
Abstract
Camurati–Engelmann disease (CED) is an autosomal dominant bone dysplasia characterized by progressive hyperostosis of the skull base and diaphyses of the long bones. CED is further divided into two subtypes, CED1 and CED2, according to the presence or absence of TGFB1 mutations, respectively. In this study, we used exome sequencing to investigate the genetic cause of CED2 in three pedigrees and identified two de novo heterozygous mutations in TGFB2 among the three patients. Both mutations were located in the region of the gene encoding the straitjacket subdomain of the latency-associated peptide (LAP) of pro-TGF-β2. Structural simulations of the mutant LAPs suggested that the mutations could cause significant conformational changes and lead to a reduction in TGF-β2 inactivation. An activity assay confirmed a significant increase in TGF-β2/SMAD signaling. In vitro osteogenic differentiation experiment using iPS cells from one of the CED2 patients showed significantly enhanced ossification, suggesting that the pathogenic mechanism of CED2 is increased activation of TGF-β2 by loss-of-function of the LAP. These results, in combination with the difference in hyperostosis patterns between CED1 and CED2, suggest distinct functions between TGFB1 and TGFB2 in human skeletal development and homeostasis.
Similar content being viewed by others


Introduction
Camurati–Engelmann disease (CED; MIM #131300; also known as progressive diaphyseal dysplasia) is a rare autosomal dominant genetic disorder that is classified among the osteosclerotic disorders (Group 24) in the current International Classification of Genetic Disorders of the Skeleton [1]. CED is characterized by progressive hyperostosis of the skull base and diaphyses of the long bones [2, 3]. The onset of CED typically occurs in childhood. Symmetrical bilateral hyperostosis first occurs in the diaphyses of the femurs and tibias and then spreads to other long bones. As the disease progresses, the metaphyses may be affected, but the epiphyses are spared. Abnormal thickening of the bones, particularly in the legs and arms, can compress nerves and cause a variety of clinical problems, including stiffness and pain, cranial nerve impingement, elongated limbs with thin musculature (a marfanoid habitus), muscle weakness, easy fatigue, delayed sexual development, and bone marrow dysfunction such as anemia, leukopenia, and hepatosplenomegaly [4].
CED is clinically and genetically heterogeneous. The transforming growth factor beta 1 gene (TGFB1), which encodes TGF-β1, a pro-osteogenic growth factor, has been identified as the disease-causing gene in some CED patients [5, 6], but has been excluded in others [7]. Patients with or without TGFB1 mutations are classified as CED1 and CED2 (MIM: %606631), respectively [8]. CED2 patients have similar radiographic findings compared to CED1 patients, but may be distinguished by: (1) bone striations in the spine, pelvis, and long tubular bones; (2) epiphyseal sclerosis; (3) coarse, sclerotic trabeculae in the short tubular bones; and (4) minimal skull involvement [8]. The genetic cause of CED2 remains unknown.
In this study, we identified TGFB2 as the gene responsible for CED2. Heterozygous mutations predicted to alter the straitjacket region of the latency-associated peptide of TGF-β2 were identified in CED2, which is clinically distinct from CED1.
Materials and methods
Three CED2 pedigrees from different ethnic backgrounds were recruited in this study through the Japanese Skeletal Dysplasia Consortium. Two of them (Cases 1 and 3) were previously reported [8]. The differential diagnosis with CED1 was made based on the absence of TGFB1 mutation. The study protocol was approved by the Ethics Committee of RIKEN (Approval Number: H16-40 (12)) and the participating institutions.
