
Abstract
Poor-risk (PR) cytogenetic/molecular abnormalities generally direct pediatric patients with acute myeloid leukemia (AML) to allogeneic hematopoietic stem cell transplant (HSCT). We assessed the predictive value of cytogenetic risk classification at diagnosis with respect to post-HSCT outcomes in pediatric patients. Patients younger than 18 years at the time of their first allogeneic HSCT for AML in CR1 between 2005 and 2022 who were reported to the European Society for Blood and Marrow Transplantation registry were subgrouped into four categories. Of the 845 pediatric patients included in this study, 36% had an 11q23 abnormality, 24% had monosomy 7/del7q or monosomy 5/del5q, 24% had a complex or monosomal karyotype, and 16% had other PR cytogenetic abnormalities. In a multivariable model, 11q23 (hazard ratio [HR] = 0.66, P = 0.03) and other PR cytogenetic abnormalities (HR = 0.55, P = 0.02) were associated with significantly better overall survival when compared with monosomy 7/del7q or monosomy 5/del5q. Patients with other PR cytogenetic abnormalities had a lower risk of disease relapse after HSCT (HR = 0.49, P = 0.01) and, hence, better leukemia-free survival (HR = 0.55, P = 0.01). Therefore, we conclude that PR cytogenetic abnormalities at diagnosis predict overall survival after HSCT for AML in pediatric patients.
Similar content being viewed by others

Introduction
Advances in risk stratification, therapy intensification, and supportive care have all contributed to improving the outcomes for children with acute myeloid leukemia (AML) [1]. Genetic/molecular features such as fusion genes and molecular aberrations of the leukemic cells, as well as the response to induction therapy, play a major role in determining which patients are at highest risk of relapse with conventional chemotherapy [2, 3]. Although the overall survival (OS) of children with AML has improved over the past few decades, only 70% of these children become long-term survivors [1]. For children with high-risk AML (HR-AML), defined by a combination of poor-risk (PR) cytogenetic/molecular abnormalities or an inadequate response to chemotherapy as assessed by measurable residual disease (MRD), outcomes are inferior, with an OS of less than 50% [4]. Although an allogeneic hematopoietic stem cell transplant (HSCT) is generally recommended for patients with HR-AML with PR cytogenetics/molecular abnormalities in first complete remission (CR1) [1], it is unknown whether these cytogenetic and molecular features retain their prognostic value after HSCT. The success of an allogeneic HSCT largely depends on the graft-versus-leukemia (GVL) effect mediated by the alloreactive immune system derived from the graft, which is distinct from the cytolytic and cytostatic effects of antileukemic drugs. Additionally, AML with PR cytogenetics is a heterogeneous category that includes several distinct cytogenetic abnormalities. As a group, patients with PR cytogenetics may not benefit from HSCT or, alternatively, specific subgroups may benefit and others may not [5,6,7,8]. In this study, we evaluated whether the specific cytogenetic abnormalities present at the diagnosis of AML were predictive of post-HSCT outcomes in pediatric patients with HR-AML.
Methods
Study design and patients
This retrospective study was conducted using the data reported to the Pediatric Diseases Working Party (PDWP) of the European Society for Blood and Marrow Transplantation (EBMT) registry by 150 participating centers in 38 countries. The EBMT is a nonprofit medical and scientific organization representing more than 600 transplant centers, mainly located in Europe. Centers commit to reporting all consecutive HSCTs and follow-up data once a year. Data are entered, managed, and maintained in a central database and are validated by verification of the computer printout of the entered data. All patients gave informed consent to the use of their personal information for research purposes. This study was approved by the PDWP of the EBMT institutional review board and was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. Eligible patients were younger than 18 years at the time of HSCT for AML, had received their first allogeneic HSCT in CR1 between January 1, 2005, and December 31, 2022, and had a recorded cytogenetic assessment at diagnosis. Cytogenetic abnormalities considered as PR were monosomy 7, del(7q), monosomy 5, del(5q), 11q23 abnormalities excluding t(9;11), t(8;16), 12p13 abnormalities, del(12p), t(9;22), t(6;9), inv(3), t(3;5), t(16;21), and 11p15 abnormalities [5, 9,10,11,12,13]. Patients characterized by t(9;11), t(8;21), t(15;17), inv(16), or t(16;16) were excluded from this analysis as they were considered to have intermediate-risk or favorable-risk cytogenetic abnormalities.
