
Abstract
Increasing lines of evidence show that the malignant behavior of cancer is not exclusively attributable to cancer cells but also radically influenced by cancerous stroma activity and controlled through various mechanisms by the microenvironment. In addition to structural components, such as the extracellular matrix, stromal cells, such as macrophages, endothelial cells, and specifically cancer-associated fibroblasts (CAFs), have attracted substantial attention over recent decades. CAFs provide routes for aggressive carcinomas and contribute to invasion and metastasis through the biochemical alteration and regulation of cancer-related pathways. However, another facet of CAFs that has been neglected by numerous studies is that CAFs might serve as a negative regulator of cancer progression under certain circumstances. The various origins of CAFs, the diverse tissues in which they reside and their interactions with different cancer cells appear to be responsible for this inconsistency. This review summarizes the latest knowledge regarding CAF heterogeneity and offers a novel perspective and a beneficial approach for obtaining an improved understanding of CAFs.
Similar content being viewed by others
Introduction
Fibroblasts were first described in the 19th century according to their location and appearance and were defined as cells in tissues that synthesized collagen [1]. Specifically these cells were identified as nonepithelial, nonvascular, noninflammatory cells in connective tissues that are responsible for the synthesis of fibrillary matrix and most likely have a mesenchymal lineage [2]. In the tumor microenvironment (TME), heterogeneous populations of cells with various or overlapping functions contribute to tumorigenesis [3], and fibroblasts play a crucial role. Accordingly, cancer-associated fibroblasts (CAFs) are found in the tumor stroma of various cancers [4], and these cells can be described as spindle-shaped cells with elongated cytoplasmic processes [5]. The term CAFs is often used as a blanket term for complex populations of activated mesenchymal cells whose locations and functions are distinct from those of normal fibroblasts. The markers used to identify CAFs are as diverse as their functions and exert dynamically heterogeneous effects on cancer at different stages [6, 7]. In this review, we summarize the characteristics of CAFs mainly in terms of markers, genetic alterations, and mechanisms related to tumor progression or cancer-restraining functions with a particular emphasis on dissimilarities between normal fibroblasts and CAFs and the heterogeneities among CAFs themselves, and we also discuss future directions for certain therapies targeting CAFs.
Origins of CAFs
The existence of fibroblastic cells with contractile properties, namely, myofibroblasts (MFBs), was first described in 1971 in granulation tissues, and MFBs were hypothesized to exhibit reparative activity during wound healing [8]. Fibroblasts are derived from the primitive mesenchyme, whereas CAFs are likely derived from resident fibroblasts, bone marrow-derived mesenchymal precursor cells [9], and endothelial and epithelial cells [10], some of which are stimulated by cytokines, such as transforming growth factor-β (TGFβ). The theory that CAFs originate from epithelial cells is based on the epithelial-to-mesenchymal transition (EMT): epithelial cells exposed to matrix metalloproteinase (MMP)-driven oxidative stress undergo DNA oxidation and experience mutations, and these cells thereby undergo a specialized EMT through which they transdifferentiate into activated MFBs [4, 11]. Endothelial cells might contribute to CAFs via the endothelial-to-mesenchymal transition (EndMT), which is described as the loss of endothelial markers, such as CD31, and an abundance of mesenchymal markers, such as fibroblast-specific protein (FSP) and α-smooth muscle actin (α-SMA), in tumor stroma [12]. In addition, CAFs can also be derived from adipocytes due to the expression of mesenchymal lineage-committed marker genes, such as peroxisome proliferator-activated receptor-γ (PPARγ), Runt-related transcription factor-2 (RUNX2), and the transcription factor SOX9 [13, 14]. In addition, bone marrow-derived mesenchymal stem cells (MSCs) can differentiate into CAFs after activation by CXC receptor 6 and its ligand CXC ligand 16 [15], and osteopontin in breast cancer induces the integrin-dependent MSC expression of TGF-β1 to mediate adoption of the CAF phenotype [16]. Furthermore, CAFs are derived from specialized cells, such as stellate cells in the pancreas and liver [17, 18], myoepithelial cells in the breast [19], and pericryptal MFBs in the gastrointestinal tract [20]. This spectrum of origins at least partially explain the heterogeneity of CAFs (Fig. 1).

Origin of cancer-associated fibroblasts (CAFs). CAFs originate from a variety of tissue types through a number of different cellular processes, including resident fibroblasts via transforming growth factor-β (TGFβ) activation and epithelial and endothelial cells through the epithelial-to-mesenchymal transition (EMT) and endothelial-to-mesenchymal transition (EndMT), respectively. CAFs might also be derived from the trans-differentiation of cells such as adipocytes, which results in the upregulation of mesenchymal lineage-committed marker genes such as peroxisome proliferator-activated receptor- γ (PPARγ) and Runt-related transcription factor-2 (RUNX2). Vitamin deficiency in certain cancer stromal cells, such as stellate cells, leads to the upregulation of smooth muscle actin (SMA), which induces trans-differentiation into CAFs. In addition, mesenchymal stem cells can also differentiate into CAFs through activation by a certain receptor or ligand or TGFβ activation by osteopontin. These multiple origins of CAFs give them a wide functional spectrum
Recognition of fibroblasts and CAFs
Markers of fibroblasts
To date, researchers have identified many candidate markers of fibroblasts and CAFs, but the identification of particular markers has been challenging due to their expression in other cells. The most commonly used markers can be defined by their overlapping expression in a vast range of populations of CAFs or fibroblasts. MFBs are often known as activated fibroblasts with high expression levels of α-SMA and play a critical role in reinforcing contractility in connective tissues [21, 22]. In addition to α-SMA, several other markers have also been described in previous reports, and these include fibroblast-activation protein (FAP), which is expressed on the surfaces of fibroblasts and melanocytes, comprises p95 and p105 subunits and serves as a serine protease [23]. FSP1, which is considered an intermediate-filament-associated protein, might be a reliable marker for the detection of quiescent fibroblasts [24] and α1/β1 integrin, which has been reported to be a collagen receptor [25]. CD90, a cell surface glycoprotein, is also highly expressed in and thus serves as a marker of human fibroblasts [26]. The intermediate-filament-associated protein vimentin, which is synthesized by fibroblasts, participates in numerous cellular processes with functions related to signaling, migration, and invasion and is reportedly involved in cell proliferation and differentiation [27]. Fibroblasts are also an important source of extracellular matrix (ECM)-degrading protease-like MMPs, whose existence can reflect the function of activated fibroblasts [28]. Discoidin domain receptor 2 (DDR2) was found to be a collagen receptor in kidney fibroblasts [29].
Markers of CAFs
In addition to the important role of fibroblasts in normal tissues, increasing evidence shows that these cells are also crucial in the TME. Subtypes of fibroblasts, such as activated fibroblasts and quiescent fibroblasts, have been studied for decades and although the understanding of quiescent and activated fibroblasts in tumor tissues remains insufficient, CAFs are the most studied subtype of activated fibroblasts. CAFs are morphologically defined as spindle-shaped cells in cancer stroma [30]. In addition, these cells are activated fibroblasts that are similar to MFBs but show differences in their duration (cannot be removed by apoptosis), and these characteristics render CAFs one of the most prominent cell types in the tumor stroma of many cancers [4, 31].
Various molecular changes occur during metabolic regulation, and some of these collectively lead to the “activated state” of CAFs, which can serve as a hallmark of CAFs. Of note, due to the existence of a myofibroblastic part of CAFs, these cells share some markers with activated fibroblasts, such as α-SMA, integrin-β1, and FSP1 [32]. Podoplanin expression has been detected on the apical membranes of cancer cells in only 9 of 177 (5.1%) cases. Nonetheless, podoplanin expression has been observed in 54 of 177 populations of CAFs (30.5%), and all podoplanin + CAFs were found in invasive adenocarcinomas, whereas none were found in noninvasive adenocarcinomas. Enhanced invasive capacities were also observed in another study, and these activities can be negated by a Rho-associated protein kinase (ROCK) inhibitor [33, 34]. Moreover, platelet-derived growth factor (PDGF) signaling appears to be essential in various mechanisms of CAFs. PDGF receptor-β (PDGFRβ) is upregulated in the TME, particularly in CAFs [35], whereas PDGF-CC can strengthen tumor growth by recruiting CAFs. To investigate the molecular identity of the supporting factors provided by CAFs, the authors performed antibody arrays and generated an in vivo coinjection model, and their results identify osteopontin as the effector of the augmented tumor growth induced by PDGF-CC [36]. PDGFRα-positive CAFs have also been observed in melanoma, which suggests that CAFs should be defined as resident-activated fibroblasts that express their markers [37]. PDGFRβ can also serve as a marker of CAFs given its function of promoting cancer cell proliferation by mediating the interaction between CAFs and cancer cells. Cervical cancer cells provide PDGF-BB to upregulate heparin-binding epidermal growth factor-like growth factor (HB-EGF) through PDGFRβ activation in an adjacent fibroblast, and this effect facilitates cancer cell growth by activating EGF receptor [38].
