
Abstract
Bipolar disorder (BD) is a chronic affective disorder with extreme mood swings that include mania or hypomania and depression. Though the exact mechanism of BD is unknown, neuroinflammation is one of the numerous investigated etiopathophysiological causes of BD. This article presents a systematic review of the data regarding brain inflammation evaluating microglia, astrocytes, cytokines, chemokines, adhesion molecules, and other inflammatory markers in postmortem BD brain samples. This systematic review was performed according to PRISMA recommendations, and relevant studies were identified by searching the PubMed/MEDLINE, PsycINFO, EMBASE, LILACS, IBECS, and Web of Science databases for peer-reviewed journal articles published by March 2019. Quality of included studies appraised using the QUADAS-2 tool. Among the 1814 articles included in the primary screening, 51 articles measured inflammatory markers in postmortem BD brain samples. A number of studies have shown evidence of inflammation in BD postmortem brain samples. However, an absolute statement cannot be concluded whether neuroinflammation is present in BD due to the large number of studies did not evaluate the presence of infiltrating peripheral immune cells in the central nervous system (CNS) parenchyma, cytokines levels, and microglia activation in the same postmortem brain sample. For example, out of 15 studies that evaluated microglia cells markers, 8 studies found no effect of BD on these cells. Similarly, 17 out of 51 studies evaluating astrocytes markers, 9 studies did not find any effect of BD on astrocyte cells, whereas 8 studies found a decrease and 2 studies presented both increase and decrease in different brain regions. In addition, multiple factors account for the variability across the studies, including postmortem interval, brain area studied, age at diagnosis, undergoing treatment, and others. Future analyses should rectify these potential sources of heterogeneity and reach a consensus regarding the inflammatory markers in postmortem BD brain samples.
Similar content being viewed by others

Introduction
Bipolar disorder (BD) is a neuropsychiatric disorder that belongs to a group of severe, recurrent affective disorders that are among the leading cause of disability and death in young people [1]. Irrespective of nationality, ethnic or cultural origin, and socioeconomic status BD affects 2–3% of the world’s population [2, 3]. The etiology of this psychiatric disorder is still unknown, however several morphological, metabolic, and signaling abnormalities have been proposed, including volume decrease in the hippocampus [4] and in the frontal cortical in the Mania group [5]. Among these potential-contributing factors, number of studies evidence the role of neuroinflammation in BD pathogenesis [6,7,8] and variation in microglia, astrocytes, and oligodendrocytes markers also have been found in postmortem studies in patients with BD [9,10,11]. However, the term neuroinflammation referred for conditions that meet all the four signs including elevations in proinflammatory cytokines, microglial activation, infiltration of immune cells from the bloodstream and secondary degeneration in the brain [12, 13].
The brain has specific immunological and inflammatory adaptations, including a protective blood–brain barrier (BBB) and highly efficient glial cells [14]. The main innate immune cells that contribute to the immune response in central nervous system (CNS) includes microglia, astrocytes, macrophages, natural killer (NK) cells, mast cells as well as oligodendrocytes and neurons [15]. Microglia, the resident macrophages of the CNS, are activated after injury or stress, release of proinflammatory cytokines that generate reactive oxygen and nitrogen species and phagocytic activity [16]. Interleukin (IL)-1β released from microglia can activate astrocytes, another important cell type in the neuroimmune system, which in turn release additional cytokines and chemokines such as IL-1β, tumor necrosis factor (TNF)-α, and chemokine (c-c motif) ligand (CCL5) and express glial fibrillary acidic protein (GFAP) and S100 calcium-binding protein B (S100B) [17,18,19,20].
Evidence from preclinical and clinical studies, including neuroimaging and genetic studies, supports the role of inflammation in the etiological pathway of BD. A recent systematic review demonstrated the increased the levels of proinflammatory cytokines such as TNF-α, IL-1β, IL-6, interferon (INF)-γ, and IL-18 in serum and plasma from acute phase of BD patients. On the other hand reduction in the levels of anti-inflammatory cytokines such as IL-10 and transforming growth factor-beta 1 (TGF-β1) was observed during the manic phases of BD [21]. Another systematic review and meta-analysis revealed fluctuations in components of the TNF pathway, including soluble and membrane-bound TNF and its two receptors, in the peripheral blood of BD subjects [22]. Even genetic studies have indicated that a multitude of genes that partake in various neuroimmunological and inflammatory pathways are either up- or downregulated in BD [23,24,25]. A clear pictorial visualization of neuroinflammation in BD has been made possible through in vivo positron emission tomography (PET) imaging studies. The activation of microglia visualized in vivo using [(11)C]-(R)-PK11195 PET imaging demonstrates the focal neuroinflammation in BD [26]. Recently reported largest cohort of study from MRI findings revealed that cortical gray matter was thinner in frontal, temporal, and parietal regions of both brain hemispheres in BD patients [27]. The implications of results from previous findings it seems plausible that inflammation may have a role in BD. In this article, we systematically review the literature on brain inflammatory markers measured in BD postmortem brains to (i) identify the inflammatory markers that are elevated in the postmortem brain of BD patients, (ii) draw attention to immune cells and immunological and inflammatory pathways, and recognize neuroinflammation in postmortem brain samples of BD patients.
Methods
We performed this systematic review as stated in a prospective protocol following the PRISMA Statement guidelines [28]. The review protocol is registered at PROSPERO (registration number: CRD42017083459; http://www.crd.york.ac.uk/prospero).
Literature search strategy
This systematic review of clinical studies was conducted to evaluate inflammatory markers in postmortem BD brain studies. The studies were identified by searching the PubMed/MEDLINE (National Library of Medicine), PsycINFO, and EMBASE (Ovid) databases for peer-reviewed journal articles that were published by March 2019. To identify additional relevant citations, we conducted forward searches in the LILACS (Latin American and Caribbean Health Sciences Literature), IBECS (Bibliographical Index in Spanish in Health Sciences), and Web of Science databases. The abovementioned databases were searched with the following combinations of keywords: (“bipolar disorder” OR “bipolar patient” OR “bipolar depression” OR “manic depressive illness” OR “mania”) AND (“postmortem” OR “post-mortem” OR “brain sample” OR “autopsy”) AND (“inflammation” OR “neuroinflammation” OR “glia” OR “microglia” OR “astrocytes” OR “cytokines” OR “chemokines”).
Review of patients, interventions, comparators, outcome measures, and study designs (PICO framework)
We posed the question “Do postmortem brain samples of BD patients’ present inflammatory markers?”
Eligibility criteria
We included original peer-reviewed articles and abstracts with no language and year restriction to identify inflammatory markers in postmortem BD brains. We omitted review articles, in vitro studies, animal studies, and studies that did not present inflammatory markers in postmortem BD brain samples.
Screening
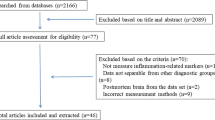
A total of 1814 articles were included in the primary screening. Reference management software (EndNote X7 for Windows from Thomson Reuters, 2013) was used for screening purposes. After the omission of 455 duplicates, a total of 1359 articles were selected for the study. The retrieved studies were first screened based on their titles and abstracts, and 1286 articles were further omitted based on the exclusion criteria (reviews, in vitro studies, animal studies, no BD, no inflammatory markers). The remaining 73 full-text articles were obtained and thoroughly evaluated in a second screening. At the end of the second screening, 51 articles were ultimately included after 22 articles were discarded based on the exclusion criteria (Fig. 1).

Flowchart of study selection
Article selection
The authors (PS, OFP, NA, VVG, and TB) screened the titles and abstracts for eligibility. Any controversies regarding the studies were resolved through simultaneous evaluation by the three primary authors. Upon agreement from the three authors, references that were valid based on the selection criteria were selected for final inclusion, and full-text PDFs were obtained and analyzed. The two authors, TB and VVG, settled issues whenever a consensus could not be reached between the first three authors.
Data extraction
The data were extracted from the comprehensively reviewed journal articles in a methodical manner. The variables extracted for our review included the study design, sample size (n), and inflammatory biomarkers as well as the way in which the inflammatory marker profile was associated with BD.
Quality assessment of the included studies
Methodological quality assessment of studies was performed according to Quality Assessment of Diagnostic Accuracy Studies-2 checklist (QUADAS-2), that consists of four key domains: patient selection, index test, reference standard and flow and timing [29]. The quality assessment was performed using Review Manager® (RevMan) version 5.3.
