
Abstract
Clinical effect of donor-derived natural killer cell infusion (DNKI) after HLA-haploidentical hematopoietic cell transplantation (HCT) was evaluated in high-risk myeloid malignancy in phase 2, randomized trial. Seventy-six evaluable patients (aged 21–70 years) were randomized to receive DNKI (N = 40) or not (N = 36) after haploidentical HCT. For the HCT conditioning, busulfan, fludarabine, and anti-thymocyte globulin were administered. DNKI was given twice 13 and 20 days after HCT. Four patients in the DNKI group failed to receive DNKI. In the remaining 36 patients, median DNKI doses were 1.0 × 108/kg and 1.4 × 108/kg on days 13 and 20, respectively. Intention-to-treat analysis showed a lower disease progression for the DNKI group (30-month cumulative incidence, 35% vs 61%, P = 0.040; subdistribution hazard ratio, 0.50). Furthermore, at 3 months after HCT, the DNKI patients showed a 1.8- and 2.6-fold higher median absolute blood count of NK and T cells, respectively. scRNA-sequencing analysis in seven study patients showed that there was a marked increase in memory-like NK cells in DNKI patients which, in turn, expanded the CD8+ effector-memory T cells. In high-risk myeloid malignancy, DNKI after haploidentical HCT reduced disease progression. This enhanced graft-vs-leukemia effect may be related to the DNKI-induced, post-HCT expansion of NK and T cells. Clinical trial number: NCT02477787.
Similar content being viewed by others


Introduction
Acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) are heterogeneous group of diseases characterized by clonal transformation of myeloid hematopoietic precursors. Although a significant proportion of patients with AML or MDS can be cured of their disease after allogeneic hematopoietic cell transplantation (HCT), progression of the underlying disease remains the most frequent cause of treatment failure. In particular, disease progression rates exceeding 50% were reported in high-risk AML and MDS [1,2,3,4].
Natural killer (NK) cells, a subset of lymphocytes, express CD56 but not CD3 [5,6,7] and exert antitumor cytotoxicity that is modulated both by activating and inhibitory receptors, notably killer immunoglobulin-like receptors (KIR) [8]. Lack of the HLA ligand for donor-inhibitory KIR was found to be correlated with decreased myeloid leukemia progression after HLA-matched sibling [9] and unrelated donor HCT [10]. In murine HCT models, the post-transplant infusion of ligand-mismatched, donor-type NK cells produced an antitumor effect without graft-vs-host disease (GVHD) [11, 12]. Interleukin (IL)-12 and IL-15-preactivated murine NK cells were more effective than non-activated [13] or IL-2-preactivated [14] murine NK cells in terms of tumor suppression. Clinical trials of NK cell transfer from allogeneic donors to patients with malignancies have been performed after HCT [15,16,17,18,19,20,21,22,23,24,25,26] and without prior HCT [27,28,29,30,31,32]. Donor NK cells were obtained either by separation from peripheral blood mononuclear cells (PBMCs) [19, 22, 24, 27, 28, 30] or by ex vivo culture from hematopoietic progenitor cells [15, 18, 20, 21, 23, 25]. Thus far, most donor NK cell infusion (DNKI) studies involved small number of patients (≤30) [17, 19, 22, 23, 25] and limited cell doses (<1 × 108/kg) [17, 19, 22,23,24,25, 29].
Various cytokines including IL-2, IL-4, IL-9, IL-12, IL-15, IL-18, and IL-21 have been used for the ex vivo expansion and activation of NK cells [25, 33, 34]. We have previously generated donor NK cells from the CD3+ cell-depleted portion of a mobilized leukapheresis product via culturing in media containing IL-15 and IL-21 [20, 21]. The median yield from a single-day leukapheresis was 2.0 × 108 donor NK cells/kg of the patient’s body weight. These cells expressed activating receptors strongly, produced interferon-γ, and showed strong cell cytotoxicity in vitro. When the divided doses of these NK cells were administered to the patients at 2 and 3 weeks after HLA-haploidentical HCT [21], there was a significant reduction of disease progression in patients with refractory acute leukemia compared to historical patients who underwent haploidentical HCT with the same conditioning regimen, but without DNKI (cumulative incidence; study cohort, 46% vs historical cohort, 74%; P = 0.038). No significant differences were observed between our study cases and historical patients in terms of engraftment, acute and chronic GVHD, or non-relapse mortality (NRM).
We have thus here investigated the clinical effects of DNKI administered after haploidentical HCT in patients with high-risk AML and MDS in a randomized phase 2 trial. The primary endpoint was progression of the underlying disease. In addition, patient immune parameters were investigated after HCT to detect any DNKI effects.
Methods
Patient enrollment
This was a phase 2, prospective, controlled, randomized, and investigator-sponsored trial approved by the Institutional Review Board of Asan Medical Center, Seoul, Korea and the Korean Ministry of Food and Drug Safety (KMFDS), and registered at ClinicalTrial.gov (#NCT02477787). The hypothesis was that DNKI given after haploidentical HCT was associated with decreased AML/MDS progression. The initial target accrual was 90 for a statistical power of 90% with a two-sided type I error rate of 0.05 for the detection of decreased disease progression from 60% to 30%. Patient enrollment began in June 2015. By July 2018, however, there were an excess number of patients experiencing disease progression in the no-DNKI group. Hence, after review by the institutional Data Safety Monitoring Board and KMFDS, it was recommended that we close the trial early. When the study was closed in October 2018, a total of 77 patients had been enrolled. All patients and donors provided written informed consent.
Enrollment criteria
The criteria for patient enrollment are described in the Supplementary Appendix.
Randomization
The treatment schema for the study is depicted in Fig. 1A. Patient and donor HLA–A, –B, –C, and –DRB1 typing and donor KIR typing were performed using polymerase chain reaction-based methods. Eligible patients were randomly assigned either to a DNKI or no-DNKI group on the day before the initiation of conditioning therapy. Patients were stratified according to their diagnosis (refractory AML vs AML in complete remission (CR) vs MDS). Patients with refractory AML were further stratified according to primary-refractory AML vs relapse-refractory/early relapse AML, as well as according to peripheral blood blasts <5% vs ≥5%. Patients with AML in CR were further stratified to first vs second CR and MDS patients were further stratified to intermediate-2 vs high risk [35].

