
Abstract
Lithium is a first-line therapy for bipolar affective disorder. However, various adverse effects, including a Parkinson-like hand tremor, often limit its use. The understanding of the neurobiological basis of these side effects is still very limited. Nigral iron elevation is also a feature of Parkinsonian degeneration that may be related to soluble tau reduction. We found that magnetic resonance imaging T2 relaxation time changes in subjects commenced on lithium therapy were consistent with iron elevation. In mice, lithium treatment lowers brain tau levels and increases nigral and cortical iron elevation that is closely associated with neurodegeneration, cognitive loss and parkinsonian features. In neuronal cultures lithium attenuates iron efflux by lowering tau protein that traffics amyloid precursor protein to facilitate iron efflux. Thus, tau- and amyloid protein precursor-knockout mice were protected against lithium-induced iron elevation and neurotoxicity. These findings challenge the appropriateness of lithium as a potential treatment for disorders where brain iron is elevated (for example, Alzheimer’s disease), and may explain lithium-associated motor symptoms in susceptible patients.
Similar content being viewed by others


Introduction
Lithium was identified as a psychoactive drug in the mid-twentieth century,1 and remains the primary therapy for bipolar affective disorder2 (although it is often used in other psychiatric disorders also). Unfortunately, it has a narrow therapeutic window limited by adverse effects,3, 4 and neither the mechanism of action nor its associated toxicity are well understood. Lithium modulates multiple key cellular signaling molecules,3, 5 including inositol monophosphatase (IMPase) and glycogen synthase kinase-3 (GSK-3), considered likely to be therapeutic targets.3 Parkinson-like hand tremor is a common adverse effect of lithium therapy, and limited cross-sectional studies and several case reports have raised concern that lithium may increase the risk for extrapyramidal syndromes (for example, dyskinesia) not only in overdose, but possibly also at appropriate maintenance levels.6, 7, 8, 9, 10, 11, 12, 13, 14, 15 Recently, long-term lithium use was reported as associated with an increased incidence of anti-parkinson drug use or Parkinson’s disease (PD) diagnosis.16 Whether lithium could induce extrapyramidal syndromes by neurotransmitter imbalance (like first-generation antipsychotics such as haloperidol) or by damage to the basal ganglia is not known.
Although a previous study demonstrated the neurotoxic potential of lithium in mice treated with a dose considered to be within the therapeutic range (2.55 g kg−1 in food),17 lithium has been more extensively investigated for a host of neuroprotective properties in various neurological and psychiatric models including cerebral ischemia/reperfusion injury18 and other neurotoxic insults.19 A recent study19 of adolescents at ultrahigh risk for psychosis20 found that low-dose lithium treatment over 4 months lowered hippocampal T2 relaxation time (T2*), and this was taken to signify neurochemical improvements that could underlie potential neuroprotective benefits of low-dose lithium as an approach to prevent the onset of psychosis.21, 22 However, a reduction of T2* can also be caused by an elevation of brain iron, and has been used to map such changes in diseases where iron levels rise, such as cortical tissue in Alzheimer’s disease (AD).23
Lithium has attracted considerable interest as a therapeutic candidate in AD research. In neuronal cultures, lithium is reported to decrease tau phosphorylation24, 25, 26, 27 and to inhibit Aβ generation.28, 29, 30 In Aβ-overexpressing transgenic models for AD, lithium treatment consistently decreases Aβ levels, GSK-3 activity and tau phosphorylation.29, 31, 32, 33, 34 In such models, lithium is reported to prevent Aβ toxicity,32 preserve dendritic structure,31 promote neurogenesis34 and rescue Aβ-induced cognitive impairment.31, 33, 34 Similarly, in transgenic mice that overexpress pathogenic mutant tau, lithium treatment reduced levels of hyperphosphorylation.35, 36, 37, 38, 39 Despite these promising preclinical findings, lithium failed to show therapeutic efficacy for AD in a 10-week multicenter, randomized, single-blind, placebo-controlled trial, with no significant effects on cerebrospinal fluid levels of Aβ and phospho-tau or cognitive end points.40
Nevertheless, lowering total tau expression is still being explored as a potential treatment for AD, and lithium is still being considered as a treatment option as it lowers both tau protein and mRNA levels in cultured cortical neurons.41, 42 Supporting this concept, long-term (5 months) lithium treatment of PrP T44 tau transgenic mice resolved tau aggregates by promoting ubiquitination and degradation of tau, rather than altering GSK-3 activity.36
We, and others, have reported concerns that lowering levels of brain tau could induce age-dependent neurodegeneration.43, 44, 45 In tauopathies (such as AD and PD), insoluble tau aggregates are accompanied by a fall in soluble tau levels in affected tissue.43, 46, 47, 48, 49, 50, 51 We have argued that this will lead to a loss of tau function that could cause neurodegeneration, as tau functions to lower neuronal iron levels by promoting the presentation of the amyloid protein precursor (APP) to the neuronal surface,43 where it promotes the efflux of iron.52, 53, 54 Without soluble tau, neurons accumulate iron to toxic levels, and hence tau-knockout mice develop a parkinsonism with impaired-cognition phenotype that is rescued by iron chelation.43, 44 Iron accumulation in the substantia nigra (SN) is a robust feature of PD, and is a sufficient cause for parkinsonism (reviewed in Ayton and Lei55). There has been one case report of such SN iron elevation with extrapyramidal signs following long-term lithium treatment.56 We hypothesized that as lithium lowers neuronal tau, lithium could engender SN iron accumulation and induce parkinsonian neurodegeneration, explaining a cryptogenic aspect of its adverse effect profile.
Materials and methods
Reagents
Reagents were purchased from Sigma-Aldrich (Castle Hill, NSW, Australia), unless specified.
The lithium clinical imaging study
The lithium clinical imaging study has been previously described.19 The pilot study was conducted as an open-label, nonrandomized, controlled, single-center study. Participants were consecutive referrals to the Personal Assessment and Crisis Evaluation (PACE) Clinic, a specialist clinic for young people at ultrahigh risk (UHR) for psychosis.20 PACE is a subprogram of ORYGEN Youth Health, a specialist service for young people with emerging mental disorders. Inclusion criteria for PACE were: (1) age between 14 and 30 inclusive years, (2) resident of the Melbourne metropolitan area or the ability to travel to the PACE clinic at least once a week and (3) meeting PACE UHR criteria. UHR criteria were determined using the Comprehensive Assessment of the At Risk Mental State (CAARMS).57 Rationale and validity for these criteria have been fully described elsewhere.58 Exclusion criteria were: (1) medical or neurological conditions that may diminish functioning or that may account for some of the symptoms leading to the initial referral, for example, epilepsy; (2) clinically relevant biochemical or hematological abnormalities; (3) serious coexisting illnesses such as liver or renal insufficiency, significant cardiac, vascular, pulmonary, gastrointestinal, endocrine, neurological or metabolic disturbances (including patients known to be HIV positive); (4) history of previous acute psychotic episodes either treated or untreated; (5) a current score of 5 or 6 on the CAARMS Mania item or a history of a previous manic episode; (6) any previous use of neuroleptic or mood-stabilizing medications; (7) history of severe drug allergy or hypersensitivity; (8) history of intellectual disability (IQ < 70); and (9) inability to understand or communicate in English. All UHR subjects received PACE treatment as usual throughout the course of the study including psychological and medical treatment. Use of antidepressants was allowed if UHR patients met criteria for major depression. UHR subjects in the low-dose lithium group took one slow-release 450 mg tablet of lithium carbonate each night for the entire study period (3 months), intended to achieve serum lithium levels of 0.3–0.5 mM, in addition to treatment as usual.
Baseline magnetic resonance imaging scans and clinical assessments were conducted before commencement of the low-dose lithium treatment, and within 2 weeks of admission to the study. Monitoring of psychopathology and tolerability measures occurred at monthly intervals, and follow-up scanning was conducted as close as possible to 3 months after baseline (median for the control group was 3.0 months, range 0.8–27 months; for the lithium group 3.8 months, range 1.6–5.9 months, with some subjects in both groups not completing the full treatment period). Extrapyramidal side effects were not systematically assessed. The local research and ethics committee approved the study protocol. All participants gave written informed consent. Additional written informed consent was obtained from parents or guardians for participants aged 14–18 years. Participants were made aware that they could withdraw from the study at any time and that withdrawal would not compromise access to any clinical services.