Results
Areas of the brain studied
The areas of the brain studied among our reviewed papers include prefrontal cortex (PFC; Brodmann area (BA) 10), dorsolateral prefrontal cortex (DLPFC; BA 46), frontal cortex (FC), orbitofrontal cortex (OFC; BA 11), anterior cingulated cortex (ACC; BA 24), frontoparietal cortex (FPC), parietal cortex (PC; BA 40), temporal cortex (TC; BA 20), superior temporal gyrus (STG), inferior temporal cortex (ITC), entorhinal cortex (ECx), hippocampus, amygdala, thalamus, cerebellum, dorsal raphe nucleus (DRN), subventricular zone (SVZ), and caudate putamen (Tables 1 and 2; Figs. 2 and 3).

Brain areas investigated in BD postmortem brain samples

Brain areas investigated in BD postmortem brain samples
Cellular parameters
Astrocytes
Astrocytes provide structural and nutritional support to neurons and play a significant role in CNS immune responses, synaptic function, neuronal metabolism, and myelin sheath development and maintenance. Changes in the number, density, or function of astrocytes seriously influence the nervous system [30, 31]. Recently, two different subtypes of reactive astrocytes that were termed “A1” and “A2” in analogy to the “M1” and “M2” macrophage nomenclature have been reported. Microglia when activated induces A1 astrocytes by secreting IL-1α, TNF and C1q. The subtype A1 lose the ability to promote neuronal survival, outgrowth, synaptogenesis, and phagocytosis but gain neurotoxic function, rapidly killing neurons and oligodendrocytes [32]. Our database search revealed 21 studies that estimated astrocyte-related changes in postmortem BD brains including changes in astrocyte size, density, and areal fraction; astrocytic end feet; and the expression of markers such as GFAP, phosphorylated GFAP (pGFAP), and S100B (Table 1; Figs. 2 and 3).
Astrocyte markers (GFAP, pGFAP, S100B, HERV-W capsid (GAG) protein iNOS, and nNOS)
Glial fibrillary acidic protein (GFAP)
GFAP is a protein in astrocyte intermediate filaments and is an important marker for astrocytes [33, 34]. Increases in GFAP may signify astrogliosis, reactive injury, or neurodegeneration, while decreased levels indicate reduced synaptic capabilities of neurons [35,36,37,38]. GFAP expression is regulated by several factors, such as cytokines, hormones, and growth factors [39, 40]. Twelve studies measured GFAP in the form of GFAP expression, GFAP-positive astrocyte number, GFAP area fraction, or GFAP mRNA expression. Among the 13 studies, five studies documented no BD-associated variations in GFAP levels [41,42,43,44,45]. Whereas three studies observed reduced GFAP expression [46,47,48], one study noted a decrease in the GFAP area fraction [47], one study reported a decrease in GFAP messenger ribonucleic acid (mRNA) levels [49], one study reported an increase in GFAP mRNA levels [9]; and only three studies cited increased the expression of GFAP [11, 18, 50]. One study demonstrated a reduction in pGFAP protein in the FC [20], whereas another reported no reduction in pGFAP-labeled astrocytes [45]. One study demonstrated no difference in astrocyte density in amygdala [51] (Table 1). When cerebellar GFAP levels in BD brains were compared with those in normal control brains, there was a 17% reduction in GFAP levels in BD brains. These levels were influenced by treatment with antidepressants, but not with antipsychotics or mood stabilizers. However, these antidepressant-induced changes may hint towards the role of antidepressants in the synthesis or destruction of GFAP by astrocytes. In addition, as astrocytes constitute 25% of the gray matter volume and as they outnumber neurons in the brain by fivefold, they may be an important target for antidepressant action in the brain [47]. When the CA1 pyramidal layer and the alveus of the hippocampus were analyzed for presence of GFAP- and S100B-positive astrocytes, the CA1 pyramidal layers in BD brains showed an attenuation in the numerical density of S100B-positive astrocytes, with no variation in the density of GFAP-immunoreactive astrocytes, compared to those in control brains [19]. When GFAP43 and GFAP41 levels were measured alongside TNF parameters in BA 24 and BA 46 in the brain, the levels in BD samples were similar to those in controls [51]. A study conducted by Pantazopoulos and collaborators noted no difference in GFAP-positive astrocyte density in the amygdala and ECx [43]. Even ventral and dorsal SVZ analysis has not shown any astrocytic alteration or gliosis, as evidenced by normal levels of GFAP [41]. Analysis of reactive astrogliosis and GFAP levels in the hippocampus after glucocorticoid elevation in chronic (BD) and acute cases (steroid treatment) has revealed that the number of GFAP-positive astrocytes is decreased in the CA1 and CA2 regions of BD samples compared to controls [46]. The GFAP area fraction is reduced in BD, and there is a difference in astrocyte space distribution and an elevation in astrocyte clustering in PFC and DLPFC white matter [18]. In the FC of postmortem BD brain samples, IL-1R cascade activation and upregulation of nuclear factor kappa beta (NF-κβ) results in increased expression of GFAP and iNOS in BD subjects compared to the expression in controls. While GFAP-positive astrocytes display fine fibrous processes in control brains, they show hypertrophy in BD brains [11, 52]. Statistics on white and gray matter reveal that GFAP mRNA levels are significantly decreased in the white matter of all brain areas and in the gray matter of the ACC in BD brains compared to the levels in control brains. These data suggest that GFAP-positive astrocytes may contribute to the glial density reduction noted in other psychiatric disorders [49]. These disparities in the outcome may partly explained by the variations in the methodology, statistical analysis and/or samples across studies. Considering brain region is one such variable across the studies, the results from four studies showed decrease astrocyte expression in hippocampus, two studies showed increased astrocyte clustering in FC; another study showed no change in astrocyte markers in FC. The other variable to be considered is the treatment regimen, a report from Ferensztajn-Rochowiak et al., demonstrated that the mRNA expression GFAP (not reach the level of statistical significance) in peripheral blood, was higher in BD patients not taking lithium than in healthy controls [53]. Thus, the reduction in GFAP levels observed in most of the studies may partially explained by the treatment regimen.
pGFAP
Phosphorylation of GFAP occurs at 5 sites (Thr7, Ser8, Ser13, Ser17, and Ser38) [54]. One study reported a reduction in pGFAP protein in the FC [20], whereas another showed no reduction in pGFAP-labeled astrocytes [45]. Proteomic analysis of 89 frontal cortical areas has shown that there is a reduction in GFAP500 in BD brains compared to the level in controls [20]. There is no reduction in astrocytes labeled with pGFAP in BD brains compared to the number in control brains [45].
S100B
S100B is a calcium-binding protein secreted by astrocytes and oligodendrocytes [55, 56]. Increased S100B levels in serum and cerebrospinal fluid (CSF) represent a suitable marker of glial damage in mood disorders [57, 58]. When the CA1 pyramidal layer and the alveus of the hippocampus were examined for S100B positive astrocytes, the right and left CA1 pyramidal layers showed a reduction in the numerical density of S100B-positive astrocytes in BD brains compared to that in controls [19]. One study demonstrated that the levels of S100B decreased in BA 9 and increased in BA 49 [44]. In addition, no significant changes was found in S100B in amygdala of BD samples [59].
Human endogenous retrovirus (HERV)
HERV, the ancient acquired elements shown to modulate innate immune responses and has protective effect against exogenous infections [60]. On the one side, it regulates inflammatory and autoimmune disorders, while on the other side to the control of excessive immune activation through their immunosuppressive properties [61,62,63]. The expression of capsid (GAG) protein of HERV− detected in neurons as well as astroglial cells in normal brain both in the anterior cingulate cortex and in the hippocampal formation. In postmortem samples from BD patients, there was a reduction of this expression in neurons and astroglial cells. This downregulation may be a result of abnormal gene regulation, a disturbance of the normal gene expression profile, or impaired genetic mechanisms during HERV transcription at the ribonucleic acid (RNA) level [64].
Astrocyte gene expression
The expression of GFAP mRNA significantly decreased in the white matter of all brain areas and in gray matter of the ACC in BD brains compared to the levels in control brains [49]. The astrocyte genes, 10-formyltetrahydrofolate dehydrogenase (ALDH1L1) and GFAP, shows increased expression in anteroventral (AV) and mediodorsal thalamic nuclei (MDN), internal capsule (IC), and putamen (Put) areas of BD brains compared to its expression in controls [9].
Microglia
Microglia, the brain sentinels, carryout the first line of defense against injury and infections [65]. Upon activation, they release proinflammatory cytokines, which exert negative effects on neuroprotective mechanisms and result in pathophysiological disturbances in BD [16, 66, 67]. In addition to causing proinflammatory cytokine release and neurotoxic effects, microglia negatively regulate anti-inflammatory and neuroprotective mechanisms. Microglia also possess phagocytic potential characterized by the surface expression of antigen-presenting markers such as MHC class II [68, 69].