Patients
Case 1
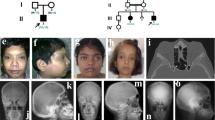
This patient was a 30-year-old Japanese woman, whose clinical and radiologic manifestations at the age of 6 years had been reported previously (Patient 2 in Nishimura et al. [8]). Briefly, she was first seen at the age of 2 years because of abnormal gait with muscle weakness, joint contractures, and occasional limb pain. Her height at 2 years of age was +0.6 SD and her weight was −0.6 SD. Limb pain was mild in the early school age. She had painful ankle joints only after long-distance walking; however, pain in thighs, knees, and ankles gradually increased from 12 to 16 years of age. At age 17, she was started on corticosteroid therapy (up to 20 mg/day), which was partially beneficial, then gradually reduced, and finally discontinued at age 23. Her height at 17 years of age was 167.8 cm (+2.0 SD) and her weight was 34.1 kg (−2.6 SD). Mitral valve prolapse was found at the age of 23 years. She underwent two bone biopsies for a small osteolytic lesion in the right fibula at age 11 years and an FDG-PET scan-positive lesion in the right radius at age 23 years. Histologic examination revealed only active bone remodeling. The radiologic manifestations at age 23 years (Fig. 1A–E) were essentially the same as those previously reported [8]. The spine, pelvis, and long tubular bones showed a coarse and striated trabecular pattern that included the epiphyses, in contrast to the CED1 phenotype. The long bones showed diaphyseal cortical hyperostosis.

Radiographic manifestations. Case 1 at age 23 years. Spine (A, B), leg (C), pelvis (D), and arm (E). Note a coarse and striated trabecular pattern including the epiphyses. The long bones exhibit diaphyseal cortical hyperostosis. Focal thickening with sclerosis in the shaft of the right fibula is likely a late consequence of a previous bone biopsy. Case 2: the right leg at age 7 months (F) showed a fracture of the distal femoral shaft and mild diaphyseal hyperostosis of the femur and tibia. The leg (G), arm (H), spine (I), skull (J), hand (K), and chest (L) at age 3–4 years: the axial and appendicular bones showed a coarse trabecular pattern including the epiphyses. The long bones showed diaphyseal cortical hyperostosis. The skull was normal. Case 3 at age 27. Mild scoliosis with minimal sclerosis in the spine and ribs (M) and hyperostosis with coarse trabecular pattern including epiphyses (N, O)
Case 2
The patient was a 4-year-old girl who presented with chronic leg pain and weakness since the age of 2.5 years. She was born normally to healthy unrelated parents of Moroccan Jewish origin. Her family history was unremarkable. She had a low-energy fracture of the right femur at 7 months of age. She had no other fractures. Mental development was normal. Her height was 95 cm (−2.0 SD) and her weight was 11 kg (−3.0 SD). She was slender and had mild dolichostenomelia. Muscle weakness and atrophy of the legs were evident. She had difficulty climbing stairs. She also had severe pain in the thighs and calves, especially when walking and at night. She did not have any joint contractures. Laboratory studies revealed slightly elevated ALP (322 U/L normal range: 38–113 U/L), low vitamin A (13.9 μg/dL; normal range: 20–100 μg/dL), and low serum IGF-1 (31.7 ng/mL; normal range: 55–248 ng/mL), but otherwise normal findings including serum creatinine, Ca, Pi, magnesium, PTH, CBC, ESR, and CRP. A bone marrow biopsy performed at the age of 3.1 years was normal. Radiographs of the right leg of patient 2 at 7 months of age showed a fracture of the distal femoral shaft and mild diaphyseal hyperostosis of the femur and tibia (Fig. 1F). Radiographs taken at 3–4 years of age (Fig. 1G–L) showed a coarse trabecular pattern of the axial and appendicular bones, including the epiphyses of the long bones. The long bones showed diaphyseal cortical hyperostosis. The skull was normal.
