
Abstract
Background
CREBBP has been extensively studied in syndromic diseases associated with skeletal dysplasia. However, there is limited research on the molecular mechanisms through which CREBBP may impact bone development. We identified a novel pathogenic CREBBP variant (c.C3862T/p.R1288W, which is orthologous to mouse c.3789 C > T/p.R1289W) in a patient with non-syndromic polydactyly.
Methods
We created a homozygous Crebbp p.R1289W mouse model and compared their skeletal phenotypes to wild-type (WT) animals. Bone marrow stem cells (BMSCs) were isolated and assessed for their proliferative capacity, proportion of apoptotic cells in culture, and differentiation to chondrocytes and osteocytes.
Results
We observed a significant decrease in body length in 8-week-old homozygous Crebbp p.R1289W mice. The relative length of cartilage of the digits of Crebbp p.R1289W mice was significantly increased compared to WT mice. BMSCs derived from Crebbp p.R1289W mice had significantly decreased cell proliferation and an elevated rate of apoptosis. Consistently, cell proliferative capacity was decreased and the proportion of apoptotic cells was increased in the distal femoral growth plate of Crebbp p.R1289W compared to WT mice. Chemical induction of BMSCs indicated that Crebbp p.R1289W may promote chondrocyte differentiation.
Conclusion
The Crebbp p.R1289W variant plays a pathogenic role in skeletal development in mice.
Impact
-
CREBBP has been extensively studied in syndromic diseases characterized by skeletal dysplasia.
-
There is limited research regarding the molecular mechanism through which CREBBP may affect bone development.
-
To our knowledge, we generated the first animal model of a novel Crebbp variant, which is predicted to be pathogenic for skeletal diseases.
-
Certain pathogenic variants, such as Crebbp p.R1289W, can independently lead to variant-specific non-syndromic skeletal dysplasia.
Similar content being viewed by others


Introduction
Skeletal malformations, such as polydactyly (extra fingers or toes), cleft palate, scoliosis, and shortening of limb bones, are rare birth defects in humans, affecting approximately 1 in 3000 individuals.1The diagnosis of skeletal defects has been increasing worldwide, with over 400 disorders of the skeletal system identified. Currently, more than 300 genes are implicated in the etiology of skeletal defects, and they play particularly important roles during embryonic or postnatal development.2 Despite substantial gains in our understanding of the genetics of skeletal diseases, the underlying causes of many skeletal disorders remain unknown. Therefore, there remains an unmet need for a more robust understanding of skeletal-disease-relevant genes and their downstream pathways to uncover potential therapeutic strategies that may improve patient outcomes.
The cyclic adenosine monophosphate (cAMP) response element-binding protein (CREBBP, OMIM#600140) gene, located on chromosome 16p13.3, is a widely expressed transcriptional coactivator, which prompts protein expression through interactions with transcription factors or acetylating specific proteins, such as histones.3 CREBBP acts as a well-known tumor suppressor by responding to cAMP response elements,4,5 and, as a core player in transcriptional regulation, defects or mutations in CREBBP have broad effects on various tissues and cellular processes that contribute to human disease. For example, studies have shown that approximately 60–70% of Rubinstein-Taybi syndrome (RSTS) cases are caused by mutations in the CREBBP gene.6 RSTS is a rare genetic disorder with autosomal dominant inheritance characterized by physical abnormalities, intellectual disability, and developmental delays.6 Acromegaly is a common limb deformity of RSTS characterized by the presence of flat and thick thumbs as well as giant toes (anterior axial multi-finger deformity), and the vast majority of patients with RSTS, approximately 96%, exhibit polydactyly and syndactyly.6 Consistently, Crebbp+/− mice were shown to have abnormal bone development,7 and single-allele deficiency of Crebbp disrupted the bone marrow (BM) microenvironment, leading to bone development defects including reduced bone volume and increased osteoclastogenesis.4 However, the mechanisms underlying the observed defects in limb and bone development in Crebbp-deficient animals have not been reported, and, despite advancements in our understanding the genetic basis of RSTS and the role of CREBBP, the specific mechanisms underlying the development of skeletal defects remain unclear. In order to more precisely reveal the direct connection between CREBBP gene mutations and non-syndromic skeletal dysplasia, we are committed to eliminating the interference of complex genetic backgrounds, aiming to provide new insights and strategies for the prevention and treatment of the diseases.
In the present study, we selected one pathogenic CREBBP mutation (c.3862 C > T/p.R1288W) identified in a patient with non-syndromic polydactyly; this mutation is homologous to the c.3789 C > T/p.R1289W mutation found in the mouse Crebbp gene. Here, we developed a novel homozygous Crebbp p.R1289W mouse model to study skeletal dysplasia. We observed that Crebbp p.R1289W mice had abnormal skeletal development compared to WT mice. Further, in vitro experiments with bone marrow stem cells (BMSCs) derived from Crebbp p.R1289W and WT mice showed that Crebbp p.R1289W BMSCs exhibited reduced proliferation and enhanced chondrocyte differentiation capacity compared to their WT counterparts. Consistently, cell proliferative capacity was decreased and the proportion of apoptotic cells was increased in the distal femoral growth plate of Crebbp p.R1289W compared to WT mice. These findings motivate further studies of the molecular mechanisms of CREBBP variants in congenital skeletal diseases.
Materials and methods
Participants
All participants in this study underwent a thorough physical examination at Shanghai Children’s Medical Center in Shanghai, China. Only individuals who met the criteria of the standard physical examination and did not exhibit syndromic clinical phenotypes were included in the study. Anomalies were defined based on standard terminology as published in relevant literature.8,9 In accordance with the Declaration of Helsinki, this study was approved by the ethics committee of Shanghai Children’s Medical Center (no. SCMCIRB-K2017008), and written informed consent was obtained from all participants or their legal guardians.