Microglial markers (MHC I, MHC II, HLA-DR, IBA-1, CD11B, c-fos, QUIN, and PrP(c))
Out of 15 studies that analyzed microglial markers in BD postmortem brain, eight studies were neutral; one study cited decreased microglial reaction due to a reduction in human leukocyte antigen-D related (HLA-DR) density in the DRN [70]; one study demonstrated decreased CD68 and CD11B in ACC [71]. One study demonstrated increased expression of c-fos and CD11B in the FC [11]; one study noted decreased QUIN in the hippocampus [72]; one study reported reduced cellular prion protein (PrP(c)) immunoreactivity in the ACC [73]; and one study reported no significant change in HLA-DR density in the DLPFC, ACC, mediodorsal thalamus and hippocampus in non-suicidal cases but reported microgliosis in BD-suicide victims [74] (Table 1). MHC induction via HLA-DR in microglia is a sensitive marker of neuroinflammation and neurodegenerative processes [75]. Examination of MHC class I protein in the DLPFC, OFC and FC has revealed that there is no difference in MHC class I levels in BD brains compared to the levels in control brains [76,77,78]. When HLA-DR positive microglia were analyzed in the DRN, a significant reduction in microglial reaction was evident from a decreased density of HLA-DR-positive microglial cells; this effect was seen only in non-suicidal depressed subjects and not in depressed suicide victims or controls. A strong correlation between antidepressant medication use and HLA-DR-positive microglial density has been noted in non-suicidal patients, supporting the protective role of the microglial reaction in these patients. A positive effect of microglia on ribosomal deoxyribonucleic acid (DNA) transcription in DRN neurons in non-suicidal depressed subjects has also been noted. DRN microglial and neuronal interactions in counteracting directions may facilitate or reduce suicidal ideation in depressed patients [70]. There are no significant differences in HLA-DR-positive microglial density between patients with BD and controls in the DLPFC, ACC, mediodorsal thalamus and hippocampus [74] and in amygdala of BD patients [59]. However, significant microgliosis has been noted in suicide patients, regardless of diagnosis, in the DLPFC, ACC and mediodorsal thalamus, and a trend towards microgliosis has been noted in the hippocampus, suggesting that increased microglial density in BD-depression is associated with suicide. In addition, in BD-1, BA 46 region, HLA-DRA decreased in both microarray and qRT-PCR [79]. Our search results showed total of 18 studies with BD suicide cases. Among these, one study reported suicide in 2 out of 7 cases due to over-dose of medication [70], one study reported suicide in 1 out of 6 cases due to intoxication [72], and one reported 3 cases out of 5 with alcohol or drug use [80]. Patients with bipolar disorder are at great risk for suicide, these findings raise the question of whether pre-suicidal stress leads to microglial activation or microglial activation triggers the release of neuroendocrine and inflammatory mediators that bring about an imbalance of noradrenergic or serotonergic neurotransmission that ultimately leads to suicidal ideation [74]. In the DLPFC, there are no significant differences in the density, area fraction or spatial distribution of ionized calcium binding adaptor molecule-1 (IBA-1)-immunoreactive microglia in BD patients compared to the corresponding parameters of controls [18]. In an analysis of microglial markers in the FC, c-fos and CD11B protein and mRNA were found to be significantly elevated in the postmortem brains of BD subjects compared to the levels in control brains. In BD tissue, HLA-DR-stained microglia display thickened processes compared to the microglia in control tissue [11]. However, in another study, CD11B levels were found not to vary in the DLPFC and ACC of BD subjects compared to the levels in controls [51]. QUIN is produced by activated microglia cells and acts as an NMDA-receptor (NMDA-R) agonist in glutamatergic neurotransmission and as a proinflammatory mediator with neurotoxic properties. When hippocampal QUIN expression and volume was analyzed, microglial QUIN expression was found to be either similar to control expression in the left CA1 and right CA2/3 areas or decreased in the right CA1 and left CA2/3 areas of the hippocampus, with no volume changes [72]. PrP(c) is a copper-binding cell surface glycosyl phosphatidylinositol (GPI)-anchored glycoprotein expressed by neurons and microglia [81, 82]. PrP(c) protects against oxidative stress, promotes neuroprotection, acts as a laminin receptor (laminin and PrP(c) together help in the learning and memory pathway), and mediates neuronal survival and differentiation. When PrP(c)-positive neurons and glial cells in the ACC were examined, the number of PrP(c)-positive glial cells was found to be significantly reduced in the white matter of BD brains compared to the number in the white matter of control brains [73]. Confounding factors, especially drugs (antipsychotics, antidepressants, and mood stabilizers), had a significant impact on the reduced expression of PrP(c) by neurons and glial cells. Overall, there is a reduction in PrP(c) immunoreactivity in white matter glial cells that significantly influenced by drugs, and there are no associated signs of oxidative stress [73]. Calprotectin is a 36 kDa calcium-binding protein of the S100 family and this protein did not present significance in BA 9 of BD patient samples [83].
Microglial gene expression
A study on DNA methylation in the HCG-9 region revealed that there is a decrease in DNA methylation in BD subjects compared to the that in controls after adjusting for the effects of discrepancies such as age, medication, and DNA sequence variation (single nucleotide polymorphism (SNP) rs1128306) [84].
Molecular parameters
Cytokines
Cytokines are a broad category of small secretory proteins that include interleukins, interferons and growth factors. They are secreted by immune cells and regulate the body’s immune responses to infection, inflammation and injury [85]. They affect neurotransmitter and neuropeptide pathways as well as the hypo-thalamic pituitary adrenal (HPA) axis. The immune system, serotonin pathway and HPA axis are all altered in BD, thus determining the roles of the cytokines that influence all these pathways in BD is crucial [86]. An assay of proinflammatory cytokines such as IL-12, IL-2, IL-4, IL-5, and IFN-γ in the PFC revealed that the levels of the type I cytokines, IFN-γ and IL-12p40, were higher in BD patients than in controls, with no differences in serum levels. However, type I cytokines showed a dissociative effect, type II cytokines exhibited no differences between central and peripheral levels. These data suggest that enhanced central, rather than peripheral, levels of type I cytokines might play a role in BD pathogenesis [87]. In another study, TNF-α, IL-1β, and IL-6 were found increased in the PFC (BA 10) of BD teenage suicide victims as compared to controls; the BD patients also had elevated levels of TNF-α and IL-1β in BA 8 of the PFC. In addition, there was an increase in the expression of toll-like receptor (TLR)-3 and TLR-4 in the PFC of depressed and non-depressed suicide victims [88]. Rao and collaborators showed elevations in the protein and mRNA levels of IL-1β, IL-1R, and MyD88 that led to IL-1R cascade activation, which in turn resulted in the upregulation of the transcription factor NF-κB and increased levels of NF-κβ subunits p50 and p65. Increases in NF-κβ levels stimulate expression of excitotoxicity markers (iNOS and c-fos) and astrocyte and microglial markers (GFAP and CD11B). The TNF family includes proinflammatory cytokines expressed by macrophages, endothelial cells, neurons, astrocytes and microglia. The family has two receptors, TNFR1 and TNFR2; TNFR2 is selectively expressed by immune cells, neurons and glial cells [89]. TNF helps to regulate synaptic transport, neurotransmission, homeostatic synaptic scaling, neurogenesis and long-term potentiation [90]. A study examining TNF parameters in BA 24 and BA 46 demonstrated that the levels of tmTNF-α are significantly increased in BA 24, but not in BA 46, in BD brain samples. sTNF-α, TNF and TNFR1 mRNA levels are unaltered in BD; in contrast, levels of TNFR2 mRNA are decreased in BA 46, but not in BA 24, in BD [51]. Levels of tmTNFα, rather than sTNFα (which is a form of TNF-α measured in blood) are altered in the cortex. The findings of that study suggest that TNF-related pathway changes in the cortex of patients with mood disorders may be a response to balance the pathophysiological processes associated with BD. As the DLPFC and the ACC are critical for cognitive processes and mood control, respectively, changes in TNF-α-related pathways in these areas could be involved in the changes in cognition and mood that occur in BD. As drugs that act on tmTNF-α have mild antidepressant activity, a better understanding of TNF pathways may even help in the development of novel drug therapies. Levels of most of the glial and inflammatory marker proteins, such as IL-1β, GFAP43, GFAP41, CD11B, and pro-IL1β, do not vary in the cortex of patients with these disorders, suggesting that these TNF-α-related changes may be different from the usual changes in the neuroinflammatory pathway [51]. A study on TNF mRNA found that there is no significant difference in the mRNA expression of the TNF pathway in the DLPFC of BD patients compared to the expression in the DLPFC of controls. However, there is an increase in plasma TNF markers, such as TNF, sTNFR1 and sTNFR2, an increased TNF/sTNFR ratio, and a reduced TNF mRNA level in plasma and whole blood without concomitant gene expression changes in PM tissue [91]. The levels of NLRP3, ASC, caspase-1, IL-1β, IL-10, and IL-6 and TNF-α increased in BA 9, 10, and 24 of BD patients’ samples [92].
