
Abstract
While a definitive understanding of schizophrenia etiology is far from current reality, an increasing body of evidence implicates perturbations in early development that alter the trajectory of brain maturation in this disorder, leading to abnormal function in early childhood and adulthood. This atypical development likely arises from an interaction of many brain cell types that follow distinct developmental paths. Because both cellular identity and development are governed by the transcriptome and epigenome, two levels of gene regulation that have the potential to reflect both genetic and environmental influences, mapping “omic” changes over development in diverse cells is a fruitful avenue for schizophrenia research. In this review, we provide a survey of human brain cellular composition and development, levels of genomic regulation that determine cellular identity and developmental trajectories, and what is known about how genomic regulation is dysregulated in specific cell types in schizophrenia. We also outline technical challenges and solutions to conducting cell type-specific functional genomic studies in human postmortem brain.
Similar content being viewed by others


Introduction
The brain holds a distinctive position in the hierarchy of human organs for generating thought, emotion, and personality, all while managing the more base functions that maintain the body—and also for being so difficult to study. The cells of the brain are so numerous and intricately commingled that long after it was known that the other organs are made of separate cells, the brain was still thought of as one large unbroken reticulum rather than many individual units. In applying Golgi staining to brain in 1888 (50 years after Theodor Schwann first extended cell theory to animals), Santiago Ramón y Cajal was first able to visualize the elaborate form of an individual neuron, debunking the prevailing theory that the brain was a continuous network and establishing the inception of modern neuroscience [1].
In the intervening century, the field of neuroscience has made many advances, but there is still a great distance to travel in understanding the regulation of the genesis of the human brain—and how this goes awry when a person develops a psychiatric syndrome such as schizophrenia. One promising avenue for interrogating the origins of psychiatric illness is to interpret the influence of the genetic code on the developmental trajectories that coalesce into a mature functioning brain, trajectories that are often highly cell type specific. Cellular function is determined by the molecular phenotype of the cell and by its connectivity. Cells are interconnected in developmentally programmed micro- and macrocircuits that are thought to be altered in schizophrenia. Technology for examining gene regulation in individual brain cells or populations has greatly improved since the Golgi stain, yet remains a challenge for high-throughput studies. This review provides an overview of selected aspects of human brain development in the context of major brain cell types, mechanisms that regulate the gene function that shapes cellular function, and what is known about how they influence schizophrenia. Finally, we end with an overview of technical challenges to achieve cell type-specific profiles of gene regulation in human postmortem brain.
Neocortical cellular structure, composition, and development
The brain is functionally organized in a regional manner, where each region is associated with a relatively specific group of functions. This review focuses on the cerebral neocortex, a 2–4.5 mm sheet of tissue that covers most of both hemispheres. Dorsally and laterally, the cerebral cortex is subdivided by functions such as vision, hearing, motor control, cognition, and perception [2]. Of particular interest in psychiatry is the prefrontal cortex, the most rostral subdivision of the neocortex, which is associated with higher cognitive functions such as decision-making, self-control, language, and social processes [3]. The dorsolateral prefrontal cortex (DLPFC) occupies Brodmann areas 9, 10, and 46 (rostral to the premotor cortex) and is associated particularly with executive functions like working memory, sensory input synthesis, attention, and goal-directed behaviors [3].
With authoritative overviews of brain development available elsewhere [4], we will briefly highlight here key events in cortical development that underlie cell fate determination and maturation. The types of cells in the six layers of the DLPFC are myriad but can broadly be distinguished as neurons and glia, with neurons representing roughly 33–39% of cells in the frontal lobe [5]. Beyond this dichotomy, the diverse shapes, electrophysiological properties, gene expression patterns, neurotransmitters, location, and projections of these cells lead to numerous potential cell classes. In simplistic terms, however, the main categories break down to six types: pyramidal neurons, interneurons, oligodendrocytes, astrocytes, microglia, and endothelial cells.
Pyramidal neurons are relatively large cells that express glutamate as their primary neurotransmitter and play an excitatory role in neuronal circuits. They are the predominant cells in the “deep” layers (5 and 6) of the cortex and have long axons that project to other nervous system regions, such as the striatum, thalamus, and spinal cord. Pyramidal cells in superficial cortical layers (e.g., layers 2 and 3) project principally within the neocortex and connect the neuronal assemblies that comprise the local and distributed cortical circuits involved in higher-order behaviors and conscious thought [6]. Interneurons are smaller cells that express GABA as their primary neurotransmitter and mostly act to inhibit circuit activity. While GABAergic neurons represent ~20% of cortical neurons, they are a much more phenotypically diverse group than their pyramidal counterparts, with subtypes characterized by distinct morphologies and molecular signatures [7]. Unlike pyramidal neurons, interneurons are relatively more abundant in layers 2 through 4. Interneuron axons also mostly do not project out of the cortex, instead ramifying around a cortical column or across neighboring columns, leading generally to a more local impact on circuit function [7]. In terms of glial types, oligodendrocytes produce myelin, a substance that ensheaths neuronal axons and insulates their electrical signaling for more efficient conduction [8]. Astrocytes are the next most abundant cortical macroglia and serve a variety of functions, from maintaining homeostasis of water and ion distribution, maintaining the blood–brain barrier, providing structural support, recycling glutamate released from pyramidal neurons, and participating in cell-to-cell signaling [8]. Microglia are the class of macrophages that reside in brain. They are critical players in inflammatory reactions within the central nervous system (CNS), early synapse pruning, maintaining homeostasis, and protecting against pathogens [9, 10]. Finally, endothelial cells play a critical role in forming the blood–brain barrier by lining brain microvessels at the blood–CNS interface [11].
The formation and composition of each cortical layer is determined by a developmental path and timeline unique to each cell type (Fig. 1a). Neurogenesis in humans begins soon after the neural folds fuse to form the neural tube roughly 4 weeks after conception [4]. Excitatory pyramidal neurons are born first in the dorsal pallium and migrate radially along radial glial processes into the cortical plate, the precursor to the cerebral cortex; inhibitory interneurons largely arise ventral to these cells in the ganglionic eminence and follow tangentially to the cortical plate [4]. Laminar positioning originates in a bottom-up formation according in part to birth order, where earlier-born pyramidal neurons first settle in layers 5 and 6, then later neurons populate the overhead layers [4]. While this occurs, pyramidal neurons help recruit specific interneuron subtypes to coexist in specific layers, depending on the subtype of each pyramidal neuron [12]. Neuronal expansion and migration continues throughout a prolonged window beginning in embryonic development and lasting until shortly before birth [13], although in some regions they last until age three [4].

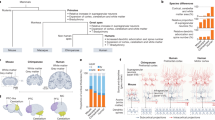
Depending on the timing of genetic or environmental influence, different cellular processes may be affected that then impact development of behavioral or cognitive systems. a Approximate timing of a selection of cell-specific developmental processes that occur during human cortical development, as well as known risk factors for schizophrenia (SCZ). a is adapted from [171]. Several cellular processes may be impacted differentially depending on the timing and magnitude of the risk factor effect; likewise, the normative patterns depicted here are also influenced by one another because of intercellular communication and coexistence in cortical circuits. b The typical time frame of achieving several developmental milestones that are often significantly delayed in children who go on to be diagnosed with schizophrenia as adults. Achievement of motor skills [172], language skills [173], and executive functioning skills [174] is impaired in children later diagnosed with schizophrenia. b is adapted from [4]. c Molecular mechanisms of gene regulation that govern cellular identity and development. The transcriptome and epigenome integrate genetic and environmental influences on developmental paths such as those highlighted in a.
Gliogenesis commences after neurogenesis begins. Oligodendrocytes first emerge in mid-gestation and continue to be generated extensively through the first 3 years of life and then at a slower rate through adulthood [4, 14]. Astrogliogenesis also is thought to begin during mid-gestation and continues abundantly through the first 3 years of postnatal life, though astrocytes can continue to divide throughout life [4, 15]. Interestingly, unlike neurons and the macroglia (oligodendrocytes and astrocytes), microglia are derived from erythromyeloid progenitors that migrate to the brain early in prenatal development [16]. Once the blood–brain barrier is formed and microglia mature, they rely on self-renewal to maintain their population under normal physiological conditions [9, 17, 18].
Cellular, laminar, and regional identity are dictated by specific transcription factor gradients. While neurons, oligodendrocytes, and astrocytes all originate from radial glial cells [8], these gradients lead to divergent outcomes. For example, excitatory pyramidal neurons are differentiated from radial glia via a transcription factor cascade including PAX6, TBR2, NEUROD1, and TBR1 [19]. Interneurons from the medial ganglionic eminence are shaped by a cascade beginning with SHH, then followed by NKX2-1, LHX6 and LHX8, and SOX6 [20]. Oligodendrocyte lineage is established with expression of PDGFRA, SOX10, and NG2, while astrocytes are guided by the expression of ALDH1L1 [8]. Cell subtype designation is further influenced by transcription factors that specify cortical layers, such as BCL11B, FEZF2, and SOX5, which specify the identity and connectivity of layer 5 glutamatergic excitatory projection neurons [21, 22]. Knowledge of these transcription factor cascades can help in identifying cell types and stages in human postmortem brain tissue, as further discussed below.
