
Abstract
The process of blood formation, haematopoiesis, depends upon a small number of haematopoietic stem cells (HSCs) that reside in the bone marrow. Differentiation of HSCs is characterised by decreased expression of genes associated with self-renewal accompanied by a stepwise activation of genes promoting differentiation. Lineage branching is further directed by groups of cooperating and counteracting genes forming complex networks of lineage-specific transcription factors. Imbalances in such networks can result in blockage of differentiation, lineage reprogramming and malignant transformation. CCAAT/enhancer-binding protein-α (C/EBPα) was originally identified 30 years ago as a transcription factor that binds both promoter and enhancer regions. Most of the early work focused on the role of C/EBPα in regulating transcriptional processes as well as on its functions in key differentiation processes during liver, adipogenic and haematopoietic development. Specifically, C/EBPα was shown to control differentiation by its ability to coordinate transcriptional output with cell cycle progression. Later, its role as an important tumour suppressor, mainly in acute myeloid leukaemia (AML), was recognised and has been the focus of intense studies by a number of investigators. More recent work has revisited the role of C/EBPα in normal haematopoiesis, especially its function in HSCs, and also started to provide more mechanistic insights into its role in normal and malignant haematopoiesis. In particular, the differential actions of C/EBPα isoforms, as well as its importance in chromatin remodelling and cellular reprogramming, are beginning to be elucidated. Finally, recent work has also shed light on the dichotomous function of C/EBPα in AML by demonstrating its ability to act as both a tumour suppressor and promoter. In the present review, we will summarise the current knowledge on the functions of C/EBPα during normal and malignant haematopoiesis with special emphasis on the recent work.
Similar content being viewed by others

Introduction
Proper haematopoietic differentiation requires a strict pattern of spatiotemporal gene transcription, which is orchestrated by intricate interactions of proteins with promoter and enhancer regions. In particular, binding of transcription factors to enhancer regions has been shown to facilitate the recruitment of RNA polymerase II to the target gene promoter and to induce epigenetic modifications that make the chromatin more accessible.1 Work performed in embryonic stem cells has shown that transcription factors bind enhancers in a sequential manner and that embryonic stem cell-specific transcription factors establish an active epigenetic state at tissue-specific regulatory elements.2 These factors are subsequently replaced by lineage-specific transcription factors that are able to induce the proper gene expression pattern of differentiating cells. Thus, dynamic binding of transcription factors appears to be mediated by the cell type-specific chromatin environment where extracellular cues are routed to intracellular transcription in a manner dependent on the recruitment of lineage-specific transcription factors to regulatory elements.
CCAAT/enhancer-binding protein-α (C/EBPα) is the founding member of a family of six transcription factors: C/EBP-α, -β, -γ, -δ, -ɛ and -ζ, which all share a basic region leucine zipper C-terminal domain. The C-terminal DNA-binding domain consists of an 86-residue α-helical structure with a leucine-rich hydrophobic part that allows for homo- or heterodimerization with family members and other proteins with a similar structure,3, 4 and simultaneously positions the basic region in the major groove enabling efficient binding to its cognate site on the DNA.5, 6
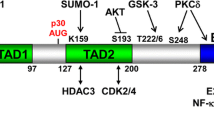
CEBPA is an intronless gene located on chromosome 19q13.1 in humans. The single mRNA transcribed from the CEBPA gene is translated into two isoforms (Figure 1), due to alternative start site usage resulting in the full-length C/EBPα (42 kDa; p42) and a shorter, N-terminal truncated isoform (30 kDa; p30). P30 lacks the first 117 amino acids of full-length p42, which includes the transactivation domain 1 (TAD1). TAD1 has been shown to regulate transcriptional activation through interactions with components of the RNA polymerase II preinitiation complex, including the TATA-box-binding protein (TBP) and the transcription factor IIB.7 TAD1 has also been implicated in C/EBPα-mediated repression of E2F activity, which has a role in the ability of C/EBPα to control cell cycle progression.8, 9, 10 The ratio between p30 and p42 is regulated at the level of translation by eIF2a/eIF3E. Abundance of nutrients and growth factors results in the activation of eIF2a/eIF3E, increasing the p30/p42 ratio through the use of alternative translational start site usage. Thus, increased translation of p30 promotes proliferation in favourable conditions.11

The functional domains of C/EBPα. The C-terminal basic region leucine zipper (BR-LZ) domain mediates DNA-binding and protein–protein interactions with transcription factors associated with differentiation and cell cycle control. Domains involved in transcriptional activation and E2F repression are lost in the truncated form of C/EBPα, whereas binding sites for proteins such as SWI/SNF and CDK2/CDK4 are retained in p30.
