
Abstract
Tumor microenvironment is composed of all the untransformed elements in the vicinity of tumor, mainly including a large number of stromal cells and extracellular matrix proteins, which play an active role in most solid tumor initiation and progression. Carcinoma-associated fibroblasts (CAFs), one of the most common stromal cell types in the tumor microenvironment, have been demonstrated to be involved in tumor growth, invasion, and metastasis. Therefore, they are becoming a promising target for anti-cancer therapies. In this review, we firstly summarize the current understandings of CAFs’ molecular biology, including the heterogeneous cellular origins and molecular markers, and then, we focus on reviewing their various tumor-promoting phenotypes involved in complex mechanisms, which can be summarized to the CAF-conveyed paracrine signals in tumor cells, cancer stem cells, and metastasis-initiating cancer cells, as well as the CAF-enhanced extrinsic tumor-promoting processes including angiogenesis, extracellular matrix remodeling, and tumor-related inflammation; finally, we describe the available directions of CAF-based target therapy and suggest research areas which need to be further explored so as to deepen the understanding of tumor evolution and provide new therapeutic targets for cancer treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The “seed and soil” hypothesis postulates that an appropriate host microenvironment (the soil) is needed for the optimal growth of tumor cells (the seed). In the past four decades, researchers have focused primarily on tumor cells. However, emerging evidences indicate that human carcinomas frequently exhibit significant stroma reactions, which are characterized by the existence of abundant stromal cells (including immune cells, fibroblasts, myofibroblasts, vascular cells, mesenchymal cells, etc) and extracellular matrix proteins—all the untransformed elements in the vicinity of the tumor make up the tumor microenvironment [1, 2]. It has been gradually realized that the tumor microenvironment plays an active role in the initiation, development, and progression of most solid tumors, and therein, the stromal cells are usually associated with high tumor malignancy and poor prognosis [3].
Carcinoma-associated fibroblasts (CAFs) are the most frequent component of the tumor stroma, a kind of particular fibroblasts possessing tumor-promoting properties. CAFs consist of activated and non-activated fibroblasts, containing distinct subsets with multiple tumor-promoting functions from various cellular sources. It has been demonstrated that CAFs can accelerate the development and progression of primary tumors as well as serve as a metastatic niche for supporting the secondary malignancies. The possible molecular mechanisms are complex and multiple, which can be summarized that CAFs could not only promote tumor cell proliferation, invasion, and metastasis via direct CAF-tumor cell communication, but also enhance several extrinsic tumor-promoting processes such as angiogenesis, extracellular matrix (ECM) remodeling, and tumor-related inflammation. CAFs are likewise important for the clinical behavior of tumor, thereby becoming a promising target.
In this review, we retrospect as well as record the latest research progress of CAFs in the tumor microenvironment and present recent studies addressing the tumor-promoting roles of CAFs, with a particular emphasis on their possible molecular mechanisms. Also, we discuss the therapeutic potential of targeting CAFs. Finally, we highlight perceived knowledge gaps and suggest research areas that need to be further explored so as to understand the tumor nature better and provide new targets for anti-cancer therapies.
The heterogeneity of CAFs
Up to now, CAF populations have displayed a high degree of heterogeneity, with a variety of morphological features and physiological functions. Tracing the sources, different CAF subsets have diverse cellular origins and express distinct biomarkers, consequently showing different biological traits.
The cellular origins of CAFs
The cellular origins of CAFs are still not fully clear. Resolving this problem may be very helpful for understanding the distinct traits of CAFs with various molecular markers and multiple tumor-promoting phenotypes. Lots of studies have concluded that CAFs mainly originate from five kinds of precursor cells.
Firstly, resident fibroblasts are the most immediate sources of CAFs. Analysis of the phenotypical characteristics of CAFs in liver metastases of colorectal cancer suggests that most CAFs display a vimentin+, α-SMA+, and Thy-1+ phenotype similar to resident liver fibroblasts, indicating that those CAFs originate from resident fibroblasts [4, 5]. Meanwhile, data have shown that CAFs’ tumor-promoting activities are partially mediated through an altered expression profile that overlaps significantly with the senescence-associated secretory phenotype (SASP). And it is argued that the senescent fibroblasts could as well drive malignant transformation and impact tumor progression. Given the significant molecular and phenotypic similarities between senescent cells and CAFs, it is speculated that senescent fibroblasts provide another possible source of CAFs [6].
In addition, bone marrow-derived mesenchymal stem cells (BMSCs) can travel to tumor stroma, where they differentiate into CAFs. Lawrenson et al. and Peng et al. recently reveal that MSCs can exhibit CAF-like characteristics with an increase of α-smooth muscle actin (SMA), vimentin expression, and tumor-promoting ability when treated with conditioned media of epithelial ovarian cancer cells in vitro or co-injection with epithelial ovarian cancer cells in vivo [5, 7]. Moreover, Peng et al. identifies in colon cancer that cancer cell-derived Notch signaling mediates the transformation of BMSCs to CAFs through activating the downstream TGF-β/Smad signaling pathway [7].
Also, the conversion of differentiated cells contributes to other origins of CAFs, such as epithelial-mesenchymal transition (EMT) and endothelial-mesenchymal transition (EndMT) [8, 9]. TGF-β is a potent inducer of both transition processes. Petersen et al. show that a population of α-SMA+ mesenchymal cells share the same chromosome aberrations with adjacent cancer cells in breast cancer tissues, expressing the mesenchymal markers Thy-1 and vimentin, and concomitantly losing the expression of keratins, which indicates the possibility that malignant epithelial cells could transform into CAFs [8]. Zeisberg et al. illustrate that proliferating endothelial cells could also contribute to CAFs via EndMT, with an emergence of fibroblast-specific protein 1 (FSP-1) and downregulation of CD31 [9].