Tenascin-C (TNC), an ECM protein that has been described in tumors, has been proposed to modulate cell signaling and has been used to identify two convergent proinvasive agents secreted by MFBs, namely, scatter factor/hepatocyte growth factor (SF/HGF) and the TGFβ-upregulated ECM glycoprotein TNC, which are both necessary though not sufficient for invasion and result in the promotion of invasion and metastasis in pathological connective tissues [39]. In addition, caveolin-1 is reportedly involved in tumor invasion and metastasis by regulating p190RhoGAP and thus stiffens the microenvironment [40]. High expression of CD90 has been found in prostate CAFs by immunohistochemistry, qRT-PCR, and proteomic analysis, and these results show that CD90 can distinguish cancer-associated stroma from “benign” stroma in the prostate and is thus a potential therapeutic target [41]. PDGFRα, PDGFRβ, FAP, FSP1, and SMA expression in esophageal squamous cell carcinoma (ESCC) has been examined by immunohistochemical staining, and the results led to the histological classification of a CAF phenotype that serves as a reliable and significant prognostic predictor in ESCC [42]. Despite the increasing lines of evidence showing that markers of CAFs are related to the aggressive behaviors of tumors in many cases, there is heterogeneity in the functions and influences of these markers. For instance, the loss of caveolin-1 in CAFs is considered predictive of a poor clinical outcome in breast cancer because these CAFs enhance the growth of triple-negative (TN) breast cancer cells [43], but this finding conflicts with the results of another study of the enhanced invasive and metastatic potency conferred by caveolin-1 in breast CAFs [40]. These results might indicate that caveolin-1 plays completely different roles in different histological types of cancer.
In addition to the well-known markers mentioned above, some newly identified markers have also come to our attention. A gene expression analysis revealed that Yes-associated protein (YAP) is highly expressed in CAFs. The remodeling of the ECM and the promotion of cancer cell invasion require the actomyosin cytoskeleton. YAP mediates the expression of several cytoskeletal regulators, including ANLN and DIAPH3, and controls the protein levels of MYL9 (also known as MLC2); therefore, YAP might also serve as a CAF marker [44]. A proteomic analysis of human breast cancer tissues revealed that galectin-1 is upregulated in cancer-associated stroma tissue [45]. Compared with normal ovarian stroma, natriuretic peptide B (NPPB) is significantly increased in ovarian cancer stroma, and NPPB has therefore been identified as a potential candidate marker for CAFs [46].
The potential markers of fibroblasts and CAFs are summarized in Table 1. Although markers of CAFs have been identified for decades, none of these markers have been found to be specific for CAF; for instance, we can also observe PDGFRβ in perivascular cells [47]. FSP1 also serves as a marker of macrophages and other immune cells and can be found in some cancer cells [48]. Relatively different descriptions of definitions of CAFs have been obtained in different studies, and these differences are likely due to the origin of their precursor fibroblasts and thus reflect the heterogeneous functions of CAFs in diverse situations. The combination of these markers might be a better strategy for distinguishing the heterogeneous populations of CAF in future investigations.
Plasticity of CAFs
In recent years, an increasing number of studies have investigated the subtypes of CAFs as researchers aim to obtain a more in-depth understanding of the TME. CAFs can gain a tumor-like phenotype by reprogramming the lipid metabolism pathway and amplifying microtubule-organizing centers (MTOCs) through pigment epithelium-derived factor (PEDF)-dependent lipid-MTOC signaling [49]. Subpopulations of pro-tumorigenic CAFs have been described as having undergone metabolic reprogramming induced by the loss of caveolin-1 [50], whereas the upregulation of caveolin-1 results in the promotion of cancer invasion and metastasis [40]. In addition, leukemia inhibitory factor (LIF) initiates epigenetic changes that lead to the activation of Janus kinase 1/signal transducer and activator of transcription 3 (JAK1/STAT3) signaling and consequently induces CAFs to switch into a proinvasive state [51]. A recent study demonstrated that a CD10+ G-protein-coupled receptor 77 (GPR77)+ subtype of CAFs promotes tumor formation and chemoresistance by providing a survival niche for cancer stem cells (CSCs). Mechanistically, CD10+GPR77+ CAFs are constantly activated by NF-κB via p65 phosphorylation and acetylation and via GPR77, a C5a receptor, which leads to continuous activation through complement signaling. In addition, CD10+GPR77+ CAFs facilitate the successful engraftment of patient-derived xenografts (PDXs), whereas the targeting of these CAFs with a neutralizing anti-GPR77 antibody abolishes tumor formation and restores tumor chemosensitivity [52]. Moreover, different subtypes of CAFs in breast cancer are defined by their expression of CD146. CD146-negative CAFs suppress estrogen receptor expression in estrogen receptor-positive breast cancer cells, decrease tumor cell sensitivity to estrogen, and increase tumor cell resistance to tamoxifen therapy, whereas CD146-positive CAFs provide estrogen-dependent tamoxifen sensitivity [53]. More generally, CAFs are divided into myofibroblastic CAFs and inflammatory CAFs, and it has been demonstrated that IL-1 induces LIF expression and downstream JAK/STAT activation to generate inflammatory CAFs. These results show that TGFβ antagonizes this process by downregulating IL-1R1 expression and promoting differentiation into MFBs [54]. The definition of distinct subtypes provides a comprehensive understanding of CAFs: these subtypes reflect the flexibility and instability of CAF but can also, as more in-depth knowledge of the TME is obtained, highly contribute to the integrity of the knowledge regarding CAFs.
Major molecular profiles of fibroblasts and CAFs
Because CAFs are positionally and functionally different from fibroblasts, the molecular alterations causing such dissimilarities remain controversial, and various studies have revealed conflicting results.
One of the initial studies reported that the transcriptional patterns displayed by fibroblasts from different anatomic sites are distinct and characteristic and suggested that fibroblasts from different organs can be considered distinct differentiated cell types [55]. In a subsequent study, the same research group defined a common transcriptomic profile for fibroblasts stimulated with serum and reported that the pattern observed during the wound healing process is similar to that associated with tumor progression [56]. Additional studies have revealed distinguishable features and gene expression profiles of CAFs and fibroblasts in breast cancer; for instance, AKR1C1 and AKR1C2, two closely related genes involved in progesterone metabolism, are expressed at lower levels in CAFs. A bifunctional transcription factor, KLF4, is also expressed at high levels in normal mammary gland stroma and is not found in CAFs [57]. These results indicate that molecular alterations might have occurred during the transition from fibroblasts to CAFs. In fact, transcriptome and secretome analyses revealed two distinct CAF subtypes that exhibit a functional difference in supporting tumor formation and invasion in oral squamous cell carcinoma (OSCC) [58]. Similarly, a gene expression analysis of CAFs and paired normal fibroblasts showed that similar transcriptomic programs might function in the transition from normal fibroblasts to CAFs in the colonic mucosa [59]. The interaction between CAFs and cancer cells can affect the activation of fibroblasts. A functional deficiency in the tumor-suppressor gene p53 in cancer cells activates fibroblasts and contributes to malignant angiogenesis [60]. Particular genes related to adhesion molecules, growth factors, and enzymes have also been discovered to be upregulated in CAFs or cancer cells through reciprocal interactions [61].
Distinct epigenetic changes have been identified in breast carcinoma and play a role in establishing the TME and enhancing tumorigenesis [62]. Several major approaches, such as posttranscriptional modification, DNA (promotor) methylation, and local nucleosome remodeling, have been thoroughly investigated. A major mechanism of posttranscriptional modification is regulated by microRNAs (miRNAs), which are involved in numerous cellular processes, including cell differentiation, the stress response, proliferation, and apoptosis [63]. MiRNAs targeting IL-6, TGFβ, and HGF, which can affect proliferation, secretion, migration, and intracellular adhesion, are reportedly upregulated or downregulated in breast CAFs [64, 65]. miR-26b expression is downregulated in breast CAFs and contributes to cancer cell migration and invasion [66]. Of the miRNAs investigated, miR-16 and miR-320 are upregulated in bladder CAFs, whereas miR-16 and miR-130a are downregulated in normal bladder fibroblasts [67]. In contrast, DNA methylation in CAFs has also been widely investigated [68]. Promoter hypermethylation might also be responsible for the inactivation of another candidate tumor-suppressor gene and the opioid binding protein/cell adhesion molecule-like gene in invasive cervical cancer CAFs [69].