Both C/EBPα isoforms share the TAD2 that mediates antimitotic effects through direct interactions with p21(ref. 12) and regulates proliferation through interactions with the chromatin remodelling complex SWI/SNF.13 In addition, proliferative arrest has been suggested to be mediated through inhibition of Cdk2/Cdk4.14 However, more recent work has shown that deletion of the Cdk2/Cdk4-binding domains of Cebpa in mice has no effect neither on proliferation nor liver development.15
The transcriptional regulation mediated by C/EBPα is essential for proper differentiation of cells in various tissues. Wang et al.16 were the first to demonstrate how non-conditional disruption of Cebpa affects liver development in the foetus. Thus, Cebpa deletion is perinatal lethal as a consequence of imbalanced energy homeostasis due to perturbed hepatic glycogenesis.16 Furthermore, newborn Cebpa−/− mice exhibit impaired adipocyte maturation, accumulate immature myeloid cells and lack mature granulocytes.17 In the haematopoietic system, C/EBPα is most prominently expressed in myeloid progenitor cells and its expression is subsequently downregulated as the cells differentiate (Figure 2). Hence, conditional deletion of Cebpa in the adult haematopoietic system of mice disrupts the transition between common myeloid progenitors and granulocyte monocyte progenitors (GMPs). Notably, although Cebpa−/−mice accumulate immature myeloid progenitors, they do not develop acute myeloid leukaemia (AML).18

The role of C/EBPα in normal haematopoietic differentiation. (a) The haematopoietic hierarchy showing the expression of Cebpa. Expression data is indicated in colour (low green; red high) and derives from http://servers.binf.ku.dk/hemaexplorer/.103 (b) The function of C/EBPα in cellular plasticity exemplified by the transdifferentiation of B/T cells towards monocytes induced by overexpression C/EBPα as well as by the enhancement of induced pluripotent stem cell (iPSC) generation, similarly mediated by overexpression C/EBPα (see main text for additional details).
C/EBPα in myeloid differentiation
Myelopoiesis is the process by which myeloid progenitor cells differentiate into myeloid cells such as granulocytes (eosinophilic, basophilic and neutrophilic) and monocytes. The above-mentioned essential role of C/EBPα in this developmental process has spurred an extensive interest in deciphering the mechanisms by which C/EBPα mediates this process in cooperation with other myeloid transcription factors (reviewed in Friedman19).
Myeloid differentiation is primed at an early stage by PU.1(refs 20, 21) and RUNX1(ref. 22) and is further directed by C/EBP family members.17, 18 Additional factors such as IRF8,23 GFI1(ref. 24) and SCL/TAL1(ref. 25) have been shown to have a role in myeloid development. Terminal myeloid differentiation is partly mediated by extrinsic signalling through the cytokines granulocyte colony-stimulating factor and macrophage colony-stimulating factor, which instruct monocyte and granulocyte differentiation, respectively.26
The E-twenty-six- family member PU.1 is a transcription factor expressed in early progenitor cells and is required for the generation of both common myeloid progenitors and common lymphoid progenitors, as well as the terminal differentiation of macrophages and B cells. Interestingly, PU.1 possesses the rare ability of binding to tightly packed chromatin and DNA in nucleosome structures and induces changes in the chromatin conformation. Thus, PU.1 acts as a priming factor (pioneer factor) during haematopoietic maturation by generating cell-type-specific regions of open chromatin in cis-regulatory elements that serve as beacons for binding additional transcription factors and chromatin remodellers. Moreover, myeloid specification is mediated by colocalization of PU.1 with C/EBPα.27 However, this effect appears to be dose-dependent as PU.1 and C/EBPα reciprocally regulate the expression of one another.28, 29, 30, 31 Specifically, C/EBPα enhances PU.1 expression, by directly binding to the SPI1 promoter and the −14 kb enhancer,28, 29 and represses PU.1 activity by displacing its cofactor c-Jun.30 The PU.1:C/EBPα ratio has also been proposed to instruct terminal differentiation of myeloid progenitor cells. Indeed, inducible expression of PU.1 and C/EBPα in Sfpi1−/− cells (PU.1 deficient) shows that a high PU.1:C/EBPα ratio induces macrophage development, whereas a lower ratio directs the cells towards granulocytic maturation.31 Furthermore, heterodimerization of C/EBPα with the AP-1 proteins c-Jun and c-Fos weakens the affinity for several canonical C/EBPα target genes while retaining specificity for PU.1, ultimately leading to skewing towards terminal monocytic differentiation.3 Hence, C/EBPα appears to affect myeloid differentiation through several layers of regulation.
Studies aiming at investigating the specific function and regulation of C/EBPα during differentiation have been complicated by disparate results in different model systems. Studies of lineage determination of myeloid cells have primarily been performed in a bipotent myeloid leukaemic cell lines, and similar experiments performed in vivo have often shown diverging results. As an example, post-translational modifications of C/EBPα residues are thought to induce conformational changes of the protein that would alter the activity of the transactivation domain. However, whereas serine 248 phosphorylation was found to induce terminal granulocytic differentiation in myeloid cell lines,32 substitution of serine 248 to alanine in knock-in mice does not affect terminal myeloid differentiation in young mice.33 Similarly, exogenous C/EBPα directs granulocytic maturation of bipotent myeloid cell lines,34 whereas ectopically expressed Cebpa in primary murine myeloid35 or lymphoid36, 37 progenitor cells induce monocytic maturation. Collectively, differentiation is a complex process, dependent on an intricate network of intrinsic and extrinsic factors, and there is a need for caution when drawing conclusions solely based on work performed in one model system. Thus, the ability of C/EBPα to drive context-specific differentiation of cells in diverse tissues is not only a matter of proper dosing but also depends upon the collaborating actions of additional transcription factors.