Case 3
This patient was a 38-year-old Japanese woman whose clinical and radiographic features up to the age of 17 years have been reported previously (Patient 1 in Nishimura et al. [8]). Briefly, she was born to healthy parents with an uneventful obstetric history. Early development was uneventful until the age of 4 when she presented with anorexia and a thin habitus. She was referred to us at the age of 14. Due to delayed puberty, she was started on estrogen therapy from age 14 until she reached her adult height of 168.5 cm, which was within her target range. Menstruation began spontaneously around age 20, although her cycle was irregular for a few years. At age 25, her menstrual cycle became regular. X-rays at age 27 showed mild scoliosis with minimal sclerosis in the spine and ribs (Fig. 1M). Bone striations were seen in both the pelvic and tubular bones (Fig. 1N). Hyperostosis of all long bones, including the epiphyses, was evident, which showed coarse and thick trabeculae and apparent increased cortical thickness (Fig. 1N, O). She was also seen by a pediatric cardiologist once a year for mild mitral valve regurgitation and mild annulo-aortic ectasia. She became pregnant without assisted reproductive therapy. No deterioration of her cardiac status was observed during her pregnancy. She gave birth to a healthy boy at the age of 37 years. Skeletal examination of the baby boy showed no significant findings. Currently, she complains of lower limb edema and pain, especially in the dorsal area of the foot. Although she has occasional pain around her hip, elbow, and hand joints, she can walk and climb stairs without assistance. On physical examination, she cannot make a tight fist or open her hands with straight fingers, suggesting either muscle weakness or joint stiffness.
Exome sequencing and variant calling
Peripheral blood was collected from patients and/or their family members with informed consent. Genomic DNA was extracted from the blood using the QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany). Exome sequencing was performed as previously described [9]. Briefly, DNA concentration was measured using a Qubit V.2.0 Fluorometer (Life Technologies, Carlsbad, CA, USA). DNA (3 µg) was sheared using an S2 Focused-ultrasonicator (Covaris, Woburn, MA, USA) and processed using the SureSelectXT Human All Exon V5 Kit (Agilent Technologies, Santa Clara, CA, USA). The captured DNA was sequenced using a HiSeq 2000 (Illumina, San Diego, CA, USA) with 101 bp pair-end reads and 7 indices. Image analysis and base calling were performed using HCS, RTA, and CASAVA software (Illumina, San Diego, CA, USA). Reads were aligned to the reference human genome (hg19) using Novoalign-3.02.04. Aligned reads were processed using Picard to remove polymerase chain reaction (PCR) duplicates. Variants were called by GATK v2.7-4 following the GATK Best Practice Workflow v3 [10] and annotated by ANNOVAR [11].
Evaluation of the mutation
All three pedigrees were nuclear trio families, so we considered all possible inheritance models, including the possibility of de novo mutations. We used the following databases for population frequency filtering: 1000 Genomes, ESP6500, gnomAD, ToMMo, HGVD, HLI, and an in-house control dataset containing exome data from 575 unrelated Japanese individuals. Variants with a population frequency >0.005 or 0.001 were removed for the recessive and dominant models, respectively. We evaluated the pathogenicity of the variants using SIFT, PolyPhen2, and MutationTaster for supporting evidence. Parameters were kept as default.
PCR and Sanger sequencing
The coding exons of TGFB2 were amplified by PCR and sequenced using the corresponding primers (Table S1). A 3730 DNA analyzer (Life Technologies, Carlsbad, CA, USA) was used for Sanger sequencing. Sequencher® v.4.7 (Gene Codes, Ann Arbor, MI, USA) was used to align sequencing chromatographs to reference sequences. Genetyx (Genetyx, Tokyo, Japan) was used for further sequence analysis.
In silico protein modeling
De novo structural prediction of the wild-type or mutant LAP monomers was performed using I-Tasser [12]. In silico tetra-structural evaluation of the identified variants was performed using PyMOL with the crystal structure of dimeric porcine pro-TGF-β1 (PDB: 3rjr) [13] as the homologous structure of human pro-TGF-β2. The free energy change for the missense mutation was calculated using FoldX software (version 4.0) [14].
TGF-β2 activity assay
We developed a TGF-β2 activity assay based on the previous protocols for TGF-β1 [6, 15]. Briefly, the full-length coding sequence (CDS) of wild-type TGFB2 was cloned into a pcDNA3.1(+) expression vector (V79020, Life Technologies, Carlsbad, CA, USA) using the primers KpnI_TGFB2_F and TGFB2_EcoRI_R (Table S2). The mutations of interest were introduced using the KOD-Plus mutagenesis kit (SMK-101, TOYOBO, Osaka, Japan) with the corresponding primers (Table S2).
