have reported that, α-tubulin mutations in cells that are resistant to microtubule-depolymerizing agents, causes decreased drug accumulation even when P-gp overexpression is not present [181]. If P-gp causes resistance to certain drugs therefore those agents that inhibit P-gp activity, are able to bring back the sensitivity of the cell to those certain drugs [174]. A recent drug, tariquidar is a P-gp inhibitor which showed an ability to bring back the sensitivity to paclitaxel in 17 women with stage III-IV breast cancer but final result was lack of efficacy [182]. Therapeutic industry needs more offer on P-gp inhibitors more specific those drugs that increase the efficacy of taxanes. Also more MTAs are required to be introduced with less susceptibility to P-gp-mediated resistance .
Tau: Intracellular Competitor of Paclitaxel
Tau protein was first time described in 1975 to be a product of a gene located in chromosome 17 (17q21) [183]. Tau is one of microtubule-associated proteins (MAPs) and attaches to tubulin both exterior and interior surfaces of microtubule and also it binds in paclitaxel-binding site therefore practically tau competes with paclitaxel. When tau attaches to paclitaxel-binding site, in the same way stabilizes the microtubule as paclitaxel does but with much more reversibility [184]. Studies revealed that those breast cancer patients that have genetically low tau expression, benefit more from paclitaxel treatment due to competition of paclitaxel and tau in attaching to microtubule. One study was experimented with paclitaxel-based neo-adjuvant chemotherapy on 82 breast cancer patients. Those patients with low tau mRNA expression level, succeeded to achieve total pathologic response (P < 0.001) [185]. Furthermore, there is a correlation between tau and ER in breast cancer patients. Those with high tau expression were reported to be more among ER-positive patients (57%) than those with ER-negative (15%) [186] and this is due to oestrogen regulation of tau gene. Oestrogen induces tau expression. Therefore co-expression of ER and tau is considered a good sign for prognosis in the breast cancer therapy and it is related to better overall survival. ER-positive patients with high tau expression are more sensitive to hormone therapy than paclitaxel therapy. On the other hand, ER-negative patients had better response to paclitaxel therapy [187]. In 2005 a group of researchers silenced tau protein in breast cancer cells and they achieved higher sensitivity to taxanes [188]. Thus, tau expression level could be an important biomarker of resistance to paclitaxe l [187].
HER2 Overexpression
HER2 is a transmembrane receptor and it has a tyrosine kinase activity [189]. HER2 belongs to a family with four members namely: EGFR/HER1, HER2, HER3 and HER4 receptors that are involved in cell growth regulation, differentiation and survival through interlink with some of survival pathways including PI3K/AKT pathway and Ras/Raf/MEK/MAPK pathway. Therefore, when HER2 is expressed, there is signaling to downstream pathways [190]. HER2 is always in active form and ready to interact with its ligands [191]. Amplification and/or overexpression of HER2 were detected in 20% of early stage breast cancer patients [192]. Breast cancer patients with HER2-negative ER-positive, had no significant benefit from paclitaxel treatment while patients with HER2-positive whether ER-positive or ER-negative showed significant improved response to therapy including 5 years of disease-free survival. Also patients with overexpressed HER2 seemed to receive even better benefit compared to HER2-negative. Therefore, HER2 amplification and overexpression is a marker of potential to emerge better response to taxane therapy. Interestingly, overexpression of HER2 is associated with lower expression of tau protein which is somehow a brief explanation for increased sensitivity to taxane therapy [193].
Determinants of Sensitivity and Resistance to MTAs
The major logic behind continuous production of MTAs is their lack of tissue selectivity and perpetual drug resistance against these drugs. For instance paclitaxel and vinca alkaloid are active against breast, ovarian and lung cancers but inactive against colon and kidney cancers and many sarcomas. Seems these drugs are more effective against haematological type of cancers rather than solid ones. The determinants of being sensitive or resistance to specific drug is inside the cell and their level of access to drugs pharmacological benefits [15]. Different levels of resistance were reported against MTAs. Those cells that ultimately resist against MTAs, oftentimes display overexpression of a class of a membrane transporter named ABC-transporters (ATP-dependent drug efflux pumps or ATP-binding cassettes). These pumps reduce intracellular drug accumulation by pumping them out of cell. Since, they may pump various drugs at the same time, they cause cross-resistance or MDR. Many of these transmembrane pumps have been identified such as P-gp which is explained earlier in this chapter [194]. To remove this obstacle considerable efforts are in the way to produce MTAs which are not removed by transmembrane pumps [195]. However, the cell’s host factors are also determine to pose the cell as sensitive or resistance to drugs. Such as abnormal expression of regulatory proteins, the different level of expression of tubulin isotypes and their post-translational modifications. There are various studies reported the correlation between high level of a specific tubulin isotype and resistance to a specific MTA [15, 19, 159, 196]. The cellular host determinants can be divided into two groups. Those that affect microtubule polymer level and therefore they also affect microtubule dynamics. Microtubule polymer level is a significant host determinant considering most organelles and components move inside the cell by attaching and moving along microtubules. Therefore, those cells that have the ability of preserve their microtubule mass in required level for organelles and components to move, survive longer under treatment of destabilizers such as vinca alkaloids. On the other hand, under paclitaxel treatment and consequently appearance of increased microtubule polymers, motor-proteins such as dynein and kinesin are not able to support the transportations anymore. Certain cancer cells that overexpress endogenous microtubule-depolymerizing factors, develop resistance against stabilizers like paclitaxel.
The importance of microtubule dynamics as host determinant was showed that those MTAs that suppress microtubule dynamics without any significant change in microtubule-polymer level, prevent metaphase to anaphase transition, such as in paclitaxel-resistance/dependent A549 lung cancer cell-line. After omitting paclitaxel in these cells, results show 57% to 167% faster microtubule dynamics compared to paclitaxel-sensitive A549 cells [147]. Fast microtubule dynamics behavior cost this group of cells disrupted spindle assembly and they were unable to transit from metaphase to anaphase. Addition of paclitaxel to this group of cells caused microtubule dynamics slow down and they went through successful mitosis. This shows their resistance and at the same time dependence to paclitaxel. This group of A549 cells undergo several endogenous changes such as overexpression of βIII-tubulin isotype, As mutated α-tubulin, overexpression of endogenous microtubule-destabilizer protein stathmin and inactivation of endogenous microtubule-stabilizer protein MAP4 [150, 197]. The point that we can understand here is microtubules destiny is affected by many endogenous determinants . Therefore, there are many undiscovered potential determinants which could be targets of chemotherapeutic drug design.
Natural or Synthetized MTAs? Which Are More Beneficial?