Novel functions for C/EBPα in HSCs
Although C/EBPα is mainly considered a lineage-instructive factor crucial for myeloid differentiation, recent data has shown that it also has an important role in haematopoietic stem cells (HSCs). HSCs exhibits a low but robust expression of Cebpa, and consistently Cebpa−/− mice display HSC phenotypes. Specifically, the Tenen Lab reported that the conditional loss of Cebpa in adult HSCs leads to an immediate expansion of functional HSCs, which was associated with an increase in proliferation.38 Further, it was shown that Cebpa−/− HSCs upregulated a foetal HSC gene expression programme as well as N-Myc, and that the latter was responsible for the increase in HSC proliferation. Using a similar system, i.e. Mx1-Cre mediated deletion of Cebpa, we recently showed that C/EBPα deletion was associated with a dramatic loss in HSC self-renewal.39 In fact, Cebpa−/− HSCs were lost in secondary recipients and Cebpa−/− bone marrow donor cells failed to rescue irradiated mice in non-competitive serial transplantation experiments. Further, we were able to show that Cebpa−/− HSCs displayed a marked increase in markers of DNA damage and apoptosis. Finally, using chromatin immunoprecipitation and sequencing analysis in HSCs and multipotent progenitor cells, we demonstrated that C/EBPα bound to genes destined for expression later during myeloid differentiation. As these regions are considered inaccessible in HSC/multipotent progenitor cells, this could potentially reflect the ability of C/EBPα to function as a pioneer factor.
Recently developed Cebpa reporter mouse lines, driven by the entire endogenous Cebpa regulatory regions or a +37 kb myeloid-specific enhancer, have also been informative with respect to the function of C/EBPα in HSCs.40, 41 In both lines, only a fraction of long-term HSCs express Cebpa (4–20%), suggesting that Cebpa expression marks a minor subset of LT-HSCs. Interestingly, one of these reports demonstrated that essentially all reconstituting activity was found in the Cebpa-expressing compartment, suggesting that C/EBPα is indeed essential for long-term HSC function, at least during haematopoietic reconstitution.40
The above-mentioned discrepancies between the Cebpa−/− phenotypes reported by our group and the Tenen group may reflect, in part, differences in the timing of analysis relative to the polyinosinic:polycytidylic acid-mediated activation of the Mx1-Cre driver. Specifically, because of the earlier time point generally used in the Tenen report, their analyses are focused on the effects of Cebpa deletion in actively cycling HSCs as a consequence of the impact of interferon signalling on HSCs.42 Indeed, loss of Cebpa in this scenario may convert adult HSCs to a more foetal-like state as suggested by the Tenen group.38 In any event, both reports clearly demonstrate the importance of intact C/EBPα function for the proper control of HSC numbers and functions.
C/EBPα levels or function is perturbed in human AML
AML is a clonal disorder that arises through the acquisition of genetic and epigenetic alterations ultimately leading to changes in the transcriptional wiring of the leukaemic cells and/or loss of cell identity. In line with this, mutations in transcription factors and epigenetic regulators with roles in normal haematopoietic development are among the most frequent aberrations detected in human AML patients.43, 44
Mutations in CEBPA in human AML were first reported by Pabst et al.45 and have later been described in numerous studies.46, 47, 48, 49, 50, 51, 52, 53, 54, 55 CEBPA mutations mainly fall into two classes. The first class involves the C-terminal part of C/EBPα, and consistently these mutations interfere with the DNA-binding and/or dimerisation properties of the protein.56, 57 The second class is located in the 5′ end of the gene and frequently disturbs the open reading frame. Most of these mutations reside between the two ATGs initiating the respective synthesis of the p42 and p30 isoforms, and, consequently, lead to the exclusive expression of the p30 isoform from the affected allele.
CEBPA mutations in human AML have been shown to be either mono- or biallelic. Monoallelic CEBPA mutations are associated with a plethora of other genetic lesions and are generally heterogeneous with respect to gene expression and prognosis.49, 58, 59, 60 In contrast, biallelic CEBPA mutant AML constitutes a distinct subtype associated with good prognosis and was recognised as such in the recent World Health Organisation classification.61 Further support for this comes from gene expression studies, which also classify biallelic CEBPA mutant AML as a distinct disease entity.48, 49, 58, 59, 60, 62 Biallelic CEBPA mutant AMLs preferentially combine an N-terminal mutation on one allele (sustaining the expression of p30 only) with a C-terminal mutation on the other (deficient in dimerization/DNA binding).54 As C/EBPα functions as a dimer, this implies that the only C/EBPα dimers able to bind DNA will be p30/p30 homodimers in AML with this combination of mutations. The remaining biallelic CEBPA mutant AMLs either combine two N-terminal mutations or an N-terminal mutation with a frameshift/nonsense mutation in the central part of CEBPA. As the latter mutations encode C/EBPα variants lacking the basic region leucine zipper domain, these combinations will result in the formation of p30/p30 homodimers as the only C/EBPα entities capable of binding DNA. Hence, these findings underscore the importance of both the presence of the p30 isoform and lack of functional p42 isoform in CEBPA mutant AML (Figure 3).

Differential actions of the p42 and p30 isoforms of C/EBPα. (a) Generally, the p42 isoform is more abundant than the p30 isoform in normal cells. Hence, the most prevalent C/EBPα entities are p42/p42 homodimers (as indicated by size). The presence of the TAD1 domain allows this isoform to interact with, and repress the activity of, E2F family members, which is key to its ability to repress proliferation. (b) In biallelic CEBPA mutant AML, the selective loss of functional p42 leads to the preferential formation of p30/p30 homodimers. (c) The potential molecular consequences of p42/p42 or p30/p30 expression (see main text for additional details).