Finally, other mesenchymal cells within tumor microenvironment could also transdifferentiate into CAFs. Strong experimental evidences indicate that adipocyte-derived fibroblasts can also be activated by nearby cancer cells and show high expression of fibronectin, type I collagen, and FSP-1, displaying an elevated invasion capability [10]. And it is speculated that vascular smooth muscle cells and pericytes may migrate from basement membrane to the tumor stroma and then transdifferentiate into CAFs, as they behave similarly in fibrosis [11].
The biomarkers of CAFs
Normal fibroblasts usually express markers of vimentin, desmin, type I collagen, fibronectin, and prolyl-4-hydroxylase [12]. Activated CAFs specifically upregulate the expression of α-SMA, fibroblast-specific protein 1 (FSP-1/S100A4), NG2, platelet-derived growth factor receptor (PDGFR)-α/β [13], fibroblast activation protein (FAP) [14], uPA [15], periostin [16], tenascin-C (TN-C) [17], palladin [18], podoplanin [19], and natriuretic peptide B (NPPB) [5]. There also exist evidences that some molecules are specifically downregulated in CAFs, such as caveolin-1 [20] and laminin [21], while epithelial cell markers (cytokeratin) and endothelial cell markers (CD31) disappear.
Unfortunately, none of the current molecule markers is unique to CAFs because they are also expressed by other stromal cells, which make it challenging to identify or isolate these heterogeneous CAFs via single molecular marker, but the combination of multiple biomarkers may help. For example, α-SMA+ myofibroblasts could highly express NG2 and PDGFR-β synchronously [13], whereas FSP-1 or PDGFR-α-expressing fibroblasts are mainly defined as non-myofibroblasts subpopulation [17, 10]. Distinct CAF subpopulations carry different biomarkers and may exhibit various biological traits and tumor-promoting functions. The heterogeneity in marker expressions and cellular origins of CAFs warrants more investigation based on tumor phenotypes.
The activation of CAFs and their tumor-promoting phenotypes
In normal situations, fibroblasts are in a quiescent state. Fibroblasts become activated when tissues need to be remodeled in wound healing and fibrosis. Similarly, in the tumor microenvironment, fibroblasts’ differentiation and activation are evoked by various stimuli, mainly including growth factors such as TGF-β, IL-1α, EGF, PDGF, and FGF2, which are released from tumor cells or infiltrating immune cells. Simultaneously, fibroblasts elongate into a fusiform shape and remain perpetually activated, resulting in the “wounds that never heal” phenotype associated with most cancers. Such activated fibroblasts, different morphologically and functionally from quiescent fibroblasts, are termed CAFs. Activated CAFs could release pools of cytokines and ECM constituents, forming an irreversibly activated stroma, which promotes tumor cell malignancy [12, 22–24].
Accumulating evidences suggest that activated CAFs significantly influence most of the hallmark capabilities of cancer [25–27]. Hence, we are here to review the tumor-promoting actions of CAFs in the context of the hallmarks of cancer, attempting to conclude diverse functional phenotypes of CAFs, along with their contributory functions to the essential hallmarks of cancer. Co-culture studies in vitro have shown that CAFs could accelerate the proliferation of tumor cells [5, 28], as well as induce the cancer stem cells’ self-renewal and differentiation phenotypes [29], suggesting CAFs’ tumorigenic potential. Several studies show that normal fibroblasts may lose their ability to inhibit epithelial cell proliferation with cancer development and their conversion into CAFs, indicating that CAFs could evade tumor growth restriction from normal microenvironments [30]. Co-implantation studies in vivo also illustrate that xenografts with CAF co-injection have increased microvessel density when compared with their counterparts, demonstrating the capability of CAFs to induce neoangiogenesis. Furthermore, CAFs have also been strongly involved in the processes of cell migration or invasion, and CAFs serve as metastasis niche to support the metastasis colonization. Besides, CAFs produce pro-inflammatory factors to induce tumor-related inflammation and immune suppression. Interestingly, it has been demonstrated that there probably exists a remarkable symbiotic relationship in energy metabolism between CAFs and cancer cells, in which CAFs in different tumor microenvironments can exchange energy sources with cancer cells to optimize metabolic efficiency and tumor growth, involving the alternative use of glucose and lactate and other energy-rich molecules [31]. All the activated tumor-promoting phenotypes of CAFs point to their contributory functions to the hallmarks of cancer.
Researchers speculated that the activated phenotypes of CAFs are mainly maintained by means of three ways: (1) persistent environmental effects—tumor cells or the stroma could produce autocrine and paracrine signals constantly to stimulate and sustain CAF phenotypes. For example, TGF-β and CXCL12 produced by CAFs could activate their receptors TβR and CXCR4 expressed by CAFs themselves, forming the TGF-β-TβR-Smad2/3 and CXCL12-CXCR4 autocrine signaling pathways to induce and sustain the differentiation of fibroblasts to CAFs and their coincident activated phenotypes [32]; for another example, sonic hedgehog (shh) secreted from transformed pancreatic epithelia can induce the differentiation of human pancreatic stellate cells into myofibroblasts, and this paracrine signaling facilitates the desmoplastic stroma in pancreatic cancer [33]. (2) genetic mutations—the tumor stroma is generally considered as genetically stable, especially when compared to the dominant tumor cells. However, several reports have documented genomic modifications in the tumor stroma, including amplifications, smaller mutations, and loss of heterozygosity (LOH). It is proposed that distinct cell origins of CAFs may contribute to explaining the discrepancy—the genetically instability stroma may represent a subset of CAFs originated from neoplastic epithelium underwent EMT [2]. (3) epigenetic alterations—somatically heritable changes which are not caused by changes in DNA sequence contribute largely to tumor initiation and progression. Certain gene-specific methylation alterations are found specific for the tumor stroma, and the global genomic hypomethylation occurs even earlier in CAFs, as compared to that both in normal stroma and malignant epithelium. These observations also suggest that CAFs can be targeted specifically, as they are more susceptible to hypomethylating drugs to produce a “hypomethylation crisis” [34].