Assignment to risk groups
Patients were assigned to one of the following subgroups according to their cytogenetic assessment at diagnosis: (a) patients with monosomy 7/del7q or monosomy 5/del5q, irrespective of the presence of any other cytogenetic abnormality; (b) patients with 11q23 abnormalities; (c) patients with a complex or monosomal karyotype; and (d) patients with other PR cytogenetic abnormalities. A complex karyotype was defined as one with three or more structural abnormalities. A monosomal karyotype was defined as a monosomy with one or more structural abnormalities or two or more autosomal monosomies. The subgroup for patients with other PR cytogenetic abnormalities included those with t(6;9), t(3;5), t(9;22), t(8;16), inv(3) or t(3;3), t(16;21), abn(11p15), or del(12p) or abn(12p13).
Statistical analysis
Qualitative variables are reported as frequencies and percentages. Quantitative variables are reported as the median, quartile 1 and quartile 3, or minimum and maximum values. The difference in the distribution of qualitative or quantitative variables among the cytogenetic groups was evaluated with chi-square tests or Fisher exact tests and with Kruskal–Wallis tests, respectively.
The primary endpoint of the study was OS, defined as the time from HSCT to death. Secondary endpoints were the leukemia-free survival (LFS), defined as the time from HSCT to relapse or death, whichever occurred first, and the relapse incidence and non-relapse mortality (NRM), defined, respectively, as the time from HSCT to relapse and the time from HSCT to death without relapse. Other secondary endpoints were the incidence of grade II–IV and grade III or IV acute graft-versus-host disease (GVHD) and the incidence of chronic GVHD. Finally, the GVHD-free relapse-free survival (GRFS) was estimated, defined as the time from HSCT to the first occurrence of grade III or IV acute GVHD, extensive chronic GVHD, relapse, or death. All of the outcomes were censored at last follow-up. OS, LFS, and GRFS curves were estimated by the Kaplan–Meier method. The cumulative incidence function was used to estimate outcomes with competing events. NRM and relapse were mutually competing events. Relapse and death were competing events for acute and chronic GVHD. Median follow-up was estimated using the reverse Kaplan–Meier method. Because of the shorter median follow-up time in one group, outcomes were censored at 3 years. Multivariable analyses were performed by fitting Cox regression models, and they included clinically relevant variables, including cytogenetic subgroup, age at HSCT, donor type, whether the HSCT was from a female donor to a male recipient, patient/donor CMV status, time from AML diagnosis to HSCT, and year of HSCT. Center effect was taken into account as frailty. Point estimates of the outcomes and hazard ratio (HR) are given with their 95% confidence intervals (CIs). Two-sided P values less than 0.05 were considered to indicate statistical significance. Analyses were performed using R software version 4.0.2.
Results
Patient characteristics
We included 845 pediatric patients from 150 participating centers in this study. Three hundred and sixty (42.7%) of the HSCT recipients were female. The median age at HSCT was 8.6 years (inter-quartile range [IQR]: 2.5–13.8 years), and the median follow-up after HSCT was 4.1 years (95% confidence interval [CI]: 3.7–4.5). Three hundred and four patients (36.0%) had 11q23 abnormalities, 199 (23.6%) had monosomy 7/del7q or monosomy 5/del5q, 207 (24.5%) had a complex or monosomal karyotype, and 135 (16.0%) had other PR cytogenetic abnormalities. Only 62 patients (7.3%) had secondary AML. Table 1 and the Supplemental Tables present the clinical characteristics of these 845 patients, stratified by the different subgroups. Pre-HSCT MRD data were available for 413 patients (48.9% of the total), 352 (85.2%) of whom were MRD negative at the time of HSCT. Patients received grafts from a matched related donor (n = 222, 26.3%), a mismatched related donor (n = 102, 12.1%), an unrelated donor (n = 385, 45.6%), or an unrelated cord blood donor (n = 136, 16.1%). Four hundred and sixty patients (54.4%) received bone marrow, 249 (29.5%) received peripheral blood–derived stem cells, and the rest (n = 136, 16.1%) received cord blood as the stem cell source. Myeloablative conditioning was used in 820 patients (97.0%). Any regimens containing intravenous Busulfan greater than 9.6 mg/kg or oral Busulfan greater than 12 mg/kg, total body irradiation greater than 8 Gy, or any Treosulfan containing regimens were considered as myeloablative.