As these cells find their way to their destinations and continue to mature, the cortex undergoes an explosive period of forming cell-to-cell connections. Thalamocortical axons reach the cortex amidst prolific dendritic arborization and synaptogenesis in late mid-fetal development; at this time, the first evidence of electrical signaling occurs and the cortical plate begins to resemble the characteristic six layers of the mature cortex [4]. This extensive connective proliferation continues through the first and second year of life, depending on the cortical region, although the expansion of pyramidal neuron size and arborization persists at a slower rate until age five [23]. Myelination, meanwhile, begins to occur in early postnatal life and continues throughout adulthood, reaching a relative plateau in DLPFC in the third decade of life [4, 24].
Following this period of synaptic overproduction, the cortex undergoes extensive pruning of primarily excitatory synapses beginning in early childhood, plateauing in early adolescence, and continuing to a lesser extent into the third decade of life [25]. While much attention has been devoted to the pruning of selected excitatory synapses in adolescence, there are also changes in synaptic architecture that appear progressive during this period of postnatal life. For example, dendritic arbors undergo extensive refinement during adolescence, including becoming more elaborate and complex, and modulatory inputs from brainstem systems, such as the dopamine system, become much more extensive [26]. The timing of maturation for different circuits is in general consistent with the developmental timing of the cognitive skills associated with their function; for instance, humans do not fully develop executive functioning until early adulthood, commensurate with the slow maturation of the DLPFC that continues well beyond the second decade of postnatal life [25].
Schizophrenia: a developmental brain disorder
Despite the adult age of typical first diagnosis, evidence from both genetic and epidemiological studies implicate aberrant brain development as a component of the etiology of schizophrenia. Schizophrenia is a highly heritable disorder, with estimates of heritability as high as 80% [27]. A large genome-wide association study (GWAS) comparing schizophrenia cases and healthy controls identified 145 genomic loci marked by common single-nucleotide polymorphisms (SNPs) that were associated with schizophrenia risk [28]. Examining the expression of genes within genomic risk loci showed that they were relatively enriched for expression in fetal compared with postnatal DLPFC [29]. A complementary study of de novo variants in schizophrenia patients found that genes containing these variants formed a network of co-expression and protein interaction in fetal DLPFC, suggesting that cortical development may be altered during gestation in schizophrenia [30].
Likewise, several early life experiences have been linked to schizophrenia development. Women whose mothers experienced severe nutritional deficits during the first trimester of pregnancy had more than doubled odds of developing schizophrenia in adulthood [31]. Obstetric complications have also been associated with developing schizophrenia [32], as well as living in an urban environment early in life [33]. Interestingly, combining genetic and environmental risk factors can greatly amplify schizophrenia risk. It was shown recently that polygenic risk score, a metric measuring inheritance of risk-associated alleles identified in GWAS, was five times more predictive of schizophrenia diagnosis in individuals that had experienced early life complications during pregnancy, labor, or delivery than in individuals without those experiences [34]. Further reinforcing the developmental component to causation is that children who grow up to be diagnosed with schizophrenia in adulthood are more likely to have delays in achieving classic developmental milestones, including those involving motor, language and cognitive skills (Fig. 1b) [35,36,37].
Although the incrimination of developmental processes in schizophrenia risk is convincing, the specific neuropathology or molecular mechanisms are not understood. Several developmental trajectories have been implicated, and due to the phenotypic heterogeneity of schizophrenia, it is likely that at least several of these play a role. For instance, neural oscillations particularly in the beta- and gamma-band frequency observed with electroencephalographic techniques are abnormal in individuals with schizophrenia [38]. Gamma oscillations emerge late in brain development, are associated with executive functioning, and are attributed to the activity of fast-spiking parvalbumin interneurons in cortical layers 2 and 3 of the DLPFC [39]. Evidence from animal studies suggesting that cortical interneurons do not develop fast-spiking characteristics until late adolescence has encouraged speculation that early developmental genetic factors bias this population of late maturing GABAergic neurons to malfunction, but the malfunction phenotype is masked until the full maturation of these cells and the neural circuitries that they subserve [40]. One hypothesis is that N-methyl-D-aspartic acid-type glutamate receptor hypofunctioning in these interneurons leads to reduced gamma-oscillation production; however, it is unclear if this imbalance is the result of a primary reduction in interneuron inhibition, or a result of upstream dysfunction in pyramidal neuron excitation [41].
A smaller body of evidence also implicates dysregulation of glial processes in schizophrenia. A possible avenue leading to cognitive dysfunction in schizophrenia is aberrant myelination, which can alter synaptic formation and function [42]. Several studies have found schizophrenia to be associated with pathways related to myelination and oligodendrocyte function [43, 44]. Another popular hypothesis is that schizophrenia may result from aberrant synaptic pruning in adolescence, a process that relies heavily on the activity of glial cells such as astrocytes and microglia [45]. Recent work identifying schizophrenia risk-associated polymorphisms within the C4 gene, a part of the complement immune system in the MHC locus, implicated a potential role for complement activation as a mechanism of the putative synaptic loss in schizophrenia [46]. Another study using co-cultured fibroblasts reprogrammed to neuronal and microglial fates found increased synaptic engulfment by microglia in patient-derived samples and further association of C4 risk alleles with increased synapse uptake, although C4 genotype did not explain all disease-associated increased pruning in their in vitro model [47]. However, while there is evidence of reduced synapses in some studies of schizophrenia postmortem brain in selective DLPFC layers (e.g., layers 2 and 3), there is no direct evidence that this reduction is a consequence of abnormal pruning rather than an epiphenomena of chronic illness and associated morbidities. A recently developed positron emission tomography technique using a radiotracer specific for SV2A, a synaptic protein that can be used as a synaptic density marker [48], may allow for better resolution studies of in vivo schizophrenia synaptic dysfunction in the future.
Given the current lack of consensus, further work to elucidate the “when” and “where” of schizophrenia pathology in terms of brain development is required, particularly for understanding how these alternative narratives of developmental perturbation fit together. Given the highly complex and synergistic developmental processes undertaken by different brain cells, it is likely that more than one cell class is implicated. It is also worth noting that the same principles that apply to schizophrenia here also apply to other neurodevelopmental psychiatric disorders, such as autism spectrum disorder and bipolar affective disorder. Identifying the timing of and cell type(s) affected by a known genetic or environmental risk factor can illuminate the path forward for rectifying the altered developmental trajectories in many related disorders, perhaps placing the brain back on a healthy path.
Functional genomics of brain development
The mechanisms that govern developmental decisions within a cell begin fundamentally with the genetic code and how the code is utilized in each cell (Fig. 1c). The central dogma of molecular biology provides a skeletal view of how this occurs: DNA is transcribed in the nucleus to mRNA, which is exported to the cytoplasm and translated to a protein that performs some cellular function. Further scrutiny drapes layers of nuance over these bones. Noncoding RNAs such as miRNA and lncRNA, for example, are not translated; rather, they can be a part of feedback loops regulating later transcription or translation of other genes or transcripts, or act themselves within the cell as signaling molecules, scaffolding structures, or regulators of protein activity [49, 50]. Posttranscriptional modifications such as alternative splicing and RNA editing can also affect how and when a transcript is used, as well as the localization of a transcript within the cell. Proteins, particularly transcription factors as discussed above, also participate in regulatory cascades affecting transcription and translation. Finally, the epigenome regulates DNA use by controlling DNA accessibility and factor recruitment, altering if and how the DNA can be used [51, 52]. Profiling the transcriptome and epigenome is particularly useful because both integrate genetic and environmental information and provide a means to interpret the functional readout of that information. Here we briefly delve further into these facets of gene regulation.
Ribonucleic acid
The transcriptome is defined as the total RNA in a sample; however, the term masks a great deal of diversity in both form and function. In addition to the mRNA expressed from the roughly 20,000 protein-coding genes in the human genome, a plethora of other noncoding RNA species exist. For instance, as mentioned above, miRNAs are roughly 22 base noncoding RNAs that influence translation of mRNA [53]. lncRNA are transcribed from noncoding genes longer than 200 bases and perform a variety of functions, from regulating transcription of neighboring cis genes, to acting as signaling molecules [54, 55]. Noncoding RNA transcribed from repetitive DNA has been shown to be an important structural component of the nuclear matrix [56]. Other RNA species are increasingly being characterized, including piRNAs, snoRNAs, circular RNAs, and others, each with their own set of functions [57]. As technology improves, the list of known RNA species expands and augments our understanding of the diverse roles transcriptional products play within the cell.