A Eligible patients were assigned randomly to a donor natural killer cell infusion (DNKI) group or a no-DNKI group on day–8 of hematopoietic cell transplantation (HCT), day 0 being the first day of HCT. Between days –7 and –1, the patients received conditioning therapy for HCT with busulfan, fludarabine, and anti-thymocyte globulin. Hematopoietic cell donors received a daily dose of granulocyte colony-stimulating factor (G-CSF) at 10 μg/kg subcutaneously for 4 days beginning on day–3 and underwent large-volume leukapheresis (Amicus, Fenwal, Inc., Lake Zurich, IL) on day 0 with a goal of collecting at least 5 × 106/kg CD34+ cells. About 20% of the donors required an extra day of G-CSF administration and leukapheresis. These cells were administered to patients through a central venous catheter on the same day for the purposes of HCT. The donors for the patients who were randomized to the DNKI group received an extra day of G-CSF administration and underwent extra leukapheresis for NK cell production. B A total of 77 patients were enrolled. One patient assigned to the no-DNKI group withdrew from the study. The remaining 76 patients were included in the intention-to-treat analysis. Four patients who had been randomized to the DNKI group did not eventually receive this therapy for the reasons listed.
Haploidentical HCT procedure
The haploidentical HCT methodology and supportive care for HCT are described in the Supplementary Appendix.
DNKI
Patients in the DNKI group received NK cells on days 13 (DNKI-1) and 20 (DNKI-2) after HCT. A delay of DNKI for up to 2 days was allowed, depending on the patient condition, delayed NK cell production, or holidays. The DNKI cell dose was based on the quantity of cells available. For DNKI-1, 1 × 108 donor NK cells/kg or about half of cell culture products were administered. For DNKI-2, the remaining cell culture products were administered. Donor NK cells were infused over 1 hour through central venous catheter, with pheniramine 45.5 mg administered by intravenous infusion 30 min earlier. Since we administered anti-thymocyte globulin (ATG) as a part of conditioning regimen for HCT, we measured the ATG levels in the patients’ serum on the day of DNKI [36].
Donor NK cell generation
The methods for donor NK cell generation are described in the Supplementary Appendix.
Patient monitoring and evaluation
The patient monitoring and evaluation methods are described in the Supplementary Appendix.
Post-HCT immune parameter monitoring and single-cell RNA sequencing
To investigate DNKI effect on immune reconstitution, the immunologic status of the patients was monitored by measuring peripheral blood lymphocyte subsets, cytokine levels, and cell cytotoxicity before conditioning and 1, 3, 6, and 12 months after HCT. After observing significantly better expansion of NK and T cells at 3 months after HCT in patients who received DNKI, we performed single-cell RNA sequencing (scRNA-seq) in the PBMCs in seven patients (four DNKI and three in no-DNKI patients) to better delineate the expanded lymphocyte populations. The methods for these analyses are described in the Supplementary Appendix.
Statistical analysis
Statistical methodology used in the study is described in the Supplementary Appendix.
Results
Patients and donors
Of the 77 enrolled patients, one in the no-DNKI group withdrew from the study after randomization (Fig. 1B). The remaining 76 patients were included in the intention-to-treat analysis and their characteristics are summarized in Table 1 according to the treatment group. More female patients were assigned to the DNKI group (53% vs 31%, P = 0.065). Otherwise, the two groups were well-balanced. Two-thirds of the study patients had refractory AML.
Donor NK cell infusion
Among the 40 patients in the DNKI group, 4 did not receive DNKI, due to cell production failure, early death, grade-4 acute GVHD, and subdural hemorrhage in one case each (Fig. 1B). Additionally, four patients received only one dose of DNKI due to production failure (N = 2), early death (N = 1), and engraftment syndrome (N = 1). The characteristics of the donor NK cells infused on day 13 (DNKI-1, N = 35) and day 20 (DNKI-2, N = 33) are summarized in Table 2. The median cell dose was 1.0 (range, 0.5–1.0) × 108/kg (of patient body weight) for DNKI-1 and 1.4 (range, 0.5–4.0)×108/kg for DNKI-2. Immunophenotype analyses showed a median percentage of CD56+ cells of 84% for DNKI-1 and 89% for DNKI-2. On the other hand, the median percentage of CD3+ cell dose was low (<1% for both DNKI-1 and DNKI-2). All patients tolerated DNKI well without acute infusion-related toxicity. The median serum ATG levels were 0.99 μg/mL (range, 0.45–5.17) and 0.88 μg/mL (range, 0.24–1.62) on DNKI-1 and DNKI-2 days, respectively.
CR, disease progression, NRM, and patient survival
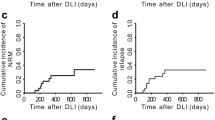
Of the 57 patients with active disease (refractory AML or MDS) at the time of HCT, 23/30 patients (77%) in the DNKI group and 14/27 (52%) in the no-DNKI group achieved complete remission (CR; P = 0.049). As of September 2020, among all 76 study patients, 15/40 patients in the DNKI group and 21/36 in the no-DNKI group experienced disease progression (30-month cumulative incidences, 35% (95% confidence interval (CI), 21–50%) and 61% (95% CI, 42–76%), respectively; P = 0.040; Table 3; Fig. 2A). One patient in the DNKI group experienced primary engraftment failure (cumulative incidence, 2.5%). Twelve/40 patients in the DNKI group and 10/36 no-DNKI subjects died without disease progression (30-month cumulative incidence of NRM, 30% and 28%, respectively; Fig. 2B). The causes of NRM are listed in Table 3.

A. B. E–H The cumulative incidences of disease progression, non-relapse mortality, neutrophil engraftment, platelet recovery, grade 2–4 acute graft-vs-host disease (GVHD), and moderate-to-severe chronic GVHD, respectively. C, D Kaplan-Meier plots of progression-free survival and overall survival, respectively. I The cumulative incidences of cytomegalovirus (CMV) reactivation, determined by the detection of either CMV pp65 antigenemia or CMV DNA copies >103/mL. J The cumulative incidences of Epstein-Barr virus (EBV) reactivation by detection of EBV DNA copies >103/mL.
To identify patients who benefitted from DNKI, we performed subgroup analyses for disease progression. The DNKI group showed a significantly reduced rate of disease progression in patients with primary-refractory AML (subdistribution hazard ratio (sHR), 0.29; 95% CI, 0.10–0.81; Fig. 3A), in patients with refractory AML with peripheral blood blast <5% (sHR, 0.29; 95% CI, 0.10–0.81), as well as in patients with AML with normal/intermediate-risk cytogenetics (sHR, 0.09; 95% CI, 0.01–0.73).