Moreover, previous studies have demonstrated little or no genetic changes in CAFs. In fact, it has been demonstrated that somatic alterations are extremely rare in CAFs and unlikely to influence cancer progression [70]. Similarly, p53 mutation and genomic changes associated with cell abnormalities rarely occur in CAFs compared with their counterpart fibroblasts [71], and we discovered several nonsynonymous mutations, although none recurred or affected the biological behaviors of CAFs [72]. The views on the genetic changes in CAFs remain contradictory, but based on the studies conducted to date, epigenetic changes rather than genetic changes appear to be more predominant for the characterization of CAFs.
Roles of CAFs in cancer progression
Based on the concept that CAFs are distinct from fibroblasts, the mechanisms through which they function and operate and their impact remain to be determined. In fact, numerous pathways and signals are involved in the tumor-enhancing process, and various studies have shown that CAFs contribute to tumorigenesis in diverse manners, particularly proliferation, invasion, and metastasis through the secretion of cytokines, chemokines, exosomes, and the ECM [4]. Recent studies indicate that CAFs might also modulate the vascular and immune system and enhance metabolic remodeling in cancer.
CAFs contribute to cancer proliferation and stemness
The promotion of cancer proliferation is considered one of the features of CAFs, and the crucial role of CAFs in tumorigenesis is likely related to the fact that fibroblasts have already been activated to transform into CAFs in the stroma at the precancerous stage of epithelial cells [73]. Fibroblasts derived from tumors and colon polyps demonstrate potential potency for proliferation, which indicates that fibroblasts might be capable of intensifying tumorigenesis in vivo [74]. CAFs can alter the epithelial morphology, decrease cell death and enhance cell proliferation in human prostate cancer, normal human prostate fibroblasts, and normal human prostate epithelial cells immortalized with SV40-large T antigen (Tag-HPE cells) [75]. In addition, various growth factors and cytokines from CAFs can enhance cancer cell proliferation through different mechanisms; for example, the injection of breast cancer cells with CAFs isolated from human breast cancer tissues into nude mice caused tumors to grow much faster than the injection of cells with fibroblasts due to the upregulated secretion of stromal cell-derived factor 1 (SDF-1), which leads to the recruitment of epithelial precursor cells and the promotion of epithelial tumorigenesis [76]. The status of fork-head box F1 (FoxF1) has been shown to regulate the ability of fibroblasts to stimulate xenograft tumor growth [77]. Proliferative tumor activity is induced in hormone-independent mice via the fibroblast growth factor-2 (FGF2)–FGF2 receptor axis, and the inhibition of PDGF secreted by CAFs can reduce angiogenesis and tumor cell proliferation [78, 79].
The CSC theory highlights a self-renewing subpopulation of cancer cells that fuels tumor growth. CAFs also play a critical role in sustaining cancer stemness. Of relevance, CAFs in breast luminal cancer tissues with high autophagy activity show increased tumorigenicity. Specifically, autophagic CAFs release high-mobility group box 1 (HMGB1), and this protein activates its receptor, Toll-like receptor (TLR) 4, which is expressed in luminal breast cancer cells, to enhance their stemness [80]. Similarly, CAFs can sustain cancer cell stemness or even induce the reacquisition of stem-like properties by expressing insulin-like growth factor (IGF)-II [81]. Furthermore, CAF-conditioned medium is able to increase the number of spheroid colonies and the expression level of the CSC markers of OCUM-12/SP and OCUM-2MD3/SP scirrhous gastric cancer cells, which can be blocked by TGFβ inhibitors, and this finding indicates that CAFs mediate cancer cell stemness through TGFβ signaling [82]. CAFs can clearly influence cancer proliferation and stemness in diverse manners, which indicates their essential role in regulating the TME and favoring cancer progression.
CAFs facilitate invasion, migration, and EMT in cancer
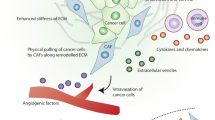
The crosstalk between different cell types and cancerous stroma is recognized to be essential in cancer progression [4, 83]. CAFs also closely and actively interact with cancer cells, and increasing lines of evidence show that CAFs can facilitate tumor invasion and migration through these interactions [84], which are also considered characteristic of CAFs in various studies. The association between cancer cells and CAFs has been well depicted by comparing their interactions with cancer cells in diffuse-type gastric cancer (diffuse-type GC) to those in intestinal-type GC (Fig. 2). Consequently, these interactions lead to the malignancy of cancer cells. Among these relationships, CAFs reportedly synthesize massive amounts of soluble tumor-promoting factors, such as SDF-1/CXCL12, which elevate invasiveness by promoting integrin beta1 clustering [85]. Moreover, the co-culture of CAFs with melanoma induces a considerably higher amount of cytokines, such as IL-6, IL-8, and IL-1β, and inhibits the expression of molecules that weaken the invasive capacity of cancer cells [86, 87]. MMP1 can activate PAR1 by reducing its extracellular region, which enhances the invasion and metastasis of tumor cells via PAR1-dependent Ca2+ signaling [88]. Under certain conditions, CAFs can work as a supporter or carrier by leading to collective invasion in squamous cell carcinoma (SCC) [89]. Similarly, highly motile CAFs, which are converted via the expression of rhomboid 5 homolog 2 (RHBDF2), can facilitate tumor cell invasion in the ECM, and this mechanism is named the “tugboat mechanism” [72]. CAFs are able to mediate directional migration by improving nonmuscle myosin II (MyoII), α5β1 integrin, and PDGFRα, which leads to the assembly of fibronectin in prostatic and pancreatic carcinoma samples [90]. CAFs have also been demonstrated to promote a cooperative collective invasion or comigration of CAFs and cancer cells through a heterophilic adhesion between CAFs and cancer cells [91].

Cancer–stromal interactions in each histological type. Images showing the immunofluorescent staining of cancer cells (AE1/AE3) and cancer-associated fibroblasts (CAFs) (α-smooth muscle actin (α-SMA)) are shown. The interactions between CAFs and cancer cells tend to be more intimate in diffuse-type gastric cancer than in intestinal-type gastric cancer. In intestinal-type cancer, an observable distance is detected between CAFs and cancer cells, and a certain stromal structure is require for maintenance of the glandular tube. In contrast, in diffuse-type cancer, CAFs closely interact with cancer cells, resulting in scattered morphological features
The EMT is one of the most pivotal mechanisms through which CAFs favor tumorigenesis, and through this process, tumor cells acquire the capacity of movement and invasion into the adjacent tissues, which results in the scattering of cancer cells and metastatic spread. Fibroblasts are converted into CAFs due to a deficiency in p85α, and paracrine Wnt10 from these CAFs activate Wnt signaling, which leads to the EMT and the upregulation of cancer cell proliferation and cell motility [92]. Activation of the TGFβ/Smad signaling pathway in cancer cells triggered by paracrine TGFβ from CAFs induces the EMT, which results in an enhancement of the metastatic potential of cancer cells [93]. As demonstrated through immunohistochemical evaluations in tongue SCC and metastatic tumors in regional lymph nodes (RLNs), the expression of EMT markers is commonly observed in both primary and metastatic tumors, whereas CAFs are commonly found in both primary and metastatic SCC, which indicates that CAFs are related to EMT induction and metastasis [94]. CAFs can also interact with tumorigenesis by secreting vast amounts of MMPs. For instance, MMP3 in the mammary epithelium triggers a cascade of events, including the cleavage of E-cadherin, which results in the EMT and facilitates the invasion of tumor cells [95]. Taken together, the results indicate that a comprehensive understanding of the underlying signaling through which CAFs promote cancer invasion, metastasis, and the EMT is crucial for the development of new therapeutic strategies that would induce signaling rather than target CAFs themselves and thereby avoid risks, such as mistargeting.