Identification of candidate pathogenic CREBBP mutations
Previously identified variants found in patients with non-syndromic polydactyly were evaluated to identify potential pathogenic variants within the coding region of the CREBBP gene.10,11,12,13 We focused on candidate CREBBP variants annotated as loss of function (including splicing, frameshift, and stop–gain mutations) as well as non-synonymous single-nucleotide variants (SNVs) with a CADD score greater than 20. A minor allele frequency threshold of 0.001 in East Asian populations was used to classify variants as pathogenic.
Mice
Crebbp R1289W knock-in mice were generated at Cyagen Biosciences (Suzhou, China). The targeting vectors containing the point mutation R1289W (CGG to TGG) also contain a neomycin (NEO) selectable marker cassette flanked by SDA (self-deletion anchor) sites and a diphtheria toxin A (DTA) selectable marker cassette for negative selection. The vector was electroporated into embryonic stem (ES) cells from C57BL/6 N mice. Correctly targeted ES cells were injected into blastocysts, which were then implanted into pseudo-pregnant females to generate chimeric mice, which were identified by genotyping. The chimeras were bred with wild-type mice to test germline transmission and F1 animal generation. Heterozygous mice were intercrossed to generate homozygous mice. The genotype of Crebbp R1289W knock-in mice was confirmed by PCR using a pair of primers (F1: 5ʹ-GGTTACTCAGCACATGCATGATGTC-3ʹ, R1: 5ʹ-AAATTTAGAACCAACTGCCACCTC-3ʹ) and sequencing. And the genotype of the inter-cross heterozygous targeted mice to generate homozygous targeted mice was confirmed by PCR using a pair of primers (F2: 5ʹ-AAATAGCAAAAGGTTTGACCCTGG-3ʹ, R2:5ʹ-GATGATAGCAGCTCTTGTCAGTCT-3ʹ). The lengths of the PCR products for the wild-type Crebbp, the heterozygous Crebbp +/p.R1289W, and the homozygous Crebbp p.R1289W mutation are 192 bp, 336 bp/192 bp (indicating a mixture of wild-type and mutant alleles), and 336 bp respectively. All animals used in the study were of the C57BL/6 N background, and WT littermates were used as controls. All mice were housed in the animal residence of Shanghai Children’s Medical Center in a pathogen-free environment. Throughout the study, the mice received long-term care and maintenance in accordance with the animal care and use policy established by the research institute. All animal procedures were approved by the animal ethics committee of Shanghai Children’s Medical Center (no. SCMC-LAWEC-2019-015).
Alcian blue/Alizarin Red staining
The mice were euthanized by asphyxiation with carbon dioxide and subsequently frozen before further processing. Starting from the abdomen, the cavity was opened and internal organs were removed. Subsequently, the chest was opened from bottom to top to ensure thorough access. The frozen samples were thawed and delicately peeled using surgical forceps. Then, the samples were subjected to an overnight wash with 95% ethanol, followed by another overnight wash with 100% acetone. Finally, the samples were washed with water. Once the washing process was complete, the samples were immersed in Alcian blue (80% ethanol + 20% acetic acid + 150 mg/L alcian blue) overnight. Subsequently, the samples were thoroughly rinsed with 70% ethanol for 1 h and then treated with 2% potassium hydroxide solution for 24 h. Next, the samples were placed in Alizarin red solution (50 mg/L Alizarin red was dissolved in 1% KOH) for overnight staining, followed by another 24-hour treatment with 2% KOH. After that, the samples underwent clarification treatment, first soaking in Fluid I for Transparency(consisting of a 20:77:3 ratio of glycerol, distilled water, and 2% KOH) for 48 hours, and then in Fluid II for Transparency(consisting of a 75:25 ratio of glycerol and distilled water) for 24 h. Finally, samples were stored in a preservation solution consisting of a 1:1 ratio of glycerol and ethanol at room temperature.
Measurement of the relative lengths of cartilage and ossification
After euthanasia of the 10-day-old mice and subsequent Alcian Blue/Alizarin Red staining, the lengths of cartilage and mineralized bone were measured in the five digits of the forelimbs and hindlimbs of mice carrying the heterozygous and homozygous mutations of Crebbp p.R1289W and WT mice. The measurements were normalized to the corresponding positions in the WT mouse group. The relative lengths of cartilage and ossification in Crebbp p.R1289W mice or Crebbp +/p.R1289W mice were calculated by comparing the mean measurements of cartilage and ossification in the digits of Crebbp p.R1289W mice or Crebbp +/p.R1289W mice to the corresponding mean measurements in WT mice.
Microcomputed tomography data analysis
The 8-week-old mice were euthanized by carbon dioxide asphyxiation, and the bilateral tibia tissues were rapidly dissected and stripped. The tibia bones were then rinsed clean with physiological saline and stored in 4% paraformaldehyde. Mouse tibias were subjected to microcomputed tomography (microCT) analysis using a high-resolution scanner (μCT 100, SCANCO Medical AG, Brüttisellen, Switzerland) at Numira Biosciences. Image data were acquired at a 10-μm isometric voxel resolution with continuous microCT planar imaging (70 kvp, 200 uA). After the scanning procedure, the region of interest (ROI) for three-dimensional image reconstruction of bone trabecula was selected as proximal tibia on the host. The primary parameters used for detection and analysis were as follows: bone volume (BV), bone volume/total volume (BV/TV), trabecular number (TB.N) and trabecular separation/spacing (TB.Sp).