Brain imaging and SN T2 relaxometry
All scans were performed on a 3T GE LX Horizon scanner (GE Healthcare, Milwaukee, WI, USA) at the Brain Research Institute (Melbourne, Victoria, Australia). Eight tilted, coronal T2-weighted images (perpendicular to the long axis of the hippocampus) were obtained over a range of echo times (29 to 231 ms; TR=4 s, slice thickness= 6 mm with 1.5 mm gap, matrix 256 × 256, field of view 24 cm). Single exponentials were then fitted to the image data of corresponding voxels from these eight echoes using iBrain (Brain Research Institute). This created a series of T2 maps, one for each coronal slice. The brightness of each individual voxel on this map represents its calculated T2 relaxation time. The images were then analyzed in various regions of interest with reference to a standard neuroanatomical atlas by two raters who were blinded to treatment group, and quantified using ANALYZE 7.2 software (Mayo Clinic, Rochester, MN, USA), as previously described.19
Primary neuron culture
Primary neurons were prepared as previously described.43 Briefly, cortices of wild-type and tau-knockout embryos (day 14) were dissected and dissociated in 0.025% (w/v) trypsin. The neurons were plated (Nunc, Mt Waverley, VIC, Australia) at a density of 600 000 cells per cm2 in plating medium (Dulbecco’s modified Eagle’s medium with 10% fetal calf serum, 5% house serum and 10 mg l–1 gentamycin sulfate; Invitrogen, Mt Waverley, VIC, Australia). After 2 h of incubation, the neurons were changed to Neurobasal supplemented medium (with B27, 500 μM glutaMAX and 10 μg ml–1 gentamycin sulfate). Experiments were performed 7 days later. Confluent cells were incubated in Neurobasal medium solution before treatment with various concentrations of lithium chloride for 18 h with or without radioactive 59Fe or 65Zn, or various compounds (L690,330; 6-bromoindirubin-3'-oxime (BIO)) at concentrations indicated. The cells were then harvested, with half the batch lysed using lysis buffer (1% Triton X-100, 50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, pH 7.4, together with 1:50 proteinase inhibitor and 1:1000 kinase inhibitors I and II), and the other half retained for metal analysis (see below), or assayed for radioactive tracers by γ-counter.
59Fe-loaded transferrin and 65Zn preparation
Purified human apo-transferrin (Tf) was loaded with 59Fe (Perkin Elmer, Glen Waverley, VIC, Australia) to form Tf(59Fe)2 as previously described.43, 52 Neurons were incubated with 1.0 × 10-6 M Tf(59Fe)2 in serum-free Neurobasal supplemented medium for periods indicated. At the conclusions of the treatments, neurons were washed thoroughly with phosphate-buffered saline (PBS) and harvested with trypsin. All media and cell lysates were measured by γ-counter (Wizard 3, Perkin Elmer) for counts per min. For iron efflux assay, cells were incubated at 37 °C and medium was removed at select time points. A comparable time-course control study was performed at 4 °C to account for nonspecific binding of 59Fe to the membrane surface.
The 65Zn experiments were adapted from a previous publication.59 Briefly, serum-free Neurobasal supplemented medium was mixed with 0.04 MBq 65Zn (Oak Ridge Laboratory, Oak Ridge, TN, USA) and unlabeled ZnCl2 (20 μM). After 18 h of treatment, the reaction was stopped by washing cell monolayers three times with ice-cold nonlabeled Hanks’ balanced salt solution (with 2 mM L-histidine and 5 mM EDTA). The cells were then harvested and measured for 65Zn retention by γ-counter (1282 CompuGamma; LKB Wallac, Mount Waverley, VIC, Australia).
Mouse tissue preparation
All mice were housed in a conventional animal facility according to standard animal care protocols and fed standard laboratory chow (Meat Free Rat and Mouse Diet, Specialty Feeds, Glen Forrest, WA, Australia) and tap water ad libitum. All animal procedures were approved by the Florey Institute animal ethics committee (10-017) and were performed in accordance with the National Health and Medical Research Council guidelines. Tau-knockout,60 APP-knockout52 and background C57Bl6/SV129 mice stocks raised under the same conditions were maintained homozygously, with backcrossing to the parental inbred strain every three generations. The mouse was killed with an overdose of sodium pentobarbitone (Lethabarb, Clifford Hallam Healthcare, Dandenong South, VIC, Australia, 100 mg kg−1) and perfused with ice-cold saline before tissues were collected. Body weight and total brain wet weight were recorded. Once extracted, the right brain hemisphere was microdissected and stored at −80 °C until required. The left brain hemisphere was fixed in 4% paraformaldehyde for 24 h, and then transferred to 30% sucrose+PBS (pH 7.4) and kept at 4 °C overnight for tyrosine hydroxylase(TH) immunohistochemistry and brain section analysis.
Lithium treatment to mice
The 3-month-old male Bl6/C57J mice (used in Figure 2) as well as age-matched tau-knockout, APP-knockout and background control (BL6/129sv) mice (used in Figure 4) were randomly assigned (by Excel) and orally gavaged with lithium chloride (3.6 mg kg−1 per day, freshly dissolved in 0.9% NaCl, 0.5% Na-carboxymethylcellulose, 0.5% benzyl alcohol and 0.4% Tween-80, with the use of an oroesophageal needle, continuously for 21 days) or the solution alone. Behavioral tests were performed at days 18–21, 2 h after the daily lithium dose. To control for effects of potential acute sedation, drug-naive Bl6/C57J mice were treated with a single dose of diazepam (3 mg kg−1) or lithium (3.6 mg kg−1) and the performance tests carried out 2 h later.
Behavioral measurements
Pole test assessment was performed as previously described.43 Briefly, mice were placed vertically on a 30 cm vertical, 1 cm diameter pole placed within in their home cage, where mice make an 180° turn and return to the base of the pole. On the day before testing (day 1), the animals were habituated to the pole and allowed five consecutive trials. Animals were then recorded via digital video on the test day (day 2). The interval time for the mouse to turn toward the ground (‘time to turn’) and to reach the ground (‘time to finish’) were both recorded. Each mouse underwent five trials and the average was used in analysis. When mice were unable to turn and move down the pole, the trial was determined as incomplete. Rotarod assessment was performed as previously described.43 Briefly, mice were assessed using a Panlab Rotarod apparatus (Barcelona, Spain) in an accelerating model with triplicate measurements (maximum time of 2.5 min; speed increases every 8 s). The time on the rod as well as the final speed of the rod was recorded and the triplicate averaged for analysis. Open field test: Spontaneous motor activity of mice was measured using a photo-beam activity system (Truscan 2.0, Coulbourn Instruments, Holliston, MA, USA) as previously described.43 The test area was 25.4 cm wide by 25.4 cm deep by 40.6 cm high. The mouse was placed in the chamber for 1 h to allow acclimatization to its surroundings. Parameters of movements were calculated from the interception of beams to provide an XY coordinate. For Y-maze test, a Y-shaped gray-painted timber with arms 29.5 cm long × 7.5 cm wide × 15.5 cm high was used as previously described.43 All mice were subjected to a two-trial Y-maze test separated by a 1-h intertrial interval to assess spatial recognition memory, with all testing performed during the light phase of the circadian cycle. The three identical arms were randomly designated start arm, novel arm and other arm. Visual cues were placed on the walls of the maze. The first trial (training) was for 10 min, and the mice were allowed to explore only 2 arms (starting arm and other arm). For the second trial (retention), mice were placed back in the maze in the same starting arm, and allowed to explore for 5 min with free access to all 3 arms. Behaviors were recorded on video during a 5-min trial and the Ethovision video-tracking system (Noldus, Wageningen, The Netherlands) was used for analysis. Data are expressed as the percentage of frequency and duration for novel arm entries made during the 5-min second trial. All behavioral tests were performed by an investigator blinded to the experimental group.
Anatomical morphology from brain slice
The anatomical morphology was examined as previously described.43 Briefly, caudate/putamen (CPu) and cerebellum sections from mice were sectioned using a Leica Cryostat (North Ryde, NSW, Australia) set at 50 μm thickness and areas of interest were measured using Image-J (v1.49b, NIH, Bethesda, MD, USA), using landmarks (anterior commissure for CPu and flocculus for cerebellum) to identify the depth of coronal sectioning (bregma +0.26±0.01 mm and bregma −6.12±0.01 mm). Two sections per mouse per area were analyzed. CPu area was defined by the boundaries of corpus callosum, lateral ventricle and anterior commissure. Corpus callosum, neocortical and cerebellar cortical thicknesses were averaged from five measurements. All quantifications were blinded.
TH immunohistochemistry
TH immunohistochemistry was performed as previously described.43 On a calibrated Leica Cryostat, 30 μm sections (1:3 series) were collected through the SN pars compacta (anteroposterior −2.92 to −3.64 mm from bregma, Mouse Atlas Figures 55–61), generating 8 sections per mouse (the second of the three sections was analyzed). After a further brief fixation (4% paraformaldehyde for 30 s), sections were blocked in 3% normal goat serum (Millipore, Macquarie Park, NSW, Australia) and incubated with primary anti-TH rabbit polyclonal (1:3000, Millipore) overnight. The sections were then incubated with goat anti-rabbit secondary horseradish peroxidase-conjugated antibody for 3 h (Millipore), followed by diaminobenzidine visualization (1% w/v in PBS+1% w/v CoCl2, 1% w/v NiSO4)+3% w/v hydrogen peroxide. TH immunostained sections were counterstained with Neutral Red to visualize the Nissl substance in all neurons before mounting on Superfrost-Plus slides (Trajan Scientific and Medical, Ringwood, VIC, Australia).