CAFs and exosomes
Cell-to-cell communication involves more than soluble factors, and recent studies have shown that secreted microvesicles, including exosomes, carry biologically active molecules, and activated growth factor receptors, which can be horizontally transferred and function in recipient cells [96]. Notably, exosomes can promote chemoresistance in cancers. For instance, treatment with gemcitabine-exposed CAF-conditioned medium significantly increases the survival of co-cultured epithelial cells in pancreatic cancer, whereas treatment with an inhibitor of exosome release, GW4869, reduces the proliferation of cocultured cancer cells. These exosomes from CAFs increase chemoresistance-inducing factor, Snail, in recipient epithelial cells and promote proliferation and drug resistance [97]. Similarly, when cultured in fibroblast-derived conditioned medium, colorectal CSCs show increases in their percentage, clonogenicity, and tumor growth upon treatment with 5-fluorouracil or oxaliplatin, and the inhibition of CAF exosomes decreases this phenomenon [98]. These studies demonstrate an important role for CAF exosomes in chemotherapeutic drug resistance. In addition, CAF exosomes can also influence cancer through other effects. In breast cancer, the loss of p85α can lead to the conversion of fibroblasts into CAFs, and exosomes delivered by CAFs further activate Wnt10b signaling, which results in the promotion of cancer cell proliferation and metastasis [92]. In addition, it has been reported that miR-34a-5p binds to its direct downstream target AXL to suppress cancer cell proliferation and metastasis in OSCC, and CAF-derived exosomes significantly reduce miR-34a-5p expression, which results in aggressiveness in OSCC [99]. Exosomes from CAFs can also be carriers of miRNAs that influence cancer progression in diverse manners. For example, exosomes containing miRNA21 are delivered from CAFs to cancer-associated adipocytes, where it suppresses ovarian cancer apoptosis and confers chemoresistance by binding to its direct novel target, APAF1 [100]. Similarly, in head and neck cancer (HNC), exosomal miR-196a binds novel targets, CDKN1B and ING5, to endow HNC cells with cisplatin resistance and hence regulates HNC cell survival and proliferation [101]. This evidence showing that CAF exosomes serve as positive mediators of cancer makes these exosomes noticeable in the TME.
CAFs control vascular and immune systems
Previous studies on various cancers have focused on vascularization and the immune response in the TME due to their potential to be targeted in treatment. A transgenic mouse model expressing the A. victoria green fluorescent protein (GFP) under the control of the promoter for vascular endothelial growth factor (VEGF) was used to show the early evidence of tumor vascularization by CAFs. In this model, cancer cell-induced VEGF expression in intra- or peri‐tumoral fibroblasts and these VEGF‐expressing regions are vascularized [102]. The role of CAFs in tumor vascularization has been confirmed in other models. For example, an HPV cervical carcinogenesis model, PDGFR-expressing CAFs induce cancer cell proliferation and angiogenesis. The pro-angiogenic effect is due to FGF2 secretion, which is induced by cancer cell-derived PDGF, and this effect is abrogated by pharmacological treatment to block stromal PDGFR signaling [79]. After deletion of the tumor-suppressor Pten, angiogenesis, and tumorigenesis are enhanced in MMTV-ErbB2/neu and MMTV-PyMT breast cancer models. In this tumor model, stromal Pten depletion prior to tumor onset initiates the Ets2-dependent expression of ECM remodeling, wound healing, and chronic inflammation factors in CAFs and the intratumoral recruitment of macrophages. Stromal Ets2 depletion in stromal Pten-null mice reduces growth, vascularization, and macrophage recruitment in tumors [103]. Moreover, the subcutaneously transplantation of PyMT CAFs embedded in Matrigel plugs induces vascularization of the plug, and this pro-angiogenic capacity can be degraded by silencing YAP1 [44].
The cytokines secreted by CAFs also contribute to the recruitment and polarization of immune cells. Of note, macrophage polarization has been observed in pancreatic cancer, in which CAFs produce high amounts of macrophage colony-stimulating factor and induce a tumor-promoting tumor-associated macrophage (TAM) phenotype [104]. A 3D co-culture model that includes cancer cells, CAFs, and monocytes has shown the production of various immunosuppressive cytokines that are known to promote the polarization of M2-like macrophages and myeloid-derived suppressive cells [105]. TAMs and CAFs can also work synergistically in prostate cancer, whereas CAFs produce cytokines such as IL-6 and SDF-1 to polarize macrophages toward the M2 phenotype [106]. CAFs derived from a murine model of HPV skin carcinogenesis promote tumor growth, vascularization, and macrophage recruitment when cotransplanted with murine PDSC5 skin carcinoma cells [107]. Previous studies have shown that in an orthotopic 4T1 model, FAP-positive CAF-depleted tumors upregulate IL-2 and IL-7 and downregulate IL-6 and IL-4, which consequently enhances the recruitment of antitumor immune cells (DCs and CD8+ T cells) and inhibits the recruitment of pro-tumorigenic cells. As a result, the depletion of FAP-positive CAFs inhibits lung cancer metastasis [108]. In addition, CAFs can produce various factors, such as CXCL12, which can limit T-cell movement and/or recruitment into tumor tissue [109]. This potential capacity of modulating vascular and immune cells further highlights the complexity of CAFs and the possibilities of targeting CAFs in anticancer treatment.
CAFs support cancer progression through metabolic changes
As cancer cells grow, they recruit stromal cells to induce the complex formation of the TME. These reactive stromal cells, including CAFs, coevolve and continually interact with cancer cells. During this period, CAFs produce various nutrients and undergo diverse metabolic changes, which can also serve as a nonignorable factor favoring cancer progression. Extracellular vesicle carries miR-105 from breast cancer cells and induces a metabolic program in CAFs that allows the CAFs to gain the capability of changing the metabolic environment according to different situations. In the presence of sufficient nutrients, miR-105-reprogrammed CAFs enhance glucose and glutamine metabolism to fuel adjacent cancer cells. In contrast, if the nutrient levels are low and metabolic byproducts accumulate, CAFs can scavenge metabolic wastes by converting them into energy-rich metabolites [110]. Another study on breast cancer showed that the translocation of cytoplasmic G-protein-coupled estrogen receptor (GPER) in CAFs activates the estrogen/GPER/cAMP/PKA/CREB signaling axis, which triggers the aerobic glycolysis switch in CAFs. Glycolytic CAFs feed the extra pyruvate and lactate to tumor cells and eventually induce tumor drug resistance [111]. Metabolic changes in CAFs might also directly support cancer cell proliferation; for example, ketone production by CAFs due to a deficiency in breast cancer type 1 susceptibility protein contributes to tumor growth in mice [112]. Moreover, CAFs undergo metabolic reprogramming toward a Warburg phenotype by contacting prostate cancer cells and thereby triggering increases in the expression of the glucose transporter GLUT1 and lactate production. In contrast, prostate cancer cells are reprogrammed toward aerobic metabolism after coming into contact with CAFs, resulting in a decrease in GLUT1 expression and an increase in lactate upload through the lactate transporter monocarboxylate transporter 1, which gradually results in the establishment of an anabolic pathway based on lactate upload and the maintenance of cell growth [113]. Heterocellular interactions shape the metabolic nature of the TME to support tumors in multifarious manners, and the pathways and activities mediating these processes are gaining considerable attention from the basic science community as a novel perspective for comprehending how stromal cells facilitate the rapid progression of cancer.
CAFs as tumor suppressors
Despite the overwhelming theories that CAFs are positive regulators of cancer, some researchers have proposed that CAFs or fibroblasts exert a negative influence on cancer that involves several aspects, such as proliferation, stemness, differentiation states, and the immune response. Various subtypes of CAFs, namely, CD146-positive and CD146-negative, have been described in breast cancer, and CD146-positive CAFs confer sustained estrogen-dependent proliferation and tamoxifen sensitivity to luminal breast cancer cells [53]. Moreover, CAFs have been defined as C1 and C2 types according to low and high α-SMA expression, respectively, and C1-type CAFs suppress the self-renewal of oral stem-like cancer cells through bone morphogenetic protein 4 (BMP4) [114]. The downregulation of TGFβ signaling in activated fibroblasts can induce prostatic intraepithelial neoplasia, which indicates that fibroblasts have the potential to suppress the initiation of tumors through the modulation of TGFβ signaling [115]. Some studies have also suggested that CAFs can restrain pancreatic ductal adenocarcinoma (PDAC) in a mouse model of PDAC through either the direct elimination of α-SMA+ CAFs or the suppression of Hedgehog signaling, which sustains stromal fibroblasts and leads to an undifferentiated phenotype of cancer through a mechanism related to angiogenesis. This finding indicates that the Hedgehog-driven stroma suppresses tumor growth in part by restraining tumor angiogenesis [116]. In addition, CAFs can secrete immunomodulatory cytokines, such as IL-10, TGFβ, TNF, IFNγ, and IL-6, and help recruit and polarize macrophages, T lymphocytes and natural killer cells, which highlights the possibility of a tumor-negative effect of CAFs interacting with immune cells [117]. Fibroblasts and CAFs expressing the ligand Slit2 inhibit the tumorigenicity of breast cancer cells expressing the corresponding Robo1-receptor on their surface. Ligand-induced Robo1 activation interferes with phosphoinositide 3-kinase (PI3K)- and β-catenin signaling in cancer cells and diminishes their malignant potential. Previous studies showed that Slit-stimulated signaling also inhibits the pro-tumorigenic SDF-1/CXCR4-signaling pathway [118, 119].