Chemokines
Chemokines are chemoattractive cytokines that recruit peripheral leukocytes to sites of inflammation. They are of importance in neuropsychiatric diseases such as BD as they play a role in neuronal and glial cell apoptosis, angiogenesis, and neurogenesis [93]. Abnormalities in immune mediators in the periphery may be associated with analogous immune abnormalities in the brain. Among different chemokine tested, decreased mRNA levels of CCR-5 reported in BD patients’ serum [94]. MCP-1 also known as CCL-2 a chemokine produced by microglia, astrocytes and neurons found to be higher in CSF from euthymic patients with BD compared with controls [95]. Another study also reported the increased CSF levels of MCP-1 in euthymic patients with BD and these differences remained after controlling for confounding factors, such as age, sex, smoking, blood-CSF barrier function, acute-phase proteins and body mass index [96]. A study by Barbosa et al. reported that BD patients presented higher plasma levels of CCL11, CCL24, CXCL10 and decreased plasma levels of CXCL8 [97]. When chemotactic cytokine CXCL5 levels were measured in the PFC, BD patients were found to have higher CXCL5 levels in the serum and brain compared to controls. In addition, there was an association between serum, brain and CSF levels in BD patients, suggesting a role of CXCL5 in BD and a correlation between central brain-related and peripheral serological immune activation [98]. In addition, in BD-1, BA 46 region, CCL3 decreased in both microarray and qRT-PCR [79].
Cell adhesion molecules (ICAM-1, VCAM-1, L1, and Thy-1)
Cell adhesion molecules play a major role in brain development, and consequently, their imbalance may result in psychiatric illness [99]. Intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 are expressed by endothelial cells, monocytes, and astrocytes and help in leukocyte-endothelial cell adhesion and other immune and inflammatory mechanisms [100]. Microvascular immunoreactivity of ICAM-1 is higher in the gray and white matter of the ACC in BD patients than in that of controls and schizophrenics and is higher in white matter of BD patients than in that of patients with unipolar depression (UD). There is no difference within the DLPFC. In BD, the gray and white matter of the ACC have significantly higher levels of ICAM-1 immunoreactivity than that of the DLPFC. VCAM-1 levels are too low for analysis. ICAM-1 is a marker of cerebral inflammation and ischemia. Ischemia is indicated by the presence of white matter hyperintensities in magnetic resonance imaging (MRI) in BD-associated depression [101]. The inflammatory response in the ACC and the elevation in ICAM-1 are parallel to each other and may be associated with the HPA axis [102].
Kynurenine pathway metabolites
Activation of the kynurenine pathway is a common feature of many psychiatric disorders. Linking glutamatergic neurotransmission and immune mononuclear phagocyte system (MPS) activation with the neurodegenerative hypothesis of depression in UD and BD, kynurenine metabolites generated through the indoleamine-2,3-dioxygenase (IDO) pathway are of special interest. IDO is expressed by activated microglia and induces the production of QUIN, an endogenous NMDA-R agonist with neurotoxic properties. The neurotoxicity of QUIN arises from several mechanisms, such as its agonizing effect on glutamate receptors sensitive to NMDA, its pro-oxidant properties, and its proinflammatory capacity, which results from its ability to increase the IFN-γ/IL-10 ratio as well as corticosterone and cytokine levels [72]. An increase in QUIN expression and QUIN-immunopositive microglia has been observed in the subgenual and supracallosal ACC in depressed patients [103]. However, when hippocampal QUIN expression and volume were analyzed, microglial QUIN expression in BD patients was found to be either similar to that of controls in the left CA1 and right CA2/3 areas or decreased in the right CA1 and left CA2/3 areas of the hippocampus, with no volume changes. This reduction in QUIN expression may have resulted from anti-inflammatory and neuroprotective compensatory mechanisms. Overall, the changes in QUIN levels signify the importance of NMDA-R signaling, glutamate transmission and MPS in BD-depression [72]. In the tryptophan 2,3-dioxygenase (TDO2) pathway of kynurenine, TDO2 catalyzes the first step. When the regulation of TDO2 activity was analyzed at the mRNA, protein and metabolic product levels in the ACC, an increase in kynurenine level was seen in the BD group, primarily in the BD group with psychosis, compared to the level in the control group. The ratio of KA to kynurenine was lower in the BD group than in the control group, as KA levels were unchanged. There was also an elevation in the density and intensity of both TDO2-positive white matter glia and TDO2-positive gray matter glia in the BD group [104].
AA cascade markers (cPLA2, sPLA2, COX-2, LOX-2, and cytochrome P (CYP) 450 epoxygenase)
In the CNS binding of microglial-derived cytokines to calcium channel coupled receptors on astrocytes results in activations of phospholipase enzymes that liberate AA from membrane lipoproteins. Thus, the mobilization of AA has been suggested to be a useful biomarker of neuroinflammation [105]. Results from multiple sclerosis postmortem brain samples and CSF demonstrated that arachidonic acid metabolism activated via cyclooxygenases (COXs) and lipoxygenases (LOXs). It has been postulated that motor dysfunction the hallmark of multiple sclerosis is triggered by arachidonic acid–mediated brain inflammation [106]. One study analyzed AA cascade markers in the FC and found an elevation in the mean protein and mRNA levels of phospholipases (cPLA2 type IVA, and sPLA2 type IIA) and COX-2 in BD brains compared to the levels in control brains [52]. BD brains show some epigenetic similarities to AD brains, such as global DNA methylation and histone H3 phosphorylation in the FC. Hypo- and hypermethylation of CpG islands in COX-2 and brain derived neurotrophic factor (BDNF) promoter regions, respectively, is proof of this. There is no significant epigenetic modification of 12-lipoxygenase (12-LOX) or p450 epoxygenase in AD or BD. Treatment of BD and AD with mood stabilizers and antipsychotics not targeting epigenetic regulation may not provide full recovery, as disease progression may reintroduce pathological changes due to epigenetic regulation [10].
Gene expression of inflammatory markers
When long intergenic non-coding RNAs (lincRNAs) were analyzed in the BA 11, 24, and 9 regions of BD brains, one billion reads were generated, among which 73.3% were known lincRNAs, 87.6% were novel lincRNAs and 95.7% were protein-coding genes (PCGs) expressed in all three brain regions. The lincRNAs have a significantly lower coding potential and ORF (open reading frame) ratio (ratio of ORF size to transcript length) and are relatively shorter and less conserved than the PCGs. There are many differentially expressed PCGs (DEPCGs) in BD; for example, the BD-related BDNF and gamma amino butyric acid type A receptor alpha 1 subunit (GABRA1) genes are dysregulated in BA 11. Additionally, these DEPCGs are involved in the pathology of certain functions such as synaptic transmission, CNS development and oligodendrocyte differentiation. There are 20 differentially expressed lincRNAs (DELincRNAs) in BA 11 that show brain region-specific patterns in BD. A weighted gene coexpression network analysis demonstrated that DELincRNAs, along with other genes, function as modulators of some functions, such as immune system development in BA 24 and oligodendrocyte differentiation in BA 9. Dysregulation of lincRNAs resulting from DNA methylation changes and other causes might lead to BD pathology. There is potential clinical value in investigating gene expression levels in peripheral blood samples of patients with major psychosis, so future studies should focus on investigating the dysfunctional lincRNAs in peripheral blood samples of patients with major psychosis to identify potential lincRNA biomarkers, which could help guide the diagnosis and treatment of major psychosis [107]. In a study involving the transcriptome sequencing of the dorsal striatum (caudate and putamen), 47,886 genes were studied, among which 1468 were DE genes of nominal significance; of those, 14 DE genes remained significant after multiple-testing correction at a 5% false discovery rate. These included a few immune response genes such as NLR family CARD domain containing 5 (NLRC5), S100 calcium binding protein A12 (S100A12), leukocyte immunoglobulin like receptor A4 (LILRA4) and Fc fragment of IgG binding protein (FCGBP). In a study involving gene set enrichment analysis of functional pathways, an enrichment of upregulated BD genes across immune/inflammation pathways was noted. Gene co-expression analysis by a weighted gene co-expression network analysis (WGCNA) method revealed 20 modules of highly interconnected genes for controls and BD patients, among which two modules (M11 and M13) were significantly enriched for BD susceptibility SNPs obtained from a large GWAS dataset. In addition, one of these two modules (M11) was also highly enriched in regional striatal markers and medium spiny neuronal (MSN) markers. On further analysis, it was determined that the genetic risk for BD associated with the M11 module was mainly distributed across MSN-specific genes. This revealed, to an extent, that BD etiology at the gene level is somehow linked to the striatal signaling pathway [108]. The key genes of the TGFβ-signaling pathway was upregulated in BA 46 of BD patient samples [109], however in another study TGF-β decreased in FC brain samples [110] (Table 2).