Stereological estimation of nigra neurons
SN neuron number was estimated as previously described.43 Briefly, both TH-positive and TH-negative Nissl-positive neurons were scored according to the optical fractionator rules by an investigator blinded to the experimental group. Using an unbiased counting frame (x=35 μm, y=45 μm) at regular intervals on a sampling grid (x=140 μm, y=140 μm), viewed through a 60 × 1.3 NA oil objective (DMLB Leica Microscope), cells were detected with a morphometry and design-based stereology software package (Stereo Investigator 10.04, Microbrightfield, Colchester, VT, USA). The coefficients of error and coefficients of variance were calculated as estimates of precision and only values of <0.1 were accepted.
Dopamine and DOPAC measurement
Dopamine metabolites were measured as previously described.43 Briefly, CPu tissues were homogenized in high-performance liquid chromatography (HPLC) sample buffer (0.4 M perchloric acid, 0.15% sodium metabisulfite and 0.05% EDTA) before centrifugation at 10 000 g at 4 °C for 10 min. Supernatants were used for dopamine measurement by a HPLC system (ESA Biosciences, Chelmsford, MA, USA; model 584) coupled to an electrochemical detector (ESA Biosciences; Coulochem III detector) (E1: −150 mV, E2: +220 mV, and guard cell: +250 mV). Then, 50 μl was injected onto a MD-150 reverse phase C18 column (ESA Biosciences) and elution was performed at a flow rate of 0.6 ml min−1 in the mobile phase (75 mM sodium dihydrogen phosphate, 1.7 mM 1-octanesulfonic acid sodium salt, 100 ml l−1 triethylamine, 25 mM EDTA, 10% acetonitrile, pH 3). Peaks were identified by retention times set to known standards. Data were normalized to wet weight tissue.
Metal analysis
Metal content was measured as previously described.43 Briefly, samples from each experimental condition were freeze-dried and then resuspended in 69% nitric acid (ultraclean grade, Aristar, VWR Chemicals, Radnor, PA, USA) overnight. The samples were then heated for 20 min at 90 °C, and an equivalent volume of hydrogen peroxide (30%, Merck, South Granville, NSW, Australia) was added for a further 15-min incubation at 70 °C. The samples were diluted in double-distilled water and assayed by inductively coupled plasma mass spectrometer (Ultramass 700, Varian, Palo Alto, CA, USA). Each sample was measured in triplicate and the concentrations determined from the standard curve were normalized to tissue wet weight.
Western blot
Samples from each experiment were homogenized in PBS (pH 7.4) with EDTA-free protease inhibitor cocktail (1:50, Roche, Dee Why, NSW, Australia)+phosphatase inhibitors I and II (1:1000) and centrifuged at 100 000 g for 30 min. Protein concentration of the supernatant following centrifugation was determined by BCA protein assay (Pierce, Mt Waverley, VIC, Australia). Aliquots of soluble tissue fraction with equal protein concentrations were separated in 4–12% Bis-Tris gels with NuPAGE MES running buffer (Invitrogen), and transferred to nitrocellulose membranes by iBlot (Invitrogen). The membranes were blocked with milk (10% w/v) and probed with appropriate primary and secondary IgG-horseradish peroxidase-conjugated antibodies (Dako, North Sydney, NSW, Australia). Enhanced chemiluminescence detection system (GE Healthcare) with the Fujifilm LAS-3000 (Campbellfield, VIC, Australia) was used for visualization. Densitometric quantification of immunoreactive signals was performed by ImageJ (1.49b, NIH) and normalized to the relative amount of β-actin and expressed as a percentage of the mean of the control group. The following primary antibodies were used in the current study: anti-β-actin (Sigma), anti-tau (Dako) and anti-pTau396 (Invitrogen).
Statistics
Statistical analysis was carried out in Prism 6 (GraphPad Software, La Jolla, CA, USA) and power calculations were performed using PASS 13 (trial version, NCSS Statistical Software, Kaysville, UT, USA) based on the variance of results in our previous publication10 and in pilot experiments. Equivalence of variance between experimental groups was confirmed using Levene’s test, when applicable. Normal distribution was assumed for all statistical analyses. All tests were two tailed, with the level of significance set at 0.05. Specific types of test used for each experiment are described in the figure legends.
Results
Low-dose lithium treatment induces reversible T2* changes in the SN consistent with iron elevation
To determine whether lithium treatment has the potential to raise nigral iron levels, we analyzed this nucleus in scans from a previous clinical trial.19 The impact of low-dose lithium treatment on hippocampal tissue integrity in individuals at UHR for psychosis was assessed by T2 relaxometry in an open-labeled, nonrandomized, controlled, single-center pilot study. Participants were treated with lithium or as usual for 3 months, and repeated 3T magnetic resonance images were taken from participants before commencement of the study (scan one) and after drug withdrawal (scan two). Lithium treatment induced a decrease in hippocampal T2 relaxation time19 that could not only indicate an improvement in various neuropathologies (for example, edema), but also reflect an increase in tissue iron.61, 62, 63, 64, 65
As iron elevation in the SN could contribute to Parkinson-like side effects such as the commonly observed hand tremor, we revisited this study and measured T2 relaxation in this region, as well as other comparison regions in the same coronal planes (as marked in Supplementary Figure 1a, with reference to a standard neuroanatomical atlas). We found that the average T2 relaxation time in the SN of the lithium group at 3 months (scan two) was significantly reduced from baseline (scan one) (−5%, P=0.007), and significantly lower than the control (treatment as usual) group at the same time point (−7%, P<0.001; Figures 1a and b), consistent with iron accumulation or edema. The pattern of changes was similar in both hemispheres. Other regions analyzed, caudate and lenticular nucleus, showed no change in T2 relaxation time (P=0.18 for caudate and P=0.29 for lenticular nucleus; Supplementary Figure 1b) compared with the baseline scan. The lateral ventricular cross-sectional area was also unchanged from baseline (P=0.177; Supplementary Figure 1b).

T2 relaxation time reductions in the substantia nigra (SN) of participants treated with lithium. (a) Representative magnetic resonance imaging (MRI) images from treatment as usual (top) and lithium (bottom) ultrahigh-risk groups. Arrow indicates the SN. (b) Significant reduction in T2 relaxation time was found in lithium-treated subjects (P<0.001 compared with treatment as usual controls; P=0.007 compared with pretreated scan, two-tailed t-test). Dotted line indicates values of baseline scan; n (treatment as usual)=9, n (lithium)=11. Mean±s.e.m. are shown. ***P<0.001 (versus treatment as usual), ##P<0.01 (versus prescan).
Iron accumulation and parkinsonism in a mouse model of lithium therapy
To test whether lithium-induced relaxometry changes could be due to brain iron accumulation, 3-month-old C57/Bl6J mice were orally administered lithium chloride at a low therapeutic dose40 (3.6 mg kg−1 body weight per day) for 21 days. The dose was well tolerated and body weight was unaltered during the treatment period (P=0.188; Figure 2a). Lithium levels were elevated in all tested tissues (P<0.001 for cortex, SN, cerebellum and liver), with the greatest proportional elevation in cortex (+263%) and SN (+152%; Figure 2b). Iron levels were increased in cortex (+40%, P=0.008) and SN (+19%, P=0.039), but not cerebellum, liver or plasma (Figure 2c). The effect of lithium on biological metals was specific to iron, with copper and zinc unaltered in all tested tissues (Supplementary Figures 2a and b). Lithium levels significantly correlated with iron levels in SN (P=0.007, R2=0.62; Figure 2d) and cortex (P=0.026, R2=0.439; Supplementary Figure 3a) in lithium-treated mice, consistent with elevated lithium causing an elevation in iron. The correlation was seen neither in sham-treated SN (R2=0.02; Figure 2d) nor cortex (R2=0.03; Supplementary Figure 3a), nor observed in the cerebellum of lithium- (R2=0.01) or sham-treated (R2=0.05) animals, a region that did not accumulate iron with lithium treatment (Supplementary Figure 3b). Neither copper nor zinc correlated with lithium in these tissues (Supplementary Figures 3c and d).

Lithium (Li) induces brain iron elevation and parkinsonism in mice. (a) Li treatment did not induce a change in mouse weight, monitored daily as an index of animal health. (b) Li treatment elevated lithium levels in mouse brain and liver (P<0.001 for cortex (Ctx), substantia nigra (SN), cerebellum (Cereb) or liver, two-tailed t-test). (c) Li treatment induced iron elevation in cerebral regions (P=0.008 for Ctx and P=0.039 for SN; two-tailed t-test) but not Cereb, liver or plasma. (d) Iron levels in SN correlate with lithium levels in Li-treated mice (R2=0.62, P=0.007, linear regression) but not sham-treated mice (R2=0.02, P=0.702, linear regression). (e) Quantification of western blots showed reduction of tau in cortex (P=0.032, two-tailed t-test) and SN (P<0.001, two-tailed t-test) of Li-treated mice compared with sham-treated mice. (f) Following 21 days of exposure, Li-treated mice took longer to turn (P=0.008, two-tailed t-test) in the Pole test. (g) Li-treated mice maintained less time on the rod compared with sham-treated mice (P=0.004, two-tailed t-test); n=12 per treatment group. (h, i) Representative tyrosine hydroxylase (TH) staining of sham-treated (top) and Li-treated (bottom) mice. Quantitative data are shown in (i). Significant reduction was found in TH-immunoreactive (TH-ir) neurons in the SN of Li-treated mice (P<0.001, two-tailed t-test); n=5 per treatment group. (j) Significant reductions were found in the striatal dopamine levels (P=0.023, two-tailed t-test), measured by high-performance liquid chromatography (HPLC) in Li-treated mice; n=12 per treatment group. Mean±s.e.m. are shown. *P<0.05, **P<0.01, ***P<0.001.
