
Abstract
The brain–gut axis, a bidirectional network between the central and enteric nervous system, plays a critical role in modulating the gastrointestinal tract function and homeostasis. Recently, increasing evidence suggests that neuronal signaling molecules can promote gastrointestinal cancers, however, the mechanisms remain unclear. Aberrant expression of neurotransmitter signaling genes in colorectal cancer supports the role of neurotransmitters to stimulate tumor growth and metastatic spread by promoting cell proliferation, migration, invasion, and angiogenesis. In addition, neurotransmitters can interact with immune and endothelial cells in the tumor microenvironment to promote inflammation and tumor progression. As such, pharmacological targeting of neurotransmitter signaling represent a promising novel anticancer approach. Here, we present an overview of the current evidence supporting the role of neurotransmitters in colorectal cancer biology and treatment.
Similar content being viewed by others
Introduction
Strong evidence supports the critical role of the brain–gut axis (BGA) in modulating the gastrointestinal (GI) tract function and homeostasis. Several neurotransmitters have been proven to play a significant role in the regulation of physiological responses such as nutrient absorption, gut motility, the intestinal innate immune response, and microbiota profile, as well as having a role in GI pathophysiology [1]. In pathological conditions, including inflammatory states such as inflammatory bowel disease (IBD), neurotransmitter levels are often dysregulated, contributing to maintaining the inflammation-associated signaling feedback and determining a wide range of GI symptoms [1, 2].
Notably, neurodegenerative disorders, such as Parkinson’s disease (PD) and Alzheimer’s disease, have been linked to cancer risk, depending on different tumor types [3]. Furthermore, mutations and altered expression of core genes associated with the development of these neurological disorders have been found to be prevalent across human malignancies, highlighting their potential role in tumorigenesis and cancer biology through their effects on cell cycle control, protein turnover, mitochondrial functions, oxidative stress, inflammation, and key oncogenic pathways such as Wnt/β-catenin, JAK/STAT3, and EGFR-AKT [4, 5].
Neurotransmitters and neurotrophic factors are released by nerve and glial cells of the central and peripheral nervous systems. Additionally, non-neural cells including cancer and immune cells also have the ability to secrete these molecules. Current evidence supports the role of neurotransmitter signaling to activate cancer cell growth and metastatic spread by pleiotropic modulation of cell proliferation, apoptosis, autophagy, migration, invasion, epithelial to mesenchymal transition (EMT), and stemness [6]. Notably, neurotransmitter receptors are overexpressed in tumor cells, but can also be found on the membrane of endothelial and immune cells. Hence, neurotransmitters can exert both autocrine and paracrine cancer-promoting effects interacting with tumor cells and different cell components in the tumor microenvironment (TME). Interaction with endothelial cells and immune cells, in fact, promotes inflammation and tumor progression through a dynamic interplay involving stimulation of angiogenesis, recruitment of immune-suppressive cells, macrophage M2 polarization, extracellular matrix remodeling, and pro-inflammatory cytokine signaling [6].
These findings have led to a new domain in cancer research focusing on dissecting the role of neurotransmitters and their receptors in cancer initiation, progression, drug resistance and the development of novel therapeutic and preventive strategies that target these networks. Herein, we review the current evidence supporting the role of neurotransmitter signaling in colorectal cancer (CRC) biology (Fig. 1) and its potential implications in cancer therapy.

Regulatory signals related to tumor growth, apoptosis, autophagy, invasion, and metastasis may be transmitted through the BGA via the parasympathetic, sympathetic and enteric nervous systems. In addition, neurotransmitters and neurotrophic factors may be secreted from non-neural cells and exert both paracrine and autocrine effects on CRC cells, as well as immune cells, endothelial, and stromal cells in the TME. The balance between stimulatory and inhibitory signals through the activation of specific receptors can affect CRC progression and metastatic spread by promoting cancer cell proliferation, migration, and invasion, tumor angiogenesis and inflammation in the TME. ACh Acetylcholine, BGA brain–gut axis, BDNF brain-derived nerve growth factor, CRC colorectal cancer, DA dopamine, E epinephrine, EMT epithelial to mesenchymal transition, GABA gamma-aminobutyric acid, 5-HT serotonin, MDSC myeloid-derived suppressor cells, NE norepinephrine, TME tumor microenvironment. [Adapted from “Gut–Brain Axis” and “Tumor Microenvironment”, by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates.].
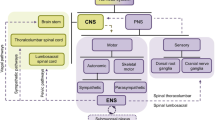
The brain–gut axis
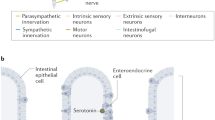
The GI tract presents a unique intrinsic nervous system, known as the enteric nervous system (ENS), which comprises several subtypes of neurons and glial cells organized in integrated circuits embedded in the walls of the digestive tract [7]. The ENS can independently modulate GI tissue dynamics and gut homeostasis while functioning in close communication with the brain. The central cognitive centers are connected with peripheral intestinal functions through the BGA, a bidirectional communication network composed of the central nervous system (CNS), the parasympathetic and sympathetic branches of the autonomic nervous system (ANS), the hypothalamic-pituitary-adrenal (HPA) system, and the gut microbiota [8]. Multiple neuroactive substances can be synthetized in the gut and affect the CNS by crossing the blood–brain barrier, whereas in exchange neuroactive molecules can affect the gut via the ANS [9]. This multidirectional crosstalk enhances the complexity of the interaction between enteric neurons and glial cells with GI mucosal cells, stromal, and immune cells in health and disease. GI cancers develop in the context of this intricate interface between ENS, CNS, gut microbiota, stromal and immune TME components. The interplay between the unique features of the BGA and tumorigenesis, progression, and metastases of GI cancers, however, remain to be fully elucidated as well as how to possibly leverage the underlying mechanisms for therapeutic purposes [10].
Electric stimulations and lesions in certain areas of the CNS have been shown to modulate peripheral natural killer (NK) cells cytotoxicity, which might in turn affect proliferation and metastasis of cancer cells. Additionally, signaling via the HPA axis in patients experiencing stress conditions or depression can impair DNA repair and increase angiogenesis through the release of catecholamine, most notably norepinephrine which increases VEGF expression via β-adrenergic receptor activation, and prostaglandins, which may result in enhanced cell survival and promote tumorigenesis [11].
The activation of the sympatho-adrenal axis of the ANS promotes GI tumorigenesis and chemical sympathectomy by means of 6-hydroxydopamin can reduce the incidence of CRC in rats [12]. Parasympathetic denervation by vagotomy and atropine administration results in significant reduction in tumor incidence, cell proliferation, tumor volume and weight, and angiogenesis mediated by downregulation of NGF, β2 adrenergic, and muscarinic M3 receptors [13].
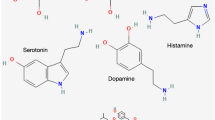
Growing evidence supports the role of neural signaling molecules, including neurotransmitters (such as dopamine, gamma-aminobutyric acid, acetylcholine, serotonin, epinephrine/norepinephrine, glutamate) and neurotrophic factors, in CRC development. Hereafter we review the main neural mediators in CRC (Table 1).
Dopamine signaling
Dopamine (DA) works as a neurotransmitter in the brain playing a critical role in several distinct pathways involved in behavioral control, motor control and in modulating the release of various hormones [14]. Outside the CNS, DA is synthesized peripherally and functions as a local chemical messenger modulating blood pressure, kidney function, and pancreatic insulin production [14]. In the gut, it reduces GI motility, modulates electrolyte exchange, and protects intestinal mucosa. Additionally, DA can inhibit the activity of lymphocytes by modulating cytokine secretion, chemotaxis and cytotoxicity [15]. DA exerts its cellular effects by binding to and activating cell surface G protein-coupled dopamine receptors (DR), classified into two families with distinct intracellular signaling pathways, known as D1-like (including receptors D1 and D5) and D2-like (including subtypes D2, D3, and D4) [16, 17]. D1-like receptor activation induces adenylyl cyclase activity translating into increased intracellular levels of cyclic AMP (cAMP) and downstream PKA signaling [16]. Conversely, D2-like receptors have inhibitory effects on adenylyl cyclase [16].