As early as discovery of natural products, they provided a rich source of compounds that were applicable in many fields of medical researches. Above half of currently used drugs in chemo therapy are natural products. Synthetized or semi-synthetized agents are either obtained from natural sources with few structural modifications (semi-synthetized), or they are totally new compounds which are designed according to a natural compound structure as a model (synthetized) [198]. The main aim of synthetizing a drug is to establish a relationship between pharmacological advantages and structure of the compound, therefore the aim is obtaining a new drug which is better than “prototype” with a view of collecting advantages from potency, less toxicity and more selectivity. Natural products which are extracted from marine products, plants and microorganisms are considered as prototype, origin, template or lead compounds [199].
To answer the question of “which one is better”, we must consider natural products provide major structural diversity which some of them have sufficient biological potency in some aspect, but some disadvantages in another aspect. For example taxanes which are great stabilizers, on the other hand they are also great substrate for plasma membrane drug-efflux pump [200]. The search for natural products is necessary and will continue to provide greater range of template [201]. These prototypes are modified to produce more improved semi-synthetized agents with better therapeutic potential by molecular modifications. Or they are template for entirely new synthetized agents as their analogues, which contain greater pharmacological activity, magnificent therapeutic possibilities and less side effects. Nowadays, drug synthesis provides huge range of improved drugs in regard of both structure and activity [198]. According to data, we can conclude both natural and synthetized drugs have different range of advantages that must be considered .
Conclusion
Critical obstacle in current cancer therapy is low selectivity of anticancer drugs against cancer cells. To differentiate between cancer and normal cells, the knowledge of cancer-specific characteristics, origin and differences of these characteristics is required. This aim directed researchers to develop molecular-based strategies. One of the main different characteristics is rapid mitosis of cancer cells. Microtubules, the main component of mitosis have two types of dynamic behaviors. The duration of each and switch rate between these two behaviors caused microtubule interesting as a potential target. MTAs are class of anticancer drugs which effect mitosis by targeting microtubules. This paradigm makes MTAs applicable to rapidly dividing cancer cells, compared to slowly dividing normal cells. Proliferation mechanisms are tightly under control in normal cells while these mechanisms are manipulated in cancer cells, due to continuous mutations. Mutations in cancer cells alter microtubule dynamic behaviors. Studying different characteristics in molecular level also clarified that mutations such as α- and β-tubulin mutations and abnormal expression of β-tubulin isotypes are associated with resistance to some of MTAs. MTAs are verity of small molecules either from natural sources or synthetized/semi-synthetized as improved analogues of natural agents, interrupt microtubule dynamic behaviors with either stabilizing or destabilizing activities. Studies showed some of MTAs beside their main task, manipulate mitotic kinases as well, or manipulate some other specific proteins that directly or indirectly boost MTAs’ main task. Moreover, MTAs suppress tumor vascularization, which avoid tumor accessibility to source of oxygen and nutrients “the blood”. No matter stabilizer or destabilizer, MTAs’ main resemblance is their final task which is suppressing microtubule dynamic behaviors. That is why more drug combination strategies were successful in emerge of synergism between different MTAs. Natural MTAs always have been main source of applicable drugs or great template in the view of structure or bioactivity. Synthetized/semi-synthetized MTAs have been improved drugs to associate between structure and bioactivity with lesser side effects and resistance to MTAs.
References
Livingston DM, Shivdasani R (2001) Toward mechanism-based cancer care. JAMA 285(5):588–593
Pecorino L (2012) Molecular biology of cancer: mechanisms, targets, and therapeutics. Oxford University Press, Oxford
Evan GI, Vousden KH (2001) Proliferation, cell cycle and apoptosis in cancer. Nature 411(6835):342–348
Sherr CJ (1996) Cancer cell cycles. Science 274(5293):1672–1677
Stocker H, Hafen E (2000) Genetic control of cell size. Curr Opin Genet Dev 10(5):529–535
Loeb LA, Loeb KR, Anderson JP (2003) Multiple mutations and cancer. Proc Natl Acad Sci 100(3):776–781
Jordan MA, Wilson L (2004) Microtubules as a target for anticancer drugs. Nat Rev Cancer 4(4):253–265
Sharp DJ, Rogers GC, Scholey JM (2000) Microtubule motors in mitosis. Nature 407(6800):41–47
Mohan R, Katrukha EA, Doodhi H, Smal I, Meijering E, Kapitein LC et al (2013) End-binding proteins sensitize microtubules to the action of microtubule-targeting agents. Proc Natl Acad Sci 110(22):8900–8905
Oakley BR (2000) An abundance of tubulins. Trends Cell Biol 10(12):537–542
Rohena CC, Mooberry SL (2014) Recent progress with microtubule stabilizers: new compounds, binding modes and cellular activities. Nat Prod Rep 31(3):335–355
Poruchynsky MS, Kim J-H, Nogales E, Annable T, Loganzo F, Greenberger LM et al (2004) Tumor cells resistant to a microtubule-depolymerizing hemiasterlin analogue, HTI-286, have mutations in α-or β-tubulin and increased microtubule stability. Biochemistry 43(44):13944–13954
Barlow SB, Gonzalez-Garay ML, Cabral F (2002) Paclitaxel-dependent mutants have severely reduced microtubule assembly and reduced tubulin synthesis. J Cell Sci 115(17):3469–3478
Amos LA (2011) What tubulin drugs tell us about microtubule structure and dynamics. Semin Cell Dev Biol 22(9):916–926