Co- and posttranscriptional RNA modifications further increase the diversity within the transcriptome. Alternative splicing, how multiple protein isoforms can arise from the same genetic sequence, is the most pivotal RNA modification [58]. RNA can also be edited by RNA editing enzymes such as ADAR to change the coding sequence or alter binding specificity in the 3′UTR [59]. Reversible modifications such as RNA methylation (i.e., m6A) and transport chaperones such as RNA binding proteins can introduce further heterogeneity and complexity [59, 60].
Beyond transcript modifications, localization of RNA can also influence RNA function. Compartmentalization by the nuclear membrane is an often-overlooked mechanism of RNA regulation. The nuclear transcriptome is populated by pre-mRNA and sequestered aberrant and activity-dependent transcripts, in contrast to cytoplasmic RNA [61,62,63,64,65,66,67]. A major role of the nuclear membrane is to act as a transcriptional noise buffer by filtering stochastic bursts of gene expression from entering the cytoplasm [68, 69]. Further, transcripts can be targeted to specific locations in the cytoplasm for immediate translation given proper environmental cues [70]. A recent study revealed that genes implicated in neurodevelopmental and neurodegenerative disorders tend to show a bias toward nuclear sequestration, implicating efficiency of translating transcripts into protein as another potential mechanism of genetic risk [71].
The brain undergoes dramatic shifts in gene and isoform expression as it develops [72], including noncoding RNA species [73]. Both coding and noncoding gene expression and alternative splicing shape the developmental trajectories defining cell identity and function within the circuits in the maturing cortex [74]. RNA editing is also developmentally regulated in human brain and is especially involved in neuronal maturation [75]. In terms of RNA localization, many of the mechanisms that regulate RNA localization across the nuclear membrane are frequently used in brain cell lineages, and have been shown to play a role in developmental programs. For instance, intron retention often occurs in weakly expressed transcripts as a signal to the nuclear surveillance machinery to sequester and degrade aberrant or superfluous transcription products via exosomes [76]. In vitro neurons as well as other cell types show increasing levels of intron retention as they differentiate from induced pluripotent stem cells, principally in lowly expressed genes involved in determining counter cell fates [77,78,79]. RNA editing has also been shown to regulate activity-dependent nuclear transcript retention, although RNA editing does not seem to globally signal sequestration [66, 80]. Overall, RNA takes on a broad assortment of shapes and roles within the cell and is highly dynamic across brain development.
Chromatin structure
Chromatin is the compacted product of DNA incorporated around proteins, reducing the length of DNA per chromosome by a factor of up to 2 × 10−5 [81]. Chromatin is composed of a series of nucleosomes—a set of eight histone proteins around which roughly 146 bases of DNA wraps twice—connected by variable length linker DNA and organized into a two-start helical 30 nm fiber [81]. Chromatin structure affects the accessibility of DNA for transcriptional or regulatory functions in a cell type-specific and activity-dependent manner as the cells develop, and can be remodeled by chromatin modifiers [82, 83].
Both histone variants themselves and modifications to the histone tails affect chromatin structure and therefore DNA use in a way that can be interpreted as a “histone code” [84]. For example, promoter sequence often occurs upstream of a nucleosome marked by trimethylation of the fourth lysine of histone 3 (H3K4me3). Monomethylation of the fourth lysine of histone 3 (H3K4me1), in conjunction with acetylation of the 27th lysine of histone 3 (H3k27ac), is a marker of active enhancer sequence. In contrast, trimethylation of the ninth lysine of histone 3 (H3K9me3) marks repressive heterochromatin [85]. These histone modifications not only label chromatin state but also actively participate in inducing them; for instance, H3K4me3 recruits the ATP-dependent chromatin remodeler CHD1 as well as other enzymes capable of repositioning nucleosomes for increased DNA accessibility and transcriptional activity [85]. H3K9me3, on the other hand, helps induce a heterochromatin state by providing a binding substrate for HP1, a protein that recruits the methyltransferase SUV39H1 that then further methylates H3K9me3 in a positive feedback loop to repress transcription [86]. Zooming out from the nucleosome to the genome scale, regions of euchromatin and heterochromatin are organized into topologically associated domains that are likewise dynamic [87].
Many studies have assessed the chromatin landscape over brain development and found chromatin patterning to reflect the rich history of cell fate decisions, experience, and genetic influence sustained by the brain region or cell population measured [88,89,90,91]. Chromatin state and chromatin remodeling are an integral facet of the epigenome that both reflect past developmental paths while being responsive to the current needs of the cell. As such, chromatin is an expression of the influence of the environment as well as genetic variation on regulation of the genome.
DNA methylation (DNAm)
DNAm, another major component of the epigenome, is the covalent addition of a methyl group to the fifth carbon of a cytosine. The methylated cytosine usually precedes a guanine (i.e., CG) but in some cell types can be followed by another base (i.e., CH, where H = A, C or T) [92].
Methylation in the CG context (mCG) is a stable modification that is faithfully copied from parent to daughter cell during DNA replication by DNMT1 [93], although de novo methylation can be added via the methyltransferases DNMT3a and DNMT3b [94]. mCG has historically been considered a repressive mark, although this view is being upended as canon [92]. In reality, the mCG landscape reads more like a map to be deciphered, where stereotyped mCG features can be interpreted to signify certain genomic conditions. In next-generation sequencing data, the ratio of methylated-to-unmethylated reads per base represents the proportion of methylation at that site across the sequenced sample. In the mammalian genome, most CGs are fully methylated, but there are notable exceptions. For example, short unmethylated regions often correspond to promoter sequence because promoters frequently contain CG islands, dense clusters of rarely methylated CGs [95]. Low-methylated regions, or short regions of less than 30% methylation, often correspond to active distal regulatory elements such as enhancers [95]. Partially methylated domains are long stretches of disordered mCG levels that correspond to heterochromatin and polycomb repressed sequence [96]. Finally, DNAm valleys (DMVs) are longer regions of hypomethylation that correspond to epigenetically regulated developmental genes. Although DMVs are unmethylated, their chromatin profile reveals a bivalent state of both activating H3K4me3 and repressing H3K27me3 signal [97]. In this way, profiling the mCG landscape can provide a picture of genomic organization and activity in the cell population being measured, although the causal relationship between DNAm levels and genomic activity is not always clear. For instance, although DNAm levels are typically reduced at transcription factor binding sites when the factor is bound, suggesting a steric hindrance effect of DNAm on binding [95, 98], actively transcribed gene bodies have increased DNAm levels in most cell types [99], and at least for the typically methylation-sensitive transcription factor CTCF, in vivo removal of DNAm does not alter CTCF binding [100].
Neurons have the unique distinction along with embryonic stems cells of having a high proportion of non-CG methylation (i.e., mCH) [101]. Unlike most mCG levels which are highly methylated, mCH levels are much lower due to the increased heterogeneity at each cytosine within a sample [102]. Unlike mCG in other cell types [99], neuronal mCH in gene bodies is anticorrelated with gene expression and is largely established de novo postnatally by DNMT3a during neuronal development [103], although low levels are detectable in prenatal human frontal cortex [104].
In terms of brain development, mCG is a significant and early player, maintaining prenatally established global mCG levels throughout the lifespan; mCH, on the other hand, increases rapidly from birth to early childhood and then continues more slowly into the 20s [101]. Postnatally, mCG and particularly mCH show the most rapid changes in the first 5 years, implicating this time of life as a period of especially high epigenetic plasticity and environmental sensitivity [105]. Changes to DNAm in both cytosine contexts also occur robustly in response to neuronal activity [106] and are critical to learning and memory and the maintenance of synaptic plasticity [107], suggesting that DNAm plays an integral role in the brain maturational processes described in the previous section. Whether the driver or passenger in establishing the epigenomic landscape, DNAm integrates the genetic and environmental influences that have paved the developmental roads.
Schizophrenia and gene regulation in brain development
Transcriptomic regulation
Many studies have identified elements of schizophrenia risk reflected in the transcriptome, particularly risk attributed to common genetic variation. For instance, an early case–control RNA-seq study concluded that at least 20% of GWAS-implicated risk loci contained variants that could influence gene expression in the DLPFC in schizophrenia [108]. A more recent study found over two-thirds of GWAS-associated risk variants to be expression quantitative trait loci (eQTLs), or variants associated with changing gene expression [109]. Another case–control transcriptomic study conducted by the PsychENCODE Consortium identified almost 5000 genes and 4000 transcripts—including many noncoding—as significantly differentially expressed in the frontal and/or temporal cortex between schizophrenic and control brains, and transcript-level changes had much greater effect sizes than gene-level expression changes [110]. However, this extent of case–control differential expression was not observed in a subsequent study [109], perhaps because of differences in how RNA quality was addressed.