Biallelic CEBPA mutant AML exhibits a distinct secondary mutational spectrum that sets this class aside from other AML subtypes.54 Although this subtype frequently exhibits mutations in well-known leukaemic players such as ASXL1, RUNX1 and TET2, these events are clearly under-represented when compared with other AML subtypes. On the other hand, mutations in WT1 and, in particular, GATA2 are highly enriched in biallelic CEBPA mutant AML, suggesting a specific collaboration between these lesions and mutated CEBPA.54 Interestingly, this pattern is also recapitulated in the rare cases of familial CEBPA mutant AML involving families of heterozygous carriers of N-terminal CEBPA mutations.63 Here, progression to AML is associated with acquisition of a C-terminal mutation on the remaining CEBPA allele as well as with frequent lesions in GATA2 or WT1.64 Collectively, these findings demonstrate that biallelic CEBPA mutant AML constitutes a distinct AML entity.
The involvement of C/EBPα in human AML is not restricted to CEBPA mutant AML. Methylation of the distal CEBPA promoter was shown to correlate inversely with the expression levels of CEBPA, but did not have any prognostic value in normal karyotype AML.65 However, the finding that CEBPA promoter methylation and mutations in the coding region of CEBPA are mutually exclusive implies that these events perturb similar cellular pathways. This is reinforced by the clustering of the transcriptional profiles derived from human CEBPA silent and biallelic mutant AML.66
Oncogenic driver proteins have also frequently been implicated in downregulating the expression of CEBPA in AML. One example is AML-ETO (t(8;21)), which downregulates the expression of CEBPA, most likely by interfering with an autoregulatory loop sustaining its expression.67, 68 At the post-transcriptional level, RNA-binding proteins such as hnRNP E2 and calreticulin expressed in blast crisis chronic myeloid leukaemia and AML1-MDS1-EVI (t(3;21))/ -MYH11 (inv16) in AML, respectively, have been shown to interfere with the translation of the CEBPA mRNA.69, 70, 71, 72 Finally, a number of oncogenic lesions also interfere with the function/levels of the C/EBPα protein (reviewed in Mueller and Pabst73). One particularly interesting case is the role of TRIB2, which was found to be overexpressed in a distinct subtype of AML frequently harbouring mutations in NOTCH1.74, 75 TRIB2 is a potent oncogene and its overexpression in mouse haematopoietic stem and progenitor cells results in AML. Gene expression analysis further revealed that the TRIB2-high human AML clustered with biallelic CEBPA mutant AML, suggesting that they operate in similar pathways.75 Indeed, TRIB2 was found to mediate the degradation of the p42 isoform of C/EBPα leading to a skewed ratio of p42/p30, resulting in the preferential formation of p30/p30 homodimers. This, in turn, explains the resemblance between biallelic CEBPA mutant and TRIB2-high AML.74, 76 Collectively, these findings demonstrate that a large fraction of human AMLs converges at downregulating the level and/or interferes with the function of C/EBPα, thereby unambiguously classifying it as a key myeloid tumour.
The requirement for C/EBPα in AML development
Despite the numerous pathways by which C/EBPα levels and functions are perturbed in human AML, cases with a complete lack of C/EBPα have yet to be reported.77 This is consistent with the lack of leukaemia development in Cebpa−/− mice and suggests that in addition to its well-established tumour-suppressive function, C/EBPα may actually also be required for the development of AML.78 Consistently, forced expression of the fusion oncogene BCR-ABL in Cebpa-deficient murine foetal liver cells results in erythroleukaemia instead of the chronic myeloid leukaemia, which normally arises in this setting.79 These findings formed the basis of a model where the block in myeloid differentiation upstream of the GMP leads to a failure of Cebpa−/− progenitors to reach a stage of myeloid identity, which may be a prerequisite of leukaemic transformation.
We have recently put this model to the test by assessing the requirement of C/EBPα in MLL-ENL-mediated transformation.80 Here we find that C/EBPα is absolutely required for the initiation of MLL fusion-driven AML and that even deletion of Cebpa at the GMP stage completely abrogates transformation. These findings suggest that it is not the lack of myeloid identity per se that underlies the requirement for C/EBPα in AML development, but more likely its ability to collaborate with MLL-ENL (and perhaps other fusion oncogenes/transcriptional regulators) in initiating the expression of a transcriptional programme required for leukaemic transformation. Strikingly, the deletion of C/EBPα in already established MLL-ENL-driven leukaemias had absolutely no impact in terms of the expression of this transcriptional programme and/or the properties of the leukaemic cells.