Dumontet C, Sikic BI (1999) Mechanisms of action of and resistance to antitubulin agents: microtubule dynamics, drug transport, and cell death. J Clin Oncol 17(3):1061
Downing KH, Nogales E (1999) Crystallographic Structure of Tubulin. Implications for Dynamics and Drug Binding. Cell Struct Funct 24(5):269–275
Snyder JP, Nettles JH, Cornett B, Downing KH, Nogales E (2001) The binding conformation of Taxol in β-tubulin: A model based on electron crystallographic density. Proc Natl Acad Sci 98(9):5312–5316
Berrieman HK, Lind MJ, Cawkwell L (2004) Do β-tubulin mutations have a role in resistance to chemotherapy? Lancet Oncol 5(3):158–164
Burkhart CA, Kavallaris M, Horwitz SB (2001) The role of β-tubulin isotypes in resistance to antimitotic drugs. Biochim Biophys Acta 1471(2):O1–O9
Igney FH, Krammer PH (2002) Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer 2(4):277–288
Zaffaroni N, Pennati M, Colella G, Perego P, Supino R, Gatti L et al (2002) Expression of the anti-apoptotic gene survivin correlates with taxol resistance in human ovarian cancer. Cell Mol Life Sci 59(8):1406–1412
O'Connor DS, Grossman D, Plescia J, Li F, Zhang H, Villa A et al (2000) Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci 97(24):13103–13107
Sparano JA (2000) Taxanes for breast cancer: an evidence-based review of randomized phase II and phase III trials. Clin Breast Cancer 1(1):32–40
Horwitz SB (1994) How to make taxol from scratch. Nature 367(6464):593
Schiff PB, Horwitz SB (1981) Taxol assembles tubulin in the absence of exogenous guanosine 5'-triphosphate or microtubule-associated proteins. Biochemistry 20(11):3247–3252
Nogales E, Wolf SG, Khan IA, Ludueña RF, Downing KH (1995) Structure of tubulin at 6.5 Å and location of the taxol-binding site. Nature 375(6530):424–427
Nogales E (2001) Structural insights into microtubule function. Annu Rev Biophys Biomol Struct 30(1):397–420
Rao S, Krauss NE, Heerding JM, Swindell CS, Ringel I, Orr GA et al (1994) 3'-(p-azidobenzamido) taxol photolabels the N-terminal 31 amino acids of beta-tubulin. J Biol Chem 269(5):3132–3134
Nogales E, Wolf SG, Downing KH (1998) Structure of the αβ tubulin dimer by electron crystallography. Nature 391(6663):199–203
Derry WB, Wilson L, Jordan MA (1995) Substoichiometric binding of taxol suppresses microtubule dynamics. Biochemistry 34(7):2203–2211
Von Hoff D (1997) The taxoids: same roots, different drugs. Semin Oncol 24(4 Suppl 13):S13-3–S13-10
Dı́az JF, Valpuesta JM, Chacón P, Diakun G, Andreu JM (1998) Changes in Microtubule Protofilament Number Induced by Taxol Binding to an easily accessible site. Internal microtubule dynamics. J Biol Chem 273(50):33803–33810
Jordan MA, Toso RJ, Thrower D, Wilson L (1993) Mechanism of mitotic block and inhibition of cell proliferation by taxol at low concentrations. Proc Natl Acad Sci 90(20):9552–9556
Pryor DE, O'Brate A, Bilcer G, Díaz JF, Wang Y, Wang Y et al (2002) The microtubule stabilizing agent laulimalide does not bind in the taxoid site, kills cells resistant to paclitaxel and epothilones, and may not require its epoxide moiety for activity. Biochemistry 41(29):9109–9115
Chen J-G, Horwitz SB (2002) Differential mitotic responses to microtubule-stabilizing and-destabilizing drugs. Cancer Res 62(7):1935–1938
Lavelle F, Bissery M, Combeau C, Riou J, Vrignaud P, Andre S (1995) Preclinical evaluation of docetaxel (Taxotere). Semin Oncol 22(2 Suppl 4):3–16
He L, Orr GA, Horwitz SB (2001) Novel molecules that interact with microtubules and have functional activity similar to Taxol™. Drug Discov Today 6(22):1153–1164
Gerth K, Bedorf N, Höfle G, Irschik H, Reichenbach H (1996) Epothilons A and B: antifungal and cytotoxic compounds from Sorangium cellulosum (Myxobacteria). Production, physico-chemical and biological properties. J Antibiot 49(6):560–563
Goodin S (2008) Novel cytotoxic agents: epothilones. Am J Health Syst Pharm 65(10 Suppl 3):S10–S15
Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J et al (1995) Epothilones, a new class of microtubule-stabilizing agents with a taxol-like mechanism of action. Cancer Res 55(11):2325–2333
Lee FY, Borzilleri R, Fairchild CR, Kim S-H, Long BH, Reventos-Suarez C et al (2001) BMS-247550 a novel epothilone analog with a mode of action similar to paclitaxel but possessing superior antitumor efficacy. Clin Cancer Res 7(5):1429–1437
Villanueva C, Vuillemin AT, Demarchi M, Bazan F, Chaigneau L, Pivot X (2009) Ixabepilone: a new active chemotherapy in the treatment of breast cancer. Womens Health (Lond) 5(2):115–121
Ghosh AK, Xu X, Kim J-H, Xu C-X (2008) Enantioselective total synthesis of peloruside A: a potent microtubule stabilizer. Org Lett 10(5):1001–1004
Hamel E, Sackett DL, Vourloumis D, Nicolaou K (1999) The coral-derived natural products eleutherobin and sarcodictyins A and B: effects on the assembly of purified tubulin with and without microtubule-associated proteins and binding at the polymer taxoid site. Biochemistry 38(17):5490–5498
Dumontet C, Jordan MA (2010) Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat Rev Drug Discov 9(10):790–803
Pineda O, Farràs J, Maccari L, Manetti F, Botta M, Vilarrasa J (2004) Computational comparison of microtubule-stabilising agents laulimalide and peloruside with taxol and colchicine. Bioorg Med Chem Lett 14(19):4825–4829
Hamel E, Day BW, Miller JH, Jung MK, Northcote PT, Ghosh AK et al (2006) Synergistic effects of peloruside A and laulimalide with taxoid site drugs, but not with each other, on tubulin assembly. Mol Pharmacol 70(5):1555–1564
Prota AE, Bargsten K, Northcote PT, Marsh M, Altmann KH, Miller JH et al (2014) Structural basis of microtubule stabilization by laulimalide and peloruside A. Angew Chem Int Ed Engl 53(6):1621–1625
Khrapunovich-Baine M, Menon V, Yang C-PH, Northcote PT, Miller JH, Angeletti RH et al (2011) Hallmarks of molecular action of microtubule stabilizing agents effects of epothilone b, ixabepilone, peloruside a, and laulimalide on microtubule conformation. J Biol Chem 286(13):11765–11778
Li J, Risinger AL, Mooberry SL (2014) Taccalonolide microtubule stabilizers. Bioorg Med Chem 22(18):5091–5096