We used the HEK293 cell line as a reporter cell because its background TGFB2 expression was very low. We evaluated whether the mutations could affect TGF-β2 secretion by measuring TGF-β2 activity in a conditioned medium (Fig. S1). The wild-type or mutant TGF-β2 expression plasmids were transfected into wells of pre-seeded HEK293 cells (TGFB2-expressing cells), the reporter vector (9xCAGA-luc) and internal control (pRL-TK) were transfected into other wells of pre-seeded HEK293 cells, using FuGENE® HD Transfection Reagent (E2311, Promega, Madison, WI, USA). One day later, the conditional medium was harvested from the TGFB2-expressing cells and exchanged for the corresponding reporter cells. The reporter cells were harvested the next day for luciferase assay. The luciferase expression of cells transfected with empty vectors represented the background level of free TGF-β. The level of free TGF-β2 released from the wild-type or mutant TGFB2 plasmids was represented by the arithmetic differences between the luciferase expression in the corresponding cells and the background. The reporter system could measure both free TGF-β1 and free TGF-β2 signals. A known CED1 mutation, p.R218C in TGFB1, was used as a positive control.
CED2 patient’s induced pluripotent stem cells (iPSCs)
We isolated lymphocytes from the peripheral blood of Case 3 and generated Epstein–Barr virus-transformed lymphoblastoid cell lines (LCL). The LCLs were then used to generate iPSC lines as previously described [16]. The patient iPSC lines were induced to become mesenchymal stem cells (MSCs) and stored in liquid nitrogen until use [17]. Osteogenic differentiation of MSCs was performed as previously described [17]. The status of osteogenic differentiation was confirmed in a time course by visualization of calcium deposits of the harvested cells using alizarin red staining [17]. Two different clones of the patient’s MSCs were used, and cells from an unrelated normal individual were used as a control. The experiment was duplicated for each clone or individual.
Results
Identification of mutations
For Case 1 and her parents, we performed exome sequencing and extracted candidate variants, considering all possible genetic models. After filtering for population frequency and removing non-flagged SNPs, we found that only one variant (a deletion of GATCGAGGC in chr1: 218520151–218520159, mapped to hg19) remained in the autosomal dominant de novo model. This in-frame deletion (c.112-120del) in exon 1 of TGFB2 (NM_003238) (Fig. 2A) was predicted to cause a loss of three amino acids (p.38_40delEAI, NP_003229; Fig. 2C). In other possible CED2 cases, we examined TGFB2 gene and found that c.106A > G (Fig. 2B; Fig. S2), a missense variant in exon 1 of TGFB2 (p.R36G; Fig. 2C), was present in two patients from unrelated families in Israel and Japan. This variant occurred de novo in each family. Both variants, c.112-120del and c.106A > G, were absent in the general population and were predicted to be disease-causing (Table S3).