Quality assessment of the included studies
QUADAS-2 showed an unclear bias in patients selection presented in most studies, 18 studies were unclear in index test and 10 studies were unclear of information about reference standard selection. The “applicability concern” was low, showing a good quality of the included studies (Figs. 4 and 5).

Quality of included studies was appraised using the QUADAS-2 tool

Summary of methodological quality of studies according to QUADAS-2 tool regarding the risk of bias and applicability in review authors’ judgments about each domain for each included study about each domain
Limitations
The discussed postmortem case-control studies are observational, and hence, causation cannot be established. The clinical articles presented a moderate amount of bias, which was expected given the designs of the included clinical studies. Most of the studies involved a relatively small number of subjects. Several studies examined only one section of the brain rather than the whole brain. The long-term influences of lithium, antidepressants, antipsychotics, and other mood stabilizers on the outcomes of the studies cannot be excluded. As the studies discussed were postmortem studies, it was also hard to track data on the history of inflammation, infections, or other chronic medical conditions across the patients’ entire life span. Differences in age between diagnostic subgroups (controls and non-suicidal and suicidal patients) is a gross limitation, mainly because of the younger age of suicidal patients, and age could also influence the inflammatory profile. The duration since death is another major limitation, as prolonged PMI could influence the analyzed results. The polymerase chain reaction (PCR) analysis in most of the studies requires brain tissue with relatively short PMI and with high-quality RNA, which is not possible in most cases. Moreover, the RNA sequencing quantification and mapping methods used may lead to potential underestimation of mRNA levels. Immunohistochemical analyses used in many of the published articles often give variable results in postmortem tissue due to variations in agonal state, PMI, fixation, and storage.
Discussion and conclusion
BD has been associated with brain inflammation for the reason that several studies have been demonstrated elevated inflammatory cytokines in the bloodstream [111,112,113], and BD patients have been presented elevated microglia activation, in the brain as measured by translocator protein (TSPO), a marker of microglial activation, using PET imaging [114, 115]. In spite of that, TSPO/PET is not a consensus of in vivo microglia activation marker and TSPO polymorphism reduces binding affinity for all second-generation tracers tested in man hitherto, in mutants relative to wild type [116]. Several studies found evidence of inflammation in BD postmortem brain samples. However, an absolute statement cannot be concluded whether neuroinflammation is present in BD due to the large number of studies did not evaluate the presence of infiltrating peripheral immune cells in the CNS parenchyma, cytokines levels, and microglia activation in the same postmortem brain sample. In addition, several studies did not find any effect of BD on brain inflammatory markers. For example, out of 15 studies that evaluated microglia cells markers, 8 studies found no effect of BD on these cells, whereas 4 studies found a decrease of markers. Similarly, 17 out of 51 studies evaluating astrocytes markers, 9 studies did not find any effect of BD on astrocyte cells, whereas 8 studies found a decrease and 2 studies presented an increase and a decrease in different brain regions. As discussed above, microglia cells are the brain-resident macrophages and these cells are key players in neurodegenerative and neuroinflammatory diseases [117]. Despite that, activated microglia cells induce neurotoxic reactive astrocytes and these cells lose the ability to promote brain homeostasis, and induce the death of neurons and oligodendrocytes [32]. An additional aspect of this systematic review about BD and neuroinflammation is a non-consistent result from glial cell markers and inflammatory mediator levels in postmortem BD brain samples. Nonetheless, without a positive correlation between these two factors we cannot affirm that BD is associated with neuroinflammation. A few studies reported increase in pro-inflammatory cytokines in BD brain samples, for example, among 7 studies, 4 presented increased IL-1β and 3 increased the expression of TNF-α levels. However, it is not possible to conclude whether these cytokines were produced in the periphery and crossed the BBB or produced in the brain of BD patients. Blood-borne cytokines might affect the function of the CNS by crossing the BBB for direct interaction with CNS tissue [118]. Mechanisms by which this process is accomplished may involve the following: (i) saturable transport systems from blood to the CNS for example IL-1α, IL-1β, IL-6, and TNF-α; (ii) simple diffusion; and (iii) areas in the brain where the BBB is incomplete. In addition, cytokines may also damage the BBB and increase its permeability without entering the brain, such as through activation and destruction of tight junctions of microvascular endothelial cells forming the BBB, for example TNF-α [118,119,120]. The diversity of studies and variations in results due to non-standardized confounding factors, whose impact may change the marker profiles, make it difficult to thoroughly compare the studies and come to an overall conclusion without discrepancies. Future analyses should rectify these potential sources of heterogeneity and reach a consensus regarding the inflammatory markers in postmortem brain samples to make them more promising for novel diagnostic and therapeutic purposes.
References
Barnett R. Bipolar disorder. Lancet. 2018;392:1510.
Merikangas KR, Jin R, He JP, Kessler RC, Lee S, Sampson NA, et al. Prevalence and correlates of bipolar spectrum disorder in the world mental health survey initiative. Arch Gen Psychiatry. 2011;68:241–51.
Bauer M, Andreassen OA, Geddes JR, Vedel Kessing L, Lewitzka U, Schulze TG, et al. Areas of uncertainties and unmet needs in bipolar disorders: clinical and research perspectives. Lancet Psychiatry. 2018;5:930–9.
Cao B, Bauer IE, Sharma AN, Mwangi B, Frazier T, Lavagnino L, et al. Reduced hippocampus volume and memory performance in bipolar disorder patients carrying the BDNF val66met met allele. J Affect Disord. 2016;198:198–205.
Abe C, Ekman CJ, Sellgren C, Petrovic P, Ingvar M, Landen M. Manic episodes are related to changes in frontal cortex: a longitudinal neuroimaging study of bipolar disorder 1. Brain. 2015;138:3440–8.
Naaldijk YM, Bittencourt MC, Sack U, Ulrich H. Kinins and microglial responses in bipolar disorder: a neuroinflammation hypothesis. Biol Chem. 2016;397:283–96.
Sigitova E, Fisar Z, Hroudova J, Cikankova T, Raboch J. Biological hypotheses and biomarkers of bipolar disorder. Psychiatry Clin Neurosci. 2017;71:77–103.
Reus GZ, Fries GR, Stertz L, Badawy M, Passos IC, Barichello T, et al. The role of inflammation and microglial activation in the pathophysiology of psychiatric disorders. Neuroscience. 2015;300:141–54.
Barley K, Dracheva S, Byne W. Subcortical oligodendrocyte- and astrocyte-associated gene expression in subjects with schizophrenia, major depression and bipolar disorder. Schizophr Res. 2009;112:54–64.
Rao JS, Keleshian VL, Klein S, Rapoport SI. Epigenetic modifications in frontal cortex from Alzheimer’s disease and bipolar disorder patients. Transl Psychiatry. 2012;2:e132.
Rao JS, Harry GJ, Rapoport SI, Kim HW. Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol Psychiatry. 2010;15:384–92.
Estes ML, McAllister AK. Alterations in immune cells and mediators in the brain: it’s not always neuroinflammation! Brain Pathol. 2014;24:623–30.
DiSabato DJ, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. 2016;139:136–53.
Muldoon LL, Alvarez JI, Begley DJ, Boado RJ, Del Zoppo GJ, Doolittle ND, et al. Immunologic privilege in the central nervous system and the blood–brain barrier. J Cereb Blood Flow Metab. 2013;33:13–21.
Stephenson J, Nutma E, van der Valk P, Amor S. Inflammation in CNS neurodegenerative diseases. Immunology. 2018;154:204–19.
Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun. 2007;21:47–59.
van Neerven S, Nemes A, Imholz P, Regen T, Denecke B, Johann S, et al. Inflammatory cytokine release of astrocytes in vitro is reduced by all-trans retinoic acid. J Neuroimmunol. 2010;229:169–79.
Hercher C, Chopra V, Beasley CL. Evidence for morphological alterations in prefrontal white matter glia in schizophrenia and bipolar disorder. J Psychiatry Neurosci. 2014;39:376–85.
Gos T, Schroeter ML, Lessel W, Bernstein HG, Dobrowolny H, Schiltz K, et al. S100B-immunopositive astrocytes and oligodendrocytes in the hippocampus are differentially afflicted in unipolar and bipolar depression: a postmortem study. J Psychiatr Res. 2013;47:1694–9.
Johnston-Wilson NL, Sims CD, Hofmann JP, Anderson L, Shore AD, Torrey EF, et al. Disease-specific alterations in frontal cortex brain proteins in schizophrenia, bipolar disorder, and major depressive disorder. The Stanley Neuropathology Consortium. Mol Psychiatry. 2000;5:142–9.