Tinley TL, Randall-Hlubek DA, Leal RM, Jackson EM, Cessac JW, Quada JC et al (2003) Taccalonolides E and A plant-derived steroids with microtubule-stabilizing activity. Cancer Res 63(12):3211–3220
Fojo T, Menefee M (2007) Mechanisms of multidrug resistance: the potential role of microtubule-stabilizing agents. Ann Oncol 18(suppl 5):v3–v8
Risinger AL, Jackson EM, Polin LA, Helms GL, LeBoeuf DA, Joe PA et al (2008) The taccalonolides: microtubule stabilizers that circumvent clinically relevant taxane resistance mechanisms. Cancer Res 68(21):8881–8888
Edler MC, Buey RM, Gussio R, Marcus AI, Vanderwal CD, Sorensen EJ et al (2005) Cyclostreptin (FR182877), an antitumor tubulin-polymerizing agent deficient in enhancing tubulin assembly despite its high affinity for the taxoid site. Biochemistry 44(34):11525–11538
Buey RM, Barasoain I, Jackson E, Meyer A, Giannakakou P, Paterson I et al (2005) Microtubule interactions with chemically diverse stabilizing agents: thermodynamics of binding to the paclitaxel site predicts cytotoxicity. Chem Biol 12(12):1269–1279
Buey RM, Calvo E, Barasoain I, Pineda O, Edler MC, Matesanz R et al (2007) Cyclostreptin binds covalently to microtubule pores and lumenal taxoid binding sites. Nat Chem Biol 3(2):117–125
Evans DC, Watt AP, Nicoll-Griffith DA, Baillie TA (2004) Drug-protein adducts: an industry perspective on minimizing the potential for drug bioactivation in drug discovery and development. Chem Res Toxicol 17(1):3–16
Mı́nguez JM, Kim S-Y, Giuliano KA, Balachandran R, Madiraju C, Day BW et al (2003) Synthesis and biological assessment of simplified analogues of the potent microtubule stabilizer (+)-discodermolide. Bioorg Med Chem 11(15):3335–3357
Minguez JM, Giuliano KA, Balachandran R, Madiraju C, Curran DP, Day BW (2002) Synthesis and High Content Cell-based Profiling of Simplified Analogues of the Microtubule Stabilizer (+)-Discodermolide 1 Supported by National Cancer Institute Grant CA78039. 1. Mol Cancer Ther 1(14):1305–1313
Martello LA, McDaid HM, Regl DL, Yang C-PH, Meng D, Pettus TR et al (2000) Taxol and discodermolide represent a synergistic drug combination in human carcinoma cell lines. Clin Cancer Res 6(5):1978–1987
Pettit GR, Cichacz ZA, Gao F, Boyd MR, Schmidt JM (1994) Isolation and structure of the cancer cell growth inhibitor dictyostatin 1. J Chem Soc Chem Commun 9:1111–1112
Kalesse M (2000) The chemistry and biology of discodermolide. ChemBioChem 1(3):171–175
Shin Y, Choy N, Balachandran R, Madiraju C, Day BW, Curran DP (2002) Discodermolide/dictyostatin hybrids: synthesis and biological evaluation. Org Lett 4(25):4443–4446
Telser J, Beumer R, Bell AA, Ceccarelli SM, Monti D, Gennari C (2001) Synthesis of a simplified sarcodictyin analogue which retains microtubule stabilising properties. Tetrahedron Lett 42(52):9187–9190
Nakao Y, Yoshida S, Matsunaga S, Fusetani N (2003) (Z)-Sarcodictyin A, a new highly cytotoxic diterpenoid from the soft coral Bellonella a lbiflora. J Nat Prod 66(4):524–527
Brocksom TJ, de Oliveira KT, Ferreira MA, Servilha BM (2015) Essential oils as raw materials in the synthesis of anticancer drugs. In: Bioactive essential oils and cancer. Springer, New York, pp 81–109
Mandhare A, Biradar S, Gurule A (2016) Azaepothilone B and its derivatives: a patent review. Expert Opin Ther Pat 26(8):891–905
Thomas ES, Gomez HL, Li RK, Chung H-C, Fein LE, Chan VF et al (2007) Ixabepilone plus capecitabine for metastatic breast cancer progressing after anthracycline and taxane treatment. J Clin Oncol 25(33):5210–5217
Noble R, Beer C, Cutts J (1959) Further biological activities of vincaleukoblastine—an alkaloid isolated from Vinca rosea (L.). Biochem Pharmacol 1(4):347–348
Johnson I, Wright H, Svoboda G 1959 Experimental basis for clinical evaluation of anti tumor principles derived from Vinca-Rosea Linn. J Lab Clin Med 54:830. Mosby-Year Book Inc 11830 Westline Industrial Dr, St Louis, MO 63146–3318
Gidding C, Kellie S, Kamps W, De Graaf S (1999) Vincristine revisited. Crit Rev Oncol Hematol 29(3):267–287
Jordan MA, Thrower D, Wilson L (1991) Mechanism of inhibition of cell proliferation by Vinca alkaloids. Cancer Res 51(8):2212–2222
Bryan J (1971) Vinblastine and microtubules: I. Induction and isolation of crystals from sea urchin oocytes. Exp Cell Res 66(1):129–136
Gigant B, Wang C, Ravelli RB, Roussi F, Steinmetz MO, Curmi PA et al (2005) Structural basis for the regulation of tubulin by vinblastine. Nature 435(7041):519–522
Jordan MA, Wilson L (1990) Kinetic analysis of tubulin exchange at microtubule ends at low vinblastine concentrations. Biochemistry 29(11):2730–2739
Na GC, Timasheff SN (1980) Stoichiometry of the vinblastine-induced self-association of calf brain tubulin. Biochemistry 19(7):1347–1354
Singer WD, Jordan M, Wilson L, Himes RH (1989) Binding of vinblastine to stabilized microtubules. Mol Pharmacol 36(3):366–370
Owellen RJ, Owens AH, Donigian DW (1972) The binding of vincristine, vinblastine and colchicine to tubulin. Biochem Biophys Res Commun 47(4):685–691
Bissinger R, Modicano P, Frauenfeld L, Lang E, Jacobi J, Faggio C et al (2013) Estramustine-induced suicidal erythrocyte death. Cell Physiol Biochem 32(5):1426–1436
Hu Z-B, Gignac SM, Quentmeier H, Pettit GR, Drexler HG (1993) Effects of dolastatins on human B-lymphocytic leukemia cell lines. Leuk Res 17(4):333–339
Ebbinghaus S, Rubin E, Hersh E, Cranmer LD, Bonate PL, Fram RJ et al (2005) A phase I study of the dolastatin-15 analogue tasidotin (ILX651) administered intravenously daily for 5 consecutive days every 3 weeks in patients with advanced solid tumors. Clin Cancer Res 11(21):7807–7816
Mitra A, Sept D (2004) Localization of the antimitotic peptide and depsipeptide binding site on β-tubulin. Biochemistry 43(44):13955–13962
Cruz-Monserrate Z, Mullaney JT, Harran PG, Pettit GR, Hamel E (2003) Dolastatin 15 binds in the vinca domain of tubulin as demonstrated by Hummel–Dreyer chromatography. Eur J Biochem 270(18):3822–3828
Lopus M (2013) Mechanism of mitotic arrest induced by dolastatin 15 involves loss of tension across kinetochore pairs. Mol Cell Biochem 382(1–2):93–102