BMSC isolation and characterization
To collect BMSCs, after euthanasia, the femurs and tibias of the mice were promptly collected (Figure S1). We then used sterile scissors to cut these bones into small pieces, which were subsequently transferred into a digestive solution composed of PBS solution containing 1 mg/ml type IV collagenase, 2 mg/ml Dispase protease, and 1% BSA. This mixture was then incubated at 37 °C with a shaking speed of 140 r.p.m. for 1–1.5 hours. To removed blood cells, an ammonium-chloride-potassium (ACK) lysis buffer was applied at room temperature for 5–10 min. Subsequently, 1×PBS was added to the cell suspension in an amount greater than its original volume, and then the suspension was centrifuged at a speed of 1,500 rpm for 5 minutes. The resulting pellet was resuspended in OriCell C57BL/6 N Mouse Bone Marrow Mesenchymal Stem Cell Growth Medium (Cyagen Biosciences, Inc., Guangzhou, China), which consisted of 440 ml of basal medium, 50 ml of OriCell Superior-Qualified Fetal Bovine Serumand Culture Supplement, 5 ml of penicillin-streptomycin, and 5 ml of L-glutamine.The cells were finally cultured at 37 °C in a 5% CO2 incubator for 3 days.
Cell viability assay
BMSCs were seeded in a 96-well culture plate at a density of 5 × 104 cells per well with 100 µl of medium. After inoculation, the cells were assessed by adding 50 µl of Promega CellTiter-Glo (CTG) reagent to each well at 24-hour, 48-hour, 72-hour, 96-hour, and 120-hour time points. The plate was then mixed on an oscillator for 2 min to induce cell lysis and ensure uniform distribution of the CTG reagent. To measure the chemiluminescence emitted by the BMSCs, a Synergy H1 microplate reader (BioTek, Winooski, Vermont) was used at room temperature. Three repetitions were performed at each time point to account for potential variations.
Cell apoptosis analysis
BMSCs were seeded on a 12-well culture plate at a density of 3×105 cells per well and allowed to incubate for 48 h. Then, the cells were detached using trypsin and subsequently collected. The collected BMSCs were stained using FITC annexin V and propidium iodide (PI) (FITC Annexin V Apoptosis Detection Kit, TransGen, China). Flow cytometry analysis was performed with a FACSCanto II instrument (Becton, Dickinson and Company, Franklin Lakes, New Jersey) to detect apoptosis in the stained BMSCs.
Immunohistochemical staining and Ki67 detection in murine femur tissue
The mice were euthanized with carbon dioxide, and their bilateral femurs were promptly dissected and stripped. The femurs were then cleaned with physiological saline and fixed in 4% paraformaldehyde for at least 24 h. Subsequently, the femurs were immersed in a decalcifying solution (#G1105, ServicebioTM, China), using a volume 15–20 times that of the tissue, and the solution was refreshed daily. The process was deemed complete when the bone tissue became elastic and could be easily bent. After rinsing with distilled water, the femurs were dehydrated in ethanol, embedded in paraffin, and sectioned. Next, the sections were placed in a 60 °C oven for ≥1 h and then deparaffinized in xylene (3×) and rehydrated. Antigen retrieval was performed, endogenous peroxidase and biotin were deactivated, non-specific binding was blocked with serum, and tissues were incubated with primary mouse Ki67 antibody (#GB121141, ServicebioTM, China), followed by goat anti-mouse IgG labeled by horseradish peroxidase (HRP, #GB23301, ServicebioTM, China). Finally, tissues were stained with diaminobenzidine (DAB) chromogenic reagent for histochemical kit (#G1212, ServicebioTM, China), counterstained with hematoxylin, dehydrated, cleared, mounted, and analyzed under a microscope (#E100, Nikon, Japan). This protocol was chosen to ensure optimal immunostaining and accurate Ki67 detection in femur sections. Ki67 positive cells were calculated using Image J software (National Institutes of Health, Bethesda, Maryland), a digital image analysis software, which is able to calculate the index accurately and efficiently.
TUNEL assay of murine femoral tissue
After decalcification treatment, the femurs were processed into frozen sections. Frozen sections were fixed and treated with Proteinase K, and then membranes were permeabilized and left to equilibrate at room temperature. TDT enzyme, dUTP, and buffer from the TUNEL assay kit (#G1502, ServicebioTM, China) were used in a 1:5:50 ratio and adjusted to the slide count and tissue size. The mixture was applied to cover the tissues, and then the slides with tissue sections were laid flat in a humid box and incubated at 37 °C for 1 h in a constant temperature chamber. Proper humidity was maintained by adding a small amount of water to the chamber. Next, tissue sections were washed three times with PBS (pH 7.4) for 5 min each. After PBS, DAPI staining solution was added dropwise to the tissues, and the slides were incubated at room temperature in the dark for 10 minutes. Slides were then submerged in PBS (pH 7.4) and agitated on a decolorizing shaker three times for 5 min each. Once sections were slightly dry, they were sealed with anti-fluorescence quenching mounting medium. Parameters related to the proportion of red positive cells were analyzed using Aipathwell software (ServicebioTM, China).
Skeletal differentiation
To assess the ability of BMSCs derived from mice to undergo skeletal differentiation, in vitro chondrogenesis and osteogenesis inductions were performed.
To induce chondrogenic differentiation, we washed the cells with 0.5 ml of a pre-mixed solution and centrifuged them at 1000 rpm for 5 min at room temperature. This solution was prepared by mixing 2 ml of OriCell® chondrogenic differentiation supplement I (#MUXMX-04041-1,Cyagen Biosciences,China) from the OriCell® Mouse BMSCs Chondrogenic Kit (#MUXMX-90041,Cyagen Biosciences,China) with 97 ml of OriCell® Basic Cell Medium (#BHDM-03011,Cyagen Biosciences,China). During the preparation of the OriCell® C57BL/6 N mouse bone marrow mesenchymal stem cell chondrogenic differentiation medium, we added 10 μl of OriCell® chondrogenic differentiation supplement II (#MUXMX-04041-2, Cyagen Biosciences, China) per 1 ml of premixed solution. Subsequently, the cells were resuspended in this culture medium. The cell suspension was gently deposited into a centrifuge tube and incubated at 37 °C with 5% CO2, while the tube cap was loosened to an appropriate degree to enhance gas exchange. The differentiation medium was changed every 2–3 days for 21–28 days. Finally, the chondrocytes were fixed in formaldehyde and embedded in paraffin to create sections. These sections were subsequently stained with Alcian blue(#ALCB-10001, Cyagen Biosciences, China).