Sayana P, Colpo GD, Simoes LR, Giridharan VV, Teixeira AL, Quevedo J, et al. A systematic review of evidence for the role of inflammatory biomarkers in bipolar patients. J Psychiatr Res. 2017;92:160–82.
Munkholm K, Brauner JV, Kessing LV, Vinberg M. Cytokines in bipolar disorder vs. healthy control subjects: a systematic review and meta-analysis. J Psychiatr Res. 2013;47:1119–33.
Wesseling H, Gottschalk MG, Bahn S. Targeted multiplexed selected reaction monitoring analysis evaluates protein expression changes of molecular risk factors for major psychiatric disorders. Int J Neuropsychopharmacol. 2014;18:1.
Mauney SPC, Goldstein JM, Petryshen T, Seidman LJ, Shenton ME, McCarley R, et al. Gene expression profiling of oligodendrocytes in dorsolateral prefrontal cortex deep white matter in bipolar disorder and schizophrenia. Schizophr Bull. 2013;39:S212.
Schmitt A, Bernstein HG, Steiner J, Schmitz C, Bogerts B, Rossner M, et al. Histological and gene expression studies of the hippocampus in schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2013;263:S40.
Haarman BC, Riemersma-Van der Lek RF, de Groot JC, Ruhe HG, Klein HC, Zandstra TE, et al. Neuroinflammation in bipolar disorder—A [(11)C]-(R)-PK11195 positron emission tomography study. Brain Behav Immun. 2014;40:219–25.
Hibar DP, Westlye LT, Doan NT, Jahanshad N, Cheung JW, Ching CRK, et al. Cortical abnormalities in bipolar disorder: an MRI analysis of 6503 individuals from the ENIGMA Bipolar Disorder Working Group. Mol Psychiatry. 2018;23:932–42.
Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. J Clin Epidemiol. 2009;62:1006–12.
Whiting P, Harbord R, Kleijnen J. No role for quality scores in systematic reviews of diagnostic accuracy studies. BMC Med Res Methodol. 2005;5:19.
Liedtke W, Edelmann W, Bieri PL, Chiu FC, Cowan NJ, Kucherlapati R, et al. GFAP is necessary for the integrity of CNS white matter architecture and long-term maintenance of myelination. Neuron. 1996;17:607–15.
Meyer-Franke A, Shen S, Barres BA. Astrocytes induce oligodendrocyte processes to align with and adhere to axons. Mol Cell Neurosci. 1999;14:385–97.
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–7.
Patanow CM, Day JR, Billingsley ML. Alterations in hippocampal expression of SNAP-25, GAP-43, stannin and glial fibrillary acidic protein following mechanical and trimethyltin-induced injury in the rat. Neuroscience. 1997;76:187–202.
Montgomery DL. Astrocytes: form, functions, and roles in disease. Vet Pathol. 1994;31:145–67.
Coyle JT, Schwarcz R. Mind glue: implications of glial cell biology for psychiatry. Arch Gen Psychiatry. 2000;57:90–3.
Rajkowska G, Miguel-Hidalgo JJ, Makkos Z, Meltzer H, Overholser J, Stockmeier C. Layer-specific reductions in GFAP-reactive astroglia in the dorsolateral prefrontal cortex in schizophrenia. Schizophr Res. 2002;57:127–38.
Moises HW, Zoega T, Gottesman II. The glial growth factors deficiency and synaptic destabilization hypothesis of schizophrenia. BMC Psychiatry. 2002;2:8.
Fatemi SH, Emamian ES, Sidwell RW, Kist DA, Stary JM, Earle JA, et al. Human influenza viral infection in utero alters glial fibrillary acidic protein immunoreactivity in the developing brains of neonatal mice. Mol Psychiatry. 2002;7:633–40.
Laping NJ, Nichols NR, Day JR, Johnson SA, Finch CE. Transcriptional control of glial fibrillary acidic protein and glutamine synthetase in vivo shows opposite responses to corticosterone in the hippocampus. Endocrinology. 1994;135:1928–33.
Norton WT, Aquino DA, Hozumi I, Chiu FC, Brosnan CF. Quantitative aspects of reactive gliosis: a review. Neurochem Res. 1992;17:877–85.
Comte I, Al-Shammari A, Szele FG. The human subventricular zone in neuropsychiatric disease. J Neurochem. 2013;125:31
Damadzic R, Bigelow LB, Krimer LS, Goldenson DA, Saunders RC, Kleinman JE, et al. A quantitative immunohistochemical study of astrocytes in the entorhinal cortex in schizophrenia, bipolar disorder and major depression: absence of significant astrocytosis. Brain Res Bull. 2001;55:611–8.
Pantazopoulos H, Woo TU, Lim MP, Lange N, Berretta S. Extracellular matrix-glial abnormalities in the amygdala and entorhinal cortex of subjects diagnosed with schizophrenia. Arch Gen Psychiatry. 2010;67:155–66.
Dean B, Gray L, Scarr E. Regionally specific changes in levels of cortical S100beta in bipolar 1 disorder but not schizophrenia. Aust N Z J Psychiatry. 2006;40:217–24.
Webster MJ, Knable MB, Johnston-Wilson N, Nagata K, Inagaki M, Yolken RH. Immunohistochemical localization of phosphorylated glial fibrillary acidic protein in the prefrontal cortex and hippocampus from patients with schizophrenia, bipolar disorder, and depression. Brain Behav Immun. 2001;15:388–400.
Muller MB, Lucassen PJ, Yassouridis A, Hoogendijk WJ, Holsboer F, Swaab DF. Neither major depression nor glucocorticoid treatment affects the cellular integrity of the human hippocampus. Eur J Neurosci. 2001;14:1603–12.
Fatemi SH, Laurence JA, Araghi-Niknam M, Stary JM, Schulz SC, Lee S, et al. Glial fibrillary acidic protein is reduced in cerebellum of subjects with major depression, but not schizophrenia. Schizophr Res. 2004;69:317–23.
Toro CT, Hallak JE, Dunham JS, Deakin JF. Glial fibrillary acidic protein and glutamine synthetase in subregions of prefrontal cortex in schizophrenia and mood disorder. Neurosci Lett. 2006;404:276–81.
Webster MJ, O’Grady J, Kleinman JE, Weickert CS. Glial fibrillary acidic protein mRNA levels in the cingulate cortex of individuals with depression, bipolar disorder and schizophrenia. Neuroscience. 2005;133:453–61.
Feresten AH, Barakauskas V, Ypsilanti A, Barr AM, Beasley CL. Increased expression of glial fibrillary acidic protein in prefrontal cortex in psychotic illness. Schizophr Res. 2013;150:252–7.
Dean B, Gibbons AS, Tawadros N, Brooks L, Everall IP, Scarr E. Different changes in cortical tumor necrosis factor-alpha-related pathways in schizophrenia and mood disorders. Mol Psychiatry. 2013;18:767–73.
Rao JS. Overlapping molecular neuropathology in post-mortem frontal cortex from bipolar disorder and schizophrenic patients. Neuropsychopharmacology. 2010;35:S47.
Ferensztajn-Rochowiak E, Tarnowski M, Samochowiec J, Michalak M, Ratajczak MZ, Rybakowski JK. Increased mRNA expression of peripheral glial cell markers in bipolar disorder: The effect of long-term lithium treatment. Eur Neuropsychopharmacol. 2016;26:1516–21.
Tardy M, Fages C, Le Prince G, Rolland B, Nunez J. Regulation of the glial fibrillary acidic protein (GFAP) and of its encoding mRNA in the developing brain and in cultured astrocytes. Adv Exp Med Biol. 1990;265:41–52.
Donato R, Sorci G, Riuzzi F, Arcuri C, Bianchi R, Brozzi F, et al. S100B’s double life: intracellular regulator and extracellular signal. Biochim Biophys Acta. 2009;1793:1008–22.
Steiner J, Bernstein HG, Bielau H, Berndt A, Brisch R, Mawrin C, et al. Evidence for a wide extra-astrocytic distribution of S100B in human brain. BMC Neurosci. 2007;8:2.
Rothermundt M, Arolt V, Wiesmann M, Missler U, Peters M, Rudolf S, et al. S-100B is increased in melancholic but not in non-melancholic major depression. J Affect Disord. 2001;66:89–93.
Schroeter ML, Abdul-Khaliq H, Sacher J, Steiner J, Blasig IE, Mueller K. Mood disorders are glial disorders: evidence from in vivo studies. Cardiovasc Psychiatry Neurol. 2010;2010:780645.
Hamidi M, Drevets WC, Price JL. Glial reduction in amygdala in major depressive disorder is due to oligodendrocytes. Biol Psychiatry. 2004;55:563–9.