Previous studies reported that peripheral DA can control tumor progression and promotes anticancer immunity in the TME by modulating the NLRP3 inflammasome, regulatory and effector T cells, myeloid-derived suppressor cells (MDSC), and tumor-associated macrophages (TAMs) [18]. Activation of DRD1 and DRD5 inhibited cancer growth across several tumor types, including CRC, by suppressing Akt/mTOR signaling [19]. A small retrospective study reported a positive prognostic value for tumor gene expression of L-DOPA decarboxylase (DDC), an enzyme that catalyzes the decarboxylation of L-DOPA to DA, on disease-free survival and overall survival in 95 patients with CRC [20]. Furthermore, functional polymorphisms of DRD2 related to reduced receptor levels were associated with increased CRC risk in a case–control study involving 370 patients [21]. More recently, germline variants in the DA pathway genes have been associated with outcome in patients with metastatic CRC (mCRC) receiving first-line targeted treatment across three randomized trials [22].
Pre-clinical experiments using a CRC cell line HT29-derived xenograft mouse model suggest that DA can inhibit VEGF-mediated vasculogenesis, and can enhance 5-fluorouracil (5-FU) efficacy via DRD2-mediated signaling, resulting in strong inhibition of tumor cell proliferation and increased apoptosis in vivo [23]. On the other hand, pimozide, a FDA-approved drug used to treat psychotic disorders which selectively blocks DRD2, has been reported to suppress CRC cell lines HCT116 and SW480 proliferation and migration via inhibition of Wnt/β-catenin signaling [24] and to exert anticancer activity in vitro and in vivo in multiple tumor types by suppressing cell proliferation, EMT, and angiogenesis. Recently, an independent study reported that DRD2 knockdown inhibited β-catenin/ZEB1 mediated CRC cell proliferation and invasion in vitro and in vivo [25]. Consistently, pimozide enhanced the cytotoxic effects of 5-FU and oxaliplatin in vitro and suppressed tumor growth and metastasis in vivo [25]. DRD2 overexpression, on the other hand, increased CRC cell growth and EMT progression [25]. The same authors showed that DRD1-4 mRNA expression was higher in CRC tissue than adjacent normal tissue with DRD2 showing the highest expression and a strong association with tumor stage. High DRD2 expression was also associated with worse patient outcome in The Cancer Genome Atlas (TCGA) database [25]. DRD2 antagonism via the antipsychotic drug trifluoperazine (TFP) also inhibits CRC cell proliferation by inducing G0/G1 cell cycle arrest as well as promoting mitochondria-mediated intrinsic apoptosis [26]. In vivo CRC cell-derived xenograft models confirmed TFP anticancer activity. Notably, both programmed death ligand 1 (PD-L1) expression in CRC cells and PD-1 expression in tumor-infiltrating T cells were increased by TFP administration in vivo, suggesting a rationale for its combination with immune checkpoint inhibitors [26].
Clarifying the precise signaling mechanisms by which DR modulators exert their anticancer effect is paramount to support the implementation of dopaminergic drugs in CRC treatment. Nevertheless, these data provide proof that targeting the DA signaling may represent a novel therapeutic strategy in CRC which warrants further exploration.
Parkinson’s disease
Parkinson’s disease (PD) affects 1–2 per 1000 individuals in the general population and up to 2% of those aged over 65 years, ranking second among the most common age-related neurodegenerative disorders [27]. Notably, the hallmark of PD is the loss of dopaminergic neurons in the substantia nigra of the brain. The genetics of sporadic and hereditary PD have been extensively studied, identifying several specific disease loci and causal genes [28]. Over the past 10 years, several epidemiological studies have consistently reported an inverse association between PD and cancer risk, although a positive association with certain cancers including melanoma, breast, and brain tumors, has also been reported [3, 29]. The biology behind this epidemiological evidence is mostly unknown, although several PD-related genes and PD-driver gene alterations (including SNCA, PARK2, LRRK2, PINK1, and DJ-1) have been linked to carcinogenesis in different tumor types [4, 5].
CRC is among the most widely reported cancer types showing a reduced incidence in PD patients, with a relative risk of 0.78 (0.66–0.91) compared to controls [30]. However, no data are available addressing the underlying mechanisms and possible biologic rationale of the inverse association between PD and CRC risk. Interestingly, stool-based methylation testing of alpha synuclein (SNCA), one of the causal genes most frequently mutated in PD, has been proposed as an effective diagnostic tool for CRC screening and early detection, and higher methylation levels have been observed in CRC patient tissue samples compared with paired controls [31]. Furthermore, recent data suggests that the SNCA protein, whose aberrant aggregation in CNS neurons leads to PD development, accumulates in the appendix of healthy subjects and a prior appendectomy has been reported to be associated with a decreased risk of PD development [32]. PD-related genes and genes variants have been linked to IBD risk and IBD phenotypes, although epidemiological evidence on this topic appears to be conflicting. More recently, SNCA genetic polymorphisms and gene expression alongside other core PD-related genes (PINK1 and LRRK2) have been associated with clinical outcome in patients with mCRC receiving first-line treatment [33]. Particularly, high SNCA expression was significantly associated with shorter progression free survival and overall survival in patients treated with anti-epidermal growth factor receptor (EGFR)-based therapy [33].
Further exploration of the interplay between PD pathophysiology and CRC may contribute to understand the role of the autonomic nervous system dysfunction in CRC development.
Monoamine oxidases
Monoamine oxidase (MAO) isoenzymes MAO-A and MAO-B are mitochondrial enzymes responsible for catalyzing the oxidative deamination of monoamines such as DA, norepinephrine, and serotonin. These isoenzymes play important functions in the metabolism of neuroactive and vasoactive monoamines in the CNS and peripheral tissues [34]. Altered expression of MAOs were found in several cancer types and have been connected to tumor development and progression. MAO-B was highly expressed in CRC compared to normal tissue in a study including 203 CRC cases [35]. High MAO-B was associated with worse disease stage, higher recurrence rates and poorer survival in CRC. Additionally, positive and negative correlations of MAO-B expression with mesenchymal-type and epithelial-type gene expression, respectively, have been reported, highlighting a potential role in EMT and invasion [35].
Notably, both MAO-A and MAO-B inhibitors (MAOI), including drugs developed for the treatment of neuropsychiatric and neurodegenerative disorders such as PD, have been reported to exert anticancer activity in in vitro and in vivo models, and phase II clinical trials are ongoing in prostate cancer (NCT02217709, NCT04586543) [36]. MAO-A has also been shown to affect TAMs immunosuppressive polarization by increasing intracellular reactive oxygen species (ROS) leading to oxidative stress, and Maoa knockout in mouse models consequently enhanced anti-tumor immunity [37]. Furthermore, MAO-A could directly regulate CD8+ T cells and suppress the tumor-infiltrating T-cell immune response by negative modulation of T-cell autocrine serotonin signaling [38]. Treatment with MAOI in combination with immune checkpoint inhibitors has been explored showing promising efficacy in pre-clinical models and suggesting that this combination may result in synergistic anticancer activity [37].
Gaba signaling
Gamma-aminobutyric acid (GABA) is one of the major inhibitory neurotransmitters in the CNS, but it also has many functions within the homeostasis of the GI tract [39]. GABA is present throughout the GI tract in enteric nerves and enteroendocrine cells (EC) and is involved in both motor and secretory activity, which is mediated via GABA receptor activation. In the context of GI diseases, and more specifically CRC, the role of GABA is less well understood. Several studies have identified GABA levels to be higher in CRC than in normal colon tissue [40]. In addition, increased GAD1 levels, the enzyme that produces GABA from glutamate, have been correlated with worse survival in patients with stage T3/T4 CRC [41]. A separate study also found ABAT, the enzyme that catabolizes GABA, to be increased in CRC as compared to normal tissue [42]. These studies provide evidence of a GABAergic environment in CRC with tumors expressing genes to both synthesize and catabolize GABA in the TME.