To induce differentiation of BMSCs into osteoblasts, the culture dishes were coated with a 0.1% gelatin solution. When cellular confluence of BMSCs was observed to reach a range of 60–70%, complete culture medium consisting of 177 ml of OriCell® C57BL/6 N Mouse Bone Marrow Mesenchymal Stem Cell Osteogenic Differentiation Basal Medium for Cell Culture(#BLDM-03011, Cyagen Biosciences, China), 20 ml of OriCell® Superior Quality Fetal Bovine Serum(#FBSSR-01021, Cyagen Biosciences, China), 3 ml of OriCell® Supplement for Mouse Bone Marrow Mesenchymal Stem Cells Osteogenic Differentiation (#MUXMX-04021, Cyagen Biosciences, China) was added and changed every 2 days. After a 14-day incubation period, the cells were stained with Alizarin red(#ALIR-10001, Cyagen Biosciences, China).
Statistical analysis
Statistical analysis was performed using the independent samples t-test to determine significant differences between the two groups. A P value of 0.05 was used as the threshold for determining statistical significance.
Results
Clinical assessment of CREBBP variants identified by targeted sequencing
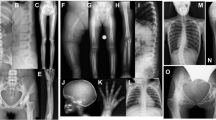
In our previous study, we collected 181 blood samples from patients with non-syndromic polydactyly, and we subjected these samples to targeted high-throughput sequencing using a panel designed to detect genetic bone diseases.10 Analysis of the raw data10,11,12,13 identified 13 deleterious CREBBP gene mutations that had not been previously reported in 13 patients. Out of the identified mutations, 11 were classified as missense mutations, 1 as a nonsense mutation, and 1 as a frameshift mutation (Table 1 and Fig. 1a). The missense mutations were assessed as deleterious by SIFT, polyphen2, and mutationTaster.10,11,12,13 Analysis of X-rays from all 13 probands revealed the clinical phenotype of non-syndromic preaxial polydactyly deformity (Fig. 1b). Among the 13 screened CREBBP gene mutations, p.R1288W is predicted to have high pathogenicity, and the R1288 residue CREBBP is highly conserved across different species screened (Fig. 1c). Therefore, we chose to construct a Crebbp p.R1289W mouse model to test whether the loss of CREBBP function resulting from this mutation would affect skeletal development.

aDistribution of the 13 variants identified on the CREBBP gene. b Polydactyly phenotype was observed in patients with CREBBP mutation. c Comparison of the partial amino acid sequence of human CREBBP with that of other species. The amino acid residue R1288 represents a conserved residue across different species. An arrow indicates the conserved amino acid residue in human CREBBP.
Crebbp p.R1289W mice have abnormal cartilage components but normal cortical and trabecular bone
In mice, the Crebbp p.R1289W variant is orthologous to human CREBBP c.C3862T/p.R1288W. Thus, to study the effect of CREBBP p.R1288W on mouse skeletal development, we generated homozygous Crebbp p.R1289W mice by crossbreeding C57BL/6 N Crebbp p.R1289W heterozygous mice, and we also used littermates of WT mice as controls(Figure S2). Interestingly, we observed a significant decrease in body length (measured from the tip of the nose to the anus on a horizontal tabletop) in 8-week-old homozygous Crebbp p.R1289W mice compared with heterozygous Crebbp +/p.R1289W mice and WT mice (Fig. 2a, b). Compared to WT mice, the femur and tibia lengths of the Crebbp +/p.R1289W heterozygous mice and Crebbp R1289W homozygous mice were significantly reduced (Fig. 2c, d). Mice were euthanized when they reached 10 days of age, and Alcian Blue and Alizarin Red staining of the cartilaginous and osteogenic components of the whole body revealed that the relative length of cartilage of the digits of Crebbp p.R1289W mice and Crebbp +/p.R1289W mice was significantly increased compared to WT mice (P < 0.05), while there was no significant difference in the relative length of ossified bone of the digits (Fig. 2e–h). Next, we compared bone morphology in Crebbp p.R1289W, Crebbp +/p.R1289W and WT mice, to ascertain the disparities among them. Histological analysis of femurs and microCT scans of tibias collected from 8-week-old mice showed no discernible differences in trabecular and cortical bone structure between Crebbp p.R1289W, Crebbp +/p.R1289W mice and WT littermate controls. (Fig. 3a, b). Moreover, quantitative analysis of trabecular parameters, including the number and thickness of trabeculae, as well as trabecular separation (BV/TV, TB.N, TB.Sp), revealed no significant differences among Crebbp p.R1289W, Crebbp +/p.R1289W and WT mice (Fig. 3c).

a Gross morphology of 8-week-old WT, Crebbp + /p.R1289W heterozygous mice and Crebbp p.R1289W homozygous mice. b The length of mice of the indicated genotypes. The number of mice included in each bar is shown. c Gross morphology of femurs and tibias of 8-week-old WT, Crebbp +/p.R1289W heterozygous mice and Crebbp R1289W homozygous mice. d The length of femurs and tibias of mice of the indicated genotypes. The number of long bones included in each bar is shown. e Alcian blue/Alizarin Red staining of 10-day-old WT, Crebbp +/p.R1289W and Crebbp p.R1289W mice. f Alcian blue/Alizarin Red staining of the back and abdomen regions in WT, Crebbp +/p.R1289W and Crebbp p.R1289W mice.*a- abdomen, b-back. g Alcian blue/Alizarin Red staining of digits in Crebbp p.R1289W, Crebbp +/p.R1289W and WT mice. All the digits of each mouse were measured at the positions corresponding to the red and green arrows indicated. h Relative length of cartilage and bone in digits of Crebbp p.R1289W, Crebbp +/p.R1289W and WT mice. Red arrow: the segment measured for cartilage length; Green arrow: the segment measured for osteogenic length. Significance was determined with the independent samples t-test, *P < 0.05, **P < 0.01, ns, no significance, P > 0.05. All data are presented as mean ± standard error of mean from each group.