Grandi N, Tramontano E. Human endogenous retroviruses are ancient acquired elements still shaping innate immune responses. Front Immunol. 2018;9:2039.
Lower R, Lower J, Kurth R. The viruses in all of us: characteristics and biological significance of human endogenous retrovirus sequences. Proc Natl Acad Sci USA. 1996;93:5177–84.
Taruscio D, Mantovani A. Human endogenous retroviral sequences: possible roles in reproductive physiopathology. Biol Reprod. 1998;59:713–24.
Obermayer-Straub P, Manns MP. Hepatitis C and D, retroviruses and autoimmune manifestations. J Autoimmun. 2001;16:275–85.
Weis S, Llenos IC, Sabunciyan S, Dulay JR, Isler L, Yolken R, et al. Reduced expression of human endogenous retrovirus (HERV)-W GAG protein in the cingulate gyrus and hippocampus in schizophrenia, bipolar disorder, and depression. J Neural Transm. 2007;114:645–55.
Thion MS, Low D, Silvin A, Chen J, Grisel P, Schulte-Schrepping J, et al. Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell. 2018;172:500–16.
Rajkowska G. Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol Psychiatry. 2000;48:766–77.
Schroeter MI, Steiner J, Mueller K. Glial pathology is modified by age in mood disorders—a systematic meta-analysis of serum 100B invivio studies. J Affect Disord. 2011;134:32–8.
Haroon E, Miller AH, Sanacora G. Inflammation, glutamate, and glia: a trio of trouble in mood disorders. Neuropsychopharmacology. 2017;42:193–215.
Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016;19:987–91.
Brisch R, Steiner J, Mawrin C, Krzyzanowska M, Jankowski Z, Gos T. Microglia in the dorsal raphe nucleus plays a potential role in both suicide facilitation and prevention in affective disorders. Eur Arch Psychiatry Clin Neurosci. 2017;267:403–15.
Seredenina T, Sorce S, Herrmann FR, Ma Mulone XJ, Plastre O, Aguzzi A, et al. Decreased NOX2 expression in the brain of patients with bipolar disorder: association with valproic acid prescription and substance abuse. Transl Psychiatry. 2017;7:e1206.
Busse M, Busse S, Myint AM, Gos T, Dobrowolny H, Muller UJ, et al. Decreased quinolinic acid in the hippocampus of depressive patients: evidence for local anti-inflammatory and neuroprotective responses? Eur Arch Psychiatry Clin Neurosci. 2015;265:321–9.
Weis S, Haybaeck J, Dulay JR, Llenos IC. Expression of cellular prion protein (PrP(c)) in schizophrenia, bipolar disorder, and depression. J Neural Transm. 2008;115:761–71.
Steiner J, Bielau H, Brisch R, Danos P, Ullrich O, Mawrin C, et al. Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J Psychiatr Res. 2008;42:151–7.
Schmitt AB, Brook GA, Buss A, Nacimiento W, Noth J, Kreutzberg GW. Dynamics of microglial activation in the spinal cord after cerebral infarction are revealed by expression of MHC class II antigen. Neuropathol Appl Neurobiol. 1998;24:167–76.
Kano S, Nwulia E, Niwa M, Chen Y, Sawa A, Cascella N. Altered MHC class I expression in dorsolateral prefrontal cortex of nonsmoker patients with schizophrenia. Neurosci Res. 2011;71:289–93.
Chen YKS, Nwulia E, Niwa M, Webster M, Torrey F, Yolken R, et al. Altered MHC class I expression in dorsolateral prefrontal cortex of nonsmoker patients with schizophrenia. Biol Psychiatry. 2011;69:225S.
Sawa AKS, Nwulia E, Niwa M, Cascella NG. Expression of immune molecules (MHC class I and complement C3) in postmortem brains of patients with schizophrenia. Schizophr Bull. 2011;37:195.
Nakatani N, Hattori E, Ohnishi T, Dean B, Iwayama Y, Matsumoto I, et al. Genome-wide expression analysis detects eight genes with robust alterations specific to bipolar I disorder: relevance to neuronal network perturbation. Hum Mol Genet. 2006;15:1949–62.
Rajkowska G, Halaris A, Selemon LD. Reductions in neuronal and glial density characterize the dorsolateral prefrontal cortex in bipolar disorder. Biol Psychiatry. 2001;49:741–52.
Kretzschmar HA, Prusiner SB, Stowring LE, DeArmond SJ. Scrapie prion proteins are synthesized in neurons. Am J Pathol. 1986;122:1–5.
Brown DR, Besinger A, Herms JW, Kretzschmar HA. Microglial expression of the prion protein. Neuroreport. 1998;9:1425–9.
Foster R, Kandanearatchi A, Beasley C, Williams B, Khan N, Fagerhol MK, et al. Calprotectin in microglia from frontal cortex is up-regulated in schizophrenia: evidence for an inflammatory process? Eur J Neurosci. 2006;24:3561–66.
Kaminsky Z, Tochigi M, Jia P, Pal M, Mill J, Kwan A, et al. A multi-tissueanalysis identifies HLA complex group 9 gene methylation differences in bipolar disorder. Mol Psychiatry. 2012;17:728–40.
Dinarello CA. Proinflammatory cytokines. Chest. 2000;118:503–8.
Ortiz-Dominguez A, Hernandez ME, Berlanga C, Gutierrez-Mora D, Moreno J, Heinze G, et al. Immune variations in bipolar disorder: phasic differences. Bipolar Disord. 2007;9:596–602.
Quinones MP, Perez-Algorta G, Jimenez F, Clark K, Bowden CL, Ahuja SS. Upregulation of type i immune responses in the brain of individuals with bipolar disorder: Uncoupling of central and peripheral responses. Biol Psychiatry. 2009;65:64S
Pandey GN. Inflammatory and innate immune markers of neuroprogression in depressed and teenage suicide brain. Mod Trends Pharmacopsychiatry. 2017;31:79–95.
Naude PJ, den Boer JA, Luiten PG, Eisel UL. Tumor necrosis factor receptor cross-talk. FEBS J. 2011;278:888–98.
Clark IA, Vissel B. A neurologist’s guide to TNF biology and to the principles behind the therapeutic removal of excess TNF in disease. Neural Plast. 2015;2015:358263.
Hoseth EZ, Ueland T, Dieset I, Birnbaum R, Shin JH, Kleinman JE, et al. A study of TNF pathway activation in schizophrenia and bipolar disorder in plasma and brain tissue. Schizophr Bull. 2017;43:881–90.
Kim HK, Andreazza AC, Elmi N, Chen W, Young LT. Nod-like receptor pyrin containing 3 (NLRP3) in the post-mortem frontal cortex from patients with bipolar disorder: a potential mediator between mitochondria and immune-activation. J Psychiatr Res. 2016;72:43–50.
Brietzke E, Kauer-Sant’Anna M, Teixeira AL, Kapczinski F. Abnormalities in serum chemokine levels in euthymic patients with bipolar disorder. Brain Behav Immun. 2009;23:1079–82.
Bellani M, Bergami A, Tomelleri L, Perlini C, Cerruti S, Ferro A, et al. Altered mRNA levels of chemokines and cytokines in schizophrenia and bipolar disorder. Schizophr Res. 2010;117:251–2.
Isgren A, Sellgren C, Ekman CJ, Holmen-Larsson J, Blennow K, Zetterberg H, et al. Markers of neuroinflammation and neuronal injury in bipolar disorder: Relation to prospective clinical outcomes. Brain Behav Immun. 2017;65:195–201.
Jakobsson J, Bjerke M, Sahebi S, Isgren A, Ekman CJ, Sellgren C, et al. Monocyte and microglial activation in patients with mood-stabilized bipolar disorder. J Psychiatry Neurosci. 2015;40:250–8.
Barbosa IG, Rocha NP, Bauer ME, de Miranda AS, Huguet RB, Reis HJ, et al. Chemokines in bipolar disorder: trait or state? Eur Arch Psychiatry Clin Neurosci. 2013;263:159–65.
Quinones MP, Jimenez F, Perez-Algorta G, Clark K, Ahuja SS, Bowden CL. Central and peripheral immune activation in bipolar illness: Possible role of the chemokine CXCL5 in disease pathogenesis. Biol Psychiatry. 2009;65:169S.
Webster MJ, Vawter MP, Freed WJ. Immunohistochemical localization of the cell adhesion molecules Thy-1 and L1 in the human prefrontal cortex patients with schizophrenia, bipolar disorder, and depression. Mol Psychiatry. 1999;4:46–52.
van de Stolpe A, van der Saag PT. Intercellular adhesion molecule-1. J Mol Med. 1996;74:13–33.
Thomas AJ, Davis S, Ferrier IN, Kalaria RN, O’Brien JT. Elevation of cell adhesion molecule immunoreactivity in the anterior cingulate cortex in bipolar disorder. Biol Psychiatry. 2004;55:652–5.