Despite this clinical evidence, mechanistic pre-clinical studies exploring the role of GABA in CRC have been conflicting. Exogenously adding GABA to CRC cells lines in vitro has produced varying effects on proliferation, migration, and invasion. One group reported that 5-FU resistant HT29 tumor cells showed reduced proliferation in the presence of GABA; interestingly, GABA had no effect on parental HT29 tumor cells [43]. An independent study showed no effects on proliferation or migration in SW480 tumor cells treated with GABA, however when the same tumor cells were stimulated with norepinephrine, GABA reduced the norepinephrine-mediated increased migration [44]. The previous two studies suggest that GABA alone is not enough to influence tumor behavior; yet, perturbations to the system, such as drug treatments or other signaling molecules, may influence how tumor cells respond to a GABAergic environment. However, others have shown that GABA alone was able to reduce proliferation of HCT116, SW620, and SW480 tumor cells, reduce migration and invasion of SW480 and SW620 tumor cells, and reduce SW480 tumor growth in a xenograft nude mouse model, suggesting GABA may have an inhibitory role on cancer progression [45]. While slight differences in the invasion and migration assays performed in the Joseph et al. and Song et al. studies may be contributing to the reported responses to GABA, more work needs to be done to understand the role of GABA in CRC progression.
Signaling of GABA can occur through two main GABA receptors: the ionotropic GABA RA and the metabotropic GABA RB [46]. One CRC study focused on GABA RA, showing that propofol, a GABA RA agonist, decreased invasion in LOVO cells [47]. However, much of the in vitro GABA receptor literature in CRC has focused on GABA RB. Activation of GABA RB via agonists, such as baclofen or nembutal, reduces CRC proliferation, invasion, and metastasis [48]. Another group showed knockdown of GABABR1 (a subunit of GABA RB) in LOVO and RKO CRC tumor cells increased proliferation, migration, invasion, and markers of EMT, suggesting subunits of GABA RB could have anti-tumor effects [49]. Alternatively, a recent study found that exogenous GABA activates GABA RB, leading to GSK-3β inhibition and increased CRC cell proliferation [40]. Additionally, they showed elevated GAD1 expression in colon adenocarcinoma cells, which led to increased GABA secretion. This research links the prior reports of elevated GAD1 and GABA levels seen in CRC patient tissues with GABA receptor activity.
While in vitro studies have focused mainly on GABA RB, analyses utilizing patient tissue found that expression of several subunits of GABA RA, including GABRD, was increased in CRC tissue and predicted worse patient outcome [50]. When Liu and Fang performed a meta-analysis of several patient cohorts, they found that GABRD is highly expressed in colon cancer patients, but those with lower GABRD expression had better overall survival and prognosis. In addition, when the authors focused on genes that were co-expressed in patients with high GABRD, they found pathways related to endothelial cell development and vasculogenesis, extracellular matrix (ECM) interactions, human papillomavirus infection, growth factor and kinase binding, and Notch signaling [51], suggesting interactions with the TME are related to the GABAergic changes within CRC.
While most GABA-related CRC research has focused on the tumor cells, a recent study demonstrated that B cells within lymph nodes secrete GABA when activated to promote a pro-tumor immune environment. Importantly, the authors showed that picrotoxin, a GABA RA antagonist, reduced tumor growth in the MC38 CRC syngeneic mouse model. In addition, knocking out GAD67 (GAD1) within the B cells lowered GABA levels and controlled tumor growth [52]. In the recent Huang et al. study described above, researchers also found that a GABAergic tumor resulted in less T-cell infiltration and that targeting GAD1 or GABA RB overcame resistance to anti-PD-1 immunotherapies in a mouse model [40]. This study begins to elucidate the role of GABA-producing immune cells within the tumor (versus in the lymph nodes as studied by Zhang et al.); however, more research is needed to understand the GABAergic crosstalk within the TME and how this connects to metastatic spread. Additionally, the presence of the microbiome in the gut adds an additional layer of complexity to the immune-tumor signaling, as the microbiome has been shown to secrete GABA and can alter tumor growth [43].
Acetylcholine signaling
Acetylcholine (ACh) functions in the ANS as a neurotransmitter at the autonomic ganglia, the parasympathetic innervated organs, and the neuromuscular junction between motor nerves and skeletal muscle. Acetylcholine receptors (AChRs) fall into one of two categories; the relatively slow activating G protein-coupled metabotropic muscarinic receptors or the faster activating ionotropic nicotinic receptors (nAChRs) [53].
Nicotinic signaling
nAChRs are composed of pentamer transmembrane protein complexes with five receptor subunits that mediate fast synaptic transmission through their ionotropic cationic nicotinic receptors. Calcium influx through these receptors facilitates signal transduction resulting in the release of neurotransmitters including catecholamine neurotransmitters norepinephrine and epinephrine, which bind to and activate β-adrenergic receptors. β-adrenergic receptors can then activate downstream signaling pathways leading to increased intracellular cAMP formation, which can have tumor-promoting effects [54]. In addition to their role in synaptic transmission in the neuronal tissues and neuromuscular junctions, nAChRs are found in cells with epithelial and endothelial origin and play a role in biological processes such as cell proliferation, with overexpression promoting tumor cell proliferation and invasion in various cancers [55]. ACh, nicotine, and nicotine-derived carcinogenic nitrosamine nicotine ketone (NNK) can activate the nAChRs. Of note, cancer cells, including CRC cells, are able to independently synthesize ACh which then acts as an autocrine/paracrine growth factor to promote tumor growth [56].
Growing evidence and interest has developed with regards to understanding the precise mechanisms of nAChRs in cancer initiation, progression, and metastasis. The alpha7-subtype of nAChR (α7nAChR) has been identified as a prominent player in cancer development by directly synthesizing autocrine growth factors and indirectly stimulating the release of norepinephrine and epinephrine, which in turn can promote cell survival, proliferation, migration and angiogenesis [55]. On the other hand, the heteromeric α4β2nAChR has been established to have anticancer effects by stimulating the release of GABA, which inhibits cAMP, thereby blocking the cancer-promoting signaling initiated by β-adrenergic receptors [55]. Notably, cancer-stimulatory nAChRs are upregulated by the chronic exposure to nicotine and nitrosamine carcinogens, whereas inhibitory receptors undergo desensitization [57].
Overexpression of α7nAChR has been found in CRC cells as well as in tumor-infiltrating immune cells [58]. Receptor activation has been shown to promote CRC cell proliferation, inhibit apoptosis and may increase CRC cell migration and metastasis through the upregulation of fibronectin [59]. Reports also highlight that NNK promotes CRC growth in vitro by increasing α7nAChR mRNA expression and enhancing NF-KB DNA binding activity, along with cyclooxygenase-2 (COX-2) and 5-lipoxygenase protein expressions [60]. Further experiments demonstrated that the use of α7nAChR antagonists and α7nAChR siRNA methods successfully inhibit nicotine-stimulated CRC cell proliferation and migration confirming α7nAChR’s critical role in nicotine and NNK pro-oncogenic signaling in CRC [61]. Emerging evidence shows that nicotine stimulates human CRC cell line HT29 proliferation and epinephrine production, mediated by β-adrenoceptors [61]. Additionally, nicotine promoted tumor growth in CRC patient-derived xenograft (PDX) models via stimulation of β-adrenoceptors and the subsequent activation of COX-2, PGE2, and VEGF expression [62]. Notably, in vitro nicotine-dependent stimulation of CRC cell invasion and migration has been reported to be mediated by the activation of p38 MAPK signaling downstream of nAChRs with subsequent increase of matrix metalloproteinases expression [63].
The α7nAChR receptor also plays a critical role in the regulation of the inflammatory response in the TME by the cholinergic anti-inflammatory pathway [64]. Evidence shows that ACh binding to α7nAChR stimulates the cholinergic anti-inflammatory pathway output, which is thought to downregulate GI inflammation through vagal signaling [65]. α7nAChR is required for Ach-mediated inhibition of TNF release from macrophages and cytokine modulation in inflammatory states [66]. As such, therapeutic approaches have tried to exploit α7nAChR’s anti-inflammatory function for the treatment of inflammation-based disorders [67]. Interestingly, a study reported that nicotine could suppress colitis-associated tumorigenesis in mice and inhibited CD4+ T cells secretion of pro-inflammatory cytokines [68]. This evidence supports a dual role for α7nAChR in CRC, possibly dependent on underlying inflammatory bowel conditions and immune TME dynamics, which may complicate its use as a therapeutic target.