a Representative histological sections of the femurs from 8-week-old Crebbp R1289W mice, Crebbp +/R1289W mice and WT mice, stained with H&E. b Representative MicroCT 2D slices of tibias from 8-week-old Crebbp p.R1289W, Crebbp +/R1289W mice and WT Mice. c MicroCT measurements of BV/TV (bone volume/ total volume), Tb.N (trabecular number), and Tb.Sp (trabecular separation) in tibias from Crebbp p.R1289W, Crebbp +/R1289W mice and WT mice. Significance was determined with the independent samples t-test. P > 0.05, ns, no significance. All data are presented as mean ± standard error of mean from each group.
Crebbp p.R1289W inhibited proliferation and promoted apoptosis of cells associated with skeletal development
We next harvested BMSCs from the spine and long bones of 4-week-old homozygous Crebbp p.R1289W and WT mice. Microscopic observation of P3-generation BMSCs derived from Crebbp p.R1289W mice exhibited slower cell proliferation compared to BMSCs derived from WT mice at the same seeding density (Fig. 4a). Moreover, in a CTG assay, proliferation of Crebbp p.R1289W BMSCs was significantly downregulated at 72 h, 96 h, and 120 h compared to WT BMSCs (P < 0.05) (Fig. 4b). Consistently, we observed a significant increase (P < 0.05) in the G0/G1 cell ratio and a decrease (P < 0.05) in the G2/S cell ratio in BMSCs from Crebbp p.R1289W mice compared to those from WT mice (Fig. 4c). Additionally, the ratio of apoptotic BMSCs from Crebbp p.R1289W mice was higher compared to WT mice (P < 0.05) (Fig. 4d). To evaluate whether these phenotypes could be observed in intact bone, we measured proliferative capacity and apoptosis in femurs collected from 8-week-old animals. We found significantly decreased cellular proliferative capacity and a higher proportion of apoptotic cells within the distal femoral growth plate region of Crebbp p.R1289W mice compared to WT mice (Fig. 4e, f). Taking into account these findings, we can speculate that Crebbp p.R1289W inhibits cell growth and induces apoptosis related to skeletal development.

a BMSCs derived from Crebbp p.R1289W and WT mice were isolated and cultured in vitro. Cell morphology was observed on day 3 and day 6 using an optical microscope. b CellTiter-Glo Luminescent Cell Viability Assay to determine the proliferative capacity of BMSCs derived from Crebbp p.R1289W and WT mice at 48 h, 72 h, 96 h, and 120 h after inoculation. c Flow cytometry analysis of cell cycle stage in BMSCs derived from Crebbp p.R1289W and WT mice. d Ratio of apoptotic BMSCs derived from Crebbp p.R1289W and WT mice. e Analysis of cell proliferation (Ki67 staining) in intact femurs from Crebbp R1289W and WT mice. f Proportion of apoptotic cells (H&E staining and TUNEL assay) in the distal femurs of Crebbp R1289W and WT mice. Significance was determined with the independent samples t-test, *P < 0.05, **P < 0.01. b–f All data are presented as mean ± standard error of mean from each group. TUNEL, TdT-mediated dUTP nick end labeling.
Crebbp p.R1289W promotes the differentiation of BMSCs derived from mice into chondrocytes
To evaluate the potential effect of Crebbp p.R1289W on BMSC differentiation, we induced BMSCs from homozygous Crebbp p.R1289W or WT mice to form chondrocytes for 21 days and then characterized the cells using Alcian blue staining. The differentiated chondrocytes from Crebbp p.R1289W BMSCs formed more compact, pelleted micromasses with less intercellular space between the chondrocytes (Fig. 5a), indicating that Crebbp p.R1289W caused BMSCs to be more effectively directionally induced. However, after 14 days of osteogenic induction, Alizarin red staining revealed no discernible difference in osteogenic differentiation between Crebbp p.R1289W and WT BMSCs (Fig. 5b). Thus, our findings suggest that the Crebbp p.R1289W variant may enhance BMSCs differentiation to chondrocytes, but it does not appear to influence osteoblast differentiation.

a BMSCs derived from Crebbp p.R1289W and WT mice were induced to form pelleted micromasses. The differentiated chondrocytes derived from Crebbp p.R1289W mice exhibited a higher proliferation efficiency compared to those from WT mice, and the pelleted micromasses of Crebbp p.R1289W BMSCs displayed a more compact structure with less space between chondrocytes. b No significant difference was observed in the osteogenic differentiation between BMSCs derived from Crebbp p.R1289W compared to WT mice. ImageJ is utilized for the analysis of the formation of the Orange-Red complex subsequent to Alizarin Red staining. Significance was determined with the independent samples t-test. P > 0.05, ns, no significance. All data are presented as mean ± standard error of mean from each group.
Discussion
Although numerous monogenic causes of skeletal disorders have been identified, the diagnostic success rate based on monogenic traits remains limited, prompting further investigation into the genetic contributions to skeletal development. In the present study, we generated, to our knowledge, the first homozygous Crebbp knock-in mouse model (c.3789 C > T/p.R1289W, the ortholog of the c.C3862T mutation in human CREBBP). These mice exhibited skeletal anomalies, making them a valuable tool for further analysis of this variant. In our mouse model, we observed that Crebbp p.R1289W was associated with abnormal cell proliferation, differentiation, chondrogenesis, and body length, suggesting that individuals carrying CREBBP variants may be at risk for skeletal disorders.