Rybakowski JK, Twardowska K. The dexamethasone/corticotropin-releasing hormone test in depression in bipolar and unipolar affective illness. J Psychiatr Res. 1999;33:363–70.
Steiner J, Walter M, Gos T, Guillemin GJ, Bernstein HG, Sarnyai Z, et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J Neuroinflamm. 2011;8:94.
Miller CL, Llenos IC, Dulay JR, Weis S. Upregulation of the initiating step of the kynurenine pathway in postmortem anterior cingulate cortex from individuals with schizophrenia and bipolar disorder. Brain Res. 2006;1073–1074:25–37.
Schain M, Kreisl WC. Neuroinflammation in neurodegenerative disorders—a review. Curr Neurol Neurosci Rep. 2017;17:25.
Palumbo S. Pathogenesis and progression of multiple sclerosis: the role of arachidonic acid-mediated neuroinflammation. In: Zagon IS, McLaughlin PJ, (eds). Multiple sclerosis: perspectives in treatment and pathogenesis. Brisbane (AU): Codon Publications Copyright: The Authors; 2017.
Hu J, Xu J, Pang L, Zhao H, Li F, Deng Y, et al. Systematically characterizing dysfunctional long intergenic non-coding RNAs in multiple brain regions of major psychosis. Oncotarget. 2016;7:71087–98.
Pacifico R, Davis RL. Transcriptome sequencing implicates dorsal striatum-specific gene network, immune response and energy metabolism pathways in bipolar disorder. Mol Psychiatry. 2017;22:441–9.
Abdolmaleky HM, Gower AC, Wong CK, Cox JW, Zhang X, Thiagalingam A, et al. Aberrant transcriptomes and DNA methylomes define pathways that drive pathogenesis and loss of brain laterality/asymmetry in schizophrenia and bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2019;180:138–49.
Bezchlibnyk YB, Wang JF, McQueen GM, Young LT. Gene expression differences in bipolar disorder revealed by cDNA array analysis of post-mortem frontal cortex. J Neurochem. 2001;79:826–34.
Rowland T, Perry BI, Upthegrove R, Barnes N, Chatterjee J, Gallacher D, et al. Neurotrophins, cytokines, oxidative stress mediators and mood state in bipolar disorder: systematic review and meta-analyses. Br J Psychiatry: J Ment Sci. 2018;213:514–25.
Siwek M, Sowa-Kucma M, Styczen K, Misztak P, Nowak RJ, Szewczyk B, et al. Associations of serum cytokine receptor levels with melancholia, staging of illness, depressive and manic phases, and severity of depression in bipolar disorder. Mol Neurobiol. 2017;54:5883–93.
Goldsmith DR, Rapaport MH, Miller BJ. A meta-analysis of blood cytokine network alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder and depression. Mol Psychiatry. 2016;21:1696–709.
Setiawan E, Attwells S, Wilson AA, Mizrahi R, Rusjan PM, Miler L, et al. Association of translocator protein total distribution volume with duration of untreated major depressive disorder: a cross-sectional study. Lancet Psychiatry. 2018;5:339–47.
van der Doef TF, Doorduin J, van Berckel BNM, Cervenka S. Assessing brain immune activation in psychiatric disorders: clinical and preclinical PET imaging studies of the 18-kDa translocator protein. Clin Transl Imaging. 2015;3:449–60.
Owen DR, Yeo AJ, Gunn RN, Song K, Wadsworth G, Lewis A, et al. An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab. 2012;32:1–5.
Borst K, Schwabenland M, Prinz M. Microglia metabolism in health and disease. Neurochem Int. (In Press, 2018). https://doi.org/10.1016/j.neuint.2018.11.006
Banks WA, Kastin AJ, Broadwell RD. Passage of cytokines across the blood–brain barrier. Neuroimmunomodulation. 1995;2:241–8.
Yarlagadda A, Alfson E, Clayton AH. The blood brain barrier and the role of cytokines in neuropsychiatry. Psychiatry. 2009;6:18–22.
Banks WA, Farr SA, Morley JE. Entry of blood-borne cytokines into the central nervous system: effects on cognitive processes. Neuroimmunomodulation. 2002;10:319–27.
Altshuler LL, Abulseoud OA, Foland-Ross L, Bartzokis G, Chang S, Mintz J, et al. Amygdala astrocyte reduction in subjects with major depressive disorder but not bipolar disorder. Bipolar Disord. 2010;12:541–9. https://doi.org/10.1111/j.1399-5618.2010.00838.x
Brauch RA, Adnan El-Masri M, Parker JC Jr., El-Mallakh RS. Glial cell number and neuron/glial cell ratios in postmortem brains of bipolar individuals. J Affective Disord. 2006;91:87–90. https://doi.org/10.1016/j.jad.2005.08.015
Cotter D, Mackay D, Landau S, Kerwin R, Everall I. Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch Gen Psychiatry. 2001;58:545–53.
Cotter D, Mackay D, Frangou S, Hudson L, Landau S. Cell density and cortical thickness in Heschl’s gyrus in schizophrenia, major depression and bipolar disorder. Br J Psychiatry: J Ment Sci. 2004;185:258–9. https://doi.org/10.1192/bjp.185.3.258
Cotter D, Hudson L, Landau S. Evidence for orbitofrontal pathology in bipolar disorder and major depression, but not in schizophrenia. Bipolar Disord. 2005;7:358–69. https://doi.org/10.1111/j.1399-5618.2005.00230.x
Gilmore JH, Bouldin TW. Analysis of ependymal abnormalities in subjects with schizophrenia, bipolar disorder, and depression. Schizophr Res. 2002;57:267–71.
Schmitt A, Steyskal C, Strocka S, Frank F, Wetzestein K, Bernstein HG, et al. Histological studies of oligodendrocytes in psychiatric diseases. Eur Arch Psychiatry Clin Neurosci. 2011;261:S45.
de Baumont A, Maschietto M, Lima L, Carraro DM, Olivieri EH, Fiorini A, et al. Innate immune response is differentially dysregulated between bipolar disease and schizophrenia. Schizophr Res. 2015;161:215–21.
Fillman SG, Sinclair D, Fung SJ, Webster MJ, Weickert CS. Markers of inflammation and stress distinguish subsets of individuals with schizophrenia and bipolar disorder. Transl Psychiatry. 2014;4:e365.
Acknowledgements
The Translational Psychiatry Program (USA) is funded by the Department of Psychiatry and Behavioral Sciences, McGovern Medical School, The University of Texas Health Science Center at Houston (UTHealth). The Laboratory of Neurosciences (Brazil) is one of the centers of the National Institute for Molecular Medicine (INCT-MM) and one of the members of the Centre of Excellence in Applied Neurosciences of Santa Catarina (NENASC). Its research is supported by grants from the National Council for Scientific and Technological Development (CNPq) (JQ and TB), the Foundation for Research and Innovation of the State of Santa Catarina (FAPESC) (JQ and TB), and the InstitutoCérebro e Mente (JQ) and UNESC (JQ and TB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
JQ: Clinical Research Support: Allergan (Clinical Trial), Janssen Pharmaceuticals, Inc. (Clinical Trial). Advisory Boards, Speaker Bureaus, Expert Witness, or Consultant. Assurex Health, Inc. (Speaker Bureau), Daiichi Sankyo (Speaker Bureau), Janssen Pharmaceuticals, Inc. (Speaker Bureau). Patent, Equity, or Royalty: Instituto de Neurociencias. Dr. Joao Quevedo (Stockholder), Other Artmed Editora (Copyright), Artmed Panamericana (Copyright), Elsevier (Copyright). The other authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Giridharan, V.V., Sayana, P., Pinjari, O.F. et al. Postmortem evidence of brain inflammatory markers in bipolar disorder: a systematic review. Mol Psychiatry 25, 94–113 (2020). https://doi.org/10.1038/s41380-019-0448-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-019-0448-7
- Springer Nature Limited
This article is cited by
-
Correlations between multimodal neuroimaging and peripheral inflammation in different subtypes and mood states of bipolar disorder: a systematic review
International Journal of Bipolar Disorders (2024)
-
Neurite outgrowth deficits caused by rare PLXNB1 mutation in pediatric bipolar disorder
Molecular Psychiatry (2023)
-
Brain Inflammatory Marker Abnormalities in Major Psychiatric Diseases: a Systematic Review of Postmortem Brain Studies
Molecular Neurobiology (2023)
-
Cerebrospinal fluid proteomic study of two bipolar disorder cohorts
Molecular Psychiatry (2022)
-
PET/CT imaging of CSF1R in a mouse model of tuberculosis
European Journal of Nuclear Medicine and Molecular Imaging (2022)