Therefore, despite significant evidence that α7nAChR blockage may be an effective anticancer strategy in CRC, targeting its downstream oncogenic effects may reveal to be challenging while maintaining a necessary balance between stimulatory and inhibitory signals involved in tumor progression and inflammatory reaction control.
Muscarinic signaling
The mAChRs are G-protein-coupled receptors classified into five subtypes: M1-M5, with distinct intracellular downstream signaling [69]. Receptors M1, M3, and M5 activation triggers the PLC pathway, eventually resulting in opening of calcium channels, leading to increased cell viability. Conversely, M2 and M4 receptors have inhibitory activity on adenylyl cyclase leading to reduced intracellular cAMP [69].
The M3 receptor subtype has been found to be overexpressed at both RNA and protein levels in CRC samples [70], and its cancer-promoting effect on tumor growth and metastasis has been established [71]. Investigations highlight that M3 receptor activation stimulates CRC cell proliferation, tumorigenesis, cell migration and invasion [72, 73]. Notably, Von Rosenvinge et. al described the role of mAChRs in EGFR transactivation in CRC cells, where M3 activation triggers matrix metalloproteinase 7 (MMP-7)-mediated cleavage of the heparin binding-EGF which in turn initiates the EGFR signaling cascade through the MEK/ERK and PI3K/Akt pathways [74]. Furthermore, vagal innervation has been shown to contribute to gastric tumorigenesis through M3 receptor-mediated activation of Wnt signaling [75].
While significant evidence highlights the ability of mAChR agonists to promote cancer growth, studies also report that selectively blocking M3-mediated signaling shows promising anticancer effects. In vitro experiments utilizing darifenacin, an M3 receptor antagonists, in CRC cell lines HT29 and SW480 resulted in a dose-dependent decrease of tumor cell proliferation and survival [76]. Darifenacin suppressed ACh-induced p38, ERK1/2, and Akt signaling, inhibiting cell invasion and MMP1 mRNA expression [76]. Additionally, it inhibited tumor growth and metastases in a xenograft mouse model [76]. M3 receptor knockout has also been shown to suppress CRC tumorigenesis in vivo, strengthening the rationale for exploring the use of M3 antagonists in CRC treatment [77].
Conversely, M1 receptors have been found to be downregulated in CRC and negatively associated with β-catenin expression [78]. In fact, despite similar receptor structures and signaling, M1 and M3 receptor activation has been reported to have opposite effects with M1 being protective against CRC tumorigenesis [79]. The mechanisms behind this divergent behavior are not clear, nevertheless, this may provide a rationale for potentially combine subtype-selective targeting of M1 and M3 receptors warranting further exploration.
In addition to ACh and muscarine, select bile acids can also interact with mAChRs, possibly due to ligand molecular mimicry, and thus initiate post-receptor signaling [80]. Evidence suggests that bile acids may promote normal colonic epithelial cells transformation into CRC stem cells through the M3 receptor and Wnt/β-catenin signaling [81]. As such, activation of M3 cancer-promoting downstream signaling may partially be responsible for the increased incidence of CRC associated to diets high in saturated fats, which are known to increase bile acids secretion [82].
Serotonin signaling
Serotonin (or 5-hydroxytryptamine, 5-HT) is one of the most potent neural, peripheral, and GI signaling molecules. Intestinal EC produce the greatest amount of 5-HT in the human body, however, serotonergic neurons in the CNS and enteric neurons also synthesize 5-HT. 5-HT receptors, comprising seven distinct classes (5-HT1–7), are G-protein-coupled receptors, excluding the ligand-gated ion channel 5-HT3, and are widely expressed within the GI tract, where 5-HT3 and 5-HT4 subtypes have been the most extensively studied and targeted for the treatment of GI motility disorders [83]. Notably, gut microbiota can promote 5-HT synthesis and gut dysbiosis affects 5-HT signaling in the GI tract through a bidirectional crosstalk with EC and the ENS [84]. 5-HT binding to its receptors promotes a range of pleiotropic functions at central and peripheral level including modulation of circadian rhythms, gastrointestinal motility, cardiovascular homeostasis, angiogenesis, neuroendocrine regulation, immunomodulation, intestinal microbiome homeostasis, epigenetics, and cancer [85].
Increased 5-HT plasma levels as well as upregulation of the expression of tryptophan hydroxylase 1 (TPH1), the rate-limiting enzyme for 5-HT biosynthesis, have been found in CRC tumor tissues from patients, CRC mouse models and CRC cell lines as compared to controls [86]. In another study, high 5-HT levels were significantly associated with advanced tumor node metastasis and had a high predictive value for poor patient recurrence-free survival and overall survival [87]. Upregulation of 5-HT1D, 5-HT3C, and 5-HT4 protein level has been reported in CRC samples, with 5-HT1D being the highest. In vitro experiments showed that 5-HT1D can promote tumor invasion by activating the Axin1/β-catenin/MMP-7 pathway. Consistently, 5-HT1D inhibition suppressed tumor metastasis in vivo through targeting of Axin1 [87]. 5-HT1B has also been found to be upregulated in CRC cell line HT29 and CRC tissue and its selective inhibition had anti-proliferative and apoptotic effects on CRC cells [88]. In addition, serotonin has been reported to modulate angiogenesis by reducing TAMs expression of matrix metalloproteinase 12 (MMP-12) and to be required for tumor growth in a CRC allografts model [89]. More recently, 5-HT receptors 5-HT1B, 1D, and 1F have been shown to be highly expressed in CRC stem cells (CSC) and to activate Wnt/β-catenin signaling in response to 5-HT stimulation resulting in CSC self-renewal and tumorigenesis [90]. Notably, in this study, 5-HT production by enteric serotoninergic neurons was promoted by isovalerate, a tumor-associated microbiota metabolite. Furthermore, inhibition of 5-HT signaling in mice models suppressed the self-renewal of CSC and exhibited anti-tumor activity against CRC by suppressing tumor progression and metastasis [90].
On the other hand, 5-HT has also been suggested to play a key role in intestinal protection from early colorectal tumorigenesis by promoting DNA repair [91]. Additionally, the use of selective 5-HT reuptake inhibitors (SSRIs) has been associated with a dose-dependent reduction of CRC risk in large patient studies [92]. Evidence on a dual role of serotonin in CRC development is further supported by the effects of SSRI fluoxetine in CRC models. This drug increases 5-HT levels and exerts anticancer activity in vivo by impairing mitochondrial reactive oxygen species production, cell cycle progression and proliferation of CRC cells, especially in hypoxic conditions, leading to reduced microvessel formation and tumor shrinkage [93].
It has recently been suggested that 5-HT-induced cancer-promoting effects are closely related to 5-HT1 and 5-HT2 signaling rather than 5-HT3, 5-HT4, 5-HT6, and 5-HT7 [94]. This is consistent with the previously discussed evidence in CRC. Notably, treatment with mirtazapine, an inhibitor of 5-HT2, resulted in reduced growth by direct modulation of immunological mechanisms in the TME of subcutaneous CRC tumor allograft models [95]. Immunological cancer-promoting effects mediated by 5-HT on the TME have been also reported in a recent independent study showing that elevated 5-HT levels activated lymphocytes cytokine release leading to a pro-inflammatory immune microenvironment permissive to CRC tumorigenesis [96].
Current evidence illustrates the complexity of serotonergic activity in CRC biology. 5-HT-mediated signaling could act protectively against early carcinogenesis in the colonic mucosa, whereas it might support CRC metastatic progression in advanced disease. The activation of differential intracellular signaling cascades triggered by different receptor subtypes could contribute to explain these findings. As such, selective targeting of serotonin receptors and other mediators of serotoninergic signaling might translate into an effective treatment approach for CRC once their specific role is fully understood.