We assessed the brains of lithium-treated mice to determine whether a drop in soluble tau could explain the iron elevation.43 As expected,36, 38, 39, 66 the fraction of brain tau phosphorylated at Ser396 was suppressed by lithium treatment (cortex: −40%, P=0.034; SN: −43%, P=0.039; Supplementary Figure 4). We also discovered that lithium administration lowered PBS-soluble total tau levels in cortex and SN (cortex: −31%, P=0.032; SN: −42%, P<0.001; Figure 2e).
As tau loss causes iron-mediated neurodegeneration with parkinsonism,43, 44, 45 we investigated whether lithium treatment impaired motor function in these mice. Lithium-treated mice compared with sham-treated mice exhibited a significant increase in time to turn (P=0.008; Figure 2f) and to finish (P=0.041; Supplementary Figure 5a) on the Pole test, and significantly impaired performance on the accelerated Rotarod test as evidenced by reduced latency to fall (P=0.004; Figure 2g) and lower average speed to fall (at speed 6, P<0.001; at speed 7, P=0.015; Supplementary Figure 5b). Rotarod impairment has previously been noted in 10-month-old mice treated for 4 weeks with lithium, but the cause was not ascertained.39 Lithium-treated mice also exhibited reduced locomotion distance (P=0.022; Supplementary Figure 5c), reduced velocity (P=0.023; Supplementary Figure 5d) and reduced average distance per movement (P=0.018; Supplementary Figure 5e), without alterations in movement duration or time spent in the corners (data not shown) in the open field test.
Control studies were performed to ascertain whether short-term sedation4, 67 could have contributed to the motor impairment induced by lithium. Diazepam, a sedative with well-characterized effects on motor behaviors,68, 69, 70, 71 was compared with lithium within 2 h after dose for acute motor behavioral effects. Single-dose lithium or diazepam did not impair mouse Rotarod performance at this interval (Supplementary Figure 6a). Diazepam, but not lithium, induced a sudden drop in spontaneous locomotion 20 min after dosing in the open field test (P=0.006; Supplementary Figure 6b). These data indicate that motor impairments induced by lithium were not due to sedation.
To determine whether the motor deficits induced by lithium could be because of Parkinson-like neuropathology, the TH-immunoreactive (dopaminergic) neurons within the SN were stereologically counted. The dopaminergic neurons were significantly decreased in mice treated with lithium for 21 days (−30%, P<0.001; Figures 2h and i), but TH-negative, Neutral Red-positive neurons were unaltered (Supplementary Figure 7a). Accompanying this loss, dopamine (−40%, P=0.023; Figure 2j) and 3,4-dihydroxyphenylacetic acid (DOPAC; −37%, P=0.024; Supplementary Figure 7b) levels in the striatum were also decreased in lithium-treated mice. Excess dopamine induces hyperactivity, a characteristic of mania,72 and is attenuated by lithium treatment.73 Lithium decreases dopaminergic release74 and suppresses levels of dopamine and its metabolites in the striatal projection field and nucleus accumbens.75 Our findings indicate that suppression of the SN–striatal dopaminergic pathway may contribute to the motor deficits induced by lithium as well as, potentially, to its long-term antimanic and antipsychosis properties.
Lithium induces tau-mediated iron accumulation in neuronal culture
Having found that by inducing nigral tau suppression lithium treatment of normal mice recapitulated a series of abnormalities that appear in tau-knockout mice (nigral iron elevation with dopaminergic neurodegeneration and parkinsonism), we investigated the mechanism in mouse primary cortical neuronal cultures. Lithium (at nontoxic concentrations29) induced significant iron accumulation within 18 h in a dose-dependent manner (5 mM Li: +48%, P=0.005; 10 mM Li: +56%, P<0.001; Figure 3a), but did not alter copper or zinc levels (Figure 3b), consistent with the changes in brain levels of these metals in mice treated with lithium (Figure 2c). Lithium (10 mM) significantly inhibited the efflux of iron (chased after 59Fe loading) into the media (P<0.001; Figure 3c), explaining the retention of iron. As expected, lithium treatment (10 mM) had no effect on zinc retention in primary neurons treated with radio-labeled 65Zn (P=0.176, Supplementary Figure 8).

Lithium (Li)-induced iron elevation is caused by tau reduction in primary neuronal culture. (a) Li treatment to neurons for 18 h induced dose-dependent 59Fe retention (P=0.005 for 5 mM Li, P<0.001 for 10 mM Li, one-way analysis of variance (ANOVA), n=4 per treatment). (b) Li treatment of neurons for 18 h induced specific iron elevation (P<0.001, two-tailed t-test, n=6 per treatment), without altering copper or zinc. (c) Li treatment (10 mM) inhibited iron export (P<0.001, nonlinear regression with extra sum-of-squares F test, n=6 per group). (d) Li treatment reduced neuronal tau levels (representative blots shown beneath) at 5 mM (P=0.007) and 10 mM (P=0.034, one-way ANOVA) after 18 h; n=4 per group. (e) Tau-knockout neurons retained more 59Fe iron (P=0.032) (18 h) but, in contrast to wild-type neurons, were resistant to Li-induced iron accumulation (P=0.038, two-way ANOVA); n=4 per group. Each experiment was independently repeated three times. Mean±s.e.m. are shown. *P<0.05, **P<0.01, ***P<0.001.
The activities of both IMPase and GSK-3 (ref. 5) are considered molecular targets for lithium’s therapeutic effect in bipolar disorder.3 L-690,330, a potent and more specific inhibitor of IMPase,76 had no effect on cellular iron level during the 18-h incubation (P=0.106; Supplementary Figure 9a), thus eliminating IMPase in the mechanism of iron accumulation induced by lithium. However, BIO, a potent and specific inhibitor of GSK-3,77 caused iron retention similar to the effect of lithium (+59%, P=0.002, Supplementary Figure 9b).
Both lithium and BIO are reported to decrease total tau protein levels in addition to their inhibitory effects of GSK in cultured neurons.42 In agreement with previous reports,41, 42 we found that lithium (⩾5 mM, 18 h) suppresses tau protein levels in primary neurons (5 mM Li: −48%, P=0.007; 10 mM Li: −35%, P=0.034; Figure 3d) and in SH-SY5Y cells (5 mM Li: −25%, P=0.003; 10 mM Li: −25%, P=0.002; Supplementary Figures 10a and b). BIO (18 h) also suppressed tau protein levels in primary neuronal culture (1 μM BIO: −25%, P=0.043; 2 μM BIO: −26%, P=0.048; Supplementary Figures 10c and d) that may be the mechanism for its ability to increase neuronal iron levels.
To demonstrate the requirement for tau in the mechanism of lithium-induced iron accumulation in neurons, lithium (10 mM, 18 h) was administered to tau-knockout primary neurons and compared with background-matched wild-type neurons (Figure 3e). As previously shown,43 tau-knockout neurons retained more iron (+20%, P=0.032) compared with wild-type neurons. As expected, lithium treatment (10 mM, 18 h) of wild-type neurons induced a significant accumulation of iron (+20%, P=0.038), but lithium treatment of tau-knockout neurons induced no further iron accumulation. Thus, lithium-induced neuronal iron accumulation requires the expression of tau protein.
Tau-knockout and APP-knockout mice are resistant to neurodegeneration caused by chronic lithium administration
Tau mediates neuronal iron efflux by facilitating the trafficking of APP to the cell surface, where it stabilizes the iron export channel ferroportin.43, 52, 53, 54 To test whether lithium-induced iron accumulation requires APP, we applied the same lithium treatment to tau-knockout mice, APP-knockout mice and their background-matched wild-type mice. We studied mice at 3 months of age, because the knockouts have yet to develop the age-dependent elevation in brain iron levels.43, 44
All three strains showed no weight reduction during 21 days of lithium treatment (Figure 4a), even though APP knockouts were lighter in weight (as reported previously78). Lithium treatment predictably caused an elevation in mouse plasma lithium levels (wild type: +6.6-fold, P<0.001; tau knockout: +6.7-fold, P<0.001; APP knockout: +4.3-fold, P=0.007; Figure 4b), although the levels in all three strains remained well below the lower limit of the therapeutic concentration range.40 As with the Bl6/C57J mice (Figure 2c), lithium treatment induced iron elevation in both the cortex and SN of the littermate Bl6/129Sv mice (cortex: +15%, P=0.031; SN: +20%, P=0.041), but despite lithium elevations in brain regions being commensurate to those in Bl6/C57J mice (Figure 2b), tau- and APP-knockout mice were unaffected (Figures 4c and d), consistent with the neuronal culture findings (Figure 3e). Brain copper or zinc levels were again unaffected (Supplementary Figures 11a–d), and iron levels in the cerebellum and liver were unaltered (Supplementary Figures 11e and f), by lithium treatment in all three strains.