Owing to its therapeutic potential, CREBBP is one of the most extensively studied proteins. Heterozygous loss-of-function variants and deletions of CREBBP are well known to be pathogenic in RSTS, a condition characterized by certain skeletal dysplasia phenotypes.6 Since 2016, numerous reports have been published on patients who exhibited distinct phenotypic features, referred to as Menke–Hennekam syndrome (MHS I, MIM#618332), resulting from mutations in specific segments of the CREBBP gene.14,15,16,17 These phenotypic features differ from those typically observed in RSTS. CREBBP haploinsufficiency has also been shown to disrupt the BM microenvironment, resulting in the loss of stem cells and excessive myelopoiesis.4 In addition, mutations in CREBBP and its closely related paralog EP300 have been implicated as molecular tools used by cancer cells to achieve and sustain an oncogenic phenotype.18 Therefore, CREBBP has emerged as an appealing target in drug design and development to treat cancer and RSTS, as well as other conditions such as Alzheimer’s disease and inflammatory diseases.19,20,21,22,23 However, to date, the association between CREBBP and several relevant diseases, including skeletal abnormalities, has been incompletely investigated.
In the present study, we determined that the CREBBP c.3862T/p.R1288W variant is predicted to be pathogenic for genetic bone diseases. Moreover, we discovered that the mouse Crebbp p.R1289W variant, which is orthologous to the human CREBBP c.C3862T/p.R1288W variant, modulates bone growth and development. This finding represents, to our knowledge, the first evidence of such an effect for this variant. Our results align with previous observations, such as the identification of a CREBBP (c.6324 C > G /p.Tyr2108*) variant, which contributes to brachydactyly and short thumbs in approximately 1 out of 108 individuals with skeletal dysplasias.24 Additionally, recent studies have found that CREBBP serves as one of the key nodes in the interaction network of differentially expressed genes and their corresponding long non-coding RNAs, shedding light on the molecular mechanisms underlying valgus-varus deformity.25 Therefore, elucidating the precise function of CREBBP in skeletal development is essential for improving our understanding of the pathogenesis of related non-syndromic skeletal diseases.
Numerous studies have demonstrated the crucial role of CREBBP in various tissues, where it regulates the activity of other genes throughout the body to control cell growth and division and promote cell differentiation.26 For instance, heterozygous loss-of-function mutations and deletions of CREBBP have been identified as causing RSTS, and it has been observed that human development is sensitive to changes in CREBBP dosage.27,28 Consistently, dysfunctional CREBBP resulting from gene modifications in animal models and mutations found in humans have been associated with the loss of stem cells and excessive myelopoiesis. Circular RNA (circRNA) is a non-coding transcript with a stable, covalently closed structure that is highly expressed in eukaryotic transcriptomes and resists RNA degradation.29 It plays a vital role in regulating messenger RNA expression, which is crucial for processes such as cell proliferation, apoptosis, invasion, migration, and metabolism.30 Recently, circCREBBP (hsa_circ_0037655) has been identified as an oncogene that enhances cell viability, growth, migration, and invasion through the activation of the phosphoinositide 3-kinase (PI3K) signaling pathway.31,32 Additionally, studies have revealed that circCREBBP impedes tumor growth in nude mice.32
The intimate link between CREBBP gene mutations and RSTS has been thoroughly investigated, yet previous literature seldom explored how this mutation independently affects abnormal skeletal development. Patients with RSTS commonly exhibit short stature, defined as a height significantly below two standard deviations of the normal range.33 Recent studies have revealed that out of nine RSTS patients carrying CREBBP gene mutations, six (66.7%) presented with short stature.34 Similarly, in another study, nineteen pathogenic variations in the CREBBP gene were identified among eighteen unrelated RSTS patients, with at least thirteen (72.2%) exhibiting short stature.35 These findings further confirm the significant association between CREBBP gene mutations and short stature. Some research has suggested that CREBBP, as an important coactivator in cell growth and proliferation,36 may indicate the pathogenic cause of short stature in patients with RSTS.37 However, the specific mechanism remains unreported, highlighting the urgency of further research in this area. In this study, we generated a novel homozygous mouse model harboring the Crebbp p.R1289W variant. Interestingly, our findings suggest that CREBBP may play a role in regulating body length in mice. Specifically, we observed a sequential decrease in body length in 8-week-old Crebbp p.R1289W homozygous mice and Crebbp +/p.R1289W heterozygous mice, when compared with WT mice. Meanwhile, we compared the lengths of femurs and tibias in mice with different genotypes and found that these results were consistent with the trend of changes in body length. Thus, our findings indicate that the Crebbp p.R1289W variant may regulate bone growth in mice in a gene-dosage-dependent manner. Furthermore, BMSCs derived from Crebbp p.R1289W mice exhibited decreased cell proliferation starting at 48 h (P < 0.01) compared to BMSCs derived from WT mice. Additionally, we observed marked differences in the distribution of cells across the G0/G1, S, and G2/M phases between BMSCs derived from Crebbp p.R1289W mice and WT mice. Under normal circumstances, cell proliferation and apoptosis are assumed to be in dynamic balance in organisms. Excessive cell proliferation or reduced apoptosis can lead to conditions characterized by cellular excess, whereas decreased proliferation or increased apoptosis can result in cytopenic diseases. Unexpectedly, we observed that, compared to WT, the proportion of Crebbp p.R1289W BMSCs in the G0/G1 phase was increased, whereas the proportions in the S and G2/M phases was substantially decreased, leading to a decrease in cell proliferation capacity. However, an increase in apoptosis was observed in BMSCs derived from mice harboring the Crebbp p.R1289W mutation. Further, in intact long-bone tissue of Crebbp R1289W mice, we observed a significant reduction in cellular proliferation within the femoral growth plate region compared to WT mice, accompanied by a relatively elevated proportion of apoptotic cells. It has been reported that apoptotic tumor cells can stimulate the repopulation of tumors from a small population of surviving cells.38 Although these results may seem paradoxical at first glance, the phenomenon of apoptosis-promoting cell proliferation has been observed in clinical tumor treatment studies involving caspase-3 activation, although the precise molecular mechanism remains unclear.38 Hence, it is possible that CREBBP plays a role in regulating the release of growth-promoting signals from dying cells, stimulating the proliferation of surviving cells to maintain a dynamic balance between viable and dead cells in the body. However, further research is necessary to verify this hypothesis and clarify the specific molecular mechanisms involved.