The molecular mechanisms underlying the tumor-promoting phenotypes of CAFs
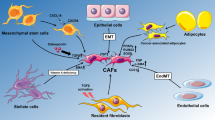
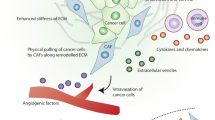
As mentioned above, increasing evidences demonstrate the tumor-promoting functions of CAFs; however, the underlying mechanisms are elusive. Herein, we make efforts to sort out and summarize their possible molecular mechanisms, so as to deepen the understanding of CAFs in cancer evolution. According to the present reports, CAFs have been shown to be a source of cytokines and ECM constituents. These secreted molecules not only directly convey intrinsic tumor-initiative, proliferative, surviving, invasive, and metastatic signals to the adjacent cancer cells (Fig. 1), but also enhance some extrinsic tumor-promoting processes such as angiogenesis, ECM remodeling, and tumor-associated inflammation (Fig. 2).

CAFs directly influence tumor cell behavior. a CAFs can exert paracrine effects on tumor cells directly, via complex signaling pathways: e.g., CXCL12 → CXCR4, CCL7 → CCR1/3, FGF1 → FGFR, Smo → Shh, CD81+ exosome → Wnt-PCP, Gal-1 → integrinβ1. b CAFs induce the cancer stem cell phenotypes and drive the EMT process, through various signaling pathways, e.g., HGF → c-Met–Wnt–β-catenin, IGF-II → IGF1R–Nanog, CCL2 → CCR2/4–Notch, MMPs → EMT. c CAFs support the ability of metastasis-initiating cancer cells and facilitate the metastasis colonization of disseminated tumor cells: POSTN can augment Wnt signaling, and activate POSTN–Integrin–PI3k–Akt pathway; TNC can also enhance Wnt and Notch signaling pathways, thereby supporting the survival of metastasis-initiating cells

CAFs enhance extrinsic tumor-promoting processes. a CAFs promote tumor angiogenesis through releasing growth factors and recruiting EPCs. b CAFs modulate the ECM via remodeling ECM architecture and promoting ECM stiffness to support the malignancy progression. c CAFs induce tumor-related inflammation and immune suppression through recruiting immune cells as well as modulating their functions in the tumor microenvironment
CAFs directly influence tumor cell behavior
As tumor progresses, CAFs coevolve with tumor cells into an activated state and then create a dynamic signaling net, composed of multiple tumor-promoting molecules and signaling pathways. The complex signaling net directly communicates with adjacent tumor populations via exerting paracrine effects on tumor cells, and meanwhile serving as niches for cancer stem cells and metastasis-initiating cells. All the signaling communication processes together influence tumor cell behavior, showing enhanced cell proliferation, survival, migration, invasion, and metastasis.
CAFs exert paracrine effects on the adjacent tumor cells
CAFs contribute to a wide variety of secreted factors in the tumor microenvironment, including multiple chemokines, growth factors, etc. CAF-derived factors could exert particular paracrine effects via interacting with their receptors or other molecules expressed in or on adjacent tumor cells, thus promoting tumor cell malignancy. CAFs secrete various chemokines, such as CXCL12 and CCL7. Orimo et al. demonstrate that CXCL12 can stimulate breast cancer growth through interacting with the cognate receptor CXCR4 expressed on cancer cells [35]; Jung et al. indicate that CCL7 may induce cytoskeletal changes in oral squamous cell carcinoma cells to enhance cell migration, via interacting with CCR1 and CCR3 on cancer cells [36]. And CAFs produce various growth factors to facilitate tumor cell malignancy. For example, Henriksson et al. find the activated FAP+ fibroblasts in colorectal cancer stroma can promote tumor cell migration through releasing FGF1 and then interacting with fibroblast growth factor receptor (FGFR) on the epithelial cells [37]. Besides, the latest research by He et al. shows that galectin-1(Gal-1) is strongly expressed in CAFs, and the upregulation of Gal-1 in CAFs can promote gastric cancer cell migration and invasion through upregulating the integrinβ1 expression in cancer cells [38].
Moreover, Luga et al. recently reveal a novel intercellular communication pathway whereby CAF-secreted exosomes facilitate breast cancer cell protrusive activity via autocrine Wnt-planar cell polarity (PCP) signaling, depending on the exosome tetraspanin, Cd81 [39]. Additionally, it is reported that prostate fibroblasts are capable of forming primary cilia, and the receptor smoothened co-localize to this structure. Smo in prostate fibroblasts is found to be competent to transduce the paracrine sonic hedgehog signaling from adjacent epithelium and elevate a signature of prostatic Hh gene expression. The smo-mediated stromal hedgehog signaling not only sustains CAF proliferation and survival but also stimulates proliferation and dedifferentiation of adjacent epithelium, suggesting a role of CAFs in driving the tumor initiation [40, 41].
CAFs induce the cancer stem cell phenotype
Emerging studies have underscored significant roles of CAFs in inducing the cancer stem cell phenotype and driving the EMT process, associated with facilitating tumor initiation, development, and metastasis [29]. It is reported that myofibroblast-secreted hepatocyte growth factor (HGF) could enhance Wnt signaling via c-Met on colon cancer cells, thereby activating β-catenin-dependent transcription and colon cancer stem cell clonogenicity, consequently reinstalling the tumor-initiating capacity in differentiated cancer cells [42]. Besides, Tsuyada et al. also prove that CAFs produce high levels of CCL2, which requires STAT3 activation by cancer cell-secreted cytokines, and CCL2 via interacting with CCR2/4 in turn induces NOTCH1 expression and the stem cell-specific, sphere-forming phenotype in breast cancer cells [43]. Chen et al. find that in vitro CAF co-cultured lung cancer cells could maintain their cancer stem cell (CSC) phenotype, showing high tumor-initiating and metastatic potential, and prove that IGF1R signaling in tumor cells activated by CAF-secreted IGF-II can induce Nanog expression, suggesting that CAF-mediated signaling pathway contributes to cancer stemness maintenance [44].