The demonstrated requirement for C/EBPα in the development of myeloid leukaemia was recently revisited.81 Consistent with the findings detailed above, leukaemic transformation mediated by MLL-AF9 was found to be strictly dependent on the presence of C/EBPα, whereas already established AMLs were refractory to C/EBPα loss. Interestingly, when mice transplanted with MLL-AF9-expressing Cebpa−/− progenitors were subjected to hydrodynamic injection of interleukin-3 and granulocyte–macrophage colony-stimulating factor-encoding vectors, Cebpa−/− AML readily developed. As interleukin-3 and granulocyte–macrophage colony-stimulating factor mediate formation of GMPs in a C/EBPα-independent manner (in a process termed emergency granulopoiesis82), the authors concluded that leukaemic transformation was dependent on the formation of GMPs and not C/EBPα per se. However, as emergency granulopoiesis upregulates the expression of Cebpb and is abrogated by Cebpb knockdown, it is likely that C/EBPβ may functionally substitute C/EBPα as has indeed been reported earlier.82, 83 Also, the report fails to explain why Cebpa−/− GMPs are refractory to leukaemic transformation.80
The findings reported above raise the possibility that C/EBPα may act as a so-called pioneer factor that facilitates access/remodelling at key regulatory elements, thereby enabling access of other factors such as MLL-ENL. Once these elements are remodelled, C/EBPα is no longer required for the function of these regulatory elements, consistent with the lack of phenotype of removing C/EBPα in already established AML.80 Consistent with this notion, C/EBPα has been reported to collaborate with HOXA9 in HOXA9-dependent AML.84 The function of C/EBPα as a potential pioneer factor also appears to extend to non-haematopoietic systems, specifically adipocyte differentiation, where C/EBPα was demonstrated to facilitate the binding of the adipogenic master regulator peroxisome proliferator-activated receptor-γ to compacted chromatin.85 This property of C/EBPα was dependent on its SWI/SNF interaction domain, which has also previously been found to be essential for C/EBPα-mediated myeloid differentiation.86
Collectively, several lines of evidence suggest that, in addition to its tumour-suppressive function, C/EBPα is also required for the development of AML, most likely through its ability to collaborate with other transcriptional regulators and by promoting chromatin remodelling.
The promotion of leukaemic transformation by mutations in CEBPA
Mutations in CEBPA are associated with leukaemic transformation, but how does these mutations mediate their effect? Mouse models have been instrumental in the efforts to answer this question, and the first Cebpa mutant lines, where an AML-like disease was reported, harboured mutations in the basic region of C/EBPα. Specifically, these mutations abrogated the interactions with members of the E2F family of cell cycle regulators without interfering with the ability of C/EBPα to bind DNA.9 These animals developed a number of conditions ranging from neutropenia, over myeloproliferation to an AML-like syndrome with limited peripheral involvement, presumably mediated by the acquisition of secondary mutations.87 Although similar mutations were not identified in human AML (in the patient studies that followed), these findings underlined the importance of C/EBPα-mediated repression of E2F activity in protecting against aberrant myelopoiesis.
As discussed above, the most common group of CEBPA mutations in human AML leads to the expression of the p30 isoform of C/EBPα. Consistently, knock-in mice homozygous for an allele of Cebpa that exclusively express this isoform (CebpaN/N) all succumb to full-blown AML within the first year of their lives.88 Remarkably, but consistent with data from retroviral transduction models, the leukaemic stem cells in these models are residing in a population of committed progenitors,89 thus challenging the previous dogma that leukaemic stem cell were residing in the HSC compartment.90 Indeed, recent efforts characterizing the cellular compartments that harbour oncogenic driver mutations in human AML have confirmed that the cell-of-origin, and consequently the leukaemic stem cell, is residing in populations downstream of HSCs.91, 92
Following the characterisation of the CebpaN/N strain, the Nerlov group developed a mouse line where they combined the CebpaN allele with an allele expressing a C-terminal mutation unable to bind DNA (CebpaC), thereby mimicking the most frequent combination of mutations in CEBPA mutant AML.93 The N/C combination promoted leukaemic development with a shortened latency as compared with the N/N combination, and this appeared to be conferred by an expansion of the HSC compartment in the preleukaemic state. Such an expansion may favour the acquisition of secondary mutations in downstream populations, thus leading to a faster developing AML.
As discussed above, both the N/N and the N/C combinations are predicted to lead to the formation of p30/p30 homodimers as the only C/EBPα entity capable of binding DNA. So what underlie the differences in latencies between N/N and N/C murine leukaemias? One potential explanation could be that, although the C-terminally mutated C/EBP variants are unable to bind DNA, they may still bind to C/EBP-interacting proteins such as PU.1 and/or SWI/SNF complexes.94 Thus, the expression of the CebpaC allele could potentially reduce the amount of C/EBP activity in the cell, thereby reducing the ability of other C/EBPs to rescue the effects of expressing the aberrant C/EBPα variants.
Recently, a more advanced model of CEBPA mutant AML was constructed by combining the CebpaN/C mice with mutations in tyrosine kinase FLT3.95 FLT3 mutations are frequent in human AML and are found in combination with biallelic CEBPA mutations, although not enriched in this subtype.54 Consistent with its leukaemogenic role, FLT3 mutations further accelerate disease development in CebpaN/C mice, underlining the importance of developing more refined models of CEBPA mutant AML. In particular, it would be interesting to combine the current model with mutations in GATA2 and WT1, both of which are particularly enriched among biallelic CEBPA mutant cases.54
Both mouse and patient studies clearly indicate a central role for the p30 form of C/EBPα in the development of AML and suggest that the formation of p30/p30 homodimers are crucial in mediating oncogenic transformation. It is well established that loss of C/EBPα-mediated E2F cell cycle control, either by the expression of basic region mutant/p30 isoforms of C/EBPα or through its complete loss, leads to a block in myeloid differentiation. However, the differences in the ability of these genetic aberrations to support leukaemic development suggest that the role of C/EBPα in cell cycle progression is not the only factor governing the tumour suppressor functions of WT C/EBPα.