Yue Q-X, Liu X, Guo D-A (2010) Microtubule-binding natural products for cancer therapy. Planta Med 76(11):1037
Niel E, Scherrmann J-M (2006) Colchicine today. Joint Bone Spine 73(6):672–678
Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A et al (2004) Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 428(6979):198–202
Lu Y, Chen J, Xiao M, Li W, Miller DD (2012) An overview of tubulin inhibitors that interact with the colchicine binding site. Pharm Res 29(11):2943–2971
Weiss C, Sammet B, Sewald N (2013) Recent approaches for the synthesis of modified cryptophycins. Nat Prod Rep 30(7):924–940
Hu Y (2013) Halichondrin B analogs. Google Patents
Pettit GR, Cragg GM, Herald DL, Schmidt JM, Lohavanijaya P (1982) Isolation and structure of combretastatin. Can J Chem 60(11):1374–1376
Kanthou C, Tozer GM (2002) The tumor vascular targeting agent combretastatin A–4-phosphate induces reorganization of the actin cytoskeleton and early membrane blebbing in human endothelial cells. Blood 99(6):2060–2069
Anderson HL, Yap JT, Miller MP, Robbins A, Jones T, Price PM (2003) Assessment of pharmacodynamic vascular response in a phase I trial of combretastatin A4 phosphate. J Clin Oncol 21(15):2823–2830
Kowalczyk JJ, Kuznetsov G, Schiller S, Seletsky BM, Spyvee M, Yang H (2006) Hemiasterlin derivatives and uses thereof. Google Patents
Kowalczyk JJ, Schiller SE, Spyvee M, Yang H, Seletsky BM, Shaffer CJ et al (2005) Synthetic analogs of the marine natural product hemiasterlin: optimization and discovery of E7974, a novel and potent antitumor agent. Cancer Res 65(9 Supplement):282
Canel C, Moraes RM, Dayan FE, Ferreira D (2000) Podophyllotoxin. Phytochemistry 54(2):115–120
Gordaliza M, Md C, Miguel del Corral J, Feliciano AS (2000) Antitumor properties of podophyllotoxin and related compounds. Curr Pharm Des 6(18):1811–1839
Alday PH, Correia JJ (2009) Macromolecular interaction of halichondrin B analogues eribulin (E7389) and ER-076349 with tubulin by analytical ultracentrifugation. Biochemistry 48(33):7927–7938
Li C-M, Wang Z, Lu Y, Ahn S, Narayanan R, Kearbey JD et al (2011) Biological activity of 4-substituted methoxybenzoyl-aryl-thiazole: an active microtubule inhibitor. Cancer Res 71(1):216–224
Hasanpourghadi M, Karthikeyan C, Pandurangan AK, Looi CY, Trivedi P, Kobayashi K et al (2016) Targeting of tubulin polymerization and induction of mitotic blockage by Methyl 2-(5-fluoro-2-hydroxyphenyl)-1 H-benzo [d] imidazole-5-carboxylate (MBIC) in human cervical cancer HeLa cell. J Exp Clin Cancer Res 35(1):1
Kang G-J, Getahun Z, Muzaffar A, Brossi A, Hamel E (1990) N-acetylcolchinol O-methyl ether and thiocolchicine, potent analogs of colchicine modified in the C ring. Evaluation of the mechanistic basis for their enhanced biological properties. J Biol Chem 265(18):10255–10259
Kuznetsov G, Tendyke K, Towle MJ, Cheng H, Liu D, Rowell C et al (2005) In vitro and in vivo antitumor activities of novel hemiasterlin analog E7974. Cancer Res 65(9 Supplement):809
Krishnamurthy G, Cheng W, Lo M-C, Aulabaugh A, Razinkov V, Ding W et al (2003) Biophysical characterization of the interactions of HTI-286 with tubulin heterodimer and microtubules. Biochemistry 42(46):13484–13495
Rowinsky EK, Calvo E (2006) Novel agents that target tublin and related elements. Semin Oncol 33(4):421–435
Nunes M, Kaplan J, Loganzo F, Zask A, Ayral-Kaloustian S, Greenberger L (2002) Two photoaffinity analogs of HTI-286, a synthetic analog of hemiasterlin, interact with alpha-tubulin. Eur J Cancer 38:S119. Pergamon-Elsevier Science Ltd The Boulevard, Langford Lane, Kidlington, Oxford OX5 1GB, England
Loganzo F, Discafani C, Annable T, Beyer C, Musto S, Hardy C et al (2002) HTI-286, a synthetic analog of the antimicrotubule tripeptide hemiasterlin, potently inhibits growth of cultured tumor cells, overcomes resistance to paclitaxel mediated by various mechanisms, and demonstrates intravenous and oral in vivo efficacy. 93rd annual meeting of American Association for Cancer Research. San Francisco, USA
Jordan MA, Wendell K, Gardiner S, Derry WB, Copp H, Wilson L (1996) Mitotic block induced in HeLa cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res 56(4):816–825
Panda D, DeLuca K, Williams D, Jordan MA, Wilson L (1998) Antiproliferative mechanism of action of cryptophycin-52: kinetic stabilization of microtubule dynamics by high-affinity binding to microtubule ends. Proc Natl Acad Sci 95(16):9313–9318
Gardner MK, Zanic M, Howard J (2013) Microtubule catastrophe and rescue. Curr Opin Cell Biol 25(1):14–22
Gardner M, Zanic M, Gell C, Bormuth V, Howard J (2011) The depolymerizing kinesins Kip3 (kinesin-8) and MCAK (kinesin-13) are catastrophe factors that destabilize microtubules by different mechanisms. Cell 147(5):1092–1103
Howard J, Hyman AA (2003) Dynamics and mechanics of the microtubule plus end. Nature 422(6933):753–758
Maurer SP, Fourniol FJ, Bohner G, Moores CA, Surrey T (2012) EBs recognize a nucleotide-dependent structural cap at growing microtubule ends. Cell 149(2):371–382
Maurer SP, Bieling P, Cope J, Hoenger A, Surrey T (2011) GTPγS microtubules mimic the growing microtubule end structure recognized by end-binding proteins (EBs). Proc Natl Acad Sci 108(10):3988–3993
Elie-Caille C, Severin F, Helenius J, Howard J, Muller DJ, Hyman A (2007) Straight GDP-tubulin protofilaments form in the presence of taxol. Curr Biol 17(20):1765–1770
Brieher WM, Yap AS (2013) Cadherin junctions and their cytoskeleton (s). Curr Opin Cell Biol 25(1):39–46
Bakhoum SF, Compton DA (2012) Kinetochores and disease: keeping microtubule dynamics in check! Curr Opin Cell Biol 24(1):64–70
Komlodi-Pasztor E, Sackett DL, Fojo AT (2012) Inhibitors targeting mitosis: tales of how great drugs against a promising target were brought down by a flawed rationale. Clin Cancer Res 18(1):51–63
Komlodi-Pasztor E, Sackett D, Wilkerson J, Fojo T (2011) Mitosis is not a key target of microtubule agents in patient tumors. Nat Rev Clin Oncol 8(4):244–250
Keen N, Taylor S (2004) Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer 4(12):927–936
Strebhardt K (2010) Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov 9(8):643–660