Lithium (Li)-induced parkinsonism is mediated by tau and amyloid protein precursor (APP). (a) Daily mean weights of the mouse cohorts revealed no significant impact of lithium treatment within each genotype group during the course of study. APP-knockout mice were lighter, as reported previously.78 (b) Li treatment significantly elevated plasma lithium levels of 3-month-old wild-type (P<0.001, two-way analysis of variance (ANOVA) with post hoc Dunnett’s test), tau-knockout (P<0.001) and APP-knockout (P=0.007) mice. These concentrations are well below the usual therapeutic range of clinical lithium therapy. (c, d) Elevated iron levels were observed in the cortex (c, P=0.031, two-way ANOVA with post hoc Dunnett’s test) and substantia nigra (SN; d, P=0.041) of Li-treated wild-type mice, but loss of tau or APP abolished the elevation. (e, f) Li treatment induced tau reduction in cortex and SN of both wild-type (e, P=0.041 for cortex, P<0.001 for SN, two-tailed t-test) and APP-knockout mice (f, P=0.006 for cortex, P=0.041 for SN, two-tailed t-test). (g, h) Li-treated wild-type mice required longer time to turn in the Pole test (g, P=0.020, two-way ANOVA with post hoc Dunnett’s test) and showed significantly reduced average distance per movement in the open field test (h, P<0.001), but loss of tau or APP protected against Li-induced motor impairment. (i) Li treatment caused SN dopaminergic neuron loss (P<0.001, two-way ANOVA with post hoc Dunnett’s test), but did not cause similar neuronal loss in tau-knockout or APP-knockout mice. (j) Li treatment impaired cognitive function of wild-type mice, evidenced by significantly reduced novel arm duration in the Y-maze test (P=0.022, two-tailed t-test). Such impairment was not evident in tau-knockout and APP-knockout mice. (k) Representative coronal section of cerebrum of wild-type mice treated with sham (left) or Li (right). Arrow indicates the landmark used to identify the section. Lines indicate the area quantified. (l) Significant enlargement of lateral ventricular (LV) was found in Li-treated wild-type mice (P=0.023, two-way ANOVA with post hoc Dunnett’s test), but not in Li-treated tau-knockout or APP-knockout mice; n=9–11 per treatment group. Mean±s.e.m. are shown. *P<0.05, **P<0.01, ***P<0.001.
Ablation of APP abolished lithium-induced iron accumulation (Figures 4c and d), despite soluble tau protein levels in cortex and SN of APP-knockout mice treated with lithium being suppressed comparably to wild type (cortex: −19%, P=0.041 for wild type and −41%, P=0.006 for APP knockout; SN: −42%, P<0.001 for wild type and 21%, P=0.041 for APP knockout; Figures 4e and f and Supplementary Figure 12). These results confirm our prediction that lithium-induced brain iron accumulation requires both tau and APP expression, with tau being upstream in the APP-associated iron homeostatic pathway.
As lithium-induced Parkinson-like symptoms such as hand tremor may be mediated by iron accumulation in the brain, disabling lithium-induced SN iron accumulation in tau- and APP-knockout mice (Figure 4d) should also prevent motor deficits and SN neuronal loss. Indeed, lithium-induced motor deficits were abolished in tau- and APP-knockout mice, as evidenced by unaffected time to turn (wild type, P=0.020; Figure 4g) and time to finish (wild type, P=0.048; Supplementary Figure 13a) in the Pole test, and unaltered average distance per movement (wild type, P=0.049; Figure 4h), distance of locomotion (wild type, P=0.020; Supplementary Figure 13b) and velocity (wild type, P=0.048; Supplementary Figure 13c) in the open field test. Untreated APP-knockout mice did show baseline motor deficits (Figures 4g and h and Supplementary Figures 13a–c), as previously reported,78 but these were not worsened by lithium. Tau or APP ablation also negated SN dopaminergic neuron loss induced by lithium treatment (wild type, P<0.001; Figure 4i and Supplementary Figure 14a). Together, these results support the conclusion that lithium-induced parkinsonism is mediated by iron accumulation that which is tau and APP dependent.
As lithium treatment also increases cortical iron levels (Figures 2c and 4c), we investigated its effect on cognitive performance and related brain anatomy. In the Y-maze test, we found the memory performance of wild-type mice was markedly impaired by 21 days of lithium treatment (P=0.022) but, as with the Pole test (Figure 4g), tau- and APP-knockout mice were unaffected by lithium (Figure 4j). Accordingly, lithium treatment caused brain atrophy in wild-type mice, evidenced by enlarged lateral ventricular area (+72%, P=0.023; Figures 4k and l), shrinkage of CPu area (−13%, P=0.011; Supplementary Figure 14b) and reduced cortical thickness (−7%, P=0.040; Supplementary Figure 14c), without affecting corpus callosum width (Supplementary Figure 14d). Again, lithium did not induce brain atrophy in tau- or APP-knockout mice (Figure 4l and Supplementary Figures 14c and d). These toxic consequences of lithium administration to cortical thickness are in agreement with a previous report that found cortical neuronal apoptosis in mice upon chronic lithium treatment.17
Discussion
Lithium has an established record of efficacy in bipolar affective disorder, despite a narrow therapeutic window limited by various toxicities of uncertain mechanism. Here we found that lithium may contribute to nigral and cortical degeneration, with motor and cognitive impairment in wild-type mice, recapitulating the neurophysiological and neurochemical phenotypes of aged tau-knockout mice.43, 44 Lithium suppresses tau protein levels in cultured neurons and in mice, causing iron accumulation through a tau- and APP-dependent interaction. The T2 relaxation time changes in the human lithium trial are consistent with iron elevation in hippocampus and SN. Although homeostasis keeps brain iron levels stringently stable in health, and geared to resist depletion,79 rodent studies have shown that nearly 10% of brain iron is in relatively rapid exchange with dietary iron,52, 80 indicating that in order to prevent iron accumulation, iron efflux from the brain must be constantly active. It is this mechanism that lithium inhibits by, at least in part, suppressing tau expression. Tau-knockout mice develop iron-induced neurodegenerative changes at 12 months of age43, 44 but lithium-induced toxic iron accumulation occurs within weeks. This is probably because the tau-knockout mice in youth have the benefit of upregulated compensatory responses from other microtubule-associated proteins45 in contrast to the more acute suppression of tau expression by lithium treatment. Our findings add to the evidence that lithium at therapeutic (and eventually even at subtherapeutic) chronic doses can cause neurodegeneration,17 and provide a possible mechanism for adverse neurological effects in susceptible individuals.
Parkinsonism is an uncommon adverse effect of lithium treatment, but hand tremor is common and may be age and dose dependent.81, 82, 83 In addition, lithium use was found to be associated with an increased incidence of anti-parkinson drug use or a PD diagnosis.16 The mechanisms underlying these motor abnormalities are not known, but could be because of persistent or transient extrapyramidal syndromes caused by damage to the basal ganglia and SN. Iron may play a critical role in promoting SN pathology in PD,55, 84 and our findings demonstrate how lithium could worsen iron-mediated SN damage. SN iron accumulation in trial subjects may have occurred after only 3 months of low-dose lithium treatment (Figure 1b), although no conspicuous motor abnormalities were reported in this group. In addition, our findings in mice support the possibility that lithium-induced parkinsonism can become irreversible in a proportion of patients.6, 7, 8, 11, 15
Unexpectedly, we found that lithium treatment at doses inducing blood levels well below the human therapeutic range caused cognitive impairment and brain atrophy in mice after 3 weeks (Figures 4j–l). It has been shown previously that lithium treatment at therapeutic doses can cause cortical neuronal apoptosis,17 and this may explain our findings. Although lithium treatment was previously reported to correct the smaller hippocampal volumes in patients with bipolar disorder,85 there is also evidence that long-term lithium treatment is associated with cognitive impairment86, 87, 88, 89 and brain atrophy.89 Neurodegeneration in mice administered lithium to achieve blood levels that are easily tolerated by humans may reflect different pharmacodynamics in the species, or that humans are less sensitive than mice to lithium-induced neurotoxicity.
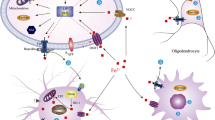
Lithium-induced motor deficits in mice are reported to involve the calcineurin/nuclear factor of activated T cells (NFAT)/Fas ligand signaling pathway, and prevented by inhibition of calcineurin or knockout of Fas.17 Iron modulates calcineurin activity90, 91 and can thereby impact the NFAT/Fas signaling pathway. Treatment with iron dramatically elevates NFAT activity,92, 93 whereas administering the iron chelator deferoxamine abolishes iron-induced NFAT activation.92, 93 Together with our current data, a proposed model pathway emerges (Figure 5) where lithium suppresses tau protein levels, thus preventing APP trafficking to the cell surface and resulting in neuronal iron retention. In turn, this activates calcineurin/NFAT/Fas signaling induces cortical and nigral neuronal apoptosis and engenders motor and cognitive disability.