TGFB2 mutations identified in this study. A Exome sequencing of Family 1. The red box showed the de novo heterozygous 9-bp deletion in Case 1. The default setting of NGS analysis was left-alignment, and the real impact of the mutation should be read as right-alignment, which was an in-frame deletion of three amino acids (EAI, marked in red) according to the HGVS nomenclature. B Sanger sequencing of Family 2. The red arrow shows the de novo heterozygous substitution. The altered amino acid is highlighted in red. C Distribution of point mutations and small indels of TGFB2. Red arrow: CED2 (Camurati–Engelmann disease, type 2) mutation; purple arrow: TAAD (thoracic aortic aneurysms and dissections) mutation. Sig. signal peptide; LAP latency-associated peptide. D The crystal structure of dimeric porcine pro-TGF-β1 (PDB code, 3rjr). Left: an overview of the pro-TGF-β1 complex, which is composed of a disulfide-bonded homodimer of the TGF-β1 proproteins, each of which contains protein regions of the LAP (magenta and cyan) and TGF-β1 (blue and yellow). Middle and right: close-up views of the altered amino acid residues, which are shown in van der Waals spheres in green (the substitution) or red (the deletion). The residues involved in a hydrophobic core around each of the variant sites (within 4 Å) are shown in translucent van der Waals spheres. The corresponding amino acid numbers of human TGF-β2 are shown in parentheses. E A schematic illustration of the large latent complex (LLC), which is composed of the small latent complex (SLC) and latent TGF-β binding protein (LTBP). SLC is composed of TGF-β and LAP produced from TGF-β pro-protein by proteolytic cleavage between the LAP and TGF-β regions. F Partial pairwise alignment of the wild type and the deletion mutant of the human TGF-β2 pre-pro-protein (Uniprot: P61812-1) containing the TGF-β2 pro-protein and its N-terminal signal peptide. The letter color: gray, the N-terminal signal peptide sequences; black, the LAP regions; orange, the cysteine residues involved in disulfide bond formation with LTBP; red, the amino acids predicted to be deleted in the variant
In silico protein modeling
The p.R36G and p.E38_I40del variants occurred within the α1 helix at the N-terminus of LAPTGFB2. The α1 helix was part of the straitjacket subdomain of LAPTGFB2, which is composed of seven α-helices. A de novo structural prediction of the LAPTGFB2 monomer showed that p.E38_I40del might cause the loss of one α-helix (Fig. S3). The pro-TGF-β proteins are normally processed with dimerization and excision to form the small latency complex (SLC) composed of a LAPTGFB dimer and a TGF-β dimer; SLC then binds to the latent TGF-β binding proteins (LTBPs) to form the large latency complex (LLC), and then be properly secreted and transported. TGF-βs are kept inactive in these latent complexes until appropriate signals cause conformational changes in the LAPs and release of TGF-βs [18]. The straitjacket subdomains on the LAPs are responsible for binding TGF-βs to form the SLCs and binding LTBPs to form the LLCs.
The crystal structure of human pro-TGF-β2 is not yet available; however, protein sequence alignment revealed extreme sequence similarity and evolutionary conservation of the straitjacket subdomains among pro-TGFBs from different species (Fig. S4). Therefore, we used the crystal structure of porcine dimeric pro-TGF-β1 (PDB: 3rjr) [13] as a model to analyze the quaternary structure of human pro-TGF-β2. The amino acids involved in the CED2 mutations, R36, E38, A39, and I40 in human pro-TGF-β2, corresponding to R45, E47, A48, and I49 in porcine pro-TGF-β1, respectively (Fig. 2D). The methylene part of the side chain of R45TGFB1/R36TGFB2 at the amino acid substitution site was involved in a hydrophobic core formed between the mature TGF-β domain and the LAP domain (Fig. S5). Thus, the R45GTGFB1/R36GTGFB2 mutation would destabilize the hydrophobic core and interfere with the formation of the SLC.
The deletion of E47_I49TGFB1/E38_I40TGFB2 created a spatial gap that would be filled by a shift of the N-terminal side of the α1 helix (Fig. 2F). As a result, the position of I49TGFB1/I40TGFB2 in the deletion mutant would be filled by I46TGFB1/I37TGFB2, and A48TGFB1/A39TGFB2 would be replaced by R45TGFB1/R36TGFB2 (Fig. 2F). However, the side chain of A48TGFB1/A39TGFB2 is involved in the formation of a hydrophobic core between mature TGF-β and the LAP (Fig. 2F). Replacement of this amino acid with the hydrophilic amino acid arginine would interfere with the interaction and disrupt the formation of the hydrophobic core. In addition, the α1 helix binds to the LTBP through a disulfide bond (as shown in Fig. 2E). The shortening of the α1 helix may affect the molecular interactions between the SLC and the LTBP. Thus, both variants are likely to destabilize the SLC and/or LLC, which we suspect would lead to the aberrant release of TGF-β.