EMT, a process by which epithelial cells lose cell–cell adhesion and gain mesenchymal properties, is essential for many developmental processes including tumor invasion and metastasis. Studies indicate that paracrine interaction between CAFs and tumor cells could induce EMT and EMT-driven CSC properties and aggressive potency [45]. Giannoni et al. identify a circuitry in which cancer cell-derived IL-6 activates fibroblasts, which in turn secrete matrix metalloproteinases (MMPs) to elicit EMT and enhance cancer stemness in prostate cancer cells, thereby culminating in tumor formation and spontaneous lung metastatic growth [46]. Additionally, CAFs are reported to express carbonic anhydrase IX (CAIX) depending on HIF-1 activation. CAIX-caused extracellular acidification can elicit activation of CAF-produced MMP-2 and MMP-9, driving EMT in prostate cancer cells, which is associated with increased motility and stemness [47].
CAFs support the ability of metastasis-initiating cancer cells
CAFs could not only confer CSCs phenotype but also serve as a permissive niche to facilitate the colonization of metastasis-initiating cancer cells. Extracellular matrix proteins POSTN and TN-C are indicated as key components of the metastatic niche, providing signal support for metastasis-initiating cells [48]. It is indicated that, in the secondary target organ—lung—stromal fibroblasts increase POSTN expression in response to infiltrating tumor cell-derived TGF-β3. The stromal POSTN augments Wnt signaling in cancer stem cells and thereby drives the colonization of disseminated breast cancer cells to the lung [49]; a latest study demonstrates that paracrine interaction between CAF-secreted POSTN and esophageal adenocarcinoma cell-expressed integrin results in PI3K/Akt activation and increases tumor cell invasion [16].
Others demonstrate the crucial role of TN-C in breast cancer metastasis to the lung. Both cancer cells and CAFs can express TN-C. Cancer cell-derived TNC can enhance their responsiveness to Wnt and Notch signaling pathways, thereby supporting the survival and fitness of metastasis-initiating cells [50]. Meanwhile, stromal TN-C from S100A4+ fibroblasts is also necessary and stimulative for the metastasis colonization [17]. Recently, Saupe et al. implicate a new mechanism underlying the TNC-promoted tumor progression in a neuroendocrine tumor model that TNC may contribute to Wnt signaling activation through downregulation of the inhibitor Dkk1 in both tumor and stromal cells [51].
CAFs enhance extrinsic tumor-promoting processes
In addition to the intrinsic signals in tumor cells, several biological processes outside of tumor cells also play crucial roles in tumor evolvement, such as tumor angiogenesis, ECM remodeling, and tumor-associated inflammation. These processes provide extrinsic forces for tumor initiation and development, and CAFs contribute to these tumor-promoting procedures via complex molecular or mechanical mechanisms.
CAFs promote tumor angiogenesis
Tumor angiogenesis is a physiopathological process essential for tumor growth and dissemination. Numerous studies have demonstrated that CAFs could promote tumor angiogenesis, mainly through releasing growth factors (VEGF, FGF, EPO) or recruiting endothelial progenitor cells (EPCs) into carcinomas [52]. For example, co-cultures of esophageal squamous cell carcinoma cells and esophageal fibroblasts suggest that the carcinoma-derived TGF-β can regulate angiogenic process through stimulating the VEGF production by fibroblasts, and the presence of fibroblasts is necessary for the formation of efficient human microvascular endothelial cell network [53]. Noma et al. also demonstrate that colon cancer CAFs produce a large amount of IL-6 and the IL-6 production enhances the release of VEGF from fibroblasts, thereby promoting angiogenesis [54]. Another study by Augsten et al. reveals the upregulation of CXCL14 in most prostate CAFs, and the upregulation of FGF-2 and VEGF-A, VEGF-B, and VEGF-C in the CXCL14-overexpressing fibroblasts, suggesting a possible mechanism of CAF-stimulated angiogenesis [55]. Besides, Xue et al. find that stromal fibroblasts in the tumor, liver, and spleen tissues specifically expressing PDGFR-β can react to tumor-derived PDGF-BB by producing erythropoietin (EPO), which is found to be a critical element driving tumor angiogenesis, growth, and metastasis [56]. In addition, CAFs can facilitate angiogenesis via recruiting endothelial progenitor cells (EPCs) into invasive breast cancers, an effect mediated partially by elevated CXCL12 secretion [35]. And recent studies indicate that activation of Ets2 in breast CAFs could also promote vessel formation in the absence of tumor cells, partially through promoting recruitment of endothelial cells [57]. Collectively, these findings highlight the crucial role of CAFs in promoting tumor angiogenesis through cytokine release or EPC recruitment.
CAFs modulate the ECM
There are increasing evidences that tumor development depends on the physical properties of the adjacent environment to a great extent, particularly the ECM architecture and stiffness. CAFs produce various ECM proteins, such as matrix metalloproteinases (MMPs), collagen, and laminin. MMPs as the key proteases capable of degrading matrix have been proven to be involved in many physiological remodeling processes [58, 59]. Collagen as the dominant structural protein in the human body, its stromal deposition and crosslinking also contribute to the matrix stiffening as well as ECM architecture alteration [60]. ECM architecture and stiffness alteration can influence mechanosensing and stimulate intracellular signalings to facilitate directional cell mobility and invasion [61].
Firstly, CAFs are indicated to generate leading paths for collective migration of cancer cells, with the aid of ECM-remodeling activities. Studies show that cytokine signaling through the receptor subunit GP130-IL6ST and the kinase JAK1 generates actomyosin contractility via Rho-kinase-dependent signaling, and the pathway generates contraction in CAFs to remodel the matrix and meanwhile generates tracks for collective migration of squamous carcinoma cells [62]; additionally, an actin-associated protein, palladin, highly expressed in CAFs of multiple solid tumors, could enhance CAFs’ ability in remodeling the extracellular matrix. It is indicated that palladin promotes the assembly of invadopodia in fibroblasts via regulating the activity of cdc42, and the matrix-degrading invadopodia meanwhile creates the extensive tunnels that are used by cancer cells follow behind [18, 63].