Survival and GVHD
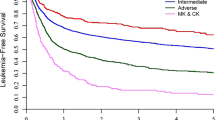
The 2-year OS and LFS for the entire cohort were 75.3% (95% CI: 72–78.3%) and 67.9% (95% CI: 64.4–71.1%), respectively (Table 2). The cumulative incidence of relapse was 23.5% (95% CI: 20.5–26.7%) and the incidence of NRM was 8.6% (95% CI: 6.7–10.7%) at 2 years after HSCT. In this cohort, 9.5% of the patients (95% CI: 7.6–11.7%) experienced grade III or IV acute GVHD within the first 100 days after HSCT and 6.7% (95% CI: 5–8.7%) experienced extensive chronic GVHD within the first 2 years after HSCT. The 2-year GVHD-free relapse-free survival (GRFS) was 57.3% (95% CI: 53.6–60.8%). Kaplan–Meier survival curves describing the OS and LFS and cumulative incidence curves showing the relapse incidence and NRM are shown in Fig. 1.

Kaplan–Meier curves showing leukemia-free survival (LFS, top left) and overall survival (OS, top right), and cumulative incidence curves showing relapse incidence (RI, bottom left) and non-relapse mortality (NRM, bottom right), with stratification by cytogenetic subgroup. CK/MK: complex or monosomal karyotype.
Effect of cytogenetic risk on post-transplant outcomes
In a multivariable model (Table 3), 11q23 (HR = 0.66 [95% CI: 0.44–0.97], P = 0.03) and other PR cytogenetic abnormalities (HR = 0.55 [95% CI: 0.33–0.91], P = 0.02) were associated with significantly better OS when compared with monosomy 7/del7q and monosomy 5/del5q. OS was not significantly different for the complex/monosomal karyotype group (HR = 0.98 [95% CI: 0.66–1.46], P = 0.94). There were no significant differences in NRM among the four subgroups (HR = 0.86 [95% CI: 0.45–1.66], P = 0.66; HR = 0.96 [95% CI: 0.48–1.94], P = 0.92; and HR = 0.73 [95% CI: 0.33–1.65], P = 0.46, for the 11q23, complex/monosomal, and other PR cytogenetics subgroups, respectively) when compared with the monosomy 7/del7q or monosomy 5/del5q subgroup. Other PR cytogenetic abnormalities were associated with a lower risk of disease relapse after HSCT (HR = 0.49 [95% CI: 0.28–0.86], P = 0.01), whereas the risk of disease relapse was not significantly different for the other groups when compared with the monosomy 7/del7q or monosomy 5/del5q subgroup (HR = 0.78 [95% CI: 0.52–1.16], P = 0.22, and HR = 1.0 [95% CI: 0.65–1.53], P = 1.0, for the 11q23 abnormality group and the complex/monosomal karyotype group, respectively). Accordingly, LFS was significantly better for the other PR cytogenetic risk group (HR = 0.55 [95% CI: 0.35–0.87], P = 0.01) but not significantly different for the two other groups when compared with the monosomy 7/del7q or monosomy 5/del5q subgroup (HR = 0.79 [95% CI: 0.56–1.12], P = 0.19, and HR = 0.97 [95% CI: 0.67–1.39], P = 0.86, for the 11q23 and complex/monosomal subgroups, respectively).
Receiving an HSCT from an unrelated donor, as opposed to a matched related donor, was associated with lower relapse incidence (HR = 0.66 [95% CI: 0.45–0.98], P = 0.04), whereas this was not significantly different for the two other groups (HR = 0.86 [95% CI: 0.51–1.46], P = 0.58, and HR = 1.10 [95% CI: 0.68–1.79], P = 0.7, for mismatched related and cord blood recipients, respectively). Lastly, older age at HSCT (12–18 years) was associated with the highest risk of NRM (HR = 2.23 [95% CI: 1.16–4.30], P = 0.02) and with worse OS (HR = 1.63 [95% CI: 1.14–2.35], P < 0.01) as compared to that of younger patients (aged 4–12 years). NRM and OS were not significantly different when very young patients (aged 0–4 years) were compared with the group aged 4–12 years (HR = 1.46 [95% CI: 0.71–3.00], P = 0.30, and HR = 1.10 [95% CI: 0.73–1.65], P = 0.66, respectively).