Neurotrophic factors
Neurotrophic factors are a family of neurotrophic molecules which can be secreted by cancer cells to promote the growth of nerves within the tumor and at the same time have autocrine and/or paracrine effects on tumor growth and metastatic spread [97]. The family includes transforming growth factors, glial cell-derived neurotrophic factors, neurotrophins, and neuropoietins [98].
Neurotrophins are classified into four types: brain‑derived neurotrophic factor (BDNF), nerve growth factor (NGF), neurotrophin‑3 (NT‑3) and NT‑4. Their downstream signaling is mediated by tyrosine kinase receptors, including TrkA, TrkB and TrkC, which can activate the PI3K/Akt, MAPK, and PLC-γ intracellular pathways [99]. BDNF and TrkB have been extensively investigated, with multiple studies consistently reporting their upregulation in several tumors, including CRC, associated with aggressive phenotypes and chemoresistance [100]. In a study, BDNF and TrkB knockdown in CRC cell lines Caco-2 and HRT18 increased apoptosis and significantly decreased cell growth and proliferation [101]. High tumor TrkB mRNA expression has been associated with poor prognosis in CRC patients and TrkB has been linked to EMT in CRC cells [102]. An independent study reported that BDNF promoted CRC cells HCT116 and SW480 migration through ERK-, p38-, and PI3K/Akt-mediated activation of heme oxygenase-1 and VEGF expression [103]. Additionally, BDNF/TrkB signaling has also been demonstrated to transactivate EGFR and to directly activate RAS [100]. Interestingly, evidence shows that BDNF can promote the release of ACh and ACh can upregulate both BDNF and NGF activating NGF/TrkA signaling, which has been reported to promote tumorigenesis, cell proliferation and survival in cancer [104, 105]. Notably, NTRK fusions, which involve rearrangements of the genes encoding for Trk receptors (i.e., NTRK1, NTRK2 and NTRK3), have emerged as rare but actionable targets in cancer, including CRC. Two small molecule inhibitors (entrectinib and larotrectinib) have already been approved by the Food and Drug Administration (FDA) for the treatment of advanced solid tumors harboring NTRK1/2/3 fusions [106]. As such, next-generation Trk inhibitors are being explored to overcome acquired resistance to first-generation agents [107].
The reelin signaling pathway is critical for neural progenitors migration during neurogenesis and impaired signaling has been implicated in the pathogenesis of numerous neuropsychiatric and neurodegenerative disorders, including autism, schizophrenia and Alzheimer’s disease [108]. Reelin belongs to the family of extracellular matrix glycoproteins and acts by initiating the activation of ApoE receptor 2 (ApoER2), very low density lipoprotein receptor (VLDLR), and the cytoplasmatic docking protein Disabled‐1 (Dab1), which control multiple intracellular pathways [109]. Increasing evidence suggests that reelin signaling may play a role in cancer development. Epigenetic silencing of reelin by promoter hypermethylation at CpG islands sites has been reported to frequently occur and to increase migration, invasiveness and reduce survival in breast, gastric and pancreatic cancers [110,111,112]. Reelin has also been shown to be able to abrogate RAS/PI3K mediated cell motility, thus potentially playing a critical role in tumor metastatic spread [113]. Notably, the Reeler mutation, which determines the loss of reelin function, compromises the intestinal barrier and promotes colitis-associated tumorigenesis in mice models [114]. On the other hand, in a small CRC study comparing genomic and transcriptional profiles of primary tumor and matched metastases, the reelin pathway was found to be differentially upregulated in metastases [115]. Based on current evidence, it appears that the reelin pathway may play a dual role in CRC where downregulation and upregulation of gene expression may alternatively promote tumor progression at different disease stages, which will need to be further addressed to define the therapeutic potential of targeting this pathway in CRC.
Expert opinion and future perspectives
Cancer neuroscience is emerging as an innovative field of research in oncology with a potential to identify novel therapeutic targets in the TME. This is particularly relevant for CRC given the unique role of the BGA in GI physiology and pathology. The increasing attention on the essential role of the TME in cancer has shed light on the complex contribution provided by neural mediators to CRC growth and progression. Further studies are needed to fully understand the underlying biology, nevertheless, this expanding knowledge is opening the door to the development of novel therapeutic strategies potentially exploiting repurposed neuroactive drugs as an anticancer approach.
A challenge that neurobiology research in CRC faces is the discrepancy between in vitro and in vivo data which may, in part, be due to the lack of physiological relevance within traditional in vitro experiments and the inability to consider the TME context in many in vitro experiments. Microenvironmental factors such as endothelial cells, biophysical forces (ECM, stiffness, and mechanical forces), immune cells, and the microbiome all need to be considered when addressing the role of neurotransmitters and neural factors within CRC progression. Physiologically relevant model systems that incorporate aspects of the TME in a tunable fashion might help elucidate the role of neurotransmitters in CRC. For instance, an organ-on-chip model that recapitulates the structure and function of the colon, such as tissue-tissue interfaces, 3D structures, and mechanical forces including fluid flow and peristalsis may address existing knowledge gaps. The potential dual role of neurotransmitters signaling in primary and metastatic disease, the differential effect based on the interaction with different receptor types and the complex interplay between stimulatory and inhibitory signals, as well as a potential organ-specific impact of these pathways should also be taken into account and carefully explored in pre-clinical studies. Finally, the safety profile and potential central and peripheral neurological adverse effects in vivo will have to be carefully assessed when using neuroactive drugs for cancer treatment.
Notably, targeting tumor neurotransmitter signaling and neurotrophic factors in the TME holds promise to be effective alone or in combination with targeted therapies. In fact, a close connection between neural signaling molecules and known druggable cancer-related pathways has been established, particularly angiogenesis, RAS/MAPK signaling and immunomodulation. Therefore, pharmacological manipulation of neurotransmitter pathways may improve the efficacy of existing targeted treatments. Dedicated studies may provide further insights on these possible synergistic effects and establish the rationale for the design of successful combination strategies advancing the therapeutic horizon in CRC treatment.
Conclusions
The BGA is a complex bidirectional signal transmission between the CNS, the ENS, and the endocrine-immune system, which has been demonstrated to play an important role in CRC tumorigenesis and development. Growing evidence supports the critical role of several neurotransmitters and neural factors in CRC biology, opening novel perspectives which warrant dedicated studies to elucidate the underlying mechanisms. The integration of a neurobiological view into CRC research may further innovative therapeutic advances by leveraging the unique interplay between neural signaling and key oncogenic pathways and the cellular crosstalk in the TME.
References
Mittal R, Debs LH, Patel AP, Nguyen D, Patel K, O’Connor G, et al. Neurotransmitters: the critical modulators regulating gut-brain axis. J Cell Physiol. 2017;232:2359–72.
Agirman G, Yu KB, Hsiao EY. Signaling inflammation across the gut-brain axis. Science. 2021;374:1087–92.
Catalá-López F, Suárez-Pinilla M, Suárez-Pinilla P, Valderas JM, Gómez-Beneyto M, Martinez S, et al. Inverse and direct cancer comorbidity in people with central nervous system disorders: a meta-analysis of cancer incidence in 577,013 participants of 50 observational studies. Psychother Psychosom. 2014;83:89–105.
Feng DD, Cai W, Chen X. The associations between Parkinson’s disease and cancer: the plot thickens. Transl Neurodegener. 2015;4:20.
Ejma M, Madetko N, Brzecka A, Guranski K, Alster P, Misiuk-Hojło M, et al. The links between Parkinson’s disease and cancer. Biomedicines. 2020;8:416.
Jiang S-H, Hu L-P, Wang X, Li J, Zhang Z-G. Neurotransmitters: emerging targets in cancer. Oncogene. 2020;39:503–15.
Furness JB. The enteric nervous system and neurogastroenterology. Nat Rev Gastroenterol Hepatol. 2012;9:286–94.
Tait C, Sayuk GS. The brain-gut-microbiotal axis: a framework for understanding functional GI illness and their therapeutic interventions. Eur J Intern Med. 2021;84:1–9.
Margolis KG, Cryan JF, Mayer EA. The microbiota-gut-brain axis: from motility to mood. Gastroenterology. 2021;160:1486–501.