Moreover, CAFs could interact with ECM proteins to promote matrix stiffening. The interaction modulates intercellular adhesion and cell contractility, and thereby increases tumor stiffness to support malignancy progression. For instance, analysis of the function and gene expression of breast CAFs reveals a positive feedback between CAF-driven matrix stiffening and mechanotransduction leading to YAP activation, which helps to maintain the CAF phenotypes. YAP regulates the expression of several cytoskeletal regulators, such as ANLN and DIAPH3, and modulates the protein levels of MYL9/MLC2, finally resulting in tumor matrix stiffening and subsequently aggressiveness [64]; caveolin-1 (Cav1) in CAFs has also been reported to modulate tissue responses through force-dependent architectural regulation of the tumor microenvironment. Cav1 is found to promote Rho- and force-dependent contractility, matrix alignment, and stiffness through regulation of p190RhoGAP, and consequently favors tumor cell invasion and increases tumor metastatic potency [65].
CAFs induce tumor-related inflammation and immune suppression
Virchow first postulated in 1863 that persistent inflammation predisposes individuals to cancer [66]. Lots of reports have demonstrated a general connection between stromal fibroblasts and tumor-related inflammation. Fibroblasts in the tumor microenvironment are activated or trained to become pro-inflammatory, mainly through paracrine signalings from tumor cells and other stromal cells, such as IL-1, TNF-a, and TGF-β. Afterwards, CAFs initiate a tumor-related inflammatory response that expresses a pro-inflammatory gene signature including IL-1β, IL-6/8, COX-2, OPN, CXCL1/2/3/12, CCL2, ICAM-1, etc [67–69]. The upregulation of pro-inflammatory cytokines not only acts on tumor cells directly but also recruits the innate and adaptive immune cells as well as modulates their functions in the tumor microenvironment [70].
Firstly, the pro-inflammatory signature in CAFs could promote recruiting immune cells into the tumor microenvironment to facilitate tumor progression [71]. Ellem et al. have proved that estrogen-induced CAFs in prostate cancer produce CXCL12, which can recruit mast cells into tumor stroma in a CXCR4-dependent manner [72]; Ksiazkiewicz et al. have also reported CCL2-CCR2A/2B pathway can promote infiltration of blood monocytes into mammary CAF areas [73]. In addition, the pro-inflammatory signature in CAFs is provided with the potential to modulate the functions of immune cells in the tumor microenvironment. CAFs express various immunosuppressive cytokines such as TGF-β1, involved in suppressing the activation of T lymphocyte functions, inhibiting the function of natural killer (NK) cells and CD8+ cytotoxic T lymphocytes (CTLs), and promoting the recruitment of tumor-promoting T cells, thus blocking anti-tumor cytotoxic activities. CAFs have also been found to affect T-cell polarization, inducing a switch from Th1- to Th2-type immunity; Monte et al. have identified the essential role for CAFs in leading to the Th2 immune deviation in pancreatic cancer through TSLP secretion, which is associated with reduced patient survival [74]. Moreover, CAFs could also enhance the immunosuppressive cell differentiation and function, thereby facilitating an immunosuppressive microenvironment. For example, pancreatic stellate cells (a subset of pancreatic CAFs) are found to produce inflammatory cytokines (IL-6, M-CSF, VEGF, SDF-1, MCP-1), and culture of the PSC supernatants with peripheral blood mononuclear cells (PBMCs) promotes PBMC differentiation into an myeloid-derived suppressor cell (MDSC, CD11b + CD33+) phenotype, which functionally suppresses autologous T lymphocyte proliferation, thereby evading immune recognition [75].
Conclusions and perspectives
As dominant cells in the tumor microenvironment, CAFs modulate the overall processes of tumor initiation and progression through complex and multiple mechanisms. CAFs are therefore emerging as a promising therapeutic target with particular advantages. Increasing targeted agents have entered the clinical tests, and the CAF-based target therapy appears to be competent to synergize the traditional anti-cancer therapy. Though some targets have not yet been applied in clinical, researchers propose a wide range of logical and feasible targets: (1) targeting TGF-β, CXCL1, Shh, PDGFs, and FGFs to suppress the activation and differentiation of CAFs directly; (2) targeting the CAF–carcinoma cell interaction through CXCL12, CXCR4, CCL7, FGF, Gal-1, Smo, HGF, CCL2, IGF-II, CAIX, TN-C, POSTN, and TGF-β [76]; (3) targeting CAF–endothelial cell interactions through VEGF, FGFs, CXCL12, and PDGFs; (4) targeting the CAF-modulated ECM remodeling by inhibiting caveolin-1 and MMPs (and someone has tried to develop new techniques to alleviate solid stress or stiffness to increase tumor perfusion) [77]; (5) targeting CAF-induced inflammation and immune escape through CXCL1, CXCL2, CXCL12, CXCL14, COX-2, CCL2, CCL5, IL-1β, and TGF-β; (6) targeting CAF-mediated anti-immune responses, the design of FAP-based therapeutic approaches to target CAFs has been developed, such as FAP neutralizing antibodies, inhibitors [78], prodrugs [79], and DNA vaccines [80]; and (7) targeting DNA methylation in CAFs may be another approach to preferentially block CAFs’ function [34]. At present, two kinds of approaches targeting DNA methylation are available: methyl donor modifiers (folate, betaine, and choline) and methyltransferase inhibitors (nucleoside inhibitors: 5-azacytidine, 5-aza-2-deoxycytidine, and zebularine; and non-nucleoside inhibitors: procaine, procainamide, EGCG, and RG108) [81].