Discussion
Allogeneic HSCT is the cornerstone of therapy for pediatric patients with AML who have adverse cytogenetic abnormalities, and it offers the best chance of long-term survival. However, the current literature lacks detail on the impact of cytogenetic abnormalities on post-HSCT outcomes in this population. Cytogenetic abnormalities remain the strongest predictors of outcomes for patients with newly diagnosed AML, but the literature on whether these abnormalities predict survival and relapse after HSCT is contradictory. Whereas some studies suggest that cytogenetic features continue to predict outcomes for adult patients with AML after HSCT [14,15,16,17,18], others have inferred that these abnormalities have no prognostic significance after HSCT [19, 20]. In this large cohort of pediatric HSCT recipients, we found that cytogenetic abnormalities remained highly predictive of outcomes even after HSCT for AML in CR1. Whereas monosomy 7/del7q or monosomy 5/del5q conferred a poor prognosis even after HSCT, 11q23 abnormalities and other PR cytogenetic abnormalities predicted a more favorable outcome. Patients with other PR cytogenetic abnormalities had a decreased incidence of relapse, leading to improved survival after HSCT.
Even though the relapse incidence was not significantly different to that of the monosomy 7/del7q or monosomy 5/del5q and complex or monosomal karyotype risk groups for patients with 11q23 abnormalities, OS was better for the latter group. This finding suggests that AML with 11q23 abnormalities is a heterogeneous disease, with some fusions and abnormalities being associated with a more favorable prognosis than others, such that the overall effect appears variable [5, 8]. Although HSCT continues to be recommended for patients with HR-AML and is often attempted for patients whose disease does not respond to traditional chemotherapy, the GVL effect of allogeneic HSCT might be insufficient to overcome the chemoresistance of the myeloblasts. Given the risk of NRM, short-term morbidity, and late effects, the role of HSCT in CR1 for some children with HR-AML requires further investigation [5,6,7,8].
We noted a statistically significant protective effect of transplants from unrelated donors, as compared with those from matched related donors, with respect to reducing the relapse incidence. The hazard ratio for the relapse incidence for recipients of transplants from mismatched related donors was favorable, but did not meet statistical significance. Advances in high-resolution human leukocyte antigen (HLA) typing, GVHD prophylaxis, and improved supportive care have expanded the use of alternative donors. Recent studies have shown comparable outcomes after allogeneic HSCT from matched unrelated and HLA-identical sibling donors [21,22,23,24]. Haploidentical-donor HSCT approaches using post-transplant cyclophosphamide and selective alpha/beta T-cell depletion of the graft have reportedly resulted in OS and LFS on par with those after HLA-matched transplants in retrospective studies, with haploidentical approaches being associated with lower rates of severe acute and chronic GVHD [25,26,27,28,29]. Prospective randomized evaluation of haploidentical donor HSCT to MUD HSCT is ongoing in a Children’s Oncology Group clinical trial (ASCT2031, NCT05457556). Our data suggest that grafts from alternative donors exert a greater immunotherapeutic effect than those from matched related donors as they provide an opportunity to leverage the alloreactivity after HSCT to target refractory leukemias. This observation should however be confirmed in larger prospective studies. Relapse remains the most common cause of treatment failure in this population, and the benefit of the GVL effect must be optimized to improve outcomes.
Limitations
This study had several limitations. First, it was a retrospective analysis and some data were missing, with information on pre- and post-HSCT chemotherapy being unavailable. Also missing was information on specific gene aberrations, such as those in FLT3, NPM1, CEBPA, and TP53, which are clearly prognostically heterogeneous [30,31,32]. By adjusting for the time from diagnosis to HSCT, we tried to avoid the potential bias of selecting patients who had undergone fewer induction cycles than others. We further included patients based on their cytogenetic abnormalities, irrespective of whether they had de novo, therapy-related, or secondary AML. Given that they historically have had a poor prognosis [33], patients with therapy-related, or secondary AML are often excluded from studies of this type. Monosomy 7/del7q or monosomy 5/del5q subgroup was relatively enriched for these patients with secondary AML and might have driven the poor outcomes in this subgroup. Third, we focused exclusively on patients in CR1 who proceeded to consolidative HSCT. Patients with PR cytogenetics with an early relapse or toxicity that precluded HSCT were excluded. Fourth, pre-HSCT MRD data were available for only half of the patients since this data is not collected mandatorily in the registry. Poor response to induction chemotherapy could have skewed the results of this study in a way unrelated to the cytogenetic risk category. Interestingly, however, despite the fact that many more patients in the other PR cytogenetic abnormality subgroup (almost 24%, as compared with just 11%–17% in the 11q23 abnormality, monosomy 7/del7q or monosomy 5/del5q, and complex or monosomal karyotype risk groups) were not in molecular remission at the time of HSCT, patients with other PR cytogenetic abnormalities had the best outcomes. Lastly, as most patients had received a myeloablative conditioning regimen, we could not explore the effect of different conditioning intensities on outcomes. However, a retrospective study has shown that relapse rates are not higher after reduced-intensity regimens, as compared with myeloablative regimens, and that myeloablative regimens are not associated with higher transplant-related mortality when compared with reduced-intensity regimens in children with AML [34].