A A forest plot of the subdistribution hazard ratios (sHR) for disease progression according to the prognostic subgroups of patients in the intention-to-treat populations. sHR (donor natural killer cell infusion (DNKI) group vs no-DNKI group) was estimated using Fine-Gray proportional hazard regression model according to patient age, gender, acute myeloid leukemia (AML) disease status, peripheral blood blast percentage in refractory AML, and AML cytogenetic risk. B, C As a part of immune parameter monitoring conducted in this study, lymphocyte subsets were counted in the peripheral blood of the patients. The median counts obtained before the initiation of conditioning therapy, and at 1 and 3 months after hematopoietic cell transplantation (HCT) were plotted. B The NK cell population as measured by CD3 ̶ /CD56+ showed cell count recovery to pre-HCT level at 1 month after HCT, when the median cell counts were similar between the DNKI and no-DNKI groups of patients (10,300/mL vs 10,500/mL). At 3 months after HCT, however, the median NK cell count was significantly higher in the DNKI group of patients (37,200/mL vs 20,550/mL, P = 0.032). All NK cell subsets were more numerous in the DNKI group with statistically significant differences in the NKG2D+, NKp46+, and CD158b+ cell numbers (see also Supplementary Table S1). C Unlike NK cells, the T and B cell count recovery was delayed until 3 months after HCT. At that time, the CD3+ T cell count was significantly higher in the DNKI group than in the no-DNKI group (90,300/mL vs 34,550/mL, P = 0.031). The higher T cell count in the DNKI group of patients was mainly due to increased CD8+, CD45RA+, and CD45RO+ cells. On the other hand, the CD4+ T cell and CD19+ B cell counts were similar between the two groups (see also Supplementary Table S2). sHR, subdistribution hazard ratio. *P < 0.05 by Mann-Whitney test. Error bars represent upper quartile range.
For the entire cohort, the median follow up time among the survivors is 33.7 months (range, 23.2–60.7 months). Progression-free survival (PFS) at 30 months was 33% and 11% (P = 0.085; Table 3; Fig. 2C) for the DNKI and no-DNKI groups, respectively. The 30-month overall survival was 35% and 19% (P = 0.456; Fig. 2D) for the DNKI and no-DNKI groups, respectively.
Engraftment, GVHD, and infections
Patients in both treatment groups showed consistent neutrophil engraftment with 37/40 patients in the DNKI group and 33/36 in the no-DNKI group achieving initial absolute neutrophil count (ANC) ≥ 500/μL at a median of 10 days after HCT (Table 3; Fig. 2E). Twenty-eight patients in the DNKI group and 22 in the no-DNKI group achieved platelet counts ≥20,000/μL at a median 15 and 13 days after HCT, respectively (Fig. 2F). At 1 month after HCT, among the cases who showed bone marrow recovery without leukemia, 32/34 patients in the DNKI group and 26/28 in the no-DNKI group showed >95% donor hematopoietic chimerism (P = 0.599).
There were no significant differences in the occurrence of grades 2–4 acute GVHD (cumulative incidences, 51% for DNKI and 47% for no-DNKI groups; Table 3; Fig. 2G), nor of grade 3 or 4 acute GVHD. There was also no significant difference in the occurrence of moderate-to-severe chronic GVHD (cumulative incidences, 20% for DNKI and 25% for no-DNKI groups; Fig. 2H) nor of severe chronic GVHD. Additionally, there were no significant differences in the cumulative incidence of either cytomegalovirus infection or Epstein-Barr virus reactivation between the treatment groups (Table 3; Fig. 2I and J). Other observed infections are listed in Table 3.
FACS analyses for immune reconstitution
At 3 months after HCT, the median peripheral blood absolute NK cell count was 1.8-fold higher in the DNKI group (37,200/mL vs 20,550/mL, P = 0.032; Fig. 3B; Supplementary Table S1). Increased cell counts were observed across all NK cell subsets. In addition, the median absolute T cell count was 2.6-fold higher in the DNKI group at 3 months (90,300/mL vs 34,550/mL, P = 0.031; Fig. 3C; Supplementary Table S2). Increases in the T cell population were mainly due to increased CD8+ cells (34,305/mL vs 12,183/mL), CD45RA+ cells (28,063/mL vs 7626/mL), and CD45RO+ cells (18,278/mL vs 8630/mL). The serum TNF-α level was higher in the DNKI group at 3 months (Supplementary Table S3). Although patients in DNKI group also tended to show higher cell cytotoxicity, the overall cytotoxicity was low (Supplementary Table S4).
Single-cell RNA-sequencing
For single-cell RNA-sequencing (scRNA-seq), stored PBMCs from seven patients (four DNKI and three no-DNKI patients) collected at three time points (before the conditioning therapy, and 1 and 3 months after HCT) were used (Supplementary Tables S5 and S6; Fig. 4A B; Supplementary Fig. S1). A total of 79,086 cell data were integrated. Subsequently, using SCTransform [37], eight major cell types were identified; B cells, CD4+ T cells, CD8+ T cells, dendritic cells, monocytes, NK cells, other T cells, and others (Fig. 4C; Supplementary Fig. S2). The cell types were validated by calculating cell type-specific signature scores and comparing them via unsupervised clustering results (Fig. 4D; Supplementary Fig. S2). A high internal correlation within each cell type and low correlation with other cell groups indicated that the cell types were annotated accurately. Furthermore, marker genes showed high expression in the corresponding cell types (Fig. 4E). When the NK cell proportions were measured according to DNKI vs no-DNKI patients, as well as according to the timing of the treatment, the NK cell proportion was significantly higher in DNKI patients at 3 months after HCT (Fig. 4F).