However, there still exist emerging fields of research on CAFs which need to be further explored. Firstly, intense disputes about the molecular biology of CAFs are urgent to be solved. We need further studies to figure out how different CAF subsets achieve their distinct tumor-promoting effect, which would meanwhile benefit to the development of more accurate target therapy. While this review mainly discusses the tumor-promoting functions of CAFs, for completeness it is crucial to point out that CAFs in some circumstances also play negative roles in tumor progression. For example, though CAF-derived TGF-β supports tumor progression in many ways as discussed above, studies based on conditional inactivation of TGF-β in fibroblasts also show tumor-promoting effects, suggesting the dual roles of TGF-β as tumor suppressor and promoter [82]. It is proposed that the tissue and cell type specificity may account for the dual roles of the stroma in tumor progression, and it is plausible that CAFs react in a context-dependent manner, which, though still ambiguous and debatable, raises another valuable research area. Besides, it is essential to further clarify the presence of genetic mutation and epigenetic alteration in CAFs, and verify the maintaining mechanism of their tumor-promoting phenotypes, so as to exploit specific genetic therapy and individualized target therapy. Moreover, exploration on the CAF-directed therapeutic approaches are mostly at the preclinical stage, yet the animal tumor models are not that authentic for evaluating the tumor-promoting functions of CAFs in the human body, so there is an urgent need to conduct extensive clinical trials for further confirmation. In conclusion, further study about CAFs will help in demonstrating the mechanisms of tumor initiation and progression, identifying specific markers of tumor diagnosis and targets of tumor therapy, and developing alternative new ways of clinical treatment, which will bring hope to the radical cure of cancer.
References
Togo S, Polanska U, Horimoto Y, Orimo A. Carcinoma-associated fibroblasts are a promising therapeutic target. Cancers. 2013;5:149–69.
Madar S, Goldstein I, Rotter V. ‘Cancer associated fibroblasts’—more than meets the eye. Trends Mol Med. 2013;19:447–53.
Paulsson J, Micke P. Prognostic relevance of cancer-associated fibroblasts in human cancer. Semin Cancer Biol. 2014;25:61–8.
Mueller L, Goumas FA, Affeldt M, Sandtner S, Gehling UM, Brilloff S, et al. Stromal fibroblasts in colorectal liver metastases originate from resident fibroblasts and generate an inflammatory microenvironment. Am J Pathol. 2007;171:1608–18.
Lawrenson K, Grun B, Lee N, Mhawech-Fauceglia P, Kan J, Swenson S, et al. NPPB is a novel candidate biomarker expressed by cancer-associated fibroblasts in epithelial ovarian cancer. Int J Cancer. 2014. doi:10.1002/ijc.29092.
Alspach E, Fu Y, Stewart SA. Senescence and the pro-tumorigenic stroma. Crit Rev Oncog. 2013;18:549–58.
Peng Y, Li Z, Yang P, Newton IP, Ren H, Zhang L, et al. Direct contacts with colon cancer cells regulate the differentiation of bone marrow mesenchymal stem cells into tumor associated fibroblasts. Biochem Biophys Res Commun. 2014;451:68–73.
Petersen OW, Nielsen HL, Gudjonsson T, Villadsen R, Rank F, Niebuhr E, et al. Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am J Pathol. 2003;162:391–402.
Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007;67:10123–8.
Bochet L, Lehuede C, Dauvillier S, Wang YY, Dirat B, Laurent V, et al. Adipocyte-derived fibroblasts promote tumor progression and contribute to the desmoplastic reaction in breast cancer. Cancer Res. 2013;73:5657–68.
Ren S, Duffield JS. Pericytes in kidney fibrosis. Curr Opin Nephrol Hypertens. 2013;22:471–80.
Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401.
Sugimoto H, Mundel TM, Kieran MW, Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer Biol Ther. 2006;5:1640–6.
Fearon DT. The carcinoma-associated fibroblast expressing fibroblast activation protein and escape from immune surveillance. Cancer Immunol Res. 2014;2:187–93.
Dano K, Behrendt N, Hoyer-Hansen G, Johnsen M, Lund LR, Ploug M, et al. Plasminogen activation and cancer. Thromb Haemost. 2005;93:676–81.
Underwood TJ, Hayden AL, Derouet M, Garcia E, Noble F, White MJ, et al. Cancer associated fibroblasts predict for poor outcome and promote periostin-dependent invasion in oesophageal adenocarcinoma. J Pathol. 2014. doi:10.1002/path.4467.
O’Connell JT, Sugimoto H, Cooke VG, MacDonald BA, Mehta AI, LeBleu VS, et al. VEGF-A and Tenascin-C produced by S100A4+ stromal cells are important for metastatic colonization. Proc Natl Acad Sci. 2011;108:16002–7.
Brentnall TA, Lai LA, Coleman J, Bronner MP, Pan S, Chen R. Arousal of cancer-associated stroma: overexpression of palladin activates fibroblasts to promote tumor invasion. PLoS ONE. 2012;7:e30219.
Inoue H, Tsuchiya H, Miyazaki Y, Kikuchi K, Ide F, Sakashita H, et al. Podoplanin expressing cancer-associated fibroblasts in oral cancer. Tumor Biol. 2014;35:11345–52.
Mercier I, Casimiro MC, Wang C, Rosenberg AL, Quong J, Minkeu A, et al. Human breast cancer-associated fibroblasts (CAFs) show caveolin-1 downregulation and RB tumor suppressor functional inactivation: Implications for the response to hormonal therapy. Cancer Biol Ther. 2008;7:1212–25.
Tlsty TD. Stromal cells can contribute oncogenic signals. Semin Cancer Biol. 2001;11:97–104.
Berdiel-Acer M, Sanz-Pamplona R, Calon A, Cuadras D, Berenguer A, Sanjuan X, et al. Differences between CAFs and their paired NCF from adjacent colonic mucosa reveal functional heterogeneity of CAFs, providing prognostic information. Mol Oncol. 2014;8:1290–305.