Transcriptome-wide association study (TWAS) analysis in the PsychENCODE study identified 64 candidate genes implicated in several different cell types as conferring risk for schizophrenia [110]. Two later TWASs identified many additional associations between disease risk and expressed features (e.g., specific exons, junctions), though interestingly, none of them were differentially expressed between cases and controls [109, 111]. Collado-Torres et al. were the first to explore the relationship between gene expression in DLPFC and hippocampus in patients and controls and revealed reduced coherence in gene expression between these regions in the brains of schizophrenia patients [109]. Jaffe et al. further utilized laser capture microdissection (LCM) to profile RNA from the granule cell layer of the human hippocampus, which identified unique molecular signatures of schizophrenia risk missed in bulk tissue [111]. However, taken together, the results largely suggest that differences in gene expression between schizophrenia cases and neurotypical controls reflect primarily state-associated factors related to the correlates, consequences, and associated phenomenology of illness.
Beyond case–control studies, several RNA sequencing studies are beginning to explore schizophrenia risk reflected in developmental transcriptomic changes. One example of a risk-associated transcript with developmental shifts in expression is a human-specific isoform of AS3MT that lacks methyltransferase activity; this isoform was found to explain at least part of the 10q24.32 locus association with schizophrenia, and its expression was regulated very early in in vitro neuronal differentiation [112]. Another study found that many expressed sequences identified as GWAS schizophrenia risk loci eQTLs mapped to genes with shifting isoform expression across the prenatal to postnatal transition—in other words, genetic risk for schizophrenia was associated with developmentally dynamic gene expression patterns [113]. In terms of noncoding RNA, several studies have also focused on miRNA as master regulators of the transcriptome that can participate in establishing the pathology of schizophrenia [114]. For example, miR-137 is critical for proper regulation of gene expression networks in human neuronal development, and both genetic and transcriptional evidence supports a role for miR-137 in dysregulation of gene expression networks in schizophrenia [115,116,117]. Importantly, genetic risk for schizophrenia has also been found to be enriched in eQTLs and splicing QTLs identified in human fetal brain [118, 119].
While a promising start, all the studies listed above share the constraint of being conducted in homogenate tissue, limiting their resolution to the average transcriptomic signal across all measured cells. Because of the technological limitations discussed in the next section, analysis of cell type-specific transcriptional effects in schizophrenia in postmortem brain has been confined mostly to examining the ontology of differentially expressed genes in these studies. However, several recent papers have attempted to identify the most relevant cell types to schizophrenia pathology by partitioning the heritability for schizophrenia captured by common variation in GWAS across cell type-specific gene expression profiles identified in mouse and human single cell or nuclei [120,121,122]. Interestingly, these papers have implicated a variety of cell types as being most culprit in the disorder. Finucane et al. found significant schizophrenia heritability enrichment in glutamatergic but not GABAergic neurons [120], while Skene et al. found enrichment in glutamatergic neurons, dopaminergic medium spiny neurons, and some cortical interneurons, with less enrichment in glial, embryonic, or progenitor cells [122]. On the other hand, Calderon et al. found significant enrichment of schizophrenia heritability in oligodendrocytes and replicating cells from fetal cortex [121]. These competing results may be influenced by the use of different mouse and human single-cell/nuclei RNA-seq human cortex datasets, as well as different statistical methods. Even so, these early studies offer new albeit preliminary evidence to support models of cell type-specific schizophrenia pathology.
Epigenomic regulation
Compared to similar transcriptomic studies, fewer significant differences have been identified between neurotypical and schizophrenic brains in genome-wide profiles of DNAm and chromatin accessibility in homogenate human cortex. Studies of DNAm using microarrays have identified 25–2104 significantly differentially methylated positions between schizophrenia cases and controls [123,124,125,126]. Likewise, a study of accessible chromatin regions in DLPFC found only three significantly differentially accessible regions between schizophrenia patients and matched neurotypical counterparts [127]. The paucity of differences identified in homogenate tissue analysis reflects the importance of accounting for cellular heterogeneity in epigenomic studies, particularly when looking for likely small changes associated with complex polygenic disorders like schizophrenia [128].
Although few strong epigenetic leads associated with diagnosis have been identified, several studies have found association between developmentally dynamic patterns of chromatin structure or DNAm and genetic schizophrenia risk. For example, in one study, DLPFC-specific peaks of H3K27ac, a histone modification associated with active regulatory elements, were strongly enriched for schizophrenia heritability in fetal and infant samples but less so in adult samples, suggesting that these putative regulatory elements specific to higher cortical areas are established and active early in the lifetime [129]. Indeed, dynamic three dimensional chromatin looping patterns identified in early neuronal development were also associated with schizophrenia genetic risk [130]. In terms of developmental DNAm patterns, two recent microarray-based DNAm studies—one in fetal cortices and one in control cortices ranging from fetal to old age—also found that GWAS risk loci were enriched for being methylation quantitative trait loci, particularly in fetal cortices [124, 131]. Interestingly, CGs that were differentially methylated between schizophrenic and control cortical tissue were enriched for sites that were also differentially methylated between prenatal and postnatal timepoints but not for timepoints around the schizophrenia age of onset between adolescence and early adulthood [124]. Assessing developmental changes associated with genetic risk for schizophrenia has so far been a more fruitful direction for illuminating genome regulation associated with schizophrenia pathology than adult case–control epigenomic measures.
An increasing number of studies are now attempting to address cellular identity in terms of the epigenome in schizophrenia, and early evidence points to a large contribution of neuron biology and developmental patterns in the etiology of the disorder. For example, although accessible chromatin regions identified in homogenate cortex are broadly highly enriched for heritability of schizophrenia [127], this is particularly true for regions that were preferentially accessible in neurons compared with non-neurons, or regions that were differentially accessible between neurons isolated from different brain areas that can be attributable to differences in neuronal subtype identity [132]. Heritability for schizophrenia was also more enriched in regions marked by H3K4me3 and H3K27ac in human neurons than non-neurons, and histone QTLs (i.e., genetic variants associated with histone modification enrichment) tagged several schizophrenia GWAS risk SNPs more strongly in neurons than non-neurons, such as a stronger H3K4me3-QTL for the miR-137 marker SNP and stronger H3K27ac-QTL for the CACNA1C marker SNP [133]. Likewise, DNA regions hypomethylated in neurons and differentially methylated in neurons between brain areas were also highly enriched for schizophrenia heritability, indicating the potential importance of neuronal function generally and different neuronal subtypes particularly in this disease [132]. Furthermore, a recent paper examining the relationship of schizophrenia risk with cell type-specific developmental DNAm patterns found that regions progressively losing DNAm over postnatal development in postmortem human neurons but not non-neurons were significantly enriched for schizophrenia heritability [105]. The majority of demethylation in these regions occurred in early life in the first 5 years, highlighting the importance of examining developmental patterns when assessing schizophrenia disease etiology [105]. Interestingly, when using prenatal samples, schizophrenia heritability was found to be enriched in regions of accessible chromatin found in the germinal zone of the developing cerebral cortex, a region in which neural progenitor cells are abundant [134]. A second study combining chromatin accessibility data with single-cell transcriptomic data from fetal brain further expanded the neuronal focus of genetic risk implicated by epigenetic studies by identifying fetal neural progenitors, oligodendrocyte precursor cells, and microglia as being enriched for schizophrenia heritability in this earlier time point [135].
While these advances have improved our understanding of the molecular pathology associated with risk for schizophrenia, there is much more to uncover before more targeted therapies or prophylactic measures can be implemented. The cell type-specific epigenomic studies in adult brain tissue largely rely on positive selection of neurons from non-neurons using NeuN antibody (further described below), meaning that heritability in the non-neuron fraction may be masked by greater heterogeneity than the neuronal fraction. However, these early leads provide foundational evidence upon which to build.
Technical considerations for working with human postmortem brain cells
Given the human-specific nature of the affected organ in schizophrenia, and the neoteny associated with the developmental trajectories of the affected brain regions, it is critical to conduct research using human postmortem brain as the substrate. Yet working with human postmortem brain is not without its unique challenges.
As described above, a major challenge in designing experiments using human brain is addressing the serious confounder of cellular heterogeneity. Previous work has shown that cell type composition is one of the largest determinants of DNAm variability in a study that uses homogenate tissue, and that age-associated DNAm changes are highly confounded by cell type composition at individual CGs [128]. However, cells of the brain are not easily untangled, particularly after the process of being flash frozen before examination. Proximate “omics” measurements taken from more easily acquired tissues such as blood, saliva, or buccal cells vary widely in their correlation to those in brain tissue at individual sites or genes of interest and still suffer from heterogeneity concerns [128, 136].
Several options now exist to address cellular heterogeneity in postmortem human brain sequencing studies. Reviews are now available that summarize options in greater detail [137, 138], but we will highlight the four most popular here. The first option is to use software that deconvolutes the homogenate signal based on known cell type-specific profiles [139, 140]. This option is the most rapid and cost-effective option; however, the results will only be an estimate from pooled DNAm or RNA signal. It is worth noting also that using this type of strategy on RNA data will estimate the proportion of the RNA that is represented by the given cell types, and not the proportion of actual cells. This computational method also relies on the existence of pure cell population references that may not be available for all cell types.