Being a transcription factor, it is likely that the normal gene regulatory properties of WT C/EBPα are affected not only by the complete loss of all C/EBPα isoforms but also by the preferential expression of p30/p30 homodimers that is observed in AML. Loss of C/EBPα was recently shown to lead to the upregulation of Cebpg, both in CEBPA-silenced AML and in murine Cebpa−/− stem and progenitor cells.96 Interestingly, knockdown of Cebpg in both these systems promoted neutrophilic differentiation demonstrating that C/EBPα-mediated repression of Cebpg has a role in the differentiation arrest induced by its loss in both CEBPA-silenced AML and during normal myeloid differentiation. Moreover, C/EBPα has also been shown to repress the expression of Sox4, a supposed oncogene in several cancer types.66 Strikingly, not only did knockdown of Sox4 inhibit the increased self-renewal and lack of myeloid differentiation observed in Cebpa−/− murine stem and progenitors but it also extinguished the self-renewal of leukaemic stem cells derived from murine CebpaN/C AML. Consistent with these findings, Sox4 expression was found to be upregulated in both human CEBPA-silenced AML and in human CEBPA mutant AML. Finally, overexpression of CebpaC in a bone marrow transplantation model has recently been shown to repress the expression of Csf1r.97 However, a potential tumour suppressor function of Csf1r was not supported by its overexpression in the CebpaC overexpression setting. On the contrary, accelerated AML development was observed. The extent to which this reflects this particular model is not clear, but the potential functional interaction between C/EBPα and CSF1R in AML warrants further investigation.
These examples aside, we know very little about global differences between p42/p42- and p30/p30-mediated transcriptional regulation, and a challenge for the future will be to address this issue using genome-wide approaches. Specifically, are we able to find genes that are deregulated through loss of p42 binding, and/or does p30 bind to sites that are also occupied by p42 in normal progenitors? Or do p42 and p30 generally share the same binding patterns, only differing in their ability to control the expression of the cognate genes? Indeed, given the recent demonstration of the selective binding of p30/p30 dimers to WDR5, differential cofactor binding of the two C/EBPα isoforms should be investigated further.98 Such approaches will lead to the identification of genes controlling leukaemic properties and potentially to genes that could be targeted therapeutically.
C/EBPα as a pioneer factor and a regulator of cell identity
Throughout this review, we have described several instances alluding to the ability of C/EBPα to drive rearrangements of closed chromatin (Figure 4). These examples include the binding of C/EBPα to myeloid lineage-affiliated genes in HSCs/multipotent progenitor cells, the ability of C/EBPα to mediate the binding of peroxisome proliferator-activated receptor-γ to compacted chromatin in mouse embryonic fibroblasts and the requirement for C/EBPα for MLL fusion proteins to access chromatin during AML development.39, 80, 85 Moreover, C/EBPα has been found to mediate robust transdifferentiation of both T and B cells into macrophages, that is, a process involving major rearrangements of both chromatin and gene expression patterns.36, 99, 100, 101 However, most strikingly, C/EBPα has been shown to enhance (>100-fold) the ability of the four Yamanaka factors to reprogramme B cells to induce pluripotent stem cells in a process mediated, in part, by the epigenetic regulator TET2.100, 102

C/EBPα as a pioneer factor. (a) C/EBPα has the potential to remodel chromatin through its ability of interacting with members of the SWI/SNF complexes.86 During adipose differentiation, this modelling allows for binding of another key adipogenic factor peroxisome proliferator-activated receptor (PPARγ).85 (b) C/EBPα is also required for MLL-ENL-mediated transformation to AML, presumably through its ability to recruit MLL-ENL to specific genomic loci.80 (c) Enforced expression of C/EBPα greatly enhances the reprogramming of haematopoietic progenitor cells to induced pluripotent stem cells (iPSCs). Presumably, C/EBPα binds to and facilitates the opening of loci, which are normally inaccessible in these progenitor cells. In addition, C/EBPα enhances the expression of TET2 leading to further transcriptional derepression through TET2-mediated hydromethylation.100, 102
Collectively, these findings suggest that C/EBPα possesses some unique properties allowing it to interact with compacted chromatin in processes with importance for normal differentiation and for the development of cancer. Molecularly, these properties have been associated with its ability to recruit SWI/SNF complexes and to induce the expression of TET2. A task for the future will be to understand the underlying mechanisms of C/EBPα-mediated chromatin remodelling in the different settings outlined above.
Concluding remarks
Here we have reviewed the current knowledge on C/EBPα in normal and malignant haematopoiesis. Strikingly, whereas the p42 isoform of this apparently uncomplicated transcription factor acts as a tumour suppressor in AML, it is equally required for the development of at least one subtype of this disease. The relative balance between context-dependent actions of C/EBPα in AML should be addressed in future studies. An equally important question to address is how the truncated p30 isoform of C/EBPα promotes AML. Does it bind selectively to some target genes, does it interact differently with various cofactors and/or does it perturb the functions of other bZIP transcription factors by heterodimerization? Finally, the exact mechanisms by which C/EBPα functions in HSCs and during myeloid differentiation still need to be fully established. Addressing all these questions has the potential of uncovering the mechanisms by which C/EBPα governs AML development and may thus potentially uncover novel targets for therapeutic intervention in AML with C/EBPα involvement.
References
Ong CT, Corces VG . Enhancer function: new insights into the regulation of tissue-specific gene expression. Nat Rev Genet 2011; 12: 283–293.
Liber D, Domaschenz R, Holmqvist PH, Mazzarella L, Georgiou A, Leleu M et al. Epigenetic priming of a pre-B cell-specific enhancer through binding of Sox2 and Foxd3 at the ESC stage. Cell Stem Cell 2010; 7: 114–126.