Melnikova I, Golden J (2004) Targeting protein kinases. Nat Rev Drug Discov 3(12):993–994
Milross CG, Mason KA, Hunter NR, Chung W-K, Peters LJ, Milas L (1996) Relationship of mitotic arrest and apoptosis to antitumor effect of paclitaxel. J Natl Cancer Inst 88(18):1308–1314
Rohena CC, Peng J, Johnson TA, Crews P, Mooberry SL (2013) Chemically diverse microtubule stabilizing agents initiate distinct mitotic defects and dysregulated expression of key mitotic kinases. Biochem Pharmacol 85(8):1104–1114
Gornstein E, Schwarz TL (2014) The paradox of paclitaxel neurotoxicity: mechanisms and unanswered questions. Neuropharmacology 76:175–183
Ferlini C, Cicchillitti L, Raspaglio G, Bartollino S, Cimitan S, Bertucci C et al (2009) Paclitaxel directly binds to Bcl-2 and functionally mimics activity of Nur77. Cancer Res 69(17):6906–6914
Gomez TS, Billadeau DD (2008) T cell activation and the cytoskeleton: you can't have one without the other. Adv Immunol 97:1–64
Giannakakou P, Sackett DL, Ward Y, Webster KR, Blagosklonny MV, Fojo T (2000) p53 is associated with cellular microtubules and is transported to the nucleus by dynein. Nat Cell Biol 2(10):709–717
Darshan MS, Loftus MS, Thadani-Mulero M, Levy BP, Escuin D, Zhou XK et al (2011) Taxane-induced blockade to nuclear accumulation of the androgen receptor predicts clinical responses in metastatic prostate cancer. Cancer Res 71(18):6019–6029
Carbonaro M, O'Brate A, Giannakakou P (2011) Microtubule disruption targets HIF-1α mRNA to cytoplasmic P-bodies for translational repression. J Cell Biol 192(1):83–99
Schwartz EL (2009) Antivascular actions of microtubule-binding drugs. Clin Cancer Res 15(8):2594–2601
Tozer GM, Kanthou C, Baguley BC (2005) Disrupting tumour blood vessels. Nat Rev Cancer 5(6):423–435
Lippert JW (2007) Vascular disrupting agents. Bioorg Med Chem 15(2):605–615
Lu H, Murtagh J, Schwartz EL (2006) The microtubule binding drug laulimalide inhibits vascular endothelial growth factor-induced human endothelial cell migration and is synergistic when combined with docetaxel (taxotere). Mol Pharmacol 69(4):1207–1215
Kamath K, Smiyun G, Wilson L, Jordan MA (2014) Mechanisms of inhibition of endothelial cell migration by taxanes. Cytoskeleton 71(1):46–60
Carbonaro M, Escuin D, O'Brate A, Thadani-Mulero M, Giannakakou P (2012) Microtubules regulate hypoxia-inducible factor-1α protein trafficking and activity implications for taxane therapy. J Biol Chem 287(15):11859–11869
Goode BL, Drubin DG, Barnes G (2000) Functional cooperation between the microtubule and actin cytoskeletons. Curr Opin Cell Biol 12(1):63–71
Mandato CA, Bement WM (2003) Actomyosin transports microtubules and microtubules control actomyosin recruitment during Xenopus oocyte wound healing. Curr Biol 13(13):1096–1105
Kanakkanthara A, Teesdale-Spittle PH, Miller JH (2013) Cytoskeletal alterations that confer resistance to anti-tubulin chemotherapeutics. Anticancer Agents Med Chem 13(1):147–158
Geraldo S, Gordon-Weeks PR (2009) Cytoskeletal dynamics in growth-cone steering. J Cell Sci 122(20):3595–3604
Tanaka E, Ho T, Kirschner MW (1995) The role of microtubule dynamics in growth cone motility and axonal growth. J Cell Biol 128(1):139–155
Gascoigne KE, Taylor SS (2008) Cancer cells display profound intra-and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 14(2):111–122
Strebhardt K, Ullrich A (2006) Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer 6(4):321–330
Slangy A, Lane HA, d'Hérin P, Harper M, Kress M, Niggt EA (1995) Phosphorylation by p34 cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell 83(7):1159–1169
Yang J, Ikezoe T, Nishioka C, Tasaka T, Taniguchi A, Kuwayama Y et al (2007) AZD1152, a novel and selective aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells in vitro and in vivo. Blood 110(6):2034–2040
Fojo AT, Adelberg DE (2010) Microtubule Targeting Agents. In: Drug Management of Prostate Cancer. Springer, New York, pp 179–194
Risinger AL, Peng J, Rohena CC, Aguilar HR, Frantz DE, Mooberry SL (2013) The bat flower: a source of microtubule-destabilizing and-stabilizing compounds with synergistic antiproliferative actions. J Nat Prod 76(10):1923–1929
Goncalves A, Braguer D, Kamath K, Martello L, Briand C, Horwitz S et al (2001) Resistance to Taxol in lung cancer cells associated with increased microtubule dynamics. Proc Natl Acad Sci 98(20):11737–11742
Richards KL, Anders KR, Nogales E, Schwartz K, Downing KH, Botstein D (2000) Structure–function relationships in yeast tubulins. Mol Biol Cell 11(5):1887–1903
Müller D, Schindler P, Towbin H, Wirth U, Voshol H, Hoving S et al (2001) Isotope-tagged cross-linking reagents. A new tool in mass spectrometric protein interaction analysis. Anal Chem 73(9):1927–1934
Martello LA, Verdier-Pinard P, Shen H-J, He L, Torres K, Orr GA et al (2003) Elevated levels of microtubule destabilizing factors in a Taxol-resistant/dependent A549 cell line with an α-tubulin mutation. Cancer Res 63(6):1207–1213
Curmi P, Nogues C, Lachkar S, Carelle N, Gonthier M, Sobel A et al (2000) Overexpression of stathmin in breast carcinomas points out to highly proliferative tumours. Br J Cancer 82(1):142
Nicoletti MI, Valoti G, Giannakakou P, Zhan Z, Kim J-H, Lucchini V et al (2001) Expression of β-Tubulin Isotypes in Human Ovarian Carcinoma Xenografts and in a Sub-Panel of Human Cancer Cell Lines from the NCI-Anticancer Drug Screen Correlation with Sensitivity to Microtubule Active Agents. Clin Cancer Res 7(9):2912–2922
Wilde C, Crowther C, Cowan N (1982) Diverse mechanisms in the generation of human beta-tubulin pseudogenes. Science 217(4559):549–552
Giannakakou P, Sackett DL, Kang Y-K, Zhan Z, Buters JT, Fojo T et al (1997) Paclitaxel-resistant human ovarian cancer cells have mutant β-tubulins that exhibit impaired paclitaxel-driven polymerization. J Biol Chem 272(27):17118–17125
Warnick CT, Dabbas B, Ford CD, Strait KA (2001) Identification of a p53 response element in the promoter region of the hMSH2 gene required for expression in A2780 ovarian cancer cells. J Biol Chem 276(29):27363–27370