A model of affected pathways for lithium-induced neurodegeneration. Lithium (LI) treatment suppresses soluble tau protein levels that prevent APP trafficking to the neuronal surface. Lack of surface amyloid protein precursor (APP) destabilizes ferroportin (Fpn), resulting in iron accumulation. Elevated iron activates calcineurin/nuclear factor of activated T cells (NFAT)/Fas signaling that then induces neuronal apoptosis, engendering motor and cognitive disability.
Lithium was an attractive drug candidate for AD as its target, GSK-3, regulates both Aβ production and tau phosphorylation.5 However, it is neither a potent nor specific inhibitor of GSK-3.94 In fact, neuronal deficits have been reported to result from both genetic reduction and specific inhibition of GSK-3,95, 96 highlighting the risk of inhibiting this enzyme.97 Lithium arrests neuropathology in some AD rodent models,36, 38, 39 but these are aggressive models driven by transgene overexpression, where the benefits of lithium inhibiting a dominant pathological target (for example, tau hyperphosphorylation) outweigh lithium-induced neurotoxicity. The lithium-induced neurotoxicity that we and others17, 39 observed might only be detected in normal mice or mice that do not express a neuropathology that is inhibited by lithium.
Our current study might help explain the failure of trials of lithium for AD40, 98 and amyotrophic lateral sclerosis,99, 100, 101 both diseases where elevated iron in the affected tissue has been implicated in pathogenesis.52, 102, 103, 104 Despite the neurotoxicity in mice that our studies have elaborated, clinical lithium pharmacotherapy has predominantly neurotrophic benefits in the treatment of bipolar disorder. It is an intriguing hypothesis to consider that these benefits might be related to the correction of iron deficiency in certain brain regions.
References
Cade JF . Lithium salts in the treatment of psychotic excitement. Med J Aust 1949; 2: 349–352.
Geddes JR, Goodwin GM, Rendell J, Azorin JM, Cipriani A, Ostacher MJ et al. Lithium plus valproate combination therapy versus monotherapy for relapse prevention in bipolar I disorder (BALANCE): a randomised open-label trial. Lancet 2010; 375: 385–395.
Phiel CJ, Klein PS . Molecular targets of lithium action. Annu Rev Pharmacol Toxicol 2001; 41: 789–813.
Grof P, Müller-Oerlinghausen B . A critical appraisal of lithium's efficacy and effectiveness: the last 60 years. Bipolar Disord 2009; 11 (Suppl 2): 10–19.
Lei P, Ayton S, Bush AI, Adlard PA . GSK-3 in neurodegenerative diseases. Int J Alzheimers Dis 2011; 2011: 189246.
Ghadirian AM, Annable L, Bélanger MC, Chouinard G . A cross-sectional study of parkinsonism and tardive dyskinesia in lithium-treated affective disordered patients. J Clin Psychiatry 1996; 57: 22–28.
Dallocchio C, Mazzarello P . A case of Parkinsonism due to lithium intoxication: treatment with Pramipexole. J Clin Neurosci 2002; 9: 310–311.
Shopsin B, Gershon S . Cogwheel rigidity related to lithium maintenance. Am J Psychiatry 1975; 132: 536–538.
Reches A, Tietler J, Lavy S . Parkinsonism due to lithium carbonate poisoning. Arch Neurol 1981; 38: 471.
Apte SN, Langston JW . Permanent neurological deficits due to lithium toxicity. Ann Neurol 1983; 13: 453–455.
Lang AE . Lithium and parkinsonism. Ann Neurol 1984; 15: 214.
Muthane UB, Prasad BN, Vasanth A, Satishchandra P . Tardive Parkinsonism, orofacial dyskinesia and akathisia following brief exposure to lithium carbonate. J Neurol Sci 2000; 176: 78–79.
Mazzini L, Oggioni GD, Nasuelli N, Servo S, Testa L, Monaco F . Disabling Parkinsonism following brief exposure to lithium carbonate in amyotrophic lateral sclerosis. J Neurol 2011; 258: 333–334.
Fallgatter AJ, Strik WK . Reversible neuropsychiatric side effects of lithium with normal serum levels. A case report. Nervenarzt 1997; 68: 586–590.
Perenyi A, Rihmer Z, Banki CM . Parkinsonian symptoms with lithium, lithium-neuroleptic, and lithium-antidepressant treatment. J Affect Disord 1983; 5: 171–177.
Marras C, Herrmann N, Fischer HD, Fung K, Gruneir A, Rochon PA et al. Lithium use in older adults is associated with increased prescribing of parkinson medications. Am J Geriatr Psychiatry 2016; 24: 301–309.
Gómez-Sintes R, Lucas JJ . NFAT/Fas signaling mediates the neuronal apoptosis and motor side effects of GSK-3 inhibition in a mouse model of lithium therapy. J Clin Invest 2010; 120: 2432–2445.
Taliyan R, Ramagiri S . Delayed neuroprotection against cerebral ischemia reperfusion injury: putative role of BDNF and GSK-3beta. J Recept Signal Transduct Res 2015; 36: 1–9.
Berger GE, Wood SJ, Ross M, Hamer CA, Wellard RM, Pell G et al. Neuroprotective effects of low-dose lithium in individuals at ultra-high risk for psychosis. A longitudinal MRI/MRS study. Curr Pharm Des 2012; 18: 570–575.
Yung AR, Phillips LJ, Yuen HP, McGorry PD . Risk factors for psychosis in an ultra high-risk group: psychopathology and clinical features. Schizophr Res 2004; 67: 131–142.
Benarous X, Consoli A, Milhiet V, Cohen D . Early interventions for youths at high risk for bipolar disorder: a developmental approach. Eur Child Adolesc Psychiatry 2016; 25: 217–233.
Berger GE, Wood S, McGorry PD . Incipient neurovulnerability and neuroprotection in early psychosis. Psychopharmacol Bull 2003; 37: 79–101.
van Rooden S, Versluis MJ, Liem MK, Milles J, Maier AB, Oleksik AM et al. Cortical phase changes in Alzheimer's disease at 7T MRI: a novel imaging marker. Alzheimers Dement 2014; 10: e19–e26.
Hong M, Chen DC, Klein PS, Lee VM . Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J Biol Chem 1997; 272: 25326–25332.
Munoz-Montano JR, Moreno FJ, Avila J, Diaz-Nido J . Lithium inhibits Alzheimer's disease-like tau protein phosphorylation in neurons. FEBS Lett 1997; 411: 183–188.
Lovestone S, Davis DR, Webster MT, Kaech S, Brion J-P, Matus A et al. Lithium reduces tau phosphorylation: effects in living cells and in neurons at therapeutic concentrations. Biol Psychiatry 1999; 45: 995–1003.
Takahashi M, Yasutake K, Tomizawa K . Lithium inhibits neurite growth and tau protein kinase I/glycogen synthase kinase-3beta-dependent phosphorylation of juvenile tau in cultured hippocampal neurons. J Neurochem 1999; 73: 2073–2083.
Sun X, Sato S, Murayama O, Murayama M, Park JM, Yamaguchi H et al. Lithium inhibits amyloid secretion in COS7 cells transfected with amyloid precursor protein C100. Neurosci Lett 2002; 321: 61–64.
Phiel CJ, Wilson CA, Lee VM-Y, Klein PS . GSK-3alpha regulates production of Alzheimer's disease amyloid-beta peptides. Nature 2003; 423: 435–439.
Su Y, Ryder J, Li B, Wu X, Fox N, Solenberg P et al. Lithium, a common drug for bipolar disorder treatment, regulates amyloid-beta precursor protein processing. Biochemistry 2004; 43: 6899–6908.
Rockenstein E, Torrance M, Adame A, Mante M, Bar-on P, Rose JB et al. Neuroprotective effects of regulators of the glycogen synthase kinase-3beta signaling pathway in a transgenic model of Alzheimer's disease are associated with reduced amyloid precursor protein phosphorylation. J Neurosci 2007; 27: 1981–1991.
Sofola O, Kerr F, Rogers I, Killick R, Augustin H, Gandy C et al. Inhibition of GSK-3 ameliorates Abeta pathology in an adult-onset Drosophila model of Alzheimer's disease. PLoS Genet 2010; 6: e1001087.
Toledo EM, Inestrosa NC . Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1DeltaE9 mouse model of Alzheimer's disease. Mol Psychiatry 2010; 15: 272–285, 228.
Fiorentini A, Rosi MC, Grossi C, Luccarini I, Casamenti F . Lithium improves hippocampal neurogenesis, neuropathology and cognitive functions in APP mutant mice. PLoS One 2010; 5: e14382.