The depletion of engineered MFB and of sonic hedgehog in a PDAC mouse model has been demonstrated to facilitate the EMT by elevating its transcription factors (Twist, Snail, and Slug), by altering immune gene expression, by increasing the amount of regulatory T cells (Tregs) and by decreasing the cytotoxic CD8+/Treg and CD3+/CD11b+ ratios; these findings demonstrate that CAF-depleted tumors exhibit a more aggressive and undifferentiated histology, which is contrary to the expected results. Even though the CAFs originated from mice, this study still unmasked a beneficial role for CAFs in cancer suppression [22]. A few studies have also shown that PDAC, breast cancer, and lung cancer patients with high desmoplasia can show improved prognosis and better survival rates than their counterparts [120]. Based on these reports, the depletion of CAFs appears to negatively affect well-differentiated cancer types, but this effect has not been observed in undifferentiated or poorly differentiated cancer types, which suggests that the significance of CAFs depends on the differentiation status of tumors. To view CAFs as a whole, the available evidence shows that CAFs can function as negative regulators in some ways, and we thus need to judge them objectively; however, the evidence tends to lean toward the conclusion that CAFs play a tumor-supporting role. As the number of studies on CAFs continues to increase, the ambiguous status of CAFs might eventually be unraveled.
Targeting CAFs in anticancer therapies
It is now clear that there is continual crosstalk not only between tumor cells and immune cells but also between immune cells and other stroma cells, such as CAFs, and anticancer therapies that target the TME have been a striking topic over the last decade. FAP is a commonly used cell surface marker of activated stromal fibroblasts and a serine protease that can modulate the recruitment, differentiation, and proliferation of MFBs [121]. It has been shown that targeting FAP in preclinical models exhibits antitumor efficacy. Mechanistically, the depletion of FAP-expressing cells grants therapeutic vaccination against tumors via interferon-γ and tumor necrosis factor-α in mice [122]. In addition, the administration of a newly developed anti-FAP monoclonal antibody, FAP5-DM1, has introduced long-lasting inhibition of tumor proliferation and even complete tumor decrease without observable toxicity in stroma-rich xenograft models of lung, pancreas, and head and neck cancers [123]. Moreover, the blockage of CXCL12 derived from FAP-positive CAFs facilitates the immunotherapy sensitivity of tumors in a human pancreatic ductal adenocarcinoma model [109]. However, in addition to CAFs, chimeric T cells recognize multipotent bone marrow stem cells that show positivity for FAP. This pleiotropic activity results in mice suffering from cachexia and lethal bone toxicities [124]. These findings lead to the conclusion that the use of systemic therapies against these targets still deserves consideration but also offers a hint for the origin of these FAP-positive CAFs in tumors.
In addition to CAF depletion, the conversion of pro-tumorigenic CAFs into quiescent-state fibroblasts is considered an appealing approach. Of relevance, vitamin A deficiency has been observed in patients with pancreatic ductal adenocarcinoma, and this deficiency induces pancreatic stellate cell (PSC) activation. In LSL-KrasG12D/+, LSL-Trp53R172H/+, and Pdx-1-Cre human pancreatic ductal adenocarcinoma model mice, all-trans retinoic acid (ATRA) reportedly stabilizes PSCs and thereby reduces the proliferation and invasive ability of cancer cells and the translocation of β-catenin to the nucleus, which increases cancer cell apoptosis [125]. Similarly, another study indicated that ATRA is capable of suppressing ECM remodeling and thus inhibiting cancer invasion and migration by downregulating the MyoII-dependent force generation and mechanosensing of PSCs and reprogramming them to a more quiescent phenotype in PDAC [126]. In contrast, vitamin D receptor has also been shown to serve as the transcriptional regulator that maintains PSCs in a quiescent state, and consequently, vitamin D sensitizes tumors to gemcitabine and reduces the tumor volume [127]. Notably, the reversible conversion of CAF phenotypes is not exclusive to PSCs because in a mouse model in which liver fibrosis spontaneously regresses, HSCs and MFBs can also revert to an inactive state characterized by the upregulation of anti-apoptotic genes [128].
Because CAFs are frequently activated, numerous cytokines and regulatory factors play crucial roles in CAF biology, and the targeting of activated signaling and effectors appears reasonable. The potential targets of this strategy can generally be divided into molecules that activate CAFs and those secreted by CAFs that promote cancer proliferation, invasion, migration, angiogenesis, and chemoresistance. Imatinib is a clinically proven kinase inhibitor that was used to block PDFGR signaling in CAFs using a mouse model of human cervical cancer, and this blockage reduced FGF2 and FGF7 production and thereby hindered cancer proliferation and angiogenesis [79]. It is known that JAK1–STAT3 signaling is greatly involved in CAF activation. A recent study revealed that cytokine signaling mediates cancer cell dissemination in force-mediated matrix remodeling by CAFs, and the findings demonstrated that a member of the IL-6 family, oncostatin M, potentiates ECM remodeling that favors cancer cell invasion by stimulating the actomyosin contractility of the CAFs through glycoprotein 130, JAK1, ROCK, and STAT3 signaling [129]. In fact, agents targeting this particular signaling in cancer are currently being investigated in a clinical trial [130].
Various anti-CAF therapies have been utilized to date, and these target different processes in tumor-promoting signaling (Fig. 3). Increasing lines of evidence from recent studies have shown that the immune response is crucial against tumors and is becoming a main field of focus. Nonetheless, certain tumors and patients are more sensitive to immunotherapies than others. One possibility is that while immunotherapies cannot be overcome by strengthening the immune system alone, and this effect might be due in part to the immunosuppressive capability of the TME, which is partially caused by CAFs. Therapies targeting the tumor stroma combined with immunotherapies could abate this influence and thereby exert an antitumor immune response.

Major anticancer therapies targeting cancer-associated fibroblasts (CAFs). Multiple strategies for targeting CAFs that support chemotherapies in anticancer treatment are shown. FAP5-DM1, a fibroblast-activation protein (FAP) inhibitor, is used to interfere with FAP function, which results in decreased tumor growth, whereas another CXCR4 inhibitor, AMD3100, can suppress the interaction between CXCL12 from CAFs and CXCR4 and thereby enhance immunotherapy sensitivity. Imatinib is a well-known platelet-derived growth factor receptor (PDGFR) inhibitor that can block PDGFR signaling, leading to the downregulation of FGF2 and FGF7 and thereby the repression of tumor growth and angiogenesis. Blockage of the IL-6 receptor using IL-6 mAb can suppress extracellular matrix (ECM) remodeling by hindering JAK/STAT3 signaling. CAF-like cells, such as stellate cells, can be converted to a quiescent state, which results in the generation of less pro-tumorigenesis factors by all-trans retinoic acid (ATRA)
Conclusions and future expectations
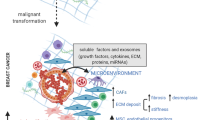
Fibroblast heterogeneity can be clearly observed in various anatomic components [131], and the different origins of CAFs result in the promotion of heterogeneous properties in tumors and functionally impact tumors through various mechanisms (Fig. 4). Studies regarding the identification of mesenchymal cells with certain functions in different tissues might also provide diverse ideas about the definition of heterogeneity [132]. Whether stable genetic alterations occur in CAFs compared with fibroblasts is difficult to determine; however, such changes might conceivably arise in CAFs because their morphology and secretory ability are distinct from those of fibroblasts.