Cai DH, Wang D, Keefer J, Yeamans C, Hensley K, Friedman AD . C/EBP alpha:AP-1 leucine zipper heterodimers bind novel DNA elements, activate the PU.1 promoter and direct monocyte lineage commitment more potently than C/EBP alpha homodimers or AP-1. Oncogene 2008; 27: 2772–2779.
Hattori T, Ohoka N, Inoue Y, Hayashi H, Onozaki K . C/EBP family transcription factors are degraded by the proteasome but stabilized by forming dimer. Oncogene 2003; 22: 1273–1280.
Ramji DP, Foka P . CCAAT/enhancer-binding proteins: structure, function and regulation. Biochem J 2002; 365: 561–575.
Miller M, Shuman JD, Sebastian T, Dauter Z, Johnson PF . Structural basis for DNA recognition by the basic region leucine zipper transcription factor CCAAT/enhancer-binding protein alpha. J Biol Chem 2003; 278: 15178–15184.
Nerlov C, Ziff EB . CCAAT/enhancer binding protein-alpha amino acid motifs with dual TBP and TFIIB binding ability co-operate to activate transcription in both yeast and mammalian cells. EMBO J 1995; 14: 4318–4328.
Johansen LM, Iwama A, Lodie TA, Sasaki K, Felsher DW, Golub TR et al. c-Myc is a critical target for c/EBPalpha in granulopoiesis. Mol Cell Biol 2001; 21: 3789–3806.
Porse BT, Pedersen TA, Xu X, Lindberg B, Wewer UM, Friis-Hansen L et al. E2F repression by C/EBPalpha is required for adipogenesis and granulopoiesis in vivo. Cell 2001; 107: 247–258.
Slomiany BA, D’Arigo KL, Kelly MM, Kurtz DT . C/EBPalpha inhibits cell growth via direct repression of E2F-DP-mediated transcription. Mol Cell Biol 2000; 20: 5986–5997.
Calkhoven CF, Muller C, Leutz A . Translational control of C/EBPalpha and C/EBPbeta isoform expression. Genes Dev 2000; 14: 1920–1932.
Timchenko NA, Wilde M, Nakanishi M, Smith JR, Darlington GJ . CCAAT/enhancer-binding protein alpha (C/EBP alpha) inhibits cell proliferation through the p21 (WAF-1/CIP-1/SDI-1) protein. Genes Dev 1996; 10: 804–815.
Muller C, Calkhoven CF, Sha X, Leutz A . The CCAAT enhancer-binding protein alpha (C/EBPalpha) requires a SWI/SNF complex for proliferation arrest. J Biol Chem 2004; 279: 7353–7358.
Wang H, Iakova P, Wilde M, Welm A, Goode T, Roesler WJ et al. C/EBPalpha arrests cell proliferation through direct inhibition of Cdk2 and Cdk4. Mol Cell 2001; 8: 817–828.
Schuster MB, Porse BT . C/EBPalpha: a tumour suppressor in multiple tissues? Biochim Biophys Acta 2006; 1766: 88–103.
Wang ND, Finegold MJ, Bradley A, Ou CN, Abdelsayed SV, Wilde MD et al. Impaired energy homeostasis in C/EBP alpha knockout mice. Science 1995; 269: 1108–1112.
Zhang DE, Zhang P, Wang ND, Hetherington CJ, Darlington GJ, Tenen DG . Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc Natl Acad Sci USA 1997; 94: 569–574.
Zhang P, Iwasaki-Arai J, Iwasaki H, Fenyus ML, Dayaram T, Owens BM et al. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity 2004; 21: 853–863.
Friedman AD . C/EBPalpha in normal and malignant myelopoiesis. Int J Hematol 2015; 101: 330–341.
Back J, Allman D, Chan S, Kastner P . Visualizing PU.1 activity during hematopoiesis. Exp Hematol 2005; 33: 395–402.
DeKoter RP, Singh H . Regulation of B lymphocyte and macrophage development by graded expression of PU.1. Science 2000; 288: 1439–1441.
Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR . AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 1996; 84: 321–330.
Tamura T, Thotakura P, Tanaka TS, Ko MS, Ozato K . Identification of target genes and a unique cis element regulated by IRF-8 in developing macrophages. Blood 2005; 106: 1938–1947.
Hock H, Hamblen MJ, Rooke HM, Traver D, Bronson RT, Cameron S et al. Intrinsic requirement for zinc finger transcription factor Gfi-1 in neutrophil differentiation. Immunity 2003; 18: 109–120.
Shivdasani RA, Mayer EL, Orkin SH . Absence of blood formation in mice lacking the T-cell leukaemia oncoprotein tal-1/SCL. Nature 1995; 373: 432–434.
Rieger MA, Hoppe PS, Smejkal BM, Eitelhuber AC, Schroeder T . Hematopoietic cytokines can instruct lineage choice. Science 2009; 325: 217–218.
Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 2010; 38: 576–589.
Kummalue T, Friedman AD . Cross-talk between regulators of myeloid development: C/EBPalpha binds and activates the promoter of the PU.1 gene. J Leukocyte Biol 2003; 74: 464–470.
Yeamans C, Wang D, Paz-Priel I, Torbett BE, Tenen DG, Friedman AD . C/EBPalpha binds and activates the PU.1 distal enhancer to induce monocyte lineage commitment. Blood 2007; 110: 3136–3142.