Another option is to perform LCM, in which single or groups of cells are excised with a laser from a thin tissue section [141]. While LCM is the only technique currently that allows for the collection of the cell cytosol from postmortem brain, the method suffers from relatively low throughput, potentially poorer RNA quality, and potential cellular contamination from 3-D sampling. Nonetheless, LCM offers the unique feature of being able to select cells a priori based on their morphology or expression of a specific marker. This approach has been applied to several different unique cell populations in the human brain, including isolating dopamine neurons from the midbrain substantia nigra and the granule cell layer from the hippocampus [111, 142].
A third option is to use fluorescence-activated nuclear sorting (FANS) to isolate cell types in a high-throughput manner [143]. In addition to a higher yield, the multiple lasers of a flow cytometer allow for multiparametric analysis that improves the yield purity and offers more flexibility in experimental design. Despite these advantages, performing FANS on frozen postmortem brain is too stressful on the tissue to isolate intact mature brain cells; therefore, sorting must be done on purified nuclei, and any antibodies used must label nuclear antigens.
While several groups have successfully used lineage-specific transcription factors to isolate select subtypes of neurons and glia [144,145,146], most studies isolate neurons from non-neurons using an antibody that targets NeuN, a splice factor that is a constitutive component of the neuronal nuclear matrix and a marker of most mature neurons [147,148,149]. Important caveats to this method include the potential for loss of immunoreactivity in postmortem brain tissue, even when using as robust an antibody as the monoclonal anti-NeuN offered by Millipore Sigma (Catalog #MAB377X). Previous work has shown that NeuN reactivity decreased in mouse cortex following cerebral ischemia, but the number of neurons was unaffected [150]. Similarly, NeuN reactivity was completely lost in the spinal cord of elderly rats, although the number of neurons remained the same [151]. These studies are reminders that interpreting the absolute yield in terms of FANS-derived populations should be undertaken with care. It is also worth noting that because FANS limits RNA studies to the nuclear fraction, understanding the compositional differences between RNA compartments over human brain development would help inform future studies using nuclear RNA without a comparable cytoplasmic fraction. Several studies have now addressed this issue and determined that at the gene level, nuclear RNA can be used as a stand in for the whole transcriptome [71, 152].
In recent years, several new technologies have been developed to profile the transcriptome and epigenome in single cells, and have been reviewed elsewhere [153, 154]. Briefly, the main two technologies emerging for this purpose are plate-based methods in which each well in a PCR plate contains a single cell or nuclei [155] and droplet-based methods in which thousands of cells can be individually sequenced by isolation in nanoliter droplets containing sequencing reagents [156]. While droplet-based sequencing strategies offer higher throughput than well-based methods, RNA sampling in each cell is much more sparse and is based on 3′ tagging, although methods for resolving full-length mRNA in droplet-based strategies are being developed [157].
These techniques are increasingly being applied to cells and nuclei isolated from human postmortem brain, furthering our understanding of intercellular variability and clarifying profiles of increasingly granular cell identity [135, 158,159,160,161,162,163,164,165,166]. Moreover, new spatially resolved single- or several-cell techniques such as RNA seqFISH+, an in situ hybridization-based method for resolving spatial gene expression patterns [167], or Slide-seq, a method that transfers RNA from a tissue section to a sheet covered in DNA-barcoded beads for sequencing [168], will add back further layers of information to sequencing data that were historically part of the cellular taxonomy [169]. As single-cell technologies improve, we will also be able to better explore genomic variation such as somatic mosaicism, a phenomenon that may preferentially affect restricted cell types and that is associated with schizophrenia [170], at the single-cell level. Assessing individual cellular landscapes of genomic regulation in developing human brains is the next frontier in parsing genetic and environmental risk for schizophrenia and will hopefully better illuminate how that risk is distributed across cell types and developmental stages.
Conclusion
The cells in a human brain develop according to genetically specified paths, influenced by environment, for well over 25 years. Faculties that become present in adulthood, such as the higher-order cognitive processes that are disrupted in schizophrenia, are built upon an infrastructure of previous cellular actions taken potentially decades earlier. These actions are reflected in the transcriptome and epigenome, along with the potential signature of genetic or environmental risk that influenced a schizophrenic brain toward that state. As noted previously, although the details of heritability, affected cell types and altered developmental trajectories may differ, the principles and approaches described here for schizophrenia apply to other neuropsychiatric diseases as well. By profiling the transcriptome and epigenome in human brain tissue—the most clinically relevant substrate to psychiatric disease—at timepoints throughout that 25-year period of risk, we can anticipate building a clearer picture of normal brain developmental infrastructure, throwing into relief the risk “stress fractures” that collapse healthy brain function in schizophrenia. As sequencing techniques advance and the field gains a finer understanding of gene regulation in individual cells and cell populations over development, we can expect our understanding of the developmental etiology of schizophrenia to improve and converge at the cellular level.
References
López-Muñoz F, Boya J, Alamo C. Neuron theory, the cornerstone of neuroscience, on the centenary of the Nobel Prize award to Santiago Ramón y Cajal. Brain Res Bull. 2006;70:391–405.
Shipp S. Structure and function of the cerebral cortex. Curr Biol. 2007;17:R443–9.
Teffer K, Semendeferi K. Human prefrontal cortex: evolution, development, and pathology. Prog Brain Res. 2012;195:191–218.
Silbereis JC, Pochareddy S, Zhu Y, Li M, Sestan N. The cellular and molecular landscapes of the developing human central nervous system. Neuron. 2016;89:248–68.
Sherwood CC, Stimpson CD, Raghanti MA, Wildman DE, Uddin M, Grossman LI, et al. Evolution of increased glia-neuron ratios in the human frontal cortex. Proc Natl Acad Sci USA. 2006;103:13606–11.
Leyva-Díaz E, López-Bendito G. In and out from the cortex: development of major forebrain connections. Neuroscience. 2013;254:26–44.
Markram H, Toledo-Rodriguez M, Wang Y, Gupta A, Silberberg G, Wu C. Interneurons of the neocortical inhibitory system. Nat Rev Neurosci. 2004;5:793–807.
Rowitch DH, Kriegstein AR. Developmental genetics of vertebrate glial-cell specification. Nature. 2010;468:214–22.
Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science. 2016;353:aad8670.
Mariani MM, Kielian T. Microglia in infectious diseases of the central nervous system. J Neuroimmune Pharm. 2009;4:448–61.
Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25.
Lodato S, Rouaux C, Quast KB, Jantrachotechatchawan C, Studer M, Hensch TK, et al. Excitatory projection neuron subtypes control the distribution of local inhibitory interneurons in the cerebral cortex. Neuron. 2011;69:763–79.
Kang HJ, Kawasawa YI, Cheng F, Zhu Y, Xu X, Li M, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–9.
Back SA, Luo NL, Borenstein NS, Volpe JJ, Kinney HC. Arrested oligodendrocyte lineage progression during human cerebral white matter development: dissociation between the timing of progenitor differentiation and myelinogenesis. J Neuropathol Exp Neurol. 2002;61:197–211.
Holst CB, Brøchner CB, Vitting-Seerup K, Møllgård K. Astrogliogenesis in human fetal brain: complex spatiotemporal immunoreactivity patterns of GFAP, S100, AQP4 and YKL-40. J Anat. 2019;235:590–615.
Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15:300–12.
Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FMV. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10:1538–43.
Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch U-K, Mack M, et al. Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci. 2007;10:1544–53.
Hevner RF, Hodge RD, Daza RAM, Englund C. Transcription factors in glutamatergic neurogenesis: conserved programs in neocortex, cerebellum, and adult hippocampus. Neurosci Res. 2006;55:223–33.
Kelsom C, Lu W. Development and specification of GABAergic cortical interneurons. Cell Biosci. 2013;3:19.
Hevner RF. Layer-specific markers as probes for neuron type identity in human neocortex and malformations of cortical development. J Neuropathol Exp Neurol. 2007;66:101–9.
Shibata M, Gulden FO, Sestan N. From trans to cis: transcriptional regulatory networks in neocortical development. Trends Genet. 2015;31:77–87.
Koenderink MJ, Uylings HB. Postnatal maturation of layer V pyramidal neurons in the human prefrontal cortex. A quantitative Golgi analysis. Brain Res. 1995;678:233–43.
Yeung MSY, Zdunek S, Bergmann O, Bernard S, Salehpour M, Alkass K, et al. Dynamics of oligodendrocyte generation and myelination in the human brain. Cell. 2014;159:766–74.
Petanjek Z, Judaš M, Šimic G, Rasin MR, Uylings HBM, Rakic P, et al. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc Natl Acad Sci USA. 2011;108:13281–6.