Leach FS, Hsieh JT, Molberg K, Saboorian MH, McConnell JD, Sagalowsky AI (2000) Expression of the human mismatch repair gene hMSH2. Cancer 88(10):2333–2341
Giannakakou P, Poy G, Zhan Z, Knutsen T, Blagosklonny MV, Fojo T (2000) Paclitaxel selects for mutant or pseudo-null p53 in drug resistance associated with tubulin mutations in human cancer. Oncogene 19(27):3078–3085
Lavarino C, Pilotti S, Oggionni M, Gatti L, Perego P, Bresciani G et al (2000) p53 gene status and response to platinum/paclitaxel-based chemotherapy in advanced ovarian carcinoma. J Clin Oncol 18(23):3936–3945
Kavallaris M, Tait AS, Walsh BJ, He L, Horwitz SB, Norris MD et al (2001) Multiple microtubule alterations are associated with Vinca alkaloid resistance in human leukemia cells. Cancer Res 61(15):5803–5809
Suzuki R, Newman S, Purohit A, Leese M, Potter B, Reed M (2003) Growth inhibition of multi-drug-resistant breast cancer cells by 2-methoxyoestradiol-bis-sulphamate and 2-ethyloestradiol-bis-sulphamate. J Steroid Biochem Mol Biol 84(2):269–278
Yen TJ, Gay DA, Pachter JS, Cleveland D (1988) Autoregulated changes in stability of polyribosome-bound beta-tubulin mRNAs are specified by the first 13 translated nucleotides. Mol Cell Biol 8(3):1224–1235
Hari M, Yang H, Zeng C, Canizales M, Cabral F (2003) Expression of class III β-tubulin reduces microtubule assembly and confers resistance to paclitaxel. Cell Motil Cytoskeleton 56(1):45–56
Hasegawa S, Miyoshi Y, Egawa C, Ishitobi M, Taguchi T, Tamaki Y et al (2003) Prediction of response to docetaxel by quantitative analysis of class I and III β-tubulin isotype mRNA expression in human breast cancers. Clin Cancer Res 9(8):2992–2997
Joe PA, Banerjee A, Ludueña RF (2008) The roles of Cys124 and Ser239 in the functional properties of human βIII tubulin. Cell Motil Cytoskeleton 65(6):476–486
Gan PP, Pasquier E, Kavallaris M (2007) Class III β-Tubulin Mediates Sensitivity to Chemotherapeutic Drugs in Non–Small Cell Lung Cancer. Cancer Res 67(19):9356–9363
Stengel C, Newman S, Leese M, Potter B, Reed M, Purohit A (2010) Class III β-tubulin expression and in vitro resistance to microtubule targeting agents. Br J Cancer 102(2):316–324
Löwe J, Li H, Downing K, Nogales E (2001) Refined structure of αβ-tubulin at 3.5 Å resolution. J Mol Biol 313(5):1045–1057
Ferlini C, Raspaglio G, Mozzetti S, Cicchillitti L, Filippetti F, Gallo D et al (2005) The seco-taxane IDN5390 is able to target class III β-tubulin and to overcome paclitaxel resistance. Cancer Res 65(6):2397–2405
Kamath K, Wilson L, Cabral F, Jordan MA (2005) βIII-tubulin induces paclitaxel resistance in association with reduced effects on microtubule dynamic instability. J Biol Chem 280(13):12902–12907
Sève P, Isaac S, Trédan O, Souquet P-J, Pachéco Y, Pérol M et al (2005) Expression of Class III β-Tubulin Is Predictive of Patient Outcome in Patients with Non–Small Cell Lung Cancer Receiving Vinorelbine-Based Chemotherapy. Clin Cancer Res 11(15):5481–5486
Leandro-García LJ, Leskelä S, Inglada-Pérez L, Landa I, de Cubas AA, Maliszewska A et al (2012) Hematologic β-tubulin VI isoform exhibits genetic variability that influences paclitaxel toxicity. Cancer Res 72(18):4744–4752
Huzil JT, Ludueña RF, Tuszynski J (2006) Comparative modelling of human β tubulin isotypes and implications for drug binding. Nanotechnology 17(4):S90
Huzil JT, Chen K, Kurgan L, Tuszynski JA (2007) The roles of [beta]2-tubulin mutations and isotype expression in acquired drug resistance. Cancer Informat 3:159–181
Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM (2006) Targeting multidrug resistance in cancer. Nat Rev Drug Discov 5(3):219–234
Leonard GD, Fojo T, Bates SE (2003) The role of ABC transporters in clinical practice. Oncologist 8(5):411–424
Fojo AT, Menefee M (2005) editors. Microtubule targeting agents: basic mechanisms of multidrug resistance (MDR). Semin Oncol 32(6 Suppl 7):S3–S8
Trock BJ, Leonessa F, Clarke R (1997) Multidrug resistance in breast cancer: a meta-analysis of MDR1/gp170 expression and its possible functional significance. J Natl Cancer Inst 89(13):917–931
Noguchi S (2006) Predictive factors for response to docetaxel in human breast cancers. Cancer Sci 97(9):813–820
Bode CJ, Gupta ML, Reiff EA, Suprenant KA, Georg GI, Himes RH (2002) Epothilone and paclitaxel: unexpected differences in promoting the assembly and stabilization of yeast microtubules. Biochemistry 41(12):3870–3874
Agrawal M, Abraham J, Balis FM, Edgerly M, Stein WD, Bates S et al (2003) Increased 99mTc-sestamibi accumulation in normal liver and drug-resistant tumors after the administration of the glycoprotein inhibitor, XR9576. Clin Cancer Res 9(2):650–656
Loganzo F, Hari M, Annable T, Tan X, Morilla DB, Musto S et al (2004) Cells resistant to HTI-286 do not overexpress P-glycoprotein but have reduced drug accumulation and a point mutation in α-tubulin. Mol Cancer Ther 3(10):1319–1327
Pusztai L, Wagner P, Ibrahim N, Rivera E, Theriault R, Booser D et al (2005) Phase II study of tariquidar, a selective P-glycoprotein inhibitor, in patients with chemotherapy-resistant, advanced breast carcinoma. Cancer 104(4):682–691
Weingarten MD, Lockwood AH, Hwo S-Y, Kirschner MW (1975) A protein factor essential for microtubule assembly. Proc Natl Acad Sci 72(5):1858–1862
Kar S, Fan J, Smith MJ, Goedert M, Amos LA (2003) Repeat motifs of tau bind to the insides of microtubules in the absence of taxol. EMBO J 22(1):70–77
Andre F, Hatzis C, Anderson K, Sotiriou C, Mazouni C, Mejia J et al (2007) Microtubule-associated protein-tau is a bifunctional predictor of endocrine sensitivity and chemotherapy resistance in estrogen receptor–positive breast cancer. Clin Cancer Res 13(7):2061–2067
Pusztai L, Jeong J-H, Gong Y, Ross JS, Kim C, Paik S et al (2009) Evaluation of microtubule-associated protein-Tau expression as a prognostic and predictive marker in the NSABP-B 28 randomized clinical trial. J Clin Oncol 27(26):4287–4292
Smoter M, Bodnar L, Duchnowska R, Stec R, Grala B, Szczylik C (2011) The role of Tau protein in resistance to paclitaxel. Cancer Chemother Pharmacol 68(3):553–557