Functional heterogeneity of cancer-associated fibroblasts (CAFs). CAFs can contribute to cancer progression through multiple mechanisms, including the promotion of proliferation, the enhancement of invasion, metastasis, and vascularization (red section), and antitumor effects (blue section). CAFs secrete numerous stromal cell growth factors, such as fibroblast growth factor (FGF), stromal cell-derived factor (SDF), and heparin-binding epidermal growth factor (HB-EGF), resulting in the promotion of cancer cell proliferation, and this effect can also be accomplished by exosomes and nutrients, such as glutamine and ketone bodies, from CAFs. Extracellular matrix (ECM) remodeling through Yes-associated protein, matrix metalloproteinase 2 (MMP-2), MMP-3 by CAFs should additionally be considered one of the main strategies for cancer promotion. CAFs also promote invasion by producing cytokines and proteinases such as IL-6, IL-8, and MMP3. Moreover, certain mechanisms causing the upregulation of transcription factors, such as RHBDF2, also contribute to cancer invasion. Furthermore, CAFs regulate angiogenesis and immune cell recruitment and polarization in a pro-tumorigenic manner by secreting growth factors and ligands such as CXCL12, whereas nutrients such as lactate or exosomes provided by CAFs induce cancer cell chemoresistance. However, CAFs do exert tumor-suppressive effects, and the inhibition of Hedgehog signaling depletes CAFs and leads to disease exacerbation with worse survival and an undifferentiated cell state, which indicates that CAFs can favor the differentiated states of cancer cells. Slit2+ and CD146+ CAFs suppress tumorigenesis, whereas molecules such as BMP4 can inhibit the self-renewal of stem-like cancer cells
Over the past few decades, CAFs have mainly been considered positive regulators of malignant tumorigenesis. Nonetheless, new evidence indicates that we should view the issue critically because in some cases, CAFs might serve as negative regulators in the process of tumor growth, and whether we should further divide these types of fibroblasts into new subtypes remains debatable. However, fibroblasts are genetically programmed to respond to wound and tissue damage, and activated fibroblasts bind to synthesize molecules that regulate inflammation and cell growth; thus, in the TME, the indirect role of fibroblasts in eventually causing tumor growth, invasion, and metastasis is not surprising. Further studies to uncover the hidden secrets of fibroblasts are worth pursuing.
References
Virchow R. Cellular pathology. As based upon physiological and pathological histology. Lecture XVI--Atheromatous affection of arteries. 1858. Nutr Rev. 1989;47:23–25.
Tarin D, Croft CB. Ultrastructural features of wound healing in mouse skin. J Anat. 1969;105:189–90.
Gascard P, Tlsty TD. Carcinoma-associated fibroblasts: orchestrating the composition of malignancy. Genes Dev. 2016;30:1002–19.
Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401.
Ishii G, Ochiai A, Neri S. Phenotypic and functional heterogeneity of cancer-associated fibroblast within the tumor microenvironment. Adv Drug Deliv Rev. 2016;99:186–96.
Ohlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med. 2014;211:1503–23.
Augsten M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front Oncol. 2014;4:62.
Gabbiani G, Ryan GB, Majne G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia. 1971;27:549–50.
Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–63.
Cirri P, Chiarugi P. Cancer associated fibroblasts: the dark side of the coin. Am J Cancer Res. 2011;1:482–97.
Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–7.
Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007;67:10123–8.
Bochet L, Lehuede C, Dauvillier S, Wang YY, Dirat B, Laurent V, et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013;73:5657–68.
Matsumoto T, Kano K, Kondo D, Fukuda N, Iribe Y, Tanaka N, et al. Mature adipocyte-derived dedifferentiated fat cells exhibit multilineage potential. J Cell Physiol. 2008;215:210–22.
Jung Y, Kim JK, Shiozawa Y, Wang J, Mishra A, Joseph J, et al. Recruitment of mesenchymal stem cells into prostate tumours promotes metastasis. Nat Commun. 2013;4:1795.
Weber CE, Kothari AN, Wai PY, Li NY, Driver J, Zapf MA, et al. Osteopontin mediates an MZF1-TGF-beta1-dependent transformation of mesenchymal stem cells into cancer-associated fibroblasts in breast cancer. Oncogene. 2015;34:4821–33.
Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest. 2007;117:50–59.
Yin C, Evason KJ, Asahina K, Stainier DY. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest. 2013;123:1902–10.
Petersen OW, Nielsen HL, Gudjonsson T, Villadsen R, Rank F, Niebuhr E, et al. Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am J Pathol. 2003;162:391–402.
De Wever O, Demetter P, Mareel M, Bracke M. Stromal myofibroblasts are drivers of invasive cancer growth. Int J Cancer. 2008;123:2229–38.
Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C. Alpha-smooth muscle actin expression upregulates fibroblast contractile activity. Mol Biol Cell. 2001;12:2730–41.
Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–34.
Ramirez-Montagut T, Blachere NE, Sviderskaya EV, Bennett DC, Rettig WJ, Garin-Chesa P, et al. FAPalpha, a surface peptidase expressed during wound healing, is a tumor suppressor. Oncogene. 2004;23:5435–46.
Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, et al. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol. 1995;130:393–405.
Gardner H, Kreidberg J, Koteliansky V, Jaenisch R. Deletion of integrin alpha 1 by homologous recombination permits normal murine development but gives rise to a specific deficit in cell adhesion. Dev Biol. 1996;175:301–13.
Kisselbach L, Merges M, Bossie A, Boyd A. CD90 expression on human primary cells and elimination of contaminating fibroblasts from cell cultures. Cytotechnology. 2009;59:31–44.
Cheng F, Shen Y, Mohanasundaram P, Lindstrom M, Ivaska J, Ny T, et al. Vimentin coordinates fibroblast proliferation and keratinocyte differentiation in wound healing via TGF-beta-Slug signaling. Proc Natl Acad Sci USA. 2016;113:E4320–4327.
Ranogajec I, Jakic-Razumovic J, Puzovic V, Gabrilovac J. Prognostic value of matrix metalloproteinase-2 (MMP-2), matrix metalloproteinase-9 (MMP-9) and aminopeptidase N/CD13 in breast cancer patients. Med Oncol. 2012;29:561–9.
Vogel W, Gish GD, Alves F, Pawson T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol Cell. 1997;1:13–23.
Strutz F. The fibroblast--a (trans-) differentiated cell? Nephrol Dial Transplant. 1995;10:1504–6.
Pietras K, Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res. 2010;316:1324–31.
Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. 2018;33:463–.e10.
Kawase A, Ishii G, Nagai K, Ito T, Nagano T, Murata Y, et al. Podoplanin expression by cancer associated fibroblasts predicts poor prognosis of lung adenocarcinoma. Int J Cancer. 2008;123:1053–9.
Neri S, Ishii G, Hashimoto H, Kuwata T, Nagai K, Date H, et al. Podoplanin-expressing cancer-associated fibroblasts lead and enhance the local invasion of cancer cells in lung adenocarcinoma. Int J Cancer. 2015;137:784–96.
Weigel MT, Banerjee S, Arnedos M, Salter J, A’Hern R, Dowsett M, et al. Enhanced expression of the PDGFR/Abl signaling pathway in aromatase inhibitor-resistant breast cancer. Ann Oncol. 2013;24:126–33.
Anderberg C, Li H, Fredriksson L, Andrae J, Betsholtz C, Li X, et al. Paracrine signaling by platelet-derived growth factor-CC promotes tumor growth by recruitment of cancer-associated fibroblasts. Cancer Res. 2009;69:369–78.
Lynch MD, Watt FM. Fibroblast heterogeneity: implications for human disease. J Clin Invest. 2018;128:26–35.
Murata T, Mizushima H, Chinen I, Moribe H, Yagi S, Hoffman RM, et al. HB-EGF and PDGF mediate reciprocal interactions of carcinoma cells with cancer-associated fibroblasts to support progression of uterine cervical cancers. Cancer Res. 2011;71:6633–42.
De Wever O, Nguyen QD, Van Hoorde L, Bracke M, Bruyneel E, Gespach C, et al. Tenascin-C and SF/HGF produced by myofibroblasts in vitro provide convergent pro-invasive signals to human colon cancer cells through RhoA and Rac. FASEB J. 2004;18:1016–8.
Goetz JG, Minguet S, Navarro-Lerida I, Lazcano JJ, Samaniego R, Calvo E, et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. 2011;146:148–63.
True LD, Zhang H, Ye M, Huang CY, Nelson PS, von Haller PD, et al. CD90/THY1 is overexpressed in prostate cancer-associated fibroblasts and could serve as a cancer biomarker. Mod Pathol. 2010;23:1346–56.
Ha SY, Yeo SY, Xuan YH, Kim SH. The prognostic significance of cancer-associated fibroblasts in esophageal squamous cell carcinoma. PLoS ONE. 2014;9:e99955.
Witkiewicz AK, Dasgupta A, Sotgia F, Mercier I, Pestell RG, Sabel M, et al. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol. 2009;174:2023–34.
Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15:637–46.
Jung EJ, Moon HG, Cho BI, Jeong CY, Joo YT, Lee YJ, et al. Galectin-1 expression in cancer-associated stromal cells correlates tumor invasiveness and tumor progression in breast cancer. Int J Cancer. 2007;120:2331–8.