A Single-cell RNA-sequencing (scRNA-seq) was performed using the stored PBMC samples from seven patients with acute myeloid leukemia [AML; four DNKI patients (numbers 2, 24, 29, and 45) and three no-DNKI patients (numbers 17, 19, 23)]. All seven patients survived for at least 4 months after HCT without AML progression. The samples were collected at 3 time points, i.e., before the conditioning therapy, and at 1 and 3 months after HCT. A total of 87,584 single-cell data were produced from the 21 thawed samples. Of those, 79,086 cell data passed the computational quality check (unique feature counts >200 and <7000; and mitochondrial read per cell <15% (Supplementary Fig. S1). Data from a total of 79,086 cells were obtained using a 10X genomics platform. B The bar plot shows the numbers of single cells, as well as the median numbers of genes obtained per cell, from each study patients. C Uniform Manifold Approximation and Projection (UMAP) plotting identified eight major cell types, i.e., B cells (n = 2575), CD4+ T cells (n = 13,618), CD8+ T cells (n = 28,047), dendritic cells (n = 1285), monocytes (n = 12,291), NK cells (n = 13,508), other T cells (n = 6038), and the others (n = 1724). In addition, 31 sub-cluster cell types were discerned. D Heatmap of the scaled average expression of marker genes in each major cell types. The marker genes for each cell type were expressed exclusively in their annotated cluster. E Evaluation of cell type marker gene expression via UMAP embedding, i.e., CD3E in T cells, KLRF1 in NK cells, CD14 in monocytes, MS4A1 in B cells, and CD1C in dendritic cells. F Proportion of NK cells according to the DNKI vs no-DNKI patients, as well as according to the timing after HCT. The proportion of NK cells was higher in the DNKI patients at 3 months after HCT. These findings corroborated our FACS data which showed higher 3-month NK cell contents in the DNKI group of patients. TCM, central memory T cell; TEM, effector memory T cell; CTL, cytotoxic T lymphocyte; Treg, regulatory T cell; dnT, double negative T cell; MAIT, mucosal-associated invariant T cell; gdT = γδ-T cell; ASDC, AXL+, and SIGLEC6+ dendritic cell; pDC, plasmacytoid dendritic cell; cDC1, conventional dendritic cell 1; cDC2, conventional dendritic cell 2; HSPC, hematopoietic stem progenitor cell; ILC, innate lymphoid cell; ns, non-significant. *P < 0.05; Error bars represent standard error of the mean.
DNKI expands memory-like and CD56bright NK cells
Via unsupervised clustering analysis we identified various differentiation states of NK cells, including memory-like, CD56bright, transient, terminally-differentiated, exhausted, and proliferating NK cells (Fig. 5A). Among these subtypes, memory-like NK cells showed exclusive expression of CCL3, CCL4, GZMA/B, and IFNG (Fig. 5B; Supplementary Fig. S3). In addition, gene ontology (GO) analysis revealed that memory-like NK cells responded to interferon-γ and had higher cytotoxic activity (Fig. 5C; Supplementary Fig. S4). CD56bright NK cells showed increased expression of SELL, IL7R, and XCL1/2 (Fig. 5B) and GO analysis indicated a tendency for cytokine and chemokine secretion. In addition, exhausted NK cells highly expressed LAG3 and ZEB2 and showed a tendency for low cytotoxicity and cell cycle arrest [38].

The memory-like NK cell expansion induced by DNKI is under the regulation of PRDM1 and TBX21. A To examine the DNKI effects on NK cell subtypes after haploidentical HCT, we performed clustering analysis of a UMAP plot which identified six NK cell subtypes, i.e., memory-like, CD56bright, transient, terminally-differentiated, exhausted, and proliferating NK cells. B Dot plot depicting the percentage of cells expressing a particular gene (size) and the average expression of that gene (color) according to the NK cell subtype. C Radar chart showing the significance scores (−log (P value)) from gene set enrichment anlaysis of eight selected gene sets and three NK cell subtypes (memory-like, CD56bright, and exhausted). D The mean normalized ratios of numbers of NK cell subtypes at 1 and 3 months after HCT over the baseline (before the conditioning). The DNKI cases showed increased ratios of memory-like and CD56bright NK cells and decreased ratio of exhaused NK cells, while no-DNKI cases showed low ratios of memory-like and CD56bright NK cells and a high ratio of exhausted NK cells. E Cell–cell interaction analysis performed with NicheNet showed an increased interaction between memory-like NK cells and CD8+ effector-memory T cells (CD8+ TEM) via IFNG and ITGB2. F Schematic representation of CD8+ TEM expansion after the DNKI therapy. DNKI augmented the number of memory-like NK cells, which then released interferon-γ and ITGB2 to clonally expand and activate CD8+ T cells into CD8+ TEM cells. G Venn diagram showing that PRDM1 and TBX21, master TFs for DNKI-specific NK cells, as well as memory-like NK cells (left panel). The violin plots show the transcriptional activity of PRDM1 and TBX21 in DNKI patients vs no-DNKI patients. The transcriptional activity of both TFs was markedly higher in the DNKI patients. H Visual representation of the reconstructed gene regulatory network of NK cells. The nodes in the network represent the two master regulator candidates (squares) and the corresponding target genes (circles). The bar plot shows the biological phenotypes of regulon regulated by PRDM1 and TBX21. TEM, effector memory T cell; n.s., non-significant. *P < 0.05; **P < 0.001; Error bars represent standard error of the mean.
In the DNKI group of patients, there was a marked increase in the population of memory-like NK cells from the baseline at 3 months after HCT (34-fold increase; Fig. 5D). Likewise, CD56bright NK cells increased as well but to a lesser degree (4-fold increases at 3 months). In the no-DNKI patients, however, there was no significant increase in the aforementioned NK cell subtypes. In contrast to memory-like and CD56bright NK cells, the exhausted NK cells decreased after HCT in the DNKI patients. Among no-DNKI patients, however, these exhausted NK cells persisted and were significantly more numerous than in DNKI patients until 3 months after HCT.
Memory-like NK cells induce the clonal expansion of CD8+ effector-memory T cells
To determine the DNKI effect on the proliferation and differentiation of CD8+ T cells, we inferred the clonality of CD8+ T cells using TRUST4 [39], which extracts T cell receptor (TCR) reads from BAM files and assembles these reads against human TCR contigs. Based on the re-aligned TCR sequences from scRNA-seq of CD8+ T cells, we annotated TCR repertories and inferred T cell clones (Supplementary Fig. S5A). By comparing the clonality of expanded CD8+ T cells, we determined that these clonally expanded T cells had the characteristics of CD8+ effector-memory T (TEM) cells (Supplementary Fig. S5B, C). Thereafter, we compared the frequency of clonally expanded CD8+ T cells in the PBMCs of the patients. The DNKI patients showed a significantly higher frequency of clonally expanded CD8+ T cells when compared to no-DNKI patients (Supplementary Fig. S5D).