Schledwitz A, Xie G, Raufman JP. Exploiting unique features of the gut-brain interface to combat gastrointestinal cancer. J Clin Investig. 2021;131:e143776.
Lutgendorf SK, Sood AK. Biobehavioral factors and cancer progression: physiological pathways and mechanisms. Psychosom Med. 2011;73:724–30.
Tatsuta M, Iishi H, Baba M, Taniguchi H. Inhibition of azoxymethane-induced experimental colon carcinogenesis in Wistar rats by 6-hydroxydopamine. Int J Cancer. 1992;50:298–301.
Sadighparvar S, Darband SG, Ghaderi-Pakdel F, Mihanfar A, Majidinia M. Parasympathetic, but not sympathetic denervation, suppressed colorectal cancer progression. Eur J Pharmacol. 2021;913:174626.
Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225.
Penedo MA, Rivera-Baltanás T, Pérez-Rodríguez D, Allen J, Borrajo A, Alonso-Crespo D, et al. The role of dopamine receptors in lymphocytes and their changes in schizophrenia. Brain Behav Immun Health. 2021;12:100199.
Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217.
Beaulieu JM, Espinoza S, Gainetdinov RR. Dopamine receptors—IUPHAR -review 13. Br J Pharm. 2015;172:1–23.
Grant CE, Flis AL, Ryan BM. Understanding the role of dopamine in cancer: past, present and future. Carcinogenesis. 2022;43:517–27.
Leng ZG, Lin SJ, Wu ZR, Guo YH, Cai L, Shang HB, et al. Activation of DRD5 (dopamine receptor D5) inhibits tumor growth by autophagic cell death. Autophagy. 2017;13:1404–19.
Kontos CK, Papadopoulos IN, Fragoulis EG, Scorilas A. Quantitative expression analysis and prognostic significance of L-DOPA decarboxylase in colorectal adenocarcinoma. Br J Cancer. 2010;102:1384–90.
Gemignani F, Landi S, Moreno V, Gioia-Patricola L, Chabrier A, Guino E, et al. Polymorphisms of the dopamine receptor gene DRD2 and colorectal cancer risk. Cancer Epidemiol Biomark Prev. 2005;14:1633–8.
Battaglin F, Cao S, Loupakis F, Stintzing S, Parikh AR, Puccini A, et al. Polymorphisms in the dopamine (DA) signaling to predict outcome in patients (pts) with metastatic colorectal cancer (mCRC): data from TRIBE, MAVERICC, and FIRE-3 phase III trials. J Clin Oncol. 2019;37:3048.
Sarkar C, Chakroborty D, Chowdhury UR, Dasgupta PS, Basu S. Dopamine increases the efficacy of anticancer drugs in breast and colon cancer preclinical models. Clin Cancer Res. 2008;14:2502–10.
Ren Y, Tao J, Jiang Z, Guo D, Tang J. Pimozide suppresses colorectal cancer via inhibition of Wnt/beta-catenin signaling pathway. Life Sci. 2018;209:267–73.
Lee H, Shim S, Kong JS, Kim MJ, Park S, Lee SS, et al. Overexpression of dopamine receptor D2 promotes colorectal cancer progression by activating the β-catenin/ZEB1 axis. Cancer Sci. 2021;112:3732–43.
Xia Y, Jia C, Xue Q, Jiang J, Xie Y, Wang R, et al. Antipsychotic drug trifluoperazine suppresses colorectal cancer by inducing G0/G1 arrest and apoptosis. Front Pharmacol. 2019;10:1029.
Tysnes OB, Storstein A. Epidemiology of Parkinson’s disease. J Neural Transm. 2017;124:901–5.
Deng H, Wang P, Jankovic J. The genetics of Parkinson disease. Ageing Res Rev. 2018;42:72–85.
Lee JYS, Ng JH, Saffari SE, Tan EK. Parkinson’s disease and cancer: a systematic review and meta-analysis on the influence of lifestyle habits, genetic variants, and gender. Aging. 2022;14:2148–73.
Fang H, Du Y, Pan S, Zhong M, Tang J. Patients with Parkinson’s disease predict a lower incidence of colorectal cancer. BMC Geriatr. 2021;21:564.
Li W-H, Zhang H, Guo Q, Wu X-D, Xu Z-S, Dang C-X, et al. Detection of SNCA and FBN1 methylation in the stool as a biomarker for colorectal cancer. Dis Markers. 2015;2015:657570.
Killinger BA, Madaj Z, Sikora JW, Rey N, Haas AJ, Vepa Y, et al. The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci Transl Med. 2018;10:eaar5280.
Battaglin F, Cao S, Puccini A, Tokunaga R, Naseem M, Arai H, et al. Gene expression and genetic variants in Parkinson’s disease (PD) genes to predict outcome in metastatic colorectal cancer (mCRC): data from FIRE-3 phase III trial. J Clin Oncol. 2019;37:3595.
Bortolato M, Chen K, Shih JC. Monoamine oxidase inactivation: from pathophysiology to therapeutics. Adv Drug Deliv Rev. 2008;60:1527–33.
Yang YC, Chien MH, Lai TC, Su CY, Jan YH, Hsiao M, et al. Monoamine oxidase B expression correlates with a poor prognosis in colorectal cancer patients and is significantly associated with epithelial-to-mesenchymal transition-related gene signatures. Int J Mol Sci. 2020;21:2813.
Aljanabi R, Alsous L, Sabbah DA, Gul HI, Gul M, Bardaweel SK. Monoamine oxidase (MAO) as a potential target for anticancer drug design and development. Molecules. 2021;26:6019.
Wang Y-C, Wang X, Yu J, Ma F, Li Z, Zhou Y, et al. Targeting monoamine oxidase A-regulated tumor-associated macrophage polarization for cancer immunotherapy. Nat Commun. 2021;12:3530.
Wang X, Li B, Kim YJ, Wang YC, Li Z, Yu J, et al. Targeting monoamine oxidase A for T cell-based cancer immunotherapy. Sci Immunol. 2021;6:eabh2383.
Auteri M, Zizzo MG, Serio R. GABA and GABA receptors in the gastrointestinal tract: from motility to inflammation. Pharm Res. 2015;93:11–21.
Huang, Wang Y, Thompson JW, Yin T, Alexander PB, Qin D, et al. Cancer-cell-derived GABA promotes β-catenin-mediated tumour growth and immunosuppression. Nat Cell Biol. 2022;24:230–41.
Yan H, Tang G, Wang H, Hao L, He T, Sun X, et al. DNA methylation reactivates GAD1 expression in cancer by preventing CTCF-mediated polycomb repressive complex 2 recruitment. Oncogene. 2016;35:3995–4008.
Qiu Y, Cai G, Zhou B, Li D, Zhao A, Xie G, et al. A distinct metabolic signature of human colorectal cancer with prognostic potential. Clin Cancer Res. 2014;20:2136–46.
An J, Seok H, Ha EM. GABA-producing Lactobacillus plantarum inhibits metastatic properties and induces apoptosis of 5-FU-resistant colorectal cancer cells via GABA. J Microbiol. 2021;59:202–16.
Joseph J, Niggemann B, Zaenker KS, Entschladen F. The neurotransmitter gamma-aminobutyric acid is an inhibitory regulator for the migration of SW 480 colon carcinoma cells. Cancer Res. 2002;62:6467–9.
Song L, Du A, Xiong Y, Jiang J, Zhang Y, Tian Z, et al. γ-Aminobutyric acid inhibits the proliferation and increases oxaliplatin sensitivity in human colon cancer cells. Tumour Biol. 2016;37:14885–94.
Ngo DH, Vo TS. An updated review on pharmaceutical properties of gamma-aminobutyric acid. Molecules. 2019;24:2678.
Miao Y, Zhang Y, Wan H, Chen L, Wang F. GABA-receptor agonist, propofol inhibits invasion of colon carcinoma cells. Biomed Pharmacother. 2010;64:583–8.
Shu Q, Liu J, Liu X, Zhao S, Li H, Tan Y, et al. GABAB R/GSK-3β/NF-κB signaling pathway regulates the proliferation of colorectal cancer cells. Cancer Med. 2016;5:1259–67.