Rouzier R, Rajan R, Wagner P, Hess KR, Gold DL, Stec J et al (2005) Microtubule-associated protein tau: a marker of paclitaxel sensitivity in breast cancer. Proc Natl Acad Sci U S A 102(23):8315–8320
Gschwind A, Fischer OM, Ullrich A (2004) The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat Rev Cancer 4(5):361–370
Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2(2):127–137
Graus-Porta D, Beerli RR, Daly JM, Hynes NE (1997) ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J 16(7):1647–1655
Ross JS, Slodkowska EA, Symmans WF, Pusztai L, Ravdin PM, Hortobagyi GN (2009) The HER-2 receptor and breast cancer: ten years of targeted anti–HER-2 therapy and personalized medicine. Oncologist 14(4):320–368
Pusztai L (2007) Markers predicting clinical benefit in breast cancer from microtubule-targeting agents. Ann Oncol 18(suppl 12):xii15–xii20
Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM (2003) P-glycoprotein: from genomics to mechanism. Oncogene 22(47):7468–7485
Geney R, Ungureanu IM, Li D, Ojima I (2002) Overcoming multidrug resistance in taxane chemotherapy. Clin Chem Lab Med 40(9):918–925
Galmarini C, Kamath K, Vanier-Viornery A, Hervieu V, Peiller E, Falette N et al (2003) Drug resistance associated with loss of p53 involves extensive alterations in microtubule composition and dynamics. Br J Cancer 88(11):1793–1799
Kavallaris M, Burkhart C, Horwitz S (1999) Antisense oligonucleotides to class III β-tubulin sensitize drug-resistant cells to Taxol. Br J Cancer 80(7):1020
Gordaliza M (2007) Natural products as leads to anticancer drugs. Clin Transl Oncol 9(12):767–776
Newman DJ, Cragg GM, Snader KM (2003) Natural products as sources of new drugs over the period 1981-2002. J Nat Prod 66(7):1022–1037
Clardy J, Walsh C (2004) Lessons from natural molecules. Nature 432(7019):829–837
Glaser KB (2007) HDAC inhibitors: clinical update and mechanism-based potential. Biochem Pharmacol 74(5):659–671
Brito DA, Yang Z, Rieder CL (2008) Microtubules do not promote mitotic slippage when the spindle assembly checkpoint cannot be satisfied. J Cell Biol 182(4):623–629
Ganguly A, Yang H, Cabral F (2010) Paclitaxel-dependent cell lines reveal a novel drug activity. Mol Cancer Ther 9(11):2914–2923
Mukhtar E, Adhami VM, Mukhtar H (2014) Targeting microtubules by natural agents for cancer therapy. Mol Cancer Ther 13(2):275–284
Hood KA, West LM, Rouwé B, Northcote PT, Berridge MV, Miller JH (2002) Peloruside A, a novel antimitotic agent with paclitaxel-like microtubule-stabilizing activity. Cancer Res 62(12):3356–3360
Huang GS, Lopez-Barcons L, Freeze BS, Smith AB, Goldberg GL, Horwitz SB et al (2006) Potentiation of taxol efficacy by discodermolide in ovarian carcinoma xenograft-bearing mice. Clin Cancer Res 12(1):298–304
Isbrucker RA, Cummins J, Pomponi SA, Longley RE, Wright AE (2003) Tubulin polymerizing activity of dictyostatin-1, a polyketide of marine sponge origin. Biochem Pharmacol 66(1):75–82
Vijayakumar S, Menakha M (2015) Pharmaceutical applications of cyanobacteria—A review. JACME 5(1):15–23
Gordaliza M, Garcıa P, Del Corral JM, Castro M, Gómez-Zurita M (2004) Podophyllotoxin: distribution, sources, applications and new cytotoxic derivatives. Toxicon 44(4):441–459
Verdier-Pinard P, Lai J-Y, Yoo H-D, Yu J, Marquez B, Nagle DG et al (1998) Structure-activity analysis of the interaction of curacin A, the potent colchicine site antimitotic agent, with tubulin and effects of analogs on the growth of MCF-7 breast cancer cells. Mol Pharmacol 53(1):62–76
Lu Y, Li C-M, Wang Z, Ross CR, Chen J, Dalton JT et al (2009) Discovery of 4-substituted methoxybenzoyl-aryl-thiazole as Novel Anticancer Agents: Synthesis, biological evaluation, and structure − activity relationships. J Med Chem 52(6):1701–1711
Hadaschik BA, Ettinger S, Sowery RD, Zoubeidi A, Andersen RJ, Roberge M et al (2008) Targeting prostate cancer with HTI-286, a synthetic analog of the marine sponge product hemiasterlin. Int J Cancer 122(10):2368–2376
Salmon HW, Siemann DW (2006) Effect of the second-generation vascular disrupting agent OXi4503 on tumor vascularity. Clin Cancer Res 12(13):4090–4094
Eskens F, Tresca P, Tosi D, Van Doorn L, Fontaine H, Van der Gaast A et al (2014) A phase I pharmacokinetic study of the vascular disrupting agent ombrabulin (AVE8062) and docetaxel in advanced solid tumours. Br J Cancer 110(9):2170–2177
Rudek MA, Dasari A, Laheru D, He P, Jin R, Walker R et al (2016) Phase 1 Study of ABT-751 in Combination With CAPIRI (Capecitabine and Irinotecan) and Bevacizumab in Patients With Advanced Colorectal Cancer. J Clin Pharmacol 56(8):966–973
Ma T, Fuld AD, Rigas JR, Hagey AE, Gordon GB, Dmitrovsky E et al (2012) A phase I trial and in vitro studies combining ABT-751 with carboplatin in previously treated non-small cell lung cancer patients. Chemotherapy 58(4):321–329
Traina T, Hudis C, Seidman A, Gajria D, Gonzalez J, Anthony S et al (2012) Abstract P6-11-10: IBL2001: Phase I/II study of a novel dose-dense schedule of oral indibulin for the treatment of metastastic breast cancer. Cancer Res 72(24 Supplement):P6-11-0-P6--0
Nahrwold M, Weiß C, Bogner T, Mertink F, Conradi J, Sammet B et al (2013) Conjugates of modified cryptophycins and RGD-peptides enter target cells by endocytosis. J Med Chem 56(5):1853–1864
Acknowledgement
The first author (Mohadeseh Hasanpourghadi) thanks University of Malaya, Kuala Lumpur, Malaysia, for providing financial support through Postgraduate grant (PG) (PG048-2015A). This study was also supported by a High-Impact Research grant from the University of Malaya (E00002–20001).
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Hasanpourghadi, M., Pandurangan, A.K., Mustafa, M.R. (2017). Microtubule Targeting Agents in Cancer Therapy: Elucidating the Underlying Molecular Mechanisms. In: Farooqi, A., Ismail, M. (eds) Molecular Oncology: Underlying Mechanisms and Translational Advancements. Springer, Cham. https://doi.org/10.1007/978-3-319-53082-6_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-53082-6_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-53081-9
Online ISBN: 978-3-319-53082-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)