Lawrenson K, Grun B, Lee N, Mhawech-Fauceglia P, Kan J, Swenson S, et al. NPPB is a novel candidate biomarker expressed by cancer-associated fibroblasts in epithelial ovarian cancer. Int J Cancer. 2015;136:1390–401.
Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215.
Osterreicher CH, Penz-Osterreicher M, Grivennikov SI, Guma M, Koltsova EK, Datz C, et al. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc Natl Acad Sci USA. 2011;108:308–13.
Nardi F, Fitchev P, Franco OE, Ivanisevic J, Scheibler A, Hayward SW, et al. PEDF regulates plasticity of a novel lipid-MTOC axis in prostate cancer-associated fibroblasts. J Cell Sci. 2018;131:jcs213579.
Guido C, Whitaker-Menezes D, Capparelli C, Balliet R, Lin Z, Pestell RG, et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-beta drives tumor growth: connecting TGF-beta signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle. 2012;11:3019–35.
Albrengues J, Bertero T, Grasset E, Bonan S, Maiel M, Bourget I, et al. Epigenetic switch drives the conversion of fibroblasts into proinvasive cancer-associated fibroblasts. Nat Commun. 2015;6:10204.
Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10(+) GPR77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell. 2018;172:841–56 e816.
Brechbuhl HM, Finlay-Schultz J, Yamamoto TM, Gillen AE, Cittelly DM, Tan AC, et al. Fibroblast subtypes regulate responsiveness of luminal breast cancer to estrogen. Clin Cancer Res. 2017;23:1710–21.
Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y. et al. IL-1-induced JAK/STAT signaling is antagonized by TGF-beta to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019;9:282–301.
Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D, et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci USA. 2002;99:12877–82.
Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, et al. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2004;2:E7.
Bauer M, Su G, Casper C, He R, Rehrauer W, Friedl A. Heterogeneity of gene expression in stromal fibroblasts of human breast carcinomas and normal breast. Oncogene. 2010;29:1732–40.
Costea DE, Hills A, Osman AH, Thurlow J, Kalna G, Huang X, et al. Identification of two distinct carcinoma-associated fibroblast subtypes with differential tumor-promoting abilities in oral squamous cell carcinoma. Cancer Res. 2013;73:3888–901.
Berdiel-Acer M, Sanz-Pamplona R, Calon A, Cuadras D, Berenguer A, Sanjuan X, et al. Differences between CAFs and their paired NCF from adjacent colonic mucosa reveal functional heterogeneity of CAFs, providing prognostic information. Mol Oncol. 2014;8:1290–305.
Hayashi Y, Tsujii M, Kodama T, Akasaka T, Kondo J, Hikita H, et al. p53 functional deficiency in human colon cancer cells promotes fibroblast-mediated angiogenesis and tumor growth. Carcinogenesis. 2016;37:972–84.
Nakagawa H, Liyanarachchi S, Davuluri RV, Auer H, Martin EW Jr., et al. Role of cancer-associated stromal fibroblasts in metastatic colon cancer to the liver and their expression profiles. Oncogene. 2004;23:7366–77.
Hu M, Yao J, Cai L, Bachman KE, van den Brule F, Velculescu V, et al. Distinct epigenetic changes in the stromal cells of breast cancers. Nat Genet. 2005;37:899–905.
Ishimoto T, Baba H, Izumi D, Sugihara H, Kurashige J, Iwatsuki M, et al. Current perspectives toward the identification of key players in gastric cancer microRNA dysregulation. Int J Cancer. 2016;138:1337–49.
Zhao L, Sun Y, Hou Y, Peng Q, Wang L, Luo H, et al. MiRNA expression analysis of cancer-associated fibroblasts and normal fibroblasts in breast cancer. Int J Biochem Cell Biol. 2012;44:2051–9.
Li P, Shan JX, Chen XH, Zhang D, Su LP, Huang XY, et al. Epigenetic silencing of microRNA-149 in cancer-associated fibroblasts mediates prostaglandin E2/interleukin-6 signaling in the tumor microenvironment. Cell Res. 2015;25:588–603.
Verghese ET, Drury R, Green CA, Holliday DL, Lu X, Nash C, et al. MiR-26b is down-regulated in carcinoma-associated fibroblasts from ER-positive breast cancers leading to enhanced cell migration and invasion. J Pathol. 2013;231:388–99.
Enkelmann A, Heinzelmann J, von Eggeling F, Walter M, Berndt A, Wunderlich H, et al. Specific protein and miRNA patterns characterise tumour-associated fibroblasts in bladder cancer. J Cancer Res Clin Oncol. 2011;137:751–9.
Vizoso M, Puig M, Carmona FJ, Maqueda M, Velasquez A, Gomez A, et al. Aberrant DNA methylation in non-small cell lung cancer-associated fibroblasts. Carcinogenesis. 2015;36:1453–63.
Ye F, Zhang SF, Xie X, Lu WG. OPCML gene promoter methylation and gene expression in tumor and stroma cells of invasive cervical carcinoma. Cancer Invest. 2008;26:569–74.
Qiu W, Hu M, Sridhar A, Opeskin K, Fox S, Shipitsin M, et al. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat Genet. 2008;40:650–5.
Hosein AN, Wu M, Arcand SL, Lavallee S, Hebert J, Tonin PN, et al. Breast carcinoma-associated fibroblasts rarely contain p53 mutations or chromosomal aberrations. Cancer Res. 2010;70:5770–7.
Ishimoto T, Miyake K, Nandi T, Yashiro M, Onishi N, Huang KK, et al. Activation of transforming growth factor beta 1 signaling in gastric cancer-associated fibroblasts increases their motility, via expression of rhomboid 5 homolog 2, and ability to induce invasiveness of gastric cancer cells. Gastroenterology. 2017;153:191–204.
Liotta LA, Kohn EC. The microenvironment of the tumour-host interface. Nature. 2001;411:375–9.
Schor SL, Haggie JA, Durning P, Howell A, Smith L, Sellwood RA, et al. Occurrence of a fetal fibroblast phenotype in familial breast cancer. Int J Cancer. 1986;37:831–6.
Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999;59:5002–11.
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–48.
Saito RA, Micke P, Paulsson J, Augsten M, Pena C, Jonsson P, et al. Forkhead box F1 regulates tumor-promoting properties of cancer-associated fibroblasts in lung cancer. Cancer Res. 2010;70:2644–54.
Giulianelli S, Cerliani JP, Lamb CA, Fabris VT, Bottino MC, Gorostiaga MA, et al. Carcinoma-associated fibroblasts activate progesterone receptors and induce hormone independent mammary tumor growth: a role for the FGF-2/FGFR-2 axis. Int J Cancer. 2008;123:2518–31.
Pietras K, Pahler J, Bergers G, Hanahan D. Functions of paracrine PDGF signaling in the proangiogenic tumor stroma revealed by pharmacological targeting. PLoS Med. 2008;5:e19.
Zhao XL, Lin Y, Jiang J, Tang Z, Yang S, Lu L, et al. High-mobility group box 1 released by autophagic cancer-associated fibroblasts maintains the stemness of luminal breast cancer cells. J Pathol. 2017;243:376–89.
Chen WJ, Ho CC, Chang YL, Chen HY, Lin CA, Ling TY, et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat Commun. 2014;5:3472.
Hasegawa T, Yashiro M, Nishii T, Matsuoka J, Fuyuhiro Y, Morisaki T, et al. Cancer-associated fibroblasts might sustain the stemness of scirrhous gastric cancer cells via transforming growth factor-beta signaling. Int J Cancer. 2014;134:1785–95.
Rupp C, Scherzer M, Rudisch A, Unger C, Haslinger C, Schweifer N, et al. IGFBP7, a novel tumor stroma marker, with growth-promoting effects in colon cancer through a paracrine tumor-stroma interaction. Oncogene. 2015;34:815–25.
Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle. 2006;5:1597–601.
Izumi D, Ishimoto T, Miyake K, Sugihara H, Eto K, Sawayama H, et al. CXCL12/CXCR4 activation by cancer-associated fibroblasts promotes integrin beta1 clustering and invasiveness in gastric cancer. Int J Cancer. 2016;138:1207–19.
Li L, Dragulev B, Zigrino P, Mauch C, Fox JW. The invasive potential of human melanoma cell lines correlates with their ability to alter fibroblast gene expression in vitro and the stromal microenvironment in vivo. Int J Cancer. 2009;125:1796–804.