Wang H, Zhang H, Sun Z, Chen W, Miao C. GABAB receptor inhibits tumor progression and epithelial-mesenchymal transition via the regulation of Hippo/YAP1 pathway in colorectal cancer. Int J Biol Sci. 2021;17:1953–62.
Yan L, Gong YZ, Shao MN, Ruan GT, Xie HL, Liao XW, et al. Distinct diagnostic and prognostic values of γ-aminobutyric acid type A receptor family genes in patients with colon adenocarcinoma. Oncol Lett. 2020;20:275–91.
Liu T, Fang Y. Research for expression and prognostic value of GABRD in colon cancer and coexpressed gene network construction based on data mining. Comput Math Methods Med. 2021;2021:5544182.
Zhang B, Vogelzang A, Miyajima M, Sugiura Y, Wu Y, Chamoto K, et al. B cell-derived GABA elicits IL-10. Nature 2021;599:471–6.
Albuquerque EX, Pereira EF, Alkondon M, Rogers SW. Mammalian nicotinic acetylcholine receptors: from structure to function. Physiol Rev. 2009;89:73–120.
Zhang H, Kong Q, Wang J, Jiang Y, Hua H. Complex roles of cAMP–PKA–CREB signaling in cancer. Exp Hematol Oncol. 2020;9:32.
Schuller HM. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat Rev Cancer. 2009;9:195–205.
Cheng K, Samimi R, Xie G, Shant J, Drachenberg C, Wade M, et al. Acetylcholine release by human colon cancer cells mediates autocrine stimulation of cell proliferation. Am J Physiol Gastrointest Liver Physiol. 2008;295:G591–7.
Kawai H, Berg DK. Nicotinic acetylcholine receptors containing alpha 7 subunits on rat cortical neurons do not undergo long-lasting inactivation even when up-regulated by chronic nicotine exposure. J Neurochem. 2001;78:1367–78.
Hajiasgharzadeh K, Somi MH, Sadigh-Eteghad S, Mokhtarzadeh A, Shanehbandi D, Mansoori B, et al. The dual role of alpha7 nicotinic acetylcholine receptor in inflammation-associated gastrointestinal cancers. Heliyon. 2020;6:e03611.
Wei PL, Kuo LJ, Huang MT, Ting WC, Ho YS, Wang W, et al. Nicotine enhances colon cancer cell migration by induction of fibronectin. Ann Surg Oncol. 2011;18:1782–90.
Ye YN, Liu ES, Shin VY, Wu WK, Cho CH. The modulating role of nuclear factor-kappaB in the action of alpha7-nicotinic acetylcholine receptor and cross-talk between 5-lipoxygenase and cyclooxygenase-2 in colon cancer growth induced by 4-(N-methyl-N-nitrosamino)-1-(3-pyridyl)-1-butanone. J Pharm Exp Ther. 2004;311:123–30.
Wong HP, Yu L, Lam EK, Tai EK, Wu WK, Cho CH. Nicotine promotes cell proliferation via alpha7-nicotinic acetylcholine receptor and catecholamine-synthesizing enzymes-mediated pathway in human colon adenocarcinoma HT-29 cells. Toxicol Appl Pharm. 2007;221:261–7.
Wong HPS, Yu L, Lam EKY, Tai EKK, Wu WKK, Cho C-H. Nicotine promotes colon tumor growth and angiogenesis through β-adrenergic activation. Toxicol Sci. 2007;97:279–87.
Xiang T, Fei R, Wang Z, Shen Z, Qian J, Chen W. Nicotine enhances invasion and metastasis of human colorectal cancer cells through the nicotinic acetylcholine receptor downstream p38 MAPK signaling pathway. Oncol Rep. 2016;35:205–10.
Pavlov VA, Wang H, Czura CJ, Friedman SG, Tracey KJ. The cholinergic anti-inflammatory pathway: a missing link in neuroimmunomodulation. Mol Med. 2003;9:125–34.
Pavlov VA, Tracey KJ. Neural circuitry and immunity. Immunol Res. 2015;63:38–57.
Parrish WR, Rosas-Ballina M, Gallowitsch-Puerta M, Ochani M, Ochani K, Yang L-H, et al. Modulation of TNF release by choline requires α7 subunit nicotinic acetylcholine receptor-mediated signaling. Mol Med. 2008;14:567–74.
Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–8.
Hayashi S, Hamada T, Zaidi SF, Oshiro M, Lee J, Yamamoto T, et al. Nicotine suppresses acute colitis and colonic tumorigenesis associated with chronic colitis in mice. Am J Physiol Gastrointest Liver Physiol. 2014;307:G968–78.
Eglen RM. Muscarinic receptor subtypes in neuronal and non-neuronal cholinergic function. Auton Autacoid Pharm. 2006;26:219–33.
Cheng K, Shang AC, Drachenberg CB, Zhan M, Raufman JP. Differential expression of M3 muscarinic receptors in progressive colon neoplasia and metastasis. Oncotarget. 2017;8:21106–14.
Tolaymat M, Larabee SM, Hu S, Xie G, Raufman JP. The role of M3 muscarinic receptor ligand-induced kinase signaling in colon cancer progression. Cancers (Basel). 2019;11:308.
Peng Z, Heath J, Drachenberg C, Raufman JP, Xie G. Cholinergic muscarinic receptor activation augments murine intestinal epithelial cell proliferation and tumorigenesis. BMC Cancer. 2013;13:204.
Said AH, Hu S, Abutaleb A, Watkins T, Cheng K, Chahdi A, et al. Interacting post-muscarinic receptor signaling pathways potentiate matrix metalloproteinase-1 expression and invasion of human colon cancer cells. Biochem J. 2017;474:647–65.
Von Rosenvinge EC, Raufman JP. Muscarinic receptor signaling in colon cancer. Cancers. 2011;3:971–81.
Zhao CM, Hayakawa Y, Kodama Y, Muthupalani S, Westphalen CB, Andersen GT, et al. Denervation suppresses gastric tumorigenesis. Sci Transl Med. 2014;6:250ra115.
Hering NA, Liu V, Kim R, Weixler B, Droeser RA, Arndt M, et al. Blockage of cholinergic signaling via muscarinic acetylcholine receptor 3 inhibits tumor growth in human colorectal adenocarcinoma. Cancers (Basel). 2021;13:3220.
Raufman JP, Samimi R, Shah N, Khurana S, Shant J, Drachenberg C, et al. Genetic ablation of M3 muscarinic receptors attenuates murine colon epithelial cell proliferation and neoplasia. Cancer Res. 2008;68:3573–8.
Alizadeh M, Schledwitz A, Cheng K, Raufman JP. Mechanistic clues provided by concurrent changes in the expression of genes encoding the M(1) muscarinic receptor, β-catenin signaling proteins, and downstream targets in adenocarcinomas of the colon. Front Physiol. 2022;13:857563.
Cheng K, Xie G, Khurana S, Heath J, Drachenberg CB, Timmons J, et al. Divergent effects of muscarinic receptor subtype gene ablation on murine colon tumorigenesis reveals association of M3R and zinc finger protein 277 expression in colon neoplasia. Mol Cancer. 2014;13:77.
Cheng K, Raufman JP. Bile acid-induced proliferation of a human colon cancer cell line is mediated by transactivation of epidermal growth factor receptors. Biochem Pharm. 2005;70:1035–47.
Farhana L, Nangia-Makker P, Arbit E, Shango K, Sarkar S, Mahmud H, et al. Bile acid: a potential inducer of colon cancer stem cells. Stem Cell Res Ther. 2016;7:181.
Raufman J-P, Dawson PA, Rao A, Drachenberg CB, Heath J, Shang AC, et al. Slc10a2-null mice uncover colon cancer-promoting actions of endogenous fecal bile acids. Carcinogenesis. 2015;36:1193–200.
Mawe GM, Hoffman JM. Serotonin signalling in the gut–functions, dysfunctions and therapeutic targets. Nat Rev Gastroenterol Hepatol. 2013;10:473–86.