Bone formation occurs primarily through the proliferation and differentiation of embryonic mesenchymal stem cells, which are regulated by metabolic hormones.39 During mammalian embryonic development, two distinct processes are involved in bone formation: intramembrane osteogenesis and endochondral osteogenesis. Most bone in the body is formed through endochondral osteogenesis. In our previous studies, we showed that the identification of novel disease-causing genes and the use of genetic models for polydactyly can provide insights into the mechanisms underlying limb patterning and bone development.10 In the case of Crebbp p.R1289W, we observed that it can enhance the differentiation of BMSCs into chondrocytes as well as promote the proliferation of chondrocytes. Additionally, we found that Crebbp p.R1289W mice exhibited increased relative length of the cartilage of the digits compared to WT mice. In a rigorous study, researchers employed osteoblast-specific Crebbp conditional knockout mouse models and discovered that, compared to normal control mice, 16-week-old experimental mice exhibited a significant reduction in bone mass at the distal femur and a notable decrease in trabecular bone number in the lumbar vertebrae.40 Intriguingly, in this study, the trabecular and cortical bone structures of Crebbp p.R1289W homozygous mice or Crebbp +/p.R1289W heterozygous mice did not show significant structural differences when compared to WT mice. Considering the variations in gene expression regulation among mice with different genetic backgrounds, identical gene expression abnormalities can lead to diverse phenotypic manifestations of skeletal developmental anomalies. This demonstrates the pivotal role of CREBBP in bone formation and the maintenance of bone homeostasis.
Our findings demonstrate the involvement of Crebbp p.R1289W in the process of bone development. The presence of Crebbp p.R1289W was associated with inhibited body growth, as evidenced by reduced body size in 8-week-old mice. Furthermore, we observed that Crebbp p.R1289W influenced cell proliferation, apoptosis, and chondroblast differentiation. It is noteworthy that 13 patients (7.2%) in a cohort of 181 patients with non-syndromic hereditary bone disease were identified as carrying a pathogenic CREBBP variant. Therefore, this study of the effect of Crebbp p.R1289W in the development of skeletal dysplasia in mice provides a preclinical rationale for further studies of CREBBP variants as potential biomarkers of isolated skeletal dysplasia.
Our study also have some limitations. Firstly, while the experimental methods have provided valuable data, they may not fully uncover the impact of the CREBBP mutation on bone metabolism and dynamic histomorphometry. Secondly, the mouse model failed to replicate the polydactyly phenotype in patients, highlighting the challenges of cross-species research and the inadequacy of mirroring the pathogenic mechanisms of human heterozygous mutations. In future studies, more diverse and multi-angled experimental methods are still needed to evaluate the impact of CREBBP mutation on bone development.
In summary, our results demonstrate that pathogenic CREBBP variants can independently lead to variant-specific non-syndromic skeletal dysplasia. This discovery holds promise for improving the accuracy and effectiveness of genetic diagnoses for various bone diseases. Our future research will prioritize investigating the mechanisms through which CREBBP regulates skeletal development.
Data availability
Data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Calder, A. D. & Foley, P. Skeletal dysplasias: an overview. Paediatr. Child Health (Oxf.) 28, 84–92 (2018).
Bonafe, L. et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am. J. Med Genet Part A 167, 2869–2892 (2015).
Raisner, R. et al. Enhancer Activity Requires CBP/P300 Bromodomain-Dependent Histone H3K27 Acetylation. Cell Rep. 24, 1722–1729 (2018).
Zimmer, S. N. et al. Crebbp haploinsufficiency in mice alters the bone marrow microenvironment, leading to loss of stem cells and excessive myelopoiesis. Blood 118, 69–79 (2011).
Gao, C. et al. Downregulating CREBBP inhibits proliferation and cell cycle progression and induces daunorubicin resistance in leukemia cells. Mol. Med. Rep. 22, 2905–2915 (2020).
Korzus, E. Rubinstein-Taybi Syndrome and epigenetic alterations. Adv. Exp. Med. Biol. 978, 39–62 (2017).
Tanaka, Y. et al. Abnormal skeletal patterning in embryos lacking a single CBP allele: a partial similarity with Rubinstein-Taybi syndrome. Proc. Natl Acad. Sci. 94, 10215 (1997).
Biesecker, L. G. et al. Elements of morphology: standard terminology for the hands and feet. Am. J. Med. Genet. Part A 149A, 93–127 (2009).
Hall, B. D. et al. Elements of morphology: standard terminology for the periorbital region. Am. J. Med. Genet. Part A 149A, 29–39 (2009).
Zu, B. et al. Identification of the genetic basis of sporadic polydactyly in China by targeted sequencing. Computational Struct. Biotechnol. J. 19, 3482–3490 (2021).
Wang, J. et al. Exome sequencing reveals a novel PTHLH mutation in a Chinese pedigree with brachydactyly type E and short stature. Clin. Chim. Acta 446, 9–14 (2015).
Wang, J. et al. Primary ovarian insufficiency collaboration. mutations in HFM1 in recessive primary ovarian insufficiency. N. Engl. J. Med. 370, 972–974 (2014).
Xiang, Y. et al. A novel nonsense GLI3 variant is associated with polydactyly and syndactyly in a family by blocking the sonic hedgehog signaling pathway. Front Genet 11, 542004 (2020).