Using NicheNet, which can infer intercellular networks by combining known cell-cell interaction and single-cell transcriptome data [40], we next evaluated the interaction between NK cell subsets and CD8+ TEM cells. In the inferred NK cell-T cell intercellular network, there was an increased number of links between memory-like NK and CD8+ TEM cells (Fig. 5E). Memory-like NK cells prominently expressed NicheNet-predicted ligands, such as IFNG, CALR, ANXA1, GZMB, and ITGB2. Among these, IFNG and ITGB2 were the expected ligands that potentially bind to IFNGR1 and CD226 proteins, both of which were abundantly expressed in CD8+ TEM cells (Fig. 5F) [41].
PRDM1 and TBX21 are dominant transcription factors that induce memory-like NK cells after haploidentical HCT and DNKI
To better understand the molecular mechanisms of memory-like NK cell expansion in AML patients undergoing haploidentical HCT and DNKI, we reconstructed gene regulatory network (GRN) from NK cell scRNA-seq data to infer transcription factor (TF) activity [42]. By comparing the TF activity of NK cells, we identified 12 TFs which showed statistically significant difference between DNKI and no-DNKI patients. These were considered as master TF candidates (Supplementary Fig. S6). Furthermore, we reconstructed the differentiation trajectory of NK cells into memory-like NK cells using Monocle3 [43], and then selected 9 TFs with a correlation with this differentiation trajectory (rho > 0.3; Supplementary Fig. S7). PRDM1 and TBX21 met the criteria of the above two selection processes (Fig. 5G). For validation, we performed GO analysis on the 84 regulons of PRDM1 and TBX21 and they were significantly related to NK cell activation, cytotoxicity, and inflammation (Fig. 5H) [44, 45].
Discussion
Thus far, DNKI trials have been hampered by limited cell doses and limited efficacy. Our donor NK cell production method involved ex vivo culture in the presence of IL-15 and IL-21 for two to three weeks. The starting cell population was a CD3-depleted portion of mononuclear cells collected from the donors after mobilization. From a single-day leukapheresis product, a median 2.0 × 108 donor NK cells/kg was generated [20, 21]. This methodology produced a much higher number of NK cells than the separation methods (CD3-depletion/CD56-selection) from mononuclear cells collected at a steady state, which yields donor NK cells in the range of 1 × 107 cells/kg (Supplementary Table S7) [17, 19, 22, 24]. In terms of the quality of the donor NK cells, cell viability was 70– 97% with a purity of 71– 97% with respect to CD56+. The median CD3+ rate was low at 0.5% (Table 2). These cells expressed known inhibitory and activating NK cell receptors, produced interferon-γ, and showed a strong in vitro cell cytotoxicity. It was shown previously that the in vitro stimulation of NK cells with cytokines, such as IL-12, IL-15, and IL-18, can generate memory-like NK cells [29, 46, 47], whose characteristics include high expression of activating receptors, strong antitumor cytotoxicity, and enhanced recall response to subsequent cytokine stimulation.
Unlike our approach, NK cells can be expanded using feeder cells [15, 26, 31, 32]. In a recent study [15], donor NK cells were expanded from 500 mL of peripheral blood using IL-21/4-1BBL-expressing K562 feeder cells. Eleven of the 12 patients with myeloid malignancy in an extension phase cohort were able to receive intended high doses of 1.0 × 108 donor NK cells/kg three times (HCT days of 2, 7, and 28). When all 24 evaluated patients in the study were compared to a contemporaneous registry cohort of patients, there was a significant reduction of disease recurrence (4% vs 38% at 24 months, P = 0.014), as well as a non-significant improvement in disease-free survival (66% vs 44%; P = 0.1).
To our knowledge, our current trial represents the first randomized study of the clinical effects of post-transplant DNKI. Our analyses showed that DNKI after haploidentical HCT in patients with high-risk AML and MDS results in a significant reduction in disease progression (35% vs 61% at 30 months; sHR, 0.50). Among the 57 patients with active disease of AML or MDS, the CR rate was significantly higher in the DNKI group than in the no-DNKI group (77% vs 52%). Other HCT outcomes, i.e., engraftment, acute and chronic GVHD, and, importantly, NRM were similar between the two groups. These data suggested that DNKI given after haploidentical HCT enhanced the graft-vs-leukemia effect without compromising GVHD. The net effect of decreased disease progression and a similar NRM resulted in an improved PFS in the DNKI group, although this difference was not statistically significant (33% vs 11% at 30 months; P = 0.085). These findings corroborated the results of earlier studies that investigated DNKI effects through comparison with registry [15] or historical cohort [21]. Subgroup analysis in our limited number of patients showed that the anti-leukemia effect of DNKI was more pronounced in patients with primary-refractory AML, with a peripheral blood blast <5%, and with normal/intermediate-risk cytogenetic changes.
In our previous DNKI study [21], we administered high-dose DNKI (median, 1.0–2.0 × 108/kg cells) and measured the NK cell contents in the peripheral blood of nine patients by FACS analysis. There was no increase in NK cell count immediately after DNKI (1, 3, and 7 days later). It was speculated that the DNKI doses we gave may be still too low to be detected in the peripheral blood. Also, a rapid extravascular redistribution of donor NK cells might have occurred. In our current study, consistent with above findings, there was no significant difference in NK cell contents between DNKI vs no-DNKI groups at 1 month after HCT (about 10 days after the DNKI-2). A 3 months after HCT, however, peripheral blood NK cell contents were significantly higher for DNKI vs no-DNKI patients (median counts, 1.8-fold higher). These findings suggested that a significant NK cell proliferation occurred in the DNKI patients between 1 and 3 months after HCT. To further characterize the expanded NK cell population, we performed scRNA-seq using the stored PBMC samples from seven of our AML patients (4 DNKI and 3 no-DNKI cases). Uniform Manifold Approximation and Projection (UMAP) plotting and clustering analysis revealed that memory-like NK cells and CD56bright NK cells represented the main population of NK cells after HCT in the DNKI patients. These cells markedly increased from baseline (i.e., prior to HCT) in the DNKI patients but not in the no-DNKI patients (Fig. 5D). Exhausted NK cells, on the other hand, showed a contrary trend with a decrement in the DNKI patients and an increment in no-DNKI patients. Similar findings of NK cell expansion after DNKI from the same HCT donor have been observed in other studies [15, 16]. Further GRN and pseudotime analyses showed that PRDM1 and TBX21, known regulators in diverse immune cell development [44, 45], were the two important TFs generating memory-like NK cells in the DNKI patients.