Fuchigami T, Kibe T, Koyama H, Kishida S, Iijima M, Nishizawa Y, et al. Regulation of IL-6 and IL-8 production by reciprocal cell-to-cell interactions between tumor cells and stromal fibroblasts through IL-1alpha in ameloblastoma. Biochem Biophys Res Commun. 2014;451:491–6.
Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. 2005;120:303–13.
Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, et al. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392–1400.
Erdogan B, Ao M, White LM, Means AL, Brewer BM, Yang L, et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J Cell Biol. 2017;216:3799–816.
Labernadie A, Kato T, Brugues A, Serra-Picamal X, Derzsi S, Arwert E, et al. A mechanically active heterotypic E-cadherin/N-cadherin adhesion enables fibroblasts to drive cancer cell invasion. Nat Cell Biol. 2017;19:224–37.
Chen Y, Zeng C, Zhan Y, Wang H, Jiang X, Li W. Aberrant low expression of p85alpha in stromal fibroblasts promotes breast cancer cell metastasis through exosome-mediated paracrine Wnt10b. Oncogene. 2017;36:4692–705.
Yu Y, Xiao CH, Tan LD, Wang QS, Li XQ, Feng YM. Cancer-associated fibroblasts induce epithelial-mesenchymal transition of breast cancer cells through paracrine TGF-beta signalling. Br J Cancer. 2014;110:724–32.
Vered M, Dayan D, Yahalom R, Dobriyan A, Barshack I, Bello IO, et al. Cancer-associated fibroblasts and epithelial-mesenchymal transition in metastatic oral tongue squamous cell carcinoma. Int J Cancer. 2010;127:1356–62.
Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67.
Taylor DD, Gercel-Taylor C. Exosomes/microvesicles: mediators of cancer-associated immunosuppressive microenvironments. Semin Immunopathol. 2011;33:441–54.
Richards KE, Zeleniak AE, Fishel ML, Wu J, Littlepage LE, Hill R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene. 2017;36:1770–8.
Hu Y, Yan C, Mu L, Huang K, Li X, Tao D, et al. Fibroblast-derived exosomes contribute to chemoresistance through priming cancer stem cells in colorectal cancer. PLoS ONE. 2015;10:e0125625.
Li YY, Tao YW, Gao S, Li P, Zheng JM, Zhang SE, et al. Cancer-associated fibroblasts contribute to oral cancer cells proliferation and metastasis via exosome-mediated paracrine miR-34a-5p. EBioMedicine. 2018;36:209–20.
Au Yeung CL, Co NN, Tsuruga T, Yeung TL, Kwan SY, Leung CS, et al. Exosomal transfer of stroma-derived miR21 confers paclitaxel resistance in ovarian cancer cells through targeting APAF1. Nat Commun. 2016;7:11150.
Qin X, Guo H, Wang X, Zhu X, Yan M, Wang X, et al. Exosomal miR-196a derived from cancer-associated fibroblasts confers cisplatin resistance in head and neck cancer through targeting CDKN1B and ING5. Genome Biol. 2019;20:12.
Fukumura D, Xavier R, Sugiura T, Chen Y, Park EC, Lu N, et al. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998;94:715–25.
Trimboli AJ, Cantemir-Stone CZ, Li F, Wallace JA, Merchant A, Creasap N, et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nature. 2009;461:1084–91.
Zhang A, Qian Y, Ye Z, Chen H, Xie H, Zhou L, et al. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017;6:463–70.
Kuen J, Darowski D, Kluge T, Majety M. Pancreatic cancer cell/fibroblast co-culture induces M2 like macrophages that influence therapeutic response in a 3D model. PLoS ONE. 2017;12:e0182039.
Comito G, Giannoni E, Segura CP, Barcellos-de-Souza P, Raspollini MR, Baroni G, et al. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene. 2014;33:2423–31.
Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. 2010;17:135–47.
Liao D, Luo Y, Markowitz D, Xiang R, Reisfeld RA. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS ONE. 2009;4:e7965.
Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. 2013;110:20212–7.
Yan W, Wu X, Zhou W, Fong MY, Cao M, Liu J, et al. Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells. Nat Cell Biol. 2018;20:597–609.
Yu T, Yang G, Hou Y, Tang X, Wu C, Wu XA, et al. Cytoplasmic GPER translocation in cancer-associated fibroblasts mediates cAMP/PKA/CREB/glycolytic axis to confer tumor cells with multidrug resistance. Oncogene. 2017;36:2131–45.
Salem AF, Howell A, Sartini M, Sotgia F, Lisanti MP. Downregulation of stromal BRCA1 drives breast cancer tumor growth via upregulation of HIF-1alpha, autophagy and ketone body production. Cell Cycle. 2012;11:4167–73.
Fiaschi T, Marini A, Giannoni E, Taddei ML, Gandellini P, De Donatis A, et al. Reciprocal metabolic reprogramming through lactate shuttle coordinately influences tumor-stroma interplay. Cancer Res. 2012;72:5130–40.
Patel AK, Vipparthi K, Thatikonda V, Arun I, Bhattacharjee S, Sharan R, et al. A subtype of cancer-associated fibroblasts with lower expression of alpha-smooth muscle actin suppresses stemness through BMP4 in oral carcinoma. Oncogenesis. 2018;7:78.
Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–51.
Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–47.
Harper J, Sainson RC. Regulation of the anti-tumour immune response by cancer-associated fibroblasts. Semin Cancer Biol. 2014;25:69–77.
Marlow R, Strickland P, Lee JS, Wu X, Pebenito M, Binnewies M, et al. SLITs suppress tumor growth in vivo by silencing Sdf1/Cxcr4 within breast epithelium. Cancer Res. 2008;68:7819–27.
Prasad A, Fernandis AZ, Rao Y, Ganju RK. Slit protein-mediated inhibition of CXCR4-induced chemotactic and chemoinvasive signaling pathways in breast cancer cells. J Biol Chem. 2004;279:9115–24.
Paulsson J, Micke P. Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin Cancer Biol. 2014;25:61–68.
Huber MA, Kraut N, Park JE, Schubert RD, Rettig WJ, Peter RU, et al. Fibroblast activation protein: differential expression and serine protease activity in reactive stromal fibroblasts of melanocytic skin tumors. J Invest Dermatol. 2003;120:182–8.
Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330:827–30.
Ostermann E, Garin-Chesa P, Heider KH, Kalat M, Lamche H, Puri C, et al. Effective immunoconjugate therapy in cancer models targeting a serine protease of tumor fibroblasts. Clin Cancer Res. 2008;14:4584–92.
Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CC, Restifo NP, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med. 2013;210:1125–35.
Froeling FE, Feig C, Chelala C, Dobson R, Mein CE, Tuveson DA, et al. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-beta-catenin signaling to slow tumor progression. Gastroenterology. 2011;141:1486–97.
Chronopoulos A, Robinson B, Sarper M, Cortes E, Auernheimer V, Lachowski D, et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat Commun. 2016;7:12630.
Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80–93.
Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci USA. 2012;109:9448–53.
Sanz-Moreno V, Gaggioli C, Yeo M, Albrengues J, Wallberg F, Viros A, et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell. 2011;20:229–45.
Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15:234–48.
Driskell RR, Lichtenberger BM, Hoste E, Kretzschmar K, Simons BD, Charalambous M, et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature. 2013;504:277–81.
Shyer AE, Huycke TR, Lee C, Mahadevan L, Tabin CJ. Bending gradients: how the intestinal stem cell gets its home. Cell. 2015;161:569–80.
Acknowledgements
This work was supported, in part, by the Japan Society for the Promotion of Science KAKENHI (grant numbers 16H06257, 17K10639, and 18K08543).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bu, L., Baba, H., Yoshida, N. et al. Biological heterogeneity and versatility of cancer-associated fibroblasts in the tumor microenvironment. Oncogene 38, 4887–4901 (2019). https://doi.org/10.1038/s41388-019-0765-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-019-0765-y
- Springer Nature Limited
This article is cited by
-
PRRX1-OLR1 axis supports CAFs-mediated lung cancer progression and immune suppression
Cancer Cell International (2024)
-
Nanoparticles in tumor microenvironment remodeling and cancer immunotherapy
Journal of Hematology & Oncology (2024)
-
Analysis of cancer-associated fibroblasts related genes identifies COL11A1 associated with lung adenocarcinoma prognosis
BMC Medical Genomics (2024)
-
Liver organoids: updates on generation strategies and biomedical applications
Stem Cell Research & Therapy (2024)
-
The molecular conversations of sarcomas: exosomal non-coding RNAs in tumor’s biology and their translational prospects
Molecular Cancer (2024)