Menke, L. A. et al. CREBBP mutations in individuals without Rubinstein-Taybi syndrome phenotype. Am. J. Med Genet A 170, 2681–2693 (2016).
Menke, L. A. et al. Further delineation of an entity caused by CREBBP and EP300 mutations but not resembling Rubinstein-Taybi syndrome. Am. J. Med Genet A 176, 862–876 (2018).
Banka, S. et al. Genotype-phenotype specificity in Menke-Hennekam syndrome caused by missense variants in exon 30 or 31 of CREBBP. Am. J. Med Genet A. 179, 1058–1062 (2019).
Nishi, E. et al. The novel and recurrent variants in exon 31 of CREBBP in Japanese patients with Menke-Hennekam syndrome. Am. J. Med Genet A. 188, 446–453 (2022).
Attar, N. & Kurdistani, S. K. Exploitation of EP300 and CREBBP lysine acetyltransferases by cancer. Cold Spring Harb. Perspect. Med. 7, a026534 (2017).
Xiao, X. et al. Targeting CREB for cancer therapy: friend or foe. Curr. Cancer Drug Targets 10, 384–391 (2010).
Carlezon, W. A. et al. The many faces of CREB. Trends Neurosci. 28, 436–445 (2005).
Tang W., et al. Association study of CREB1 and CBP genes with Alzheimer’s disease in Han Chinese. Asia Pac Psychiatry. 9. (2017)
Hadjipanayis A., et al. Thu0033 Rna profling of healthy and rheumatoid arthritis subjects treated with tofacitinib monotherapy. 284.1–284. (2019)
Akinsiku, O. E. et al. Update and potential opportunities in CBP [cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB)-binding protein] research using computational techniques. Protein J 40, 19–27 (2021).
Sentchordi-Montané, L. et al. High prevalence of variants in skeletal dysplasia associated genes in individuals with short stature and minor skeletal anomalies. Eur. Endocrinol. 185, 691–705 (2021).
Tang, H. et al. Integrative analysis of long non-coding RNA and mRNA in broilers with valgus-varus deformity. PLoS One 15, e0239450 (2020).
Deemer, B. et al. Duplication 16p13.3 and the CREBBP gene: confirmation of the phenotype. Eur. J. Med Genet 56, 26 (2013).
Petrij, F. et al. RubinsteineTaybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature 376, 348e51 (1995).
Hennekam, R. C. et al. Deletion at chromosome 16p13.3 as a cause of RubinsteineTaybi syndrome: clinical aspects. Am. J. Hum. Genet 52, 255e62 (1993).
Ebbesen, K. K. et al. Insights into circular RNA biology. RNA Biol. 14, 1035–1045 (2017).
Ambros, V. The functions of animal microRNAs. Nature 431, 350–355 (2004).
Qiao, J. et al. Microarray analysis of circRNAs expression profile in gliomas reveals that circ_0037655 could promote glioma progression by regulating miR-214/PI3K signaling. Life Sci. 245, 117363 (2020).
Zhang, L. et al. Circ-CREBBP promotes cell tumorigenesis and glutamine catabolism in glioma by regulating miR-375/glutaminase axis. Brain Res 1775, 147730 (2022).
Hutchinson, D. T. et al. Rubinstein–Taybi Syndrome. J. Hand Surg. Am. 40, 1711–1712 (2015).
Yu, P. T. et al. Rubinstein-Taybi syndrome in Chinese population with four novel mutations. Am. J. Med Genet A. 185, 267–273 (2021).
Yu, S. et al. Clinical exome sequencing identifies novel CREBBP variants in 18 Chinese Rubinstein-Taybi Syndrome kids with high frequency of polydactyly. Mol. Genet Genom. Med. 7, e1009 (2019).
Murata, T. et al. Defect of histone acetyltransferase activity of the nuclear transcriptional coactivator CBP in RubinsteinTaybi syndrome. Hum. Mol. Genet 10, 1071–1076 (2001).
Huh, R. et al. Letter to the Editor: a novel mutation in the CREBBP gene of a Korean girl with Rubinstein–Taybi syndrome. Ann. Clin. Lab Sci. 45, 458–461 (2015).
Huang, Q. et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med 17, 860–866 (2011).
Kozhemyakina, E. et al. A pathway to bone: signaling molecules and transcription factors involved in chondrocyte development and maturation. Development 142, 817–831 (2015).
Zhang, L. L. et al. Acetyltransferases CBP/p300 control transcriptional switch of β-catenin and stat1 promoting osteoblast differentiation. J. Bone Min. Res 38, 1885–1899 (2023).
Acknowledgements
We express our gratitude to the patients and their families who willingly participated in this study.
Funding
The research was financially supported by grants from the National Natural Science Foundation of China (No. 82372328), the Medicine Guide Project (Chinese and Western medicine) of the Shanghai Science and Technology Committee (No. 18411961400), and the Science and Technology Commission Fund of Shanghai (No. 20ZR1434800). The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Contributions
Conceptualization: Q.F., J.F. and Y.L.X. Formal analysis: B.W., Q.H.F., Y.X. Investigation: F.Q. and X.Q.Z. Writing-original draft preparation: Y.X. Writing-review and editing: Y.X., B.W., Y.L.X. and Q.H.F.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and informed consent
The study protocol was approved by the Ethics Committee of Shanghai Children’s Medical Center, School of Medicine, Shanghai Jiao Tong University(no. SCMCIRB-K2017008),and written informed consent was obtained from all participants or their legal guardians. All animal procedures were approved by the animal ethics committee of Shanghai Children’s Medical Center (no. SCMC-LAWEC-2019-015).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Fang, Q., Xu, Y., Feng, J. et al. Functional analysis of a novel pathogenic variant in CREBBP associated with bone development. Pediatr Res (2024). https://doi.org/10.1038/s41390-024-03490-z
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41390-024-03490-z
- Springer Nature America, Inc.