Lambe EK, Krimer LS, Goldman-Rakic PS. Differential postnatal development of catecholamine and serotonin inputs to identified neurons in prefrontal cortex of rhesus monkey. J Neurosci. 2000;20:8780–7.
Gejman PV, Sanders AR, Duan J. The role of genetics in the etiology of schizophrenia. Psychiatr Clin North Am. 2010;33:35–66.
Pardiñas AF, Holmans P, Pocklington AJ, Escott-Price V, Ripke S, Carrera N, et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet 2018;50:381–9.
Birnbaum R, Jaffe AE, Chen Q, Hyde TM, Kleinman JE, Weinberger DR. Investigation of the prenatal expression patterns of 108 schizophrenia-associated genetic loci. Biol Psychiatry. 2015;77:e43–51.
Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013;154:518–29.
Susser ES, Lin SP. Schizophrenia after prenatal exposure to the Dutch Hunger Winter of 1944-5. Arch Gen Psychiatry. 1992;49:983–8.
Cannon M, Jones PB, Murray RM. Obstetric complications and schizophrenia: historical and meta-analytic review. Am J Psychiatry. 2002;159:1080–92.
Vassos E, Pedersen CB, Murray RM, Collier DA, Lewis CM. Meta-analysis of the association of urbanicity with schizophrenia. Schizophr Bull. 2012;38:1118–23.
Ursini G, Punzi G, Chen Q, Marenco S, Robinson JF, Porcelli A, et al. Convergence of placenta biology and genetic risk for schizophrenia. Nat Med. 2018;24:792–801.
Filatova S, Koivumaa-Honkanen H, Hirvonen N, Freeman A, Ivandic I, Hurtig T, et al. Early motor developmental milestones and schizophrenia: a systematic review and meta-analysis. Schizophr Res. 2017;188:13–20.
Reichenberg A, Caspi A, Harrington H, Houts R, Keefe RSE, Murray RM, et al. Static and dynamic cognitive deficits in childhood preceding adult schizophrenia: a 30-year study. Am J Psychiatry. 2010;167:160–9.
Jones P. Child developmental risk factors for adult schizophrenia in the British 1946 birth cohort. Lancet. 1994;344:1398–402.
Uhlhaas PJ, Singer W. Abnormal neural oscillations and synchrony in schizophrenia. Nat Rev Neurosci. 2010;11:100–13.
Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35:57–67.
Weinberger DR, Levitt P. Neurodevelopmental origins of schizophrenia. In: Weinberger DR, Harrison PJ, editors. Schizophrenia. 3rd ed. West Sussex, UK: Blackwell Publishing Ltd; 2011. p. 393–412.
Nakazawa K, Zsiros V, Jiang Z, Nakao K, Kolata S, Zhang S, et al. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology. 2012;62:1574–83.
Takahashi N, Sakurai T, Davis KL, Buxbaum JD. Linking oligodendrocyte and myelin dysfunction to neurocircuitry abnormalities in schizophrenia. Prog Neurobiol. 2011;93:13–24.
Yu H, Bi W, Liu C, Zhao Y, Zhang D, Yue W. A hypothesis-driven pathway analysis reveals myelin-related pathways that contribute to the risk of schizophrenia and bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2014;51:140–5.
Duncan LE, Holmans PA, Lee PH, O’Dushlaine CT, Kirby AW, Smoller JW, et al. Pathway analyses implicate glial cells in schizophrenia. PLoS ONE. 2014;9:e89441.
Neniskyte U, Gross CT. Errant gardeners: glial-cell-dependent synaptic pruning and neurodevelopmental disorders. Nat Rev Neurosci. 2017;18:658–70.
Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530:177–83.
Sellgren CM, Gracias J, Watmuff B, Biag JD, Thanos JM, Whittredge PB, et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat Neurosci. 2019;22:374–85.
Onwordi EC, Halff EF, Whitehurst T, Mansur A, Cotel M-C, Wells L, et al. Synaptic density marker SV2A is reduced in schizophrenia patients and unaffected by antipsychotics in rats. Nat Commun. 2020;11:246.
Geisler S, Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol. 2013;14:699–712.
Schmiedel JM, Klemm SL, Zheng Y, Sahay A, Blüthgen N, Marks DS, et al. Gene expression. MicroRNA control of protein expression noise. Science. 2015;348:128–32.
Perino M, Veenstra GJC. Chromatin control of developmental dynamics and plasticity. Dev Cell. 2016;38:610–20.
Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82.
Follert P, Cremer H, Béclin C. MicroRNAs in brain development and function: a matter of flexibility and stability. Front Mol Neurosci. 2014;7:5.
Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–89.
Andersen RE, Lim DA. Forging our understanding of lncRNAs in the brain. Cell Tissue Res. 2018;371:55–71.
Hall LL, Carone DM, Gomez AV, Kolpa HJ, Byron M, Mehta N, et al. Stable C0T-1 repeat RNA is abundant and is associated with euchromatic interphase chromosomes. Cell. 2014;156:907–19.
Cech TR, Steitz JA. The noncoding RNA revolution-trashing old rules to forge new ones. Cell. 2014;157:77–94.
Iijima T, Hidaka C, Iijima Y. Spatio-temporal regulations and functions of neuronal alternative RNA splicing in developing and adult brains. Neurosci Res. 2016;109:1–8.
Li S, Mason CE. The pivotal regulatory landscape of RNA modifications. Annu Rev Genom Hum Genet. 2014;15:127–50.
Gerstberger S, Hafner M, Tuschl T. A census of human RNA-binding proteins. Nat Rev Genet. 2014;15:829–45.
Tilgner H, Knowles DG, Johnson R, Davis CA, Chakrabortty S, Djebali S, et al. Deep sequencing of subcellular RNA fractions shows splicing to be predominantly co-transcriptional in the human genome but inefficient for lncRNAs. Genome Res. 2012;22:1616–25.
Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, et al. Landscape of transcription in human cells. Nature. 2012;489:101–8.
Zaghlool A, Ameur A, Nyberg L, Halvardson J, Grabherr M, Cavelier L, et al. Efficient cellular fractionation improves RNA sequencing analysis of mature and nascent transcripts from human tissues. BMC Biotechnol. 2013;13:99.
Solnestam BW, Stranneheim H, Hällman J, Käller M, Lundberg E, Lundeberg J, et al. Comparison of total and cytoplasmic mRNA reveals global regulation by nuclear retention and miRNAs. BMC Genom. 2012;13:574.
Pandya-Jones A, Bhatt DM, Lin C-H, Tong A-J, Smale ST, Black DL. Splicing kinetics and transcript release from the chromatin compartment limit the rate of lipid A-induced gene expression. RNA. 2013;19:811–27.
Prasanth KV, Prasanth SG, Xuan Z, Hearn S, Freier SM, Bennett CF, et al. Regulating gene expression through RNA nuclear retention. Cell. 2005;123:249–63.
Mauger O, Lemoine F, Scheiffele P. Targeted intron retention and excision for rapid gene regulation in response to neuronal activity. Neuron. 2016;92:1266–78.
Bahar Halpern K, Caspi I, Lemze D, Levy M, Landen S, Elinav E, et al. Nuclear retention of mRNA in mammalian tissues. Cell Rep. 2015;13:2653–62.
Battich N, Stoeger T, Pelkmans L. Control of transcript variability in single mammalian cells. Cell. 2015;163:1596–610.
Medioni C, Mowry K, Besse F. Principles and roles of mRNA localization in animal development. Development. 2012;139:3263–76.
Price AJ, Hwang T, Tao R, Burke EE, Rajpurohit A, Shin JH, et al. Characterizing the nuclear and cytoplasmic transcriptomes in developing and mature human cortex uncovers new insight into psychiatric disease gene regulation. Genome Res. 2020;30:1–11.
Jaffe AE, Shin J, Collado-Torres L, Leek JT, Tao R, Li C, et al. Developmental regulation of human cortex transcription and its clinical relevance at single base resolution. Nat Neurosci. 2015;18:154–61.
Beveridge NJ, Santarelli DM, Wang X, Tooney PA, Webster MJ, Weickert CS, et al. Maturation of the human dorsolateral prefrontal cortex coincides with a dynamic shift in microRNA expression. Schizophr Bull. 2014;40:399–409.
Zhang X, Chen MH, Wu X, Kodani A, Fan J, Doan R, et al. Cell-type-specific alternative splicing governs cell fate in the developing cerebral cortex. Cell. 2016;166:1147–62.e15.
Hwang T, Park C-K, Leung AKL, Gao Y, Hyde TM, Kleinman JE, et al. Dynamic regulation of RNA editing in human brain development and disease. Nat Neurosci. 2016;19:1093–9.
Boutz PL, Bhutkar A, Sharp PA. Detained introns are a novel, widespread class of post-transcriptionally spliced introns. Genes Dev. 2015;29:63–80.
Wong JJ-L, Ritchie W, Ebner OA, Selbach M, Wong JWH, Huang Y, et al. Orchestrated intron retention regulates normal granulocyte differentiation. Cell. 2013;154:583–95.