TGF-β2 activity assay
To investigate the pathogenic mechanism of the identified TGFB2 mutations, we used an in vitro luciferase reporter system to evaluate downstream gene expression and quantify the level of free TGF-β2 in the cell culture medium. The level of free TGF-β2 in cells transfected with the TGFB2 plasmid containing the p.R36G or p.E38_I40del mutations was found to be 17.4-fold and 20.1-fold higher, respectively, than in cells transfected with the wild-type TGFB2 plasmid (Fig. 3). The increase in free TGF-β2 in cells transfected with TGFB2 plasmid containing the p.R36G or p.E38_I40del mutation was similar to that of the positive control (a known TGFB1 mutation causing CED1), which showed a 15.3-fold increase. This result suggested that CED2 could be caused by a similar pathogenic mechanism as CED1.

Increased TGF-β-SMAD signals of the TGFB2 mutations. Conditioned media of HEK293 cells transfected with WT or mutant TGF-β constructs were transferred to HEK293 cells transfected with a TGF-β responsive reporter construct, 9xCAGA-luc. The relative luciferase activities of the identified TGFB2 mutations were determined after 24 h. A known TGFB1 mutation (p.R218C) was used as a positive control. Vec. empty vector; WT wild type
Osteogenic differentiation of the patient’s iPS cells
We used iPS cells generated from the lymphocytes of Case 3 to validate the effect of high free TGF-β2 on osteogenesis. We compared the osteogenic differentiation process of the patient’s iPS cells with that of a control individual. The results showed that the iPS cells from the CED2 patient had significantly accelerated osteogenesis (Fig. 4), which could explain the osteopetrosis phenotype of the patient.

Osteogenic induction of the MSC of Case 3. A Schematic diagram of the manipulation and induction of the patient’s cells. PBL peripheral blood lymphocytes; LCL lymphoblastoid cell line; iPSc induced pluripotent stem cell; MSC mesenchymal stem cell. B Peripheral blood lymphocyte time course images of osteogenic differentiation of the MSC-derived lymphocytes. C-1 and C-2: MSCs derived from two independent iPSc clones. Ctrl control (MSC of a normal individual)
Discussion
In this study, we identified TGFB2 as the causative gene for CED2 and characterized the pathogenic mechanism by structure simulation, activity assays, and osteogenic differentiation of patient iPS cells. We showed that CED2 and CED1 have similar pathogenic mechanisms: i.e., the over-release of active TGF-β dimers due to mutations that affect the structure of the LAP. Mammals have three TGFB genes: TGFB1, TGFB2, and TGFB3. The C-termini of these genes encode mature (active) TGF-βs, which have similar sequences and structures (Fig. S4) and are involved in the development and maintenance of bone and other tissues. The remaining parts of the TGFB genes mostly encode LAPs, which are direct inhibitors of the corresponding TGF-βs.
Most CED1-related mutations occur in the LAP domain of pro-TGF-β1 and are thought to disrupt LAPTGFB1 function, leading to increased TGF-β1 activity and hyperostosis via the SMAD2/3 pathway [19]. Previous studies have shown two distinct pathomechanisms of the LAP mutations. One is a decreased binding capacity of the LAP with mature TGFβ1 due to its conformational change [6, 15]. Alternatively, the TGFβ1 mutation would result in the impairment of binding with LTBP, significantly lower levels of total TGF-β1, and misfolding of LAP [18].
TGF-βs initiate multiple downstream gene expressions and are involved in a variety of cellular processes, including cell growth, differentiation, and migration. Therefore, the release of active TGF-βs is well regulated, with only a small percentage being released in the activated form (<5% for TGF-β1 and <1% for TGF-β2). The SLC and LLC are a kind of safety measure to ensure correct spatio-temporal activation.