Bae JY, Kim EK, Yang DH, Zhang X, Park Y, Lee DY, et al. Reciprocal interaction between carcinoma-associated fibroblasts and squamous carcinoma cells through interleukin-1α induces cancer progression. Neoplasia. 2014;16:928–38.
Räsänen K, Vaheri A. Activation of fibroblasts in cancer stroma. Exp Cell Res. 2010;316:2713–22.
Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. 2012;21:309–22.
Pietras K, Östman A. Hallmarks of cancer: Interactions with the tumor stroma. Exp Cell Res. 2010;316:1324–31.
Hanahan D, Weinberg RA. Hallmarks of cancer: The Next Generation. Cell. 2011;144:646–74.
Lin J, Liu C, Ge L, Gao Q, He X, Liu Y, et al. Carcinoma-associated fibroblasts promotes the proliferation of a lingual carcinoma cell line by secreting keratinocyte growth factor. Tumor Biol. 2011;32:597–602.
Liao CP, Adisetiyo H, Liang M, Roy-Burman P. Cancer-associated fibroblasts enhance the gland-forming capability of prostate cancer stem cells. Cancer Res. 2010;70:7294–303.
Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. 2011;17:320–9.
Rattigan YI, Patel BB, Ackerstaff E, Sukenick G, Koutcher JA, Glod JW, et al. Lactate is a mediator of metabolic cooperation between stromal carcinoma associated fibroblasts and glycolytic tumor cells in the tumor microenvironment. Exp Cell Res. 2012;318:326–35.
Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, et al. Autocrine TGF-β and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci. 2010;107:20009–14.
Bailey JM, Swanson BJ, Hamada T, Eggers JP, Singh PK, Caffery T, et al. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin Cancer Res. 2008;14:5995–6004.
Gonda TA, Varro A, Wang TC, Tycko B. Molecular biology of cancer-associated fibroblasts: can these cells be targeted in anti-cancer therapy? Semin Cell Dev Biol. 2010;21:2–10.
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–48.
Jung D, Che ZM, Kim J, Kim K, Kim K, Williams D, et al. Tumor-stromal crosstalk in invasion of oral squamous cell carcinoma: a pivotal role of CCL7. Int J Cancer. 2010;127:332–44.
Henriksson ML, Edin S, Dahlin AM, Oldenborg P, Öberg Å, Van Guelpen B, et al. Colorectal cancer cells activate adjacent fibroblasts resulting in FGF1/FGFR3 signaling and increased invasion. Am J Pathol. 2011;178:1387–94.
He X, Tao H, Hu Z, Ma Y, Xu J, Wang H, et al. Expression of galectin-1 in carcinoma-associated fibroblasts promotes gastric cancer cell invasion through upregulation of integrin β1. Cancer Sci. 2014;105:1402–10.
Luga V, Zhang L, Viloria-Petit AM, Ogunjimi AA, Inanlou MR, Chiu E, et al. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell. 2012;151:1542–56.
Tian H, Callahan CA, DuPree KJ, Darbonne WC, Ahn CP, Scales SJ, et al. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc Natl Acad Sci U S A. 2009;106:4254–9.
Wilkinson SE, Furic L, Buchanan G, Larsson O, Pedersen J, Frydenberg M, et al. Hedgehog signaling is active in human prostate cancer stroma and regulates proliferation and differentiation of adjacent epithelium. Prostate. 2013;73:1810–23.
Vermeulen L, De Sousa E, Melo F, van der Heijden M, Cameron K, de Jong JH, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468–76.
Tsuyada A, Chow A, Wu J, Somlo G, Chu P, Loera S, et al. CCL2 mediates cross-talk between cancer cells and stromal fibroblasts that regulates breast cancer stem cells. Cancer Res. 2012;72:2768–79.
Chen WJ, Ho CC, Chang YL, Chen HY, Lin CA, Ling TY, et al. Cancer-associated fibroblasts regulate the plasticity of lung cancer stemness via paracrine signalling. Nat Commun. 2014;5:3472.
Zhou B, Chen W, Wang Y, Lin Z, Zhang D, Fan S, et al. A role for cancer-associated fibroblasts in inducing the epithelial-to-mesenchymal transition in human tongue squamous cell carcinoma. J Oral Pathol Med. 2014;43:585–92.
Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010;70:6945–56.
Fiaschi T, Giannoni E, Taddei ML, Cirri P, Marini A, Pintus G, et al. Carbonic anhydrase IX from cancer-associated fibroblasts drives epithelial-mesenchymal transition in prostate carcinoma cells. Cell Cycle. 2013;12:1791–801.
Oskarsson T, Massagué J. Extracellular matrix players in metastatic niches. EMBO J. 2012;31:254–6.
Malanchi I, Santamaria-Martínez A, Susanto E, Peng H, Lehr H, Delaloye J, et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2011;481:85–9.
Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nat Med. 2011;17:867–74.
Saupe F, Schwenzer A, Jia Y, Gasser I, Spenle C, Langlois B, et al. Tenascin-C downregulates wnt inhibitor dickkopf-1, promoting tumorigenesis in a neuroendocrine tumor model. Cell Rep. 2013;5:482–92.
Wang S, Ma N, Kawanishi S, Hiraku Y, Oikawa S, Xie Y, et al. Relationships of alpha-SMA-positive fibroblasts and SDF-1-positive tumor cells with neoangiogenesis in nasopharyngeal carcinoma. Biomed Res Int. 2014;2014:1–9.
Noma K, Smalley KSM, Lioni M, Naomoto Y, Tanaka N, El Deiry W, et al. The essential role of fibroblasts in esophageal squamous cell carcinoma-induced angiogenesis. Gastroenterology. 2008;134:1981–93.