The DNKI patients in our study also showed better expanded CD8+ T cells by FACS analysis when compared to no-DNKI subjects (median counts, 2.6-fold higher). Analysis of the clonality of TCR reads from scRNA-seq using TRUST4 [39] revealed that the main cell type of CD8+ T cells in our DNKI patients was CD8+ TEM, a subset of CD8+ memory T cells with a strong cytotoxic function [48]. Analyses using NicheNet documented a strong intercellular network between memory-like NK cells and CD8+ TEM cells mediated by interferon-γ and ITGB2. Hence, DNKI after haploidentical HCT facilitated NK cell expansion, with the dominant subsets being memory-like NK cells. These expanded memory-like NK cells, in turn, may have activated and expanded CD8+ TEM cells (Fig. 5F).
Although our finding of decreased disease progression after DNKI after haploidentical HCT is encouraging, our study is limited by small patient numbers. Furthermore, we used non-intensive preparatory regimen along with ATG/cyclosporine for GVHD prophylaxis. Therefore, our results need to be confirmed by further studies involving larger numbers of patients, using different haploidentical HCT methodologies, such as dose-intensive conditioning therapy and post-transplantation cyclophosphamide for GVHD prophylaxis [49]. In addition, the NK/CD8+ T cell expansion after DNKI needs to be confirmed and their dynamics to be better defined by more frequent lymphocyte subset measurement.
NK alloreactivity between the donor and patient [12, 50], as well as donor KIR haplotype [51] showed differential antileukemia effects after haploidentical HCT. In our study, NK alloreactivity determined by ligand-ligand and receptor-ligand models, as well as donor KIR haplotype were well-balanced between the DNKI and no DNKI patients (Table 1). There was no significant difference in disease progression according to these donor NK-related variables in our study patient cohort (data not shown). However, these variables need to be investigated further for their role in the DNKI treatment.
In vitro NK cell cytotoxicity assay performed using PBMCs collected from the patients showed no significant difference between the DNKI and no-DNKI patients (Supplementary Table S4). This may be due to different mechanisms of in vivo vs in vitro cytotoxicity. Or, the sensitivity of in vitro assay may be too low to detect the difference. Clinical utility of such assays in DNKI therapy remains to be investigated.
ATG given as a part of conditioning regimen is known to have a long serum half-life [36]. ATG was detected in varying levels at the time of DNKI in our study patients. ATG contains diverse antibodies against various immune cells including NK cells [52]. Although ATG given as a part of conditioning was shown to delay T and B cell reconstitution, NK cell recovery was not affected [53]. The significance of ATG affecting DNKI remains to be determined.
In conclusion, ex vivo culture of mobilized, CD3-depleted donor mononuclear cells using IL-15 and IL-21 produced a large number of activated NK cells. When these donor NK cells were administered 2 to 3 weeks after haploidentical HCT to patients with high-risk AML and MDS, disease progression was reduced without increased GVHD or NRM. Furthermore, this treatment seemed to facilitate immune reconstitution with higher blood contents of NK and CD8+ T cells at 3 months after HCT. The main populations of these cells were memory-like NK and CD8+ TEM cells.
Data availability
For the patient-level data generated in the study, please contact Dr. Kyoo-Hyung Lee at haploset@gmail.com.
References
Brissot E, Labopin M, Ehninger G, Stelljes M, Brecht A, Ganser A, et al. Haploidentical versus unrelated allogeneic stem cell transplantation for relapsed/refractory acute myeloid leukemia: a report on 1578 patients from the Acute Leukemia Working Party of the EBMT. Haematologica. 2019;104:524–32.
Deeg HJ, Scott BL, Fang M, Shulman HM, Gyurkocza B, Myerson D, et al. Five-group cytogenetic risk classification, monosomal karyotype, and outcome after hematopoietic cell transplantation for MDS or acute leukemia evolving from MDS. Blood. 2012;120:1398–408.
Lee KH, Lee JH, Kim DY, Seol M, Lee YS, Kang YA, et al. Reduced-intensity conditioning therapy with busulfan, fludarabine, and antithymocyte globulin for HLA-haploidentical hematopoietic cell transplantation in acute leukemia and myelodysplastic syndrome. Blood. 2011;118:2609–17.
Fuchs EJ, O’Donnell PV, Eapen M, Logan B, Antin JH, Dawson P, et al. Double unrelated umbilical cord blood vs HLA-haploidentical bone marrow transplantation: the BMT CTN 1101 trial. Blood. 2021;137:420–8.
Herberman RB, Nunn ME, Holden HT, Lavrin DH. Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells. Int J Cancer. 1975;16:230–9.
Kiessling R, Klein E, Pross H, Wigzell H. “Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell. Eur J Immunol. 1975;5:117–21.
Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:503–10.
Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. 2018;18:671–88.
Hsu KC, Keever-Taylor CA, Wilton A, Pinto C, Heller G, Arkun K, et al. Improved outcome in HLA-identical sibling hematopoietic stem-cell transplantation for acute myelogenous leukemia predicted by KIR and HLA genotypes. Blood. 2005;105:4878–84.
Miller JS, Cooley S, Parham P, Farag SS, Verneris MR, McQueen KL, et al. Missing KIR ligands are associated with less relapse and increased graft-versus-host disease (GVHD) following unrelated donor allogeneic HCT. Blood. 2007;109:5058–61.
Lundqvist A, Philip McCoy J, Samsel L, Childs R. Reduction of GVHD and enhanced antitumor effects after adoptive infusion of alloreactive Ly49-mismatched NK cells from MHC-matched donors. Blood. 2007;109:3603–6.
Ruggeri L, Capanni M, Urbani E, Perruccio K, Shlomchik WD, Tosti A, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295:2097–100.
Song Y, Hu B, Liu Y, Jin Z, Zhang Y, Lin D, et al. IL-12/IL-18-preactivated donor NK cells enhance GVL effects and mitigate GvHD after allogeneic hematopoietic stem cell transplantation. Eur J Immunol. 2018;48:670–82.