Conclusions
PR cytogenetic abnormalities at diagnosis remain predictive of OS after HSCT for AML in pediatric patients. Monosomy 7/del7q or monosomy 5/del5q, which overlapped with the therapy-related or secondary AML cohort, confer a poor prognosis even after HSCT, whereas 11q23 abnormalities and other PR cytogenetic abnormalities predict a more favorable outcome after HSCT. Our data also suggest that an HSCT from an unrelated donor offers greater protection against relapse than an HSCT from a matched related donor, but this observation requires further validation.
Data availability
Deidentified dataset maybe shared upon request to the corresponding author.
References
Rubnitz JE, Kaspers GJL. How I treat pediatric acute myeloid leukemia. Blood. 2021;138:1009–18.
Loken MR, Alonzo TA, Pardo L, Gerbing RB, Raimondi SC, Hirsch BA, et al. Residual disease detected by multidimensional flow cytometry signifies high relapse risk in patients with de novo acute myeloid leukemia: a report from Children’s Oncology Group. Blood. 2012;120:1581–8.
Pui CH, Carroll WL, Meshinchi S, Arceci RJ. Biology, risk stratification, and therapy of pediatric acute leukemias: an update. J Clin Oncol. 2011;29:551–65.
Rubnitz JE. Current management of childhood acute myeloid leukemia. Paediatr Drugs. 2017;19:1–10.
Balgobind BV, Raimondi SC, Harbott J, Zimmermann M, Alonzo TA, Auvrignon A, et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood. 2009;114:2489–96.
Horan JT, Alonzo TA, Lyman GH, Gerbing RB, Lange BJ, Ravindranath Y, et al. Impact of disease risk on efficacy of matched related bone marrow transplantation for pediatric acute myeloid leukemia: the Children’s Oncology Group. J Clin Oncol. 2008;26:5797–801.
Pollard JA, Guest E, Alonzo TA, Gerbing RB, Loken MR, Brodersen LE, et al. Gemtuzumab Ozogamicin improves event-free survival and reduces relapse in pediatric KMT2A-rearranged AML: results from the Phase III children’s oncology group trial AAML0531. J Clin Oncol. 2021;39:3149–60.
van Weelderen RE, Klein K, Harrison CJ, Jiang Y, Abrahamsson J, Arad-Cohen N, et al. Measurable residual disease and fusion partner independently predict survival and relapse risk in childhood KMT2A-rearranged acute myeloid leukemia: a study by the international Berlin-Frankfurt-Munster Study Group. J Clin Oncol. 2023;41:2963–74.
Coenen EA, Zwaan CM, Reinhardt D, Harrison CJ, Haas OA, de Haas V, et al. Pediatric acute myeloid leukemia with t(8;16)(p11;p13), a distinct clinical and biological entity: a collaborative study by the International-Berlin-Frankfurt-Munster AML-study group. Blood. 2013;122:2704–13.
Harrison CJ, Hills RK, Moorman AV, Grimwade DJ, Hann I, Webb DK, et al. Cytogenetics of childhood acute myeloid leukemia: United Kingdom Medical Research Council Treatment trials AML 10 and 12. J Clin Oncol. 2010;28:2674–81.
Marceau-Renaut A, Duployez N, Ducourneau B, Labopin M, Petit A, Rousseau A, et al. Molecular profiling defines distinct prognostic subgroups in childhood AML: a report from the french ELAM02 Study Group. Hemasphere. 2018;2:e31.