TGF-β1 and TGF-β2 are similar in sequence and structure, and function through the same receptor-signaling systems [20, 21]. However, the sequence homology of their LAPs is not so high (only 49%, Fig. S4). It is known that there are differences between the LAPs, such as the presence of a conserved RGD domain (an integrin binding site responsible for the release of TGF-β1) in LAPTGFB1 but not in LAPTGFB2. These differences may be related to the regulation of their specific activation patterns. The similarities between TGF-βs and the differences between LAPs (as well as other factors such as gene expression levels) contribute to the similar but distinct phenotypes of CED1 and CED2. Further basic research on TGFB2, such as its actual crystal structure, is needed to elucidate the detailed mechanism.
The TGFB2 mutations are a good example of how different mutation mechanisms can lead to different phenotypes. Prior to our identification of the CED2-related mutations, other heterozygous TGFB2 mutations had been reported in a number of autosomal dominant aortic aneurysm-related disorders, such as thoracic aortic aneurysms and dissections and Loeys–Dietz syndrome 4. The majority of these aortic aneurysm-related mutations are obvious loss-of-function mutations, as they include whole gene deletions, nonsense mutations, and frameshift mutations. In contrast, the CED2-related mutations may directly cause the observed phenotypes. The impaired LAP dimer cannot hold TGF-β2, leading to its pre-release during secretion and transport. As we have shown in our induction experiment using iPS cells, the pre-release of TGF-β can induce unexpected additional osteogenesis.
We confirmed the clinical and radiologic features of CED2, which may facilitate the differential diagnosis with CED1 in clinical practice. We followed up two previously reported Japanese cases (Cases 1 and 3) and recruited a new Israeli case (Case 2). Despite the large ethnic difference, the clinical and radiologic features of the new case were similar to those of previously reported CED2 cases [8]. Common clinical features included a waddling gait, muscle weakness, and severe leg pain (see Table 1). A marfanoid habitus was not evident. The radiologic findings of CED2 were similar to those of osteopathia striata with cranial sclerosis (MIM #300373): cortical thickening extending to the metaphyses of the long bones in addition to the diaphyses, coarse and thick trabeculae of the long and short tubular bones, striations in the spine, pelvis, and long tubular bones, and cranial sclerosis confined to the petromastoid regions. The differential diagnosis should also include Gaucher and related disorders and some variants of hypertrophic osteoarthropathy (defects in prostaglandin synthesis); the former is distinguished by the absence of splenomegaly and the latter by the absence of digital clubbing and normal skin.
Our research has identified the genetic cause of CED2 and characterized its pathogenic mechanisms, providing a foundation for further research on TGFB2 and its downstream genes. This could potentially aid in the development of drugs and treatments for this and other bone density disorders.
URLs
The Japanese Skeletal Dysplasia Consortium: http://www2.riken.jp/lab/OA-team/JSDC/
The 1000 Genomes Project (1000 genomes): http://www.1000genomes.org/
The NHLBI GO Exome Sequencing Project (ESP6500): https://evs.gs.washington.edu/EVS/
The Genome Aggregation Database (gnomAD): https://gnomad.broadinstitute.org
The Tohoku Medical Megabank Organization (ToMMo): https://www.megabank.tohoku.ac.jp
The Human Genetic Variation Database (HGVD): https://www.hgvd.genome.med.kyoto-u.ac.jp
The Human Longevity Inc. gene browser (HLI): http://hli-opensearch.com/
The Human Gene Mutation Database (HGMD):
https://portal.biobase-international.com/hgmd/pro/start.php
PyMOL: http://www.pymol.org/
References
Mortier GR, Cohn DH, Cormier-Daire V, Hall C, Krakow D, Mundlos S, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet Part A. 2019;179:2393–419.
Camurati M. Di un raro caso di osteite simmetrica erditaria degli arti inferiori. Chir Organ Mov. 1922;6:662–5.
Engelmann G. Ein Fall von Osteopathia Hyperosteotica (sclerotisans) multiplex infantilis. Fortschr Rontgenstr. 1929;39:1101–6.
Janssens K, Vanhoenacker F, Bonduelle M, Verbruggen L, Van Maldergem L, Ralston S, et al. Camurati–Engelmann disease: review of the clinical, radiological, and molecular data of 24 families and implications for diagnosis and treatment. J Med Genet. 2006;43:1–11.