Huber CM, Doisne JM, Colucci F. IL-12/15/18-preactivated NK cells suppress GvHD in a mouse model of mismatched hematopoietic cell transplantation. Eur J Immunol. 2015;45:1727–35.
Ciurea SO, Kongtim P, Soebbing D, Trikha P, Behbehani G, Rondon G, et al. Decrease post-transplant relapse using donor-derived expanded NK-cells. Leukemia. 2022;36:155–64.
Berrien-Elliott MM, Foltz JA, Russler-Germain DA, Neal CC, Tran J, Gang M, et al. Hematopoietic cell transplantation donor-derived memory-like NK cells functionally persist after transfer into patients with leukemia. Sci Transl Med. 2022;14:eabm1375.
Devillier R, Calmels B, Guia S, Taha M, Fauriat C, Mfarrej B, et al. Phase I trial of prophylactic donor-derived IL-2-activated NK cell infusion after allogeneic hematopoietic stem cell transplantation from a matched sibling donor. Cancers. 2021;13:2673.
Ciurea SO, Schafer JR, Bassett R, Denman CJ, Cao K, Willis D, et al. Phase 1 clinical trial using mbIL21 ex vivo-expanded donor-derived NK cells after haploidentical transplantation. Blood. 2017;130:1857–68.
Jaiswal SR, Zaman S, Nedunchezhian M, Chakrabarti A, Bhakuni P, Ahmed M, et al. CD56-enriched donor cell infusion after post-transplantation cyclophosphamide for haploidentical transplantation of advanced myeloid malignancies is associated with prompt reconstitution of mature natural killer cells and regulatory T cells with reduced incidence of acute graft versus host disease: a pilot study. Cytotherapy. 2017;19:531–42.
Choi I, Yoon SR, Park SY, Kim H, Jung SJ, Kang YL, et al. Donor-derived natural killer cell infusion after human leukocyte antigen-haploidentical hematopoietic cell transplantation in patients with refractory acute leukemia. Biol Blood Marrow Transpl. 2016;22:2065–76.
Choi I, Yoon SR, Park SY, Kim H, Jung SJ, Jang YJ, et al. Donor-derived natural killer cells infused after human leukocyte antigen-haploidentical hematopoietic cell transplantation: a dose-escalation study. Biol Blood Marrow Transpl. 2014;20:696–704.
Stern M, Passweg JR, Meyer-Monard S, Esser R, Tonn T, Soerensen J, et al. Pre-emptive immunotherapy with purified natural killer cells after haploidentical SCT: a prospective phase II study in two centers. Bone Marrow Transpl. 2013;48:433–8.
Brehm C, Huenecke S, Quaiser A, Esser R, Bremm M, Kloess S, et al. IL-2 stimulated but not unstimulated NK cells induce selective disappearance of peripheral blood cells: concomitant results to a phase I/II study. PLoS One. 2011;6:e27351.
Rizzieri DA, Storms R, Chen DF, Long G, Yang Y, Nikcevich DA, et al. Natural killer cell-enriched donor lymphocyte infusions from A 3-6/6 HLA matched family member following nonmyeloablative allogeneic stem cell transplantation. Biol Blood Marrow Transpl. 2010;16:1107–14.
Yoon SR, Lee YS, Yang SH, Ahn KH, Lee JH, Lee JH, et al. Generation of donor natural killer cells from CD34(+) progenitor cells and subsequent infusion after HLA-mismatched allogeneic hematopoietic cell transplantation: a feasibility study. Bone Marrow Transplant. 2010;45:1038–46.
Shah NN, Baird K, Delbrook CP, Fleisher TA, Kohler ME, Rampertaap S, et al. Acute GVHD in patients receiving IL-15/4-1BBL activated NK cells following T-cell-depleted stem cell transplantation. Blood. 2015;125:784–92.
Fehniger TA, Miller JS, Stuart RK, Cooley S, Salhotra A, Curtsinger J, et al. A Phase 1 trial of CNDO-109-activated natural killer cells in patients with high-risk acute myeloid leukemia. Biol Blood Marrow Transpl. 2018;24:1581–9.
Bachanova V, Sarhan D, DeFor TE, Cooley S, Panoskaltsis-Mortari A, Blazar BR, et al. Haploidentical natural killer cells induce remissions in non-Hodgkin lymphoma patients with low levels of immune-suppressor cells. Cancer Immunol Immunother. 2018;67:483–94.
Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8:357ra123.
Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051–7.
Yang Y, Lim O, Kim TM, Ahn YO, Choi H, Chung H, et al. Phase I study of random healthy donor-derived allogeneic natural killer cell therapy in patients with malignant lymphoma or advanced solid tumors. Cancer Immunol Res. 2016;4:215–24.
Szmania S, Lapteva N, Garg T, Greenway A, Lingo J, Nair B, et al. Ex vivo-expanded natural killer cells demonstrate robust proliferation in vivo in high-risk relapsed multiple myeloma patients. J Immunother. 2015;38:24–36.
Wagner J, Pfannenstiel V, Waldmann A, Bergs JWJ, Brill B, Huenecke S, et al. A two-phase expansion protocol combining interleukin (IL)-15 and IL-21 improves natural killer cell proliferation and cytotoxicity against rhabdomyosarcoma. Front Immunol. 2017;8:676.
Fehniger TA, Shah MH, Turner MJ, VanDeusen JB, Whitman SP, Cooper MA, et al. Differential cytokine and chemokine gene expression by human NK cells following activation with IL-18 or IL-15 in combination with IL-12: implications for the innate immune response. J Immunol. 1999;162:4511–20.
Greenberg P, Cox C, LeBeau MM, Fenaux P, Morel P, Sanz G, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079–88.
Kakhniashvili I, Filicko J, Kraft WK, Flomenberg N. Heterogeneous clearance of antithymocyte globulin after CD34+-selected allogeneic hematopoietic progenitor cell transplantation. Biol Blood Marrow Transpl. 2005;11:609–18.
Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, et al. Integrated analysis of multimodal single-cell data. Cell 2021;184:3573–87.e29.
Bi J, Tian Z. NK cell exhaustion. Front Immunol. 2017;8:760.
Song L, Cohen D, Ouyang Z, Cao Y, Hu X, Liu XS. TRUST4: immune repertoire reconstruction from bulk and single-cell RNA-seq data. Nat Methods. 2021;18:627–30.
Browaeys R, Saelens W, Saeys Y. NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods. 2020;17:159–62.