Masetti R, Pigazzi M, Togni M, Astolfi A, Indio V, Manara E, et al. CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood. 2013;121:3469–72.
von Neuhoff C, Reinhardt D, Sander A, Zimmermann M, Bradtke J, Betts DR, et al. Prognostic impact of specific chromosomal aberrations in a large group of pediatric patients with acute myeloid leukemia treated uniformly according to trial AML-BFM 98. J Clin Oncol. 2010;28:2682–9.
Armand P, Kim HT, DeAngelo DJ, Ho VT, Cutler CS, Stone RM, et al. Impact of cytogenetics on outcome of de novo and therapy-related AML and MDS after allogeneic transplantation. Biol Blood Marrow Transplant. 2007;13:655–64.
Armand P, Kim HT, Zhang MJ, Perez WS, Dal Cin PS, Klumpp TR, et al. Classifying cytogenetics in patients with acute myelogenous leukemia in complete remission undergoing allogeneic transplantation: a Center for International Blood and Marrow Transplant Research study. Biol Blood Marrow Transplant. 2012;18:280–8.
Chevallier P, Labopin M, Milpied N, Cornelissen JJ, Blaise D, Petersen E, et al. Impact of cytogenetics risk on outcome after reduced intensity conditioning allo-SCT from an HLA-identical sibling for patients with AML in first CR: a report from the acute leukemia working party of EBMT. Bone Marrow Transplant. 2012;47:1442–7.
Ferrant A, Labopin M, Frassoni F, Prentice HG, Cahn JY, Blaise D, et al. Karyotype in acute myeloblastic leukemia: prognostic significance for bone marrow transplantation in first remission: a European Group for Blood and Marrow Transplantation study. Acute Leukemia Working Party of the European Group for Blood and Marrow Transplantation (EBMT). Blood. 1997;90:2931–8.
Ogawa H, Ikegame K, Kawakami M, Takahashi S, Sakamaki H, Karasuno T, et al. Impact of cytogenetics on outcome of stem cell transplantation for acute myeloid leukemia in first remission: a large-scale retrospective analysis of data from the Japan Society for Hematopoietic Cell Transplantation. Int J Hematol. 2004;79:495–500.
Alloin AL, Leverger G, Dalle JH, Galambrun C, Bertrand Y, Baruchel A, et al. Cytogenetics and outcome of allogeneic transplantation in first remission of acute myeloid leukemia: the French pediatric experience. Bone Marrow Transplant. 2017;52:516–21.
Chalandon Y, Barnett MJ, Horsman DE, Conneally EA, Nantel SH, Nevill TJ, et al. Influence of cytogenetic abnormalities on outcome after allogeneic bone marrow transplantation for acute myeloid leukemia in first complete remission. Biol Blood Marrow Transplant. 2002;8:435–43.
Gupta V, Tallman MS, He W, Logan BR, Copelan E, Gale RP, et al. Comparable survival after HLA-well-matched unrelated or matched sibling donor transplantation for acute myeloid leukemia in first remission with unfavorable cytogenetics at diagnosis. Blood. 2010;116:1839–48.
Schetelig J, Bornhauser M, Schmid C, Hertenstein B, Schwerdtfeger R, Martin H, et al. Matched unrelated or matched sibling donors result in comparable survival after allogeneic stem-cell transplantation in elderly patients with acute myeloid leukemia: a report from the cooperative German Transplant Study Group. J Clin Oncol. 2008;26:5183–91.
Walter RB, Pagel JM, Gooley TA, Petersdorf EW, Sorror ML, Woolfrey AE, et al. Comparison of matched unrelated and matched related donor myeloablative hematopoietic cell transplantation for adults with acute myeloid leukemia in first remission. Leukemia. 2010;24:1276–82.
Yanada M, Kurosawa S, Yamaguchi T, Uchida N, Miyawaki S, Kanamori H, et al. Effect of related donor availability on outcome of AML in the context of related and unrelated hematopoietic cell transplantation. Bone Marrow Transplant. 2013;48:390–5.
Bashey A, Zhang X, Sizemore CA, Manion K, Brown S, Holland HK, et al. T-cell-replete HLA-haploidentical hematopoietic transplantation for hematologic malignancies using post-transplantation cyclophosphamide results in outcomes equivalent to those of contemporaneous HLA-matched related and unrelated donor transplantation. J Clin Oncol. 2013;31:1310–6.