Yap K, Lim ZQ, Khandelia P, Friedman B, Makeyev EV. Coordinated regulation of neuronal mRNA steady-state levels through developmentally controlled intron retention. Genes Dev. 2012;26:1209–23.
Braunschweig U, Gueroussov S, Plocik AM, Graveley BR, Blencowe BJ. Dynamic integration of splicing within gene regulatory pathways. Cell. 2013;152:1252–69.
Chen L. Characterization and comparison of human nuclear and cytosolic editomes. Proc Natl Acad Sci USA. 2013;110:E2741–7.
Mariño-Ramírez L, Kann MG, Shoemaker BA, Landsman D. Histone structure and nucleosome stability. Expert Rev Proteom. 2005;2:719–29.
Chen T, Dent SYR. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat Rev Genet. 2014;15:93–106.
Hota SK, Bruneau BG. ATP-dependent chromatin remodeling during mammalian development. Development. 2016;143:2882–97.
Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80.
Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–95.
Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature. 2001;410:116–20.
Delaneau O, Zazhytska M, Borel C, Giannuzzi G, Rey G, Howald C, et al. Chromatin three-dimensional interactions mediate genetic effects on gene expression. Science. 2019;364. https://doi.org/10.1126/science.aat8266.
Cheung I, Shulha HP, Jiang Y, Matevossian A, Wang J, Weng Z, et al. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc Natl Acad Sci USA. 2010;107:8824–9.
Won H, de la Torre-Ubieta L, Stein JL, Parikshak NN, Huang J, Opland CK, et al. Chromosome conformation elucidates regulatory relationships in developing human brain. Nature. 2016;538:523–7.
Su Y, Shin J, Zhong C, Wang S, Roychowdhury P, Lim J, et al. Neuronal activity modifies the chromatin accessibility landscape in the adult brain. Nat Neurosci. 2017;20:476–83.
Zhu J, Adli M, Zou JY, Verstappen G, Coyne M, Zhang X, et al. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell. 2013;152:642–54.
Rodenhiser D, Mann M. Epigenetics and human disease: translating basic biology into clinical applications. CMAJ. 2006;174:341–8.
Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14:204–20.
Feng J, Zhou Y, Campbell SL, Le T, Li E, Sweatt JD, et al. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat Neurosci. 2010;13:423–30.
Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Schöler A, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–5.
Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–22.
Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, Ray P, et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell. 2013;153:1134–48.
Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017;356. https://doi.org/10.1126/science.aaj2239.
Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT, et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320–31.
Maurano MT, Wang H, John S, Shafer A, Canfield T, Lee K, et al. Role of DNA methylation in modulating transcription factor occupancy. Cell Rep. 2015;12:1184–95.
Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905.
He Y, Ecker JR. Non-CG methylation in the human genome. Annu Rev Genom Hum Genet. 2015;16:55–77.
Guo W, Zhang MQ, Wu H. Mammalian non-CG methylations are conserved and cell-type specific and may have been involved in the evolution of transposon elements. Sci Rep. 2016;6:32207.
Perzel Mandell KA, Price AJ, Wilton R, Collado Torres L, Tao R, Eagles NJ, et al. Characterizing the dynamic and functional DNA methylation landscape in the developing human cortex. Epigenetics. 2020 (In Press).
Price AJ, Collado-Torres L, Ivanov NA, Xia W, Burke EE, Shin JH, et al. Divergent neuronal DNA methylation patterns across human cortical development reveal critical periods and a unique role of CpH methylation. Genome Biol. 2019;20:196.
Guo JU, Ma DK, Mo H, Ball MP, Jang M-H, Bonaguidi MA, et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci. 2011;14:1345–51.
Halder R, Hennion M, Vidal RO, Shomroni O, Rahman R-U, Rajput A, et al. DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat Neurosci. 2016;19:102–10.
Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci. 2016;19:1442–53.
Collado-Torres L, Burke EE, Peterson A, Shin J, Straub RE, Rajpurohit A, et al. Regional heterogeneity in gene expression, regulation, and coherence in the frontal cortex and hippocampus across development and schizophrenia. Neuron. 2019;103:203–16.e8.
Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, et al. Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science. 2018;362. https://doi.org/10.1126/science.aat8127.
Jaffe AE, Hoeppner DJ, Saito T, Blanpain L, Ukaigwe J, Burke EE, et al. Profiling gene expression in the human dentate gyrus granule cell layer reveals insights into schizophrenia and its genetic risk. Nat Neurosci. 2020. https://doi.org/10.1038/s41593-020-0604-z.
Li M, Jaffe AE, Straub RE, Tao R, Shin JH, Wang Y, et al. A human-specific AS3MT isoform and BORCS7 are molecular risk factors in the 10q24.32 schizophrenia-associated locus. Nat Med. 2016;22:649–56.
Jaffe AE, Straub RE, Shin JH, Tao R, Gao Y, Collado-Torres L, et al. Developmental and genetic regulation of the human cortex transcriptome illuminate schizophrenia pathogenesis. Nat Neurosci. 2018;21:1117–25.
Geaghan M, Cairns MJ. Microrna and posttranscriptional dysregulation in psychiatry. Biol Psychiatry. 2015;78:231–9.
Mahmoudi E, Cairns MJ. MiR-137: an important player in neural development and neoplastic transformation. Mol Psychiatry. 2017;22:44–55.
Wright C, Gupta CN, Chen J, Patel V, Calhoun VD, Ehrlich S, et al. Polymorphisms in MIR137HG and microRNA-137-regulated genes influence gray matter structure in schizophrenia. Transl Psychiatry. 2016;6:e724.
Wright C, Calhoun VD, Ehrlich S, Wang L, Turner JA, Bizzozero NIP-. Meta gene set enrichment analyses link miR-137-regulated pathways with schizophrenia risk. Front Genet. 2015;6:147.
Walker RL, Ramaswami G, Hartl C, Mancuso N, Gandal MJ, de la Torre-Ubieta L, et al. Genetic control of expression and splicing in developing human brain informs disease mechanisms. Cell. 2019;179:750–71.e22.
O’Brien HE, Hannon E, Hill MJ, Toste CC, Robertson MJ, Morgan JE, et al. Expression quantitative trait loci in the developing human brain and their enrichment in neuropsychiatric disorders. Genome Biol. 2018;19:194.
Finucane HK, Reshef YA, Anttila V, Slowikowski K, Gusev A, Byrnes A, et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat Genet. 2018;50:621–9.
Calderon D, Bhaskar A, Knowles DA, Golan D, Raj T, Fu AQ, et al. Inferring relevant cell types for complex traits by using single-cell gene expression. Am J Hum Genet. 2017;101:686–99.
Skene NG, Bryois J, Bakken TE, Breen G, Crowley JJ, Gaspar HA, et al. Genetic identification of brain cell types underlying schizophrenia. Nat Genet. 2018;50:825–33.
Montano C, Taub MA, Jaffe A, Briem E, Feinberg JI, Trygvadottir R, et al. Association of DNA methylation differences with schizophrenia in an epigenome-wide association study. JAMA Psychiatry. 2016;73:506–14.
Jaffe AE, Gao Y, Deep-Soboslay A, Tao R, Hyde TM, Weinberger DR, et al. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci. 2016;19:40–47.
Viana J, Hannon E, Dempster E, Pidsley R, Macdonald R, Knox O, et al. Schizophrenia-associated methylomic variation: molecular signatures of disease and polygenic risk burden across multiple brain regions. Hum Mol Genet. 2017;26:210–25.
Hannon E, Dempster E, Viana J, Burrage J, Smith AR, Macdonald R, et al. An integrated genetic-epigenetic analysis of schizophrenia: evidence for co-localization of genetic associations and differential DNA methylation. Genome Biol. 2016;17:176.
Bryois J, Garrett ME, Song L, Safi A, Giusti-Rodriguez P, Johnson GD, et al. Evaluation of chromatin accessibility in prefrontal cortex of individuals with schizophrenia. Nat Commun. 2018;9:3121.
Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014;15:R31.
Li M, Santpere G, Imamura Kawasawa Y, Evgrafov OV, Gulden FO, Pochareddy S, et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science. 2018;362. https://doi.org/10.1126/science.aat7615.
Rajarajan P, Borrman T, Liao W, Schrode N, Flaherty E, Casiño C, et al. Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science. 2018;362. https://doi.org/10.1126/science.aat4311.
Hannon E, Spiers H, Viana J, Pidsley R, Burrage J, Murphy TM, et al. Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat Neurosci. 2016;19:48–54.
Rizzardi LF, Hickey PF, Rodriguez DiBlasi V, Tryggvadóttir R, Callahan CM, Idrizi A, et al. Neuronal brain-region-specific DNA methylation and chromatin accessibility are associated with neuropsychiatric trait heritability. Nat Neurosci. 2019;22:307–16.