Braun M, Aguilera AR, Sundarrajan A, Corvino D, Stannard K, Krumeich S, et al. CD155 on tumor cells drives resistance to immunotherapy by inducing the degradation of the activating receptor CD226 in CD8(+) T Cells. Immunity. 2020;53:805–23.e15.
Alvarez MJ, Shen Y, Giorgi FM, Lachmann A, Ding BB, Ye BH, et al. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat Genet. 2016;48:838–47.
Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature. 2019;566:496–502.
Kallies A, Carotta S, Huntington ND, Bernard NJ, Tarlinton DM, Smyth MJ, et al. A role for Blimp1 in the transcriptional network controlling natural killer cell maturation. Blood. 2011;117:1869–79.
Gordon SM, Chaix J, Rupp LJ, Wu J, Madera S, Sun JC, et al. The transcription factors T-bet and Eomes control key checkpoints of natural killer cell maturation. Immunity. 2012;36:55–67.
Cooper MA. Natural killer cells might adapt their inhibitory receptors for memory. Proc Natl Acad Sci USA. 2018;115:11357–9.
Berrien-Elliott MM, Cashen AF, Cubitt CC, Neal CC, Wong P, Wagner JA, et al. Multidimensional analyses of donor memory-like NK cells reveal new associations with response after adoptive immunotherapy for leukemia. Cancer Discov. 2020;10:1854–71.
Martin MD, Badovinac VP. Defining memory CD8 T cell. Front Immunol. 2018;9:2692.
Ruggeri A, Sun Y, Labopin M, Bacigalupo A, Lorentino F, Arcese W, et al. Post-transplant cyclophosphamide versus anti-thymocyte globulin as graft- versus-host disease prophylaxis in haploidentical transplant. Haematologica. 2017;102:401–10.
Leung W, Iyengar R, Turner V, Lang P, Bader P, Conn P, et al. Determinants of antileukemia effects of allogeneic NK cells. J Immunol. 2004;172:644–50.
Cooley S, Trachtenberg E, Bergemann TL, Saeteurn K, Klein J, Le CT, et al. Donors with group B KIR haplotypes improve relapse-free survival after unrelated hematopoietic cell transplantation for acute myelogenous leukemia. Blood. 2009;113:726–32.
Storek J, Mohty M, Boelens JJ. Rabbit anti-T cell globulin in allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transpl. 2015;21:959–70.
Bosch M, Dhadda M, Hoegh-Petersen M, Liu Y, Hagel LM, Podgorny P, et al. Immune reconstitution after anti-thymocyte globulin-conditioned hematopoietic cell transplantation. Cytotherapy. 2012;14:1258–75.
Acknowledgements
The authors thank Drs. Koen van Besien (Weill Cornell Medicine) and Haesook Teresa Kim (Dana-Farber Cancer Institute) for their critical review of the manuscript and invaluable comments. This work was supported by grants from the following bodies: 1. Korean Health Technology R&D Project, Ministry of Health and Welfare (A121934, HI12C1788), Republic of Korea. 2. R&D Convergence Program of the National Research Council of Science & Technology (NST) (CRC-15-02-KRIBB), Republic of Korea. 3. National Research Foundation of Korea (NRF) (2019R1A2C3002034, 2019R1A2C1007906, 2020R1A2C2012467, 2020R1A2B5B03094920, and 2021M3A9I4024447), Republic of Korea. 4. Electronics and Telecommunications Research Institute (ETRI) grant (22RB1100, Exploratory and Strategic Research of ETRI-KAIST ICT Future Technology), Republic of Korea. 5. Korea Research Institute of Bioscience and Biotechnology (KRIBB) Research Initiative Program (KGM 5502221), Republic of Korea.
Author information
Authors and Affiliations
Contributions
K-HL: Conceptualization, resources, study design, project administration, supervision, manuscript writing, and editing. SRY: NK cell production, analytical experiments, and data analysis. J-RG: Single-cell RNA analysis, manuscript writing and editing. E-JC: Study patient reenrollment and care, data analysis, statistical analysis, and manuscript editing. HSK: FACS data analysis. C-JP: FACS data analysis. S-CY: Statistical consultation and analysis. S-YP: NK cell production, analytical experiments, and data analysis. S-JJ: NK cell production, analytical experiments, and data analysis. HK: NK cell production, analytical experiments, and data analysis. SYL: NK cell production, analytical experiments, and data analysis. HJ: Single-cell RNA analysis and data interpretation. J-EB: Single-cell RNA analysis and data interpretation. MK: Single-cell RNA analysis and data interpretation. S-YK: Single-cell RNA analysis and data interpretation. J-HK: Single-cell RNA analysis and data interpretation. J-HL: Study patient enrollment and care, clinical data interpretation, and data analysis. J-HL: Clinical data analysis and interpretation. YC: Clinical data analysis and interpretation. H-SP: Patient management, clinical data interpretation, and data analysis. Y-SL: HCT management and clinical study data management. Y-AK: HCT management and clinical study data management. MJ: HCT management and clinical data management. JMW: HCT management and clinical data management. HK: HCT management and clinical data management. SB: HCT management and study data management. SMK: HCT management and study data management. H-MK: Single-cell RNA analysis, manuscript writing and editing. K-HC: Study design, resources, supervision, single-cell RNA analysis, manuscript writing and editing. IC: Conceptualization, resources, study design, project administration, supervision, manuscript writing, and editing.
Corresponding authors
Ethics declarations
Competing interests
K-HL, E-JC, J-HL, J-HL, and H-SP are recipients of royalty from Ingenium Therapeutics, Daejon, Korea. K-HL and E-JC are consultants for the same company and receive consultation fees. The other authors declare no competing conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lee, KH., Yoon, S.R., Gong, JR. et al. The infusion of ex vivo, interleukin-15 and -21-activated donor NK cells after haploidentical HCT in high-risk AML and MDS patients—a randomized trial. Leukemia 37, 807–819 (2023). https://doi.org/10.1038/s41375-023-01849-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41375-023-01849-5
- Springer Nature Limited
This article is cited by
-
Targeting natural killer cells: from basic biology to clinical application in hematologic malignancies
Experimental Hematology & Oncology (2024)
-
Interleukin-15 and -21-activated, donor-derived NK cell infusion after haploidentical HCT in high-risk AML and MDS—a cohort analysis
Leukemia (2024)