Pérez M, Hernández F, Lim F, Díaz-Nido J, Avila J . Chronic lithium treatment decreases mutant tau protein aggregation in a transgenic mouse model. J Alzheimers Dis 2003; 5: 301–308.
Nakashima H, Ishihara T, Suguimoto P, Yokota O, Oshima E, Kugo A et al. Chronic lithium treatment decreases tau lesions by promoting ubiquitination in a mouse model of tauopathies. Acta Neuropathol 2005; 110: 547–556.
Noble WJ, Planel E, Zehr C, Olm V, Meyerson J, Suleman F et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo. Proc Natl Acad Sci USA 2005; 102: 6990–6995.
Engel T, Goñi-Oliver P, Lucas JJ, Avila J, Hernández F . Chronic lithium administration to FTDP-17 tau and GSK-3beta overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J Neurochem 2006; 99: 1445–1455.
Leroy K, Ando K, Héraud C, Yilmaz Z, Authelet M, Boeynaems J-M et al. Lithium treatment arrests the development of neurofibrillary tangles in mutant tau transgenic mice with advanced neurofibrillary pathology. J Alzheimers Dis 2010; 19: 705–719.
Hampel H, Ewers M, Bürger K, Annas P, Mörtberg A, Bogstedt A et al. Lithium trial in Alzheimer's disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry 2009; 70: 922–931.
Rametti A, Esclaire F, Yardin C, Cogné N, Terro F . Lithium down-regulates tau in cultured cortical neurons: a possible mechanism of neuroprotection. Neurosci Lett 2008; 434: 93–98.
Martin L, Magnaudeix A, Esclaire F, Yardin C, Terro F . Inhibition of glycogen synthase kinase-3beta downregulates total tau proteins in cultured neurons and its reversal by the blockade of protein phosphatase-2A. Brain Res 2009; 1252: 66–75.
Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med 2012; 18: 291–295.
Lei P, Ayton S, Moon S, Zhang Q, Volitakis I, Finkelstein DI et al. Motor and cognitive deficits in aged tau knockout mice in two background strains. Mol Neurodegener 2014; 9: 29.
Ma QL, Zuo X, Yang F, Ubeda OJ, Gant DJ, Alaverdyan M et al. Loss of MAP function leads to hippocampal synapse loss and deficits in the Morris water maze with aging. J Neurosci 2014; 34: 7124–7136.
Zhukareva V, Vogelsberg-Ragaglia V, Van Deerlin VM, Bruce J, Shuck T, Grossman M et al. Loss of brain tau defines novel sporadic and familial tauopathies with frontotemporal dementia. Ann Neurol 2001; 49: 165–175.
Zhukareva V, Sundarraj S, Mann D, Sjogren M, Blenow K, Clark CM et al. Selective reduction of soluble tau proteins in sporadic and familial frontotemporal dementias: an international follow-up study. Acta Neuropathol 2003; 105: 469–476.
Ksiezak-Reding H, Binder LI, Yen S-HC . Immunochemical and biochemical characterization of tau proteins in normal and Alzheimer's disease brains with Alz 50 and Tau-1. J Biol Chem 1988; 263: 7948–7953.
Shin RW, Iwaki T, Kitamoto T, Sato Y, Tateishi J . Massive accumulation of modified tau and severe depletion of normal tau characterize the cerebral cortex and white matter of Alzheimer's disease. Demonstration using the hydrated autoclaving method. Am J Pathol 1992; 140: 937–945.
Khatoon S, Grundke-Iqbal I, Iqbal K . Levels of normal and abnormally phosphorylated tau in different cellular and regional compartments of Alzheimer disease and control brains. FEBS Lett 1994; 351: 80–84.
van Eersel J, Bi M, Ke YD, Hodges JR, Xuereb JH, Gregory GC et al. Phosphorylation of soluble tau differs in Pick's disease and Alzheimer's disease brains. J Neural Transm 2009; 116: 1243–1251.
Duce JA, Tsatsanis A, Cater MA, James SA, Robb E, Wikhe K et al. Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer's disease. Cell 2010; 142: 857–867.
McCarthy RC, Park YH, Kosman DJ . sAPP modulates iron efflux from brain microvascular endothelial cells by stabilizing the ferrous iron exporter ferroportin. EMBO Rep 2014; 15: 809–815.
Wong BX, Tsatsanis A, Lim LQ, Adlard PA, Bush AI, Duce JA . beta-Amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS One 2014; 9: e114174.
Ayton S, Lei P . Nigral iron elevation is an invariable feature of Parkinson's disease and is a sufficient cause of neurodegeneration. Biomed Res Int 2014; 2014: 581256.
Campbell WG, Raskind MA, Gordon T, Shaw CM . Iron pigment in the brain of a man with tardive dyskinesia. Am J Psychiatry 1985; 142: 364–365.
Yung AR, Yuen HP, McGorry PD, Phillips LJ, Kelly D, Dell'Olio M et al. Mapping the onset of psychosis: the Comprehensive Assessment of At-Risk Mental States. Aust N Z J Psychiatry 2005; 39: 964–971.
Yung AR, Phillips LJ, Yuen HP, Francey SM, McFarlane CA, Hallgren M et al. Psychosis prediction: 12-month follow up of a high-risk ("prodromal") group. Schizophr Res 2003; 60: 21–32.
Greenough MA, Volitaskis I, Li Q-X, Laughton KM, Evin G, Ho M et al. Presenilins promote the cellular uptake of copper and zinc and maintain copper chaperone of SOD1-dependent copper/zinc superoxide dismutase activity. J Biol Chem 2011; 286: 9776–9786.
Dawson HN, Ferreira A, Eyster MV, Ghoshal N, Binder LI, Vitek MP . Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J Cell Sci 2001; 114 (Pt 6): 1179–1187.
Chen JC, Hardy PA, Clauberg M, Joshi JG, Parravano J, Deck JH et al. T2 values in the human brain: comparison with quantitative assays of iron and ferritin. Radiology 1989; 173: 521–526.
Bartzokis G, Garber HJ, Marder SR, Olendorf WH . MRI in tardive dyskinesia: shortened left caudate T2. Biol Psychiatry 1990; 28: 1027–1036.
Dhenain M, Duyckaerts C, Michot JL, Volk A, Picq JL, Boller F . Cerebral T2-weighted signal decrease during aging in the mouse lemur primate reflects iron accumulation. Neurobiol Aging 1998; 19: 65–69.
Positano V, Salani B, Pepe A, Santarelli MF, De Marchi D, Ramazzotti A et al. Improved T2* assessment in liver iron overload by magnetic resonance imaging. Magn Reson Imaging 2009; 27: 188–197.
Sun H, Walsh AJ, Lebel RM, Blevins G, Catz I, Lu JQ et al. Validation of quantitative susceptibility mapping with Perls' iron staining for subcortical gray matter. Neuroimage 2014; 105: 486–492.
Caccamo A, Oddo S, Tran LX, LaFerla FM . Lithium reduces tau phosphorylation but not A beta or working memory deficits in a transgenic model with both plaques and tangles. Am J Pathol 2007; 170: 1669–1675.
Malhi GS, Adams D, Berk M . Is lithium in a class of its own? A brief profile of its clinical use. Aust NZ J Psychiatry 2009; 43: 1096–1104.
Frank GB, Jhamandas K . Effects of drugs acting alone and in combination on the motor activity of intact mice. Br J Pharmacol 1970; 39: 696–706.
Healy TE, Lautch H, Hall N, Tomlin PJ, Vickers MD . Interdisciplinary study of diazepam sedation for outpatient dentistry. Br Med J 1970; 3: 13–17.
Tornberg J, Segerstråle M, Kulesskaya N, Voikar V, Taira T, Airaksinen MS . KCC2-deficient mice show reduced sensitivity to diazepam, but normal alcohol-induced motor impairment, gaboxadol-induced sedation, and neurosteroid-induced hypnosis. Neuropsychopharmacology 2007; 32: 911–918.
Zeller A, Crestani F, Camenisch I, Iwasato T, Itohara S, Fritschy JM et al. Cortical glutamatergic neurons mediate the motor sedative action of diazepam. Mol Pharmacol 2008; 73: 282–291.
Snyder SH, Taylor KM, Coyle JT, Meyerhoff JL . The role of brain dopamine in behavioral regulation and the actions of psychotropic drugs. Am J Psychiatry 1970; 127: 199–207.
Cox C, Harrison-Read PE, Steinberg H, Tomkiewicz M . Lithium attenuates drug-induced hyperactivity in rats. Nature 1971; 232: 336–338.
Friedman ES, Gershon S . Effect of lithium on brain dopamine. Nature 1973; 243: 520–521.
Dziedzicka-Wasylewska M, Maćkowiak M, Fijat K, Wedzony K . Adaptive changes in the rat dopaminergic transmission following repeated lithium administration. J Neural Transm 1996; 103: 765–776.
Atack JR, Cook SM, Watt AP, Fletcher SR, Ragan CI . In vitro and in vivo inhibition of inositol monophosphatase by the bisphosphonate L-690,330. J Neurochem 1993; 60: 652–658.