Ghadami M, Makita Y, Yoshida K, Nishimura G, Fukushima Y, Wakui K, et al. Genetic mapping of the Camurati–Engelmann disease locus to chromosome 19q13.1-q13.3. Am J Hum Genet. 2000;66:143–7.
Kinoshita A, Saito T, Tomita H, Makita Y, Yoshida K, Ghadami M, et al. Domain-specific mutations in TGFB1 result in Camurati–Engelmann disease. Nat Genet. 2000;26:19–20.
Hecht JT, Blanton SH, Broussard S, Scott A, Rhoades Hall C, Milunsky JM. Evidence for locus heterogeneity in the Camurati–Engelmann (DPD1) syndrome. Clin Genet. 2001;59:198–200.
Nishimura G, Nishimura H, Tanaka Y, Makita Y, Ikegawa S, Ghadami M, et al. Camurati–Engelmann disease type II: progressive diaphyseal dysplasia with striations of the bones. Am J Med Genet. 2002;107:5–11.
Wang Z, Horemuzova E, Iida A, Guo L, Liu Y, Matsumoto N, et al. Axial spondylometaphyseal dysplasia is also caused by NEK1 mutations. J Hum Genet. 2017;62:503–6.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164–e164.
Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods. 2015;12:7–8.
Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, et al. Latent TGF-β structure and activation. Nature. 2011;474:343–9.
Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005;33:W382–W388.
Janssens K, ten Dijke P, Ralston SH, Bergmann C, Van Hul W. Transforming growth factor-β1 mutations in Camurati–Engelmann disease lead to increased signaling by altering either activation or secretion of the mutant protein. J Biol Chem. 2003;278:7718–24.
Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011;8:409–12.
Fukuta M, Nakai Y, Kirino K, Nakagawa M, Sekiguchi K, Nagata S, et al. Derivation of mesenchymal stromal cells from pluripotent stem cells through a neural crest lineage using small molecule compounds with defined media. PLoS ONE. 2014;9:e112291.
Walton KL, Makanji Y, Chen J, Wilce MC, Chan KL, Robertson DM, et al. Two distinct regions of latency-associated peptide coordinate stability of the latent transforming growth factor-beta1 complex. J Biol Chem. 2010;285:17029–37.
Saito T, Kinoshita A, Yoshiura KI, Makita Y, Wakui K, Honke K, et al. Domain-specific mutations of a transforming growth factor (TGF)-β1 latency-associated peptide cause Camurati–Engelmann disease because of the formation of a constitutively active form of TGF-β1. J Biol Chem. 2001;276:11469–72.
Sporn MB, Roberts AB, Wakefield LM, Assoian RK. Transforming growth factor-β: biological function and chemical structure. Science. 1986;233:532–4.
Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–29.
Acknowledgements
We thank all the patients for their participation. We thank Dr. Iida Aritoshi for his cooperation. This study was supported by research grants from the Japan Agency for Medical Research and Development (AMED) (Contract No. 14525125), the Japan Society for the Promotion of Science (WAKATE B, No. 17K16710), and the National Key Research and Development Program of China, RIKEN-MOST (SI, ZW; 2016YFE0128400).
Author information
Authors and Affiliations
Contributions
Conceptualization: SI; investigation: ZW, MS, TF, HN, TI, JX, LG, MH, KO; resources: MK, LZ, HO, YW, AK, KY, DC, GN; writing—original draft: ZW, SI; writing—review and editing: SI. All authors read and approved the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
About this article

Cite this article
Wang, Z., Kometani, M., Zeitlin, L. et al. Heterozygous mutations in the straitjacket region of the latency-associated peptide domain of TGFB2 cause Camurati–Engelmann disease type II. J Hum Genet (2024). https://doi.org/10.1038/s10038-024-01274-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s10038-024-01274-1
- Springer Nature Singapore Pte Ltd.