Bertaina A, Zecca M, Buldini B, Sacchi N, Algeri M, Saglio F, et al. Unrelated donor vs HLA-haploidentical alpha/beta T-cell- and B-cell-depleted HSCT in children with acute leukemia. Blood. 2018;132:2594–607.
Ciurea SO, Zhang MJ, Bacigalupo AA, Bashey A, Appelbaum FR, Aljitawi OS, et al. Haploidentical transplant with posttransplant cyclophosphamide vs matched unrelated donor transplant for acute myeloid leukemia. Blood. 2015;126:1033–40.
Berger M, Lanino E, Cesaro S, Zecca M, Vassallo E, Faraci M, et al. Feasibility and outcome of haploidentical hematopoietic stem cell transplantation with post-transplant high-dose cyclophosphamide for children and adolescents with hematologic malignancies: an AIEOP-GITMO retrospective multicenter study. Biol Blood Marrow Transplant. 2016;22:902–9.
Locatelli F, Merli P, Pagliara D, Li Pira G, Falco M, Pende D, et al. Outcome of children with acute leukemia given HLA-haploidentical HSCT after alphabeta T-cell and B-cell depletion. Blood. 2017;130:677–85.
Mrozek K, Marcucci G, Nicolet D, Maharry KS, Becker H, Whitman SP, et al. Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J Clin Oncol. 2012;30:4515–23.
Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079–89.
Rollig C, Bornhauser M, Thiede C, Taube F, Kramer M, Mohr B, et al. Long-term prognosis of acute myeloid leukemia according to the new genetic risk classification of the European LeukemiaNet recommendations: evaluation of the proposed reporting system. J Clin Oncol. 2011;29:2758–65.
Sharma A, Huang S, Li Y, Brooke RJ, Ahmed I, Allewelt HB, et al. Outcomes of pediatric patients with therapy-related myeloid neoplasms. Bone Marrow Transplant. 2021;56:2997–3007.
Bitan M, He W, Zhang MJ, Abdel-Azim H, Ayas MF, Bielorai B, et al. Transplantation for children with acute myeloid leukemia: a comparison of outcomes with reduced intensity and myeloablative regimens. Blood. 2014;123:1615–20.
Acknowledgements
We would like to thank Keith A. Laycock, PhD, ELS for scientific editing of the manuscript. We would like to thank our colleagues, advanced practice providers, nurses, data managers and other healthcare professional who participated in patient care. We would also like to thank the parents who entrusted the care of our children to us. This work was supported by funds from the American Lebanese Syrian Associated Charities (ALSAC) (to Akshay Sharma).
Author information
Authors and Affiliations
Contributions
AS, NSB and SVB conceptualized and designed the study, interpreted the results, wrote the first draft of the manuscript, and made revisions. J-EG and AD provided data management and performed statistical analysis. KK and SC oversaw the study and provided supervision. Several authors were involved in caring for the patients included in this study at their respective centers. All authors critically reviewed the manuscript, provided comments, and approved the final version.
Corresponding author
Ethics declarations
Competing interests
AS has received consultant fees from Spotlight Therapeutics, Medexus Inc., Vertex Pharmaceuticals, Sangamo Therapeutics, and Editas Medicine. He is a medical monitor for an RCI BMT CSIDE clinical trial for which he receives financial compensation. He has also received research funding from CRISPR Therapeutics and honoraria from Vindico Medical Education. AS is the St. Jude Children’s Research Hospital site principal investigator of clinical trials for genome editing of sickle cell disease sponsored by Vertex Pharmaceuticals/CRISPR Therapeutics (NCT03745287), Novartis Pharmaceuticals (NCT04443907), and Beam Therapeutics (NCT05456880). The industry sponsors provide funding for the clinical trial, which includes salary support paid to AS’s institution. AS has no direct financial interest in these therapies.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sharma, A., Galimard, JE., Pryce, A. et al. Cytogenetic abnormalities predict survival after allogeneic hematopoietic stem cell transplantation for pediatric acute myeloid leukemia: a PDWP/EBMT study. Bone Marrow Transplant 59, 451–458 (2024). https://doi.org/10.1038/s41409-024-02197-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41409-024-02197-3
- Springer Nature Limited