Meijer L, Skaltsounis AL, Magiatis P, Polychronopoulos P, Knockaert M, Leost M et al. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem Biol 2003; 10: 1255–1266.
Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW et al. beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 1995; 81: 525–531.
Dallman PR, Spirito RA . Brain iron in the rat: extremely slow turnover in normal rats may explain long-lasting effects of early iron deficiency. J Nutr 1977; 107: 1075–1081.
Chen JH, Shahnavas S, Singh N, Ong WY, Walczyk T . Stable iron isotope tracing reveals significant brain iron uptake in adult rats. Metallomics 2013; 5: 167–173.
Murray N, Hopwood S, Balfour DJ, Ogston S, Hewick DS . The influence of age on lithium efficacy and side-effects in out-patients. Psychol Med 1983; 13: 53–60.
Bell AJ, Cole A, Eccleston D, Ferrier IN . Lithium neurotoxicity at normal therapeutic levels. Br J Psychiatry 1993; 162: 689–692.
Flint A . Ageing as a risk factor for lithium neurotoxicity at therapeutic serum levels. Br J Psychiatry 1993; 163: 555–556.
Hare D, Ayton S, Bush A, Lei P . A delicate balance: iron metabolism and diseases of the brain. Front Aging Neurosci 2013; 5: 34.
Hajek T, Kopecek M, Hoschl C, Alda M . Smaller hippocampal volumes in patients with bipolar disorder are masked by exposure to lithium: a meta-analysis. J Psychiatry Neurosci 2012; 37: 333–343.
Pfennig A, Alda M, Young T, MacQueen G, Rybakowski J, Suwalska A et al. Prophylactic lithium treatment and cognitive performance in patients with a long history of bipolar illness: no simple answers in complex disease-treatment interplay. Int J Bipolar Disord 2014; 2: 1.
Wingo AP, Wingo TS, Harvey PD, Baldessarini RJ . Effects of lithium on cognitive performance: a meta-analysis. J Clin Psychiatry 2009; 70: 1588–1597.
Mora E, Portella MJ, Forcada I, Vieta E, Mur M . Persistence of cognitive impairment and its negative impact on psychosocial functioning in lithium-treated, euthymic bipolar patients: a 6-year follow-up study. Psychol Med 2013; 43: 1187–1196.
Evrensel A, Unsalver BO, Ceylan ME, Comert G . Lithium-induced cortical atrophy and cognitive dysfunction. BMJ Case Rep 2014; 2014: pii: bcr2014207646.
Wang X, Culotta VC, Klee CB . Superoxide dismutase protects calcineurin from inactivation. Nature 1996; 383: 434–437.
Namgaladze D, Hofer HW, Ullrich V . Redox control of calcineurin by targeting the binuclear Fe(2+)-Zn(2+) center at the enzyme active site. J Biol Chem 2002; 277: 5962–5969.
Huang C, Li J, Zhang Q, Huang X . Role of bioavailable iron in coal dust-induced activation of activator protein-1 and nuclear factor of activated T cells: difference between Pennsylvania and Utah coal dusts. Am J Respir Cell Mol Biol 2002; 27: 568–574.
Huang X, Dai J, Huang C, Zhang Q, Bhanot O, Pelle E . Deferoxamine synergistically enhances iron-mediated AP-1 activation: a showcase of the interplay between extracellular-signal-regulated kinase and tyrosine phosphatase. Free Radic Res 2007; 41: 1135–1142.
Kremer A . GSK3 and Alzheimer’s disease: facts and fiction. Front Mol Neurosci 2011; 4: 1–10.
Hu S, Begum AN, Jones MR, Oh MS, Beech WK, Beech BH et al. GSK3 inhibitors show benefits in an Alzheimer's disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol Dis 2009; 33: 193–206.
Gómez-Sintes R, Hernández F, Bortolozzi A, Artigas F, Avila J, Zaratin P et al. Neuronal apoptosis and reversible motor deficit in dominant-negative GSK-3 conditional transgenic mice. EMBO J 2007; 26: 2743–2754.
Ayton S, Lei P . The Abeta-induced NFAT apoptotic pathway is also activated by GSK-3 inhibition: implications for Alzheimer therapeutics. J Neurosci 2012; 32: 9454–9456.
Macdonald A, Briggs K, Poppe M, Higgins A, Velayudhan L, Lovestone S . A feasibility and tolerability study of lithium in Alzheimer's disease. Int J Geriatr Psychiatry 2008; 23: 704–711.
Aggarwal SP, Zinman L, Simpson E, Mckinley J, Jackson KE, Pinto H et al. Safety and efficacy of lithium in combination with riluzole for treatment of amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol 2010; 9: 481–488.
Swash M . Lithium time-to-event trial in amyotrophic lateral sclerosis stops early for futility. Lancet Neurol 2010; 9: 449–451.
Verstraete E, Veldink JH, Huisman MH, Draak T, Uijtendaal EV, van der Kooi AJ et al. Lithium lacks effect on survival in amyotrophic lateral sclerosis: a phase IIb randomised sequential trial. J Neurol Neurosurg Psychiatry 2012; 83: 557–564.
Ayton S, Lei P, Bush AI . Biometals and their therapeutic implications in Alzheimer's disease. Neurotherapeutics 2014; 12: 109–120.
Langkammer C, Enzinger C, Quasthoff S, Grafenauer P, Soellinger M, Fazekas F et al. Mapping of iron deposition in conjunction with assessment of nerve fiber tract integrity in amyotrophic lateral sclerosis. J Magn Reson Imaging 2010; 31: 1339–1345.
Oba H, Araki T, Ohtomo K, Monzawa S, Uchiyama G, Koizumi K et al. Amyotrophic lateral sclerosis: T2 shortening in motor cortex at MR imaging. Radiology 1993; 189: 843–846.
Acknowledgements
We thank A Sedjahtera, L Lam, L Gunawan, L Bray and K Wikhe for technical assistance. We also acknowledge L Phillips and B Nelson for their contributions in lithium human trial. This study was supported by funds from the Australian Research Council, the National Health & Medical Research Council (NHMRC) of Australia, the Cooperative Research Center for Mental Health, Alzheimer’s Australia Dementia Research Foundation and National Natural Science Foundation of China (81571236). A Bush was supported by a NHMRC Australia Fellowship (AF79) and a NHMRC Senior Principal Research Fellowship (1103703). C Pantelis was supported by a NHMRC Senior Principal Research Fellowship (628386 and 1105825). The imaging work on lithium was supported by NHMRC Project Grant (145627) and NHMRC Program Grants (350241 and 566529). P McGorry currently receives research support from NHMRC of Australia, the Colonial Foundation and NARSAD. Florey Institute of Neuroscience and Mental Health acknowledges the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant.
Author contributions
PL and AIB conceived and raised funds for the study. PL, SA, JAD, RC, DIF and AIB designed and performed the experiments. ATA, SM, IV and MG and assisted with the experiments. SJW, GB, CP, PM and AY conducted the lithium human trial. PL and AIB integrated the data and wrote the drafts of the manuscript. All authors edited the manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
Dr Finkelstein is a paid scientific consultant for Prana Biotechnology. Dr Bush is a shareholder in Prana Biotechnology, Eucalyptus, Mesoblast, Brighton Biotech and Nextvet, and a paid consultant for Collaborative Medicinal Developments. Dr Pantelis has participated on Advisory Boards for Janssen-Cilag, Astra-Zeneca, Lundbeck and Servier. He has received honoraria for talks presented at educational meetings organized by Astra-Zeneca, Janssen-Cilag, Eli Lilly, Pfizer, Lundbeck and Shire. Dr McGorry receives unrestricted research funding from Astra-Zenica, Eli Lilly, Janssen-Cilag, Pfizer and Novartis, as well as honoraria for educational activities with Astra-Zenica, Eli Lilly, Janssen-Cilag, Pfizer, Bristol Myer Squibb, Roche and the Lunbeck Institute. The remaining authors declare no conflicts of interest.
Additional information
Supplementary Information accompanies the paper on the Molecular Psychiatry website
Supplementary information
Rights and permissions
About this article

Cite this article
Lei, P., Ayton, S., Appukuttan, A. et al. Lithium suppression of tau induces brain iron accumulation and neurodegeneration. Mol Psychiatry 22, 396–406 (2017). https://doi.org/10.1038/mp.2016.96
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mp.2016.96
- Springer Nature Limited
This article is cited by
-
Tau in neurodegenerative diseases: molecular mechanisms, biomarkers, and therapeutic strategies
Translational Neurodegeneration (2024)
-
Friend or foe: role of pathological tau in neuronal death
Molecular Psychiatry (2023)
-
Perturbed iron biology in the prefrontal cortex of people with schizophrenia
Molecular Psychiatry (2023)
-
Alzheimer’s Disease: Novel Targets and Investigational Drugs for Disease Modification
Drugs (2023)
-
Tau suppresses microtubule-regulated pancreatic insulin secretion
Molecular Psychiatry (2023)