Kwon YH, Khan WI. Peripheral serotonin: cultivating companionship with gut microbiota in intestinal homeostasis. Am J Physiol Cell Physiol. 2022;323:C550–5.
Kannen V, Bader M, Sakita JY, Uyemura SA, Squire JA. The dual role of serotonin in colorectal cancer. Trends Endocrinol Metab. 2020;31:611–25.
Li T, Fu B, Zhang X, Zhou Y, Yang M, Cao M, et al. Overproduction of gastrointestinal 5-HT promotes colitis-associated colorectal cancer progression via enhancing NLRP3 inflammasome activation. Cancer Immunol Res. 2021;9:1008–23.
Sui H, Xu H, Ji Q, Liu X, Zhou L, Song H, et al. 5-hydroxytryptamine receptor (5-HT1DR) promotes colorectal cancer metastasis by regulating Axin1/β-catenin/MMP-7 signaling pathway. Oncotarget. 2015;6:25975–87.
Ataee R, Ajdary S, Zarrindast M, Rezayat M, Hayatbakhsh MR. Anti-mitogenic and apoptotic effects of 5-HT1B receptor antagonist on HT29 colorectal cancer cell line. J Cancer Res Clin Oncol. 2010;136:1461–9.
Nocito A, Dahm F, Jochum W, Jang JH, Georgiev P, Bader M, et al. Serotonin regulates macrophage-mediated angiogenesis in a mouse model of colon cancer allografts. Cancer Res. 2008;68:5152–8.
Zhu P, Lu T, Chen Z, Liu B, Fan D, Li C, et al. 5-hydroxytryptamine produced by enteric serotonergic neurons initiates colorectal cancer stem cell self-renewal and tumorigenesis. Neuron. 2022;110:2268–82.e4.
Sakita JY, Bader M, Santos ES, Garcia SB, Minto SB, Alenina N, et al. Serotonin synthesis protects the mouse colonic crypt from DNA damage and colorectal tumorigenesis. J Pathol. 2019;249:102–13.
Zhang N, Sundquist J, Sundquist K, Zhang ZG, Ji J. Correction to: combined use of aspirin and selective serotonin reuptake inhibitors is associated with lower risk of colorectal cancer: a nested case-control study. Am J Gastroenterol. 2021;116:2310.
Kannen V, Garcia SB, Silva WA Jr., Gasser M, Mönch R, Alho EJ, et al. Oncostatic effects of fluoxetine in experimental colon cancer models. Cell Signal. 2015;27:1781–8.
Sarrouilhe D, Mesnil M. Serotonin and human cancer: a critical view. Biochimie 2019;161:46–50.
Fang CK, Chen HW, Chiang IT, Chen CC, Liao JF, Su TP, et al. Mirtazapine inhibits tumor growth via immune response and serotonergic system. PloS ONE. 2012;7:e38886.
Chan YL, Lai WC, Chen JS, Tseng JT, Chuang PC, Jou J, et al. TIAM2S mediates serotonin homeostasis and provokes a pro-inflammatory immune microenvironment permissive for colorectal tumorigenesis. Cancers (Basel). 2020;12:1844.
Di Y-Z, Han B-S, Di J-M, Liu W-Y, Tang Q. Role of the brain-gut axis in gastrointestinal cancer. World J Clin Cases. 2019;7:1554–70.
Liu S. Neurotrophic factors in enteric physiology and pathophysiology. Neurogastroenterol Motil. 2018;30:e13446.
Meldolesi J. Neurotrophin Trk receptors: new targets for cancer therapy. Rev Physiol Biochem Pharm. 2018;174:67–79.
Radin DP, Patel P. BDNF: an oncogene or tumor suppressor? Anticancer Res. 2017;37:3983–90.
Yang X, Martin TA, Jiang WG. Biological influence of brain-derived neurotrophic factor (BDNF) on colon cancer cells. Exp Ther Med. 2013;6:1475–81.
Fujikawa H, Tanaka K, Toiyama Y, Saigusa S, Inoue Y, Uchida K, et al. High TrkB expression levels are associated with poor prognosis and EMT induction in colorectal cancer cells. J Gastroenterol. 2012;47:775–84.
Huang SM, Lin C, Lin HY, Chiu CM, Fang CW, Liao KF, et al. Brain-derived neurotrophic factor regulates cell motility in human colon cancer. Endocr Relat Cancer. 2015;22:455–64.
Hayakawa Y, Sakitani K, Konishi M, Asfaha S, Niikura R, Tomita H, et al. Nerve growth factor promotes gastric tumorigenesis through aberrant cholinergic signaling. Cancer Cell. 2017;31:21–34.
Molloy NH, Read DE, Gorman AM. Nerve growth factor in cancer cell death and survival. Cancers. 2011;3:510–30.
Drilon A. TRK inhibitors in TRK fusion-positive cancers. Ann Oncol. 2019;30:viii23–30.
Kojadinovic A, Laderian B, Mundi PS. Targeting TRK: a fast-tracked application of precision oncology and future directions. Crit Rev Oncol Hematol. 2021;165:103451.
Fatemi SH. Reelin mutations in mouse and man: from reeler mouse to schizophrenia, mood disorders, autism and lissencephaly. Mol Psychiatry. 2001;6:129–33.
Lee GH, D’Arcangelo G. New insights into reelin-mediated signaling pathways. Front Cell Neurosci. 2016;10:122.
Dohi O, Takada H, Wakabayashi N, Yasui K, Sakakura C, Mitsufuji S, et al. Epigenetic silencing of RELN in gastric cancer. Int J Oncol. 2010;36:85–92.
Sato N, Fukushima N, Chang R, Matsubayashi H, Goggins M. Differential and epigenetic gene expression profiling identifies frequent disruption of the RELN pathway in pancreatic cancers. Gastroenterology. 2006;130:548–65.
Stein T, Cosimo E, Smith P, Simon R, Price K, Baird L, et al. Reelin expression in breast tumours is associated with increased survival and is controlled by promoter methylation. Breast Cancer Res. 2008;10:P25.
Castellano E, Molina-Arcas M, Krygowska AA, East P, Warne P, Nicol A, et al. RAS signalling through PI3-Kinase controls cell migration via modulation of Reelin expression. Nat Commun. 2016;7:11245.
Carvajal AE, Serrano-Morales JM, Vazquez-Carretero MD, Garcia-Miranda P, Calonge ML, Peral MJ, et al. Reelin protects from colon pathology by maintaining the intestinal barrier integrity and repressing tumorigenic genes. Biochim Biophys Acta Mol Basis Dis. 2017;1863:2126–34.
Vignot S, Lefebvre C, Frampton GM, Meurice G, Yelensky R, Palmer G, et al. Comparative analysis of primary tumour and matched metastases in colorectal cancer patients: evaluation of concordance between genomic and transcriptional profiles. Eur J Cancer. 2015;51:791–9.
Acknowledgements
We thank David W. Craig for critical reading of the paper.
Funding
This work was partly supported by National Cancer Institute (grant numbers P30CA014089 and R01CA241137), Gloria Borges WunderGlo Foundation, Dhont Family Foundation, Gene Gregg Pancreas Research Fund, San Pedro Peninsula Cancer Guild, Daniel Butler Research Fund, V foundation for cancer research, Victoria and Philip Wilson Research Fund, Fong research project, Ming Hsieh research fund, Boyd and Elsie Welin Professorship to JCS and USC Trustee Tsai family fund to JCS. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
FB drafted the paper with the aid of CS and AL. HJL supervised the paper. All authors directly provided their contribution, read and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Battaglin, F., Jayachandran, P., Strelez, C. et al. Neurotransmitter signaling: a new frontier in colorectal cancer biology and treatment. Oncogene 41, 4769–4778 (2022). https://doi.org/10.1038/s41388-022-02479-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-022-02479-4
- Springer Nature Limited
This article is cited by
-
Crosstalk Between the Nervous System and Colorectal Cancer
Neuroscience Bulletin (2024)
-
The Diagnostic and Prognostic Value of Neurotransmitter Receptor-Related Genes in Colon Adenocarcinoma
Molecular Biotechnology (2023)