Nagasaki T, Hara M, Nakanishi H, Takahashi H, Sato M, Takeyama H. Interleukin-6 released by colon cancer-associated fibroblasts is critical for tumour angiogenesis: anti-interleukin-6 receptor antibody suppressed angiogenesis and inhibited tumour–stroma interaction. Brit J Cancer. 2013;110:469–78.
Augsten M, Hagglof C, Olsson E, Stolz C, Tsagozis P, Levchenko T, et al. CXCL14 is an autocrine growth factor for fibroblasts and acts as a multi-modal stimulator of prostate tumor growth. Proc Natl Acad Sci U S A. 2009;106:3414–9.
Xue Y, Lim S, Yang Y, Wang Z, Jensen L, Hedlund E, et al. PDGF-BB modulates hematopoiesis and tumor angiogenesis by inducing erythropoietin production in stromal cells. Nat Med. 2011;18:100–10.
Wallace JA, Li F, Balakrishnan S, Cantemir-Stone CZ, Pecot T, Martin C, et al. Ets2 in tumor fibroblasts promotes angiogenesis in breast cancer. PLoS ONE. 2013;8:e71533.
Almholt K, Johnsen M. Stromal cell involvement in cancer. Recent Results Cancer Res. 2003;162:31–42.
Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25:9–34.
Carey SP, Kraning-Rush CM, Williams RM, Reinhart-King CA. Biophysical control of invasive tumor cell behavior by extracellular matrix microarchitecture. Biomaterials. 2012;33:4157–65.
Kharaishvili G, Simkova D, Bouchalova K, Gachechiladze M, Narsia N, Bouchal J. The role of cancer-associated fibroblasts, solid stress and other microenvironmental factors in tumor progression and therapy resistance. Cancer Cell Int. 2014;14:41.
Sanz-Moreno V, Gaggioli C, Yeo M, Albrengues J, Wallberg F, Viros A, et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell. 2011;20:229–45.
Goicoechea SM, García-Mata R, Staub J, Valdivia A, Sharek L, McCulloch CG, et al. Palladin promotes invasion of pancreatic cancer cells by enhancing invadopodia formation in cancer-associated fibroblasts. Oncogene. 2013;33:1265–73.
Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15:637–46.
Goetz JG, Minguet S, Navarro-Lérida I, Lazcano JJ, Samaniego R, Calvo E, et al. Biomechanical remodeling of the microenvironment by stromal caveolin-1 favors tumor invasion and metastasis. Cell. 2011;146:148–63.
Fitzpatrick FA. Inflammation, carcinogenesis and cancer. Int Immunopharmacol. 2001;1:1651–67.
Erez N, Glanz S, Raz Y, Avivi C, Barshack I. Cancer associated fibroblasts express pro-inflammatory factors in human breast and ovarian tumors. Biochem Biophys Res Commun. 2013;437:397–402.
Kogan-Sakin I, Cohen M, Paland N, Madar S, Solomon H, Molchadsky A, et al. Prostate stromal cells produce CXCL-1, CXCL-2, CXCL-3 and IL-8 in response to epithelia-secreted IL-1. Carcinogenesis. 2009;30:698–705.
Maxwell PJ, Neisen J, Messenger J, Waugh DJ. Tumor-derived CXCL8 signaling augments stroma-derived CCL2-promoted proliferation and CXCL12-mediated invasion of PTEN-deficient prostate cancer cells. Oncotarget. 2014;5:4895–908.
Servais C, Erez N. From sentinel cells to inflammatory culprits: cancer-associated fibroblasts in tumour-related inflammation. J Pathol. 2013;229:198–207.
Comito G, Giannoni E, Segura CP, Barcellos-de-Souza P, Raspollini MR, Baroni G, et al. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene. 2014;33:2423–31.
Ellem SJ, Taylor RA, Furic L, Larsson O, Frydenberg M, Pook D, et al. A pro-tumourigenic loop at the human prostate tumour interface orchestrated by oestrogen, CXCL12 and mast cell recruitment. J Pathol. 2014;234:86–98.
Ksiazkiewicz M, Gottfried E, Kreutz M, Mack M, Hofstaedter F, Kunz-Schughart LA. Importance of CCL2-CCR2A/2B signaling for monocyte migration into spheroids of breast cancer-derived fibroblasts. Immunobiology. 2010;215:737–47.
De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H, et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med. 2011;208:469–78.
Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013;73:3007–18.
Giannelli G, Villa E, Lahn M. Transforming growth factor-β as a therapeutic target in hepatocellular carcinoma. Cancer Res. 2014;74:1890–4.
Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, Bardeesy N, et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc Natl Acad Sci. 2012;109:15101–8.
Poplawski SE, Lai JH, Li Y, Jin Z, Liu Y, Wu W, et al. Identification of selective and potent inhibitors of fibroblast activation protein and prolyl oligopeptidase. J Med Chem. 2013;56:3467–77.
Brennen WN, Rosen DM, Wang H, Isaacs JT, Denmeade SR. Targeting carcinoma-associated fibroblasts within the tumor stroma with a fibroblast activation protein-activated prodrug. JNCI J Nat Cancer Inst. 2012;104:1320–34.
Gottschalk S, Yu F, Ji M, Kakarla S, Song X. A vaccine that co-targets tumor cells and cancer associated fibroblasts results in enhanced antitumor activity by inducing antigen spreading. PLoS ONE. 2013;8:e82658.
Stresemann C, Brueckner B, Musch T, Stopper H, Lyko F. Functional diversity of DNA methyltransferase inhibitors in human cancer cell lines. Cancer Res. 2006;66:2794–800.
Bhowmick NA, Chytil A, Chytil D, Gorska AE, Dumont N, Shappell S, et al. TGF-β signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–51.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81402299, 81372334).
Conflicts of interest
None
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Han, Y., Zhang, Y., Jia, T. et al. Molecular mechanism underlying the tumor-promoting functions of carcinoma-associated fibroblasts. Tumor Biol. 36, 1385–1394 (2015). https://doi.org/10.1007/s13277-015-3230-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-3230-8