Girdhar K, Hoffman GE, Jiang Y, Brown L, Kundakovic M, Hauberg ME, et al. Cell-specific histone modification maps in the human frontal lobe link schizophrenia risk to the neuronal epigenome. Nat Neurosci. 2018;21:1126–36.
de la Torre-Ubieta L, Stein JL, Won H, Opland CK, Liang D, Lu D, et al. The dynamic landscape of open chromatin during human cortical neurogenesis. Cell. 2018;172:289–304.e18.
Polioudakis D, de la Torre-Ubieta L, Langerman J, Elkins AG, Shi X, Stein JL, et al. A Single-Cell Transcriptomic Atlas of Human Neocortical Development during Mid-gestation. Neuron. 2019;103:785–801.e8.
Braun PR, Han S, Hing B, Nagahama Y, Gaul LN, Heinzman JT, et al. Genome-wide DNA methylation comparison between live human brain and peripheral tissues within individuals. Transl Psychiatry. 2019;9:47.
Shin J, Ming G, Song H. Decoding neural transcriptomes and epigenomes via high-throughput sequencing. Nat Neurosci. 2014;17:1463–75.
McGregor K, Bernatsky S, Colmegna I, Hudson M, Pastinen T, Labbe A, et al. An evaluation of methods correcting for cell-type heterogeneity in DNA methylation studies. Genome Biol. 2016;17:84.
Guintivano J, Aryee MJ, Kaminsky ZA. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics. 2013;8:290–302.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–7.
Rossner MJ, Hirrlinger J, Wichert SP, Boehm C, Newrzella D, Hiemisch H, et al. Global transcriptome analysis of genetically identified neurons in the adult cortex. J Neurosci. 2006;26:9956–66.
Dong X, Liao Z, Gritsch D, Hadzhiev Y, Bai Y, Locascio JJ, et al. Enhancers active in dopamine neurons are a primary link between genetic variation and neuropsychiatric disease. Nat Neurosci. 2018;21:1482–92.
Matevossian A, Akbarian S. Neuronal nuclei isolation from human postmortem brain tissue. J Vis Exp. 2008. https://doi.org/10.3791/914.
Kozlenkov A, Wang M, Roussos P, Rudchenko S, Barbu M, Bibikova M, et al. Substantial DNA methylation differences between two major neuronal subtypes in human brain. Nucleic Acids Res. 2016;44:2593–612.
Okada S, Saiwai H, Kumamaru H, Kubota K, Harada A, Yamaguchi M, et al. Flow cytometric sorting of neuronal and glial nuclei from central nervous system tissue. J Cell Physiol. 2011;226:552–8.
Ernst A, Alkass K, Bernard S, Salehpour M, Perl S, Tisdale J, et al. Neurogenesis in the striatum of the adult human brain. Cell. 2014;156:1072–83.
Sarnat HB, Nochlin D, Born DE. Neuronal nuclear antigen (NeuN): a marker of neuronal maturation in early human fetal nervous system. Brain Dev. 1998;20:88–94.
Dent MAR, Segura-Anaya E, Alva-Medina J, Aranda-Anzaldo A. NeuN/Fox-3 is an intrinsic component of the neuronal nuclear matrix. FEBS Lett. 2010;584:2767–71.
Kim KK, Adelstein RS, Kawamoto S. Identification of neuronal nuclei (NeuN) as Fox-3, a new member of the Fox-1 gene family of splicing factors. J Biol Chem. 2009;284:31052–61.
Unal-Cevik I, Kilinç M, Gürsoy-Ozdemir Y, Gurer G, Dalkara T. Loss of NeuN immunoreactivity after cerebral ischemia does not indicate neuronal cell loss: a cautionary note. Brain Res. 2004;1015:169–74.
Portiansky EL, Barbeito CG, Gimeno EJ, Zuccolilli GO, Goya RG. Loss of NeuN immunoreactivity in rat spinal cord neurons during aging. Exp Neurol. 2006;202:519–21.
Bakken TE, Hodge RD, Miller JA, Yao Z, Nguyen TN, Aevermann B, et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS ONE. 2018;13:e0209648.
Svensson V, Vento-Tormo R, Teichmann SA. Exponential scaling of single-cell RNA-seq in the past decade. Nat Protoc. 2018;13:599–604.
Schwartzman O, Tanay A. Single-cell epigenomics: techniques and emerging applications. Nat Rev Genet. 2015;16:716–26.
Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9:171–81.
Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–14.
Gupta I, Collier PG, Haase B, Mahfouz A, Joglekar A, Floyd T, et al. Single-cell isoform RNA sequencing characterizes isoforms in thousands of cerebellar cells. Nat Biotechnol. 2018. https://doi.org/10.1038/nbt.4259.
Lake BB, Ai R, Kaeser GE, Salathia NS, Yung YC, Liu R, et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science. 2016;352:1586–90.
Lacar B, Linker SB, Jaeger BN, Krishnaswami SR, Barron JJ, Kelder MJE, et al. Nuclear RNA-seq of single neurons reveals molecular signatures of activation. Nat Commun. 2016;7:11022.
Krishnaswami SR, Grindberg RV, Novotny M, Venepally P, Lacar B, Bhutani K, et al. Using single nuclei for RNA-seq to capture the transcriptome of postmortem neurons. Nat Protoc. 2016;11:499–524.
Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM, et al. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci USA. 2015;112:7285–90.
Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, et al. Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat Methods. 2017;14:955–8.
Luo C, Keown CL, Kurihara L, Zhou J, He Y, Li J, et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science. 2017;357:600–4.
Pollen AA, Nowakowski TJ, Shuga J, Wang X, Leyrat AA, Lui JH, et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat Biotechnol. 2014;32:1053–8.
Fan X, Dong J, Zhong S, Wei Y, Wu Q, Yan L, et al. Spatial transcriptomic survey of human embryonic cerebral cortex by single-cell RNA-seq analysis. Cell Res. 2018;28:730–45.
Zhong S, Zhang S, Fan X, Wu Q, Yan L, Dong J, et al. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature. 2018;555:524–8.
Eng C-HL, Lawson M, Zhu Q, Dries R, Koulena N, Takei Y, et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH. Nature. 2019;568:235–9.
Rodriques SG, Stickels RR, Goeva A, Martin CA, Murray E, Vanderburg CR, et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019;363:1463–7.
Maynard KR, Jaffe AE, Martinowich K. Spatial transcriptomics: putting genome-wide expression on the map. Neuropsychopharmacology. 2020;45:232–3.
Fullard JF, Charney AW, Voloudakis G, Uzilov AV, Haroutunian V, Roussos P. Assessment of somatic single-nucleotide variation in brain tissue of cases with schizophrenia. Transl Psychiatry. 2019;9:21.
Birnbaum R, Weinberger DR. Genetic insights into the neurodevelopmental origins of schizophrenia. Nat Rev Neurosci. 2017;18:727–40.
WHO Multicentre Growth Reference Study Group. WHO Child Growth Standards based on length/height, weight and age. Acta Paediatr Suppl. 2006;450:76–85.
Johnson JS, Newport EL. Critical period effects in second language learning: The influence of maturational state on the acquisition of English as a second language. Cogn Psychol. 1989;21:60–99.
Catts VS, Fung SJ, Long LE, Joshi D, Vercammen A, Allen KM, et al. Rethinking schizophrenia in the context of normal neurodevelopment. Front Cell Neurosci. 2013;7:60.
Acknowledgements
The authors thank Emily E. Burke for contributing to the figure in this publication.
Funding
This work is supported by the generous donations of the Lieber and Maltz families. In addition, AJP was supported by R21MH105853, R01MH112751, and U01MH106893; AEJ was supported by R21MH105853 and R01MH112751; and DRW was supported by R21MH105853 and U01MH106893.
Author information
Authors and Affiliations
Contributions
AJP: conceptualization and writing—original draft preparation, writing—review and editing. DRW: writing—review and editing. AEJ: writing—review and editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Price, A.J., Jaffe, A.E. & Weinberger, D.R. Cortical cellular diversity and development in schizophrenia. Mol Psychiatry 26, 203–217 (2021). https://doi.org/10.1038/s41380-020-0775-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-020-0775-8
- Springer Nature Limited
This article is cited by
-
Unveiling Promising Neuroimaging Biomarkers for Schizophrenia Through Clinical and Genetic Perspectives
Neuroscience Bulletin (2024)
-
The genetic relationships between brain structure and schizophrenia
Nature Communications (2023)
-
Neuroimaging and multiomics reveal cross-scale circuit abnormalities in schizophrenia
Nature Mental Health (2023)
-
Integrative omics of schizophrenia: from genetic determinants to clinical classification and risk prediction
Molecular Psychiatry (2022)
-
Multiple alterations in glutamatergic transmission and dopamine D2 receptor splicing in induced pluripotent stem cell-derived neurons from patients with familial schizophrenia
Translational Psychiatry (2021)