Reddy VA, Iwama A, Iotzova G, Schulz M, Elsasser A, Vangala RK et al. Granulocyte inducer C/EBPalpha inactivates the myeloid master regulator PU.1: possible role in lineage commitment decisions. Blood 2002; 100: 483–490.
Dahl R, Walsh JC, Lancki D, Laslo P, Iyer SR, Singh H et al. Regulation of macrophage and neutrophil cell fates by the PU.1:C/EBPalpha ratio and granulocyte colony-stimulating factor. Nat Immunol 2003; 4: 1029–1036.
Behre G, Singh SM, Liu H, Bortolin LT, Christopeit M, Radomska HS et al. Ras signaling enhances the activity of C/EBP alpha to induce granulocytic differentiation by phosphorylation of serine 248. J Biol Chem 2002; 277: 26293–26299.
Hasemann MS, Schuster MB, Frank AK, Theilgaard-Monch K, Pedersen TA, Nerlov C et al. Phosphorylation of serine 248 of C/EBPalpha is dispensable for myelopoiesis but its disruption leads to a low penetrant myeloid disorder with long latency. PLoS One 2012; 7: e38841.
Radomska HS, Huettner CS, Zhang P, Cheng T, Scadden DT, Tenen DG . CCAAT/enhancer binding protein alpha is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol Cell Biol 1998; 18: 4301–4314.
Wang D, D’Costa J, Civin CI, Friedman AD . C/EBPalpha directs monocytic commitment of primary myeloid progenitors. Blood 2006; 108: 1223–1229.
Xie H, Ye M, Feng R, Graf T . Stepwise reprogramming of B cells into macrophages. Cell 2004; 117: 663–676.
Hsu CL, King-Fleischman AG, Lai AY, Matsumoto Y, Weissman IL, Kondo M . Antagonistic effect of CCAAT enhancer-binding protein-alpha and Pax5 in myeloid or lymphoid lineage choice in common lymphoid progenitors. Proc Natl Acad Sci USA 2006; 103: 672–677.
Ye M, Zhang H, Amabile G, Yang H, Staber PB, Zhang P et al. C/EBPa controls acquisition and maintenance of adult haematopoietic stem cell quiescence. Nat Cell Biol 2013; 15: 385–394.
Hasemann MS, Lauridsen FK, Waage J, Jakobsen JS, Frank AK, Schuster MB et al. C/EBPalpha is required for long-term self-renewal and lineage priming of hematopoietic stem cells and for the maintenance of epigenetic configurations in multipotent progenitors. PLoS Genet 2014; 10: e1004079.
Guo H, Ma O, Friedman AD . The Cebpa +37-kb enhancer directs transgene expression to myeloid progenitors and to long-term hematopoietic stem cells. J Leukoc Biol 2014; 96: 419–426.
Wolfler A, Danen-van Oorschot AA, Haanstra JR, Valkhof M, Bodner C, Vroegindeweij E et al. Lineage-instructive function of C/EBPalpha in multipotent hematopoietic cells and early thymic progenitors. Blood 2010; 116: 4116–4125.
Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature 2009; 458: 904–908.
Shih AH, Abdel-Wahab O, Patel JP, Levine RL . The role of mutations in epigenetic regulators in myeloid malignancies. Nat Rev Cancer 2012; 12: 599–612.
Abdel-Wahab O, Levine RL . Mutations in epigenetic modifiers in the pathogenesis and therapy of acute myeloid leukemia. Blood 2013; 121: 3563–3572.
Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S et al. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet 2001; 27: 263–270.
Leroy H, Roumier C, Huyghe P, Biggio V, Fenaux P, Preudhomme C . CEBPA point mutations in hematological malignancies. Leukemia 2005; 19: 329–334.
Schlenk RF, Dohner K, Kneba M, Gotze K, Hartmann F, Del Valle F et al. Gene mutations and response to treatment with all-trans retinoic acid in elderly patients with acute myeloid leukemia. Results from the AMLSG Trial AML HD98B. Haematologica 2009; 94: 54–60.
Schlenk RF, Dohner K, Krauter J, Frohling S, Corbacioglu A, Bullinger L et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med 2008; 358: 1909–1918.
Patel JP, Gonen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012; 366: 1079–1089.
Benthaus T, Schneider F, Mellert G, Zellmeier E, Schneider S, Kakadia PM et al. Rapid and sensitive screening for CEBPA mutations in acute myeloid leukaemia. Br J Haematol 2008; 143: 230–239.
Diaz-Beya M, Brunet S, Nomdedeu J, Tejero R, Diaz T, Pratcorona M et al. MicroRNA expression at diagnosis adds relevant prognostic information to molecular categorization in patients with intermediate-risk cytogenetic acute myeloid leukemia. Leukemia 2014; 28: 804–812.
Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, Zimmermann M, Peeters JK, Valk PJ et al. Characterization of CEBPA mutations and promoter hypermethylation in pediatric acute myeloid leukemia. Haematologica 2011; 96: 384–392.
Frohling S, Schlenk RF, Stolze I, Bihlmayr J, Benner A, Kreitmeier S et al. Mutations in younger adults with acute myeloid leukemia and normal cytogenetics: prognostic relevance and analysis of cooperating mutations. J Clin Oncol 2004; 22: 624–633.
Fasan A, Haferlach C, Alpermann T, Jeromin S, Grossmann V, Eder C et al. The role of different genetic subtypes of CEBPA mutated AML. Leukemia 2014; 28: 794–803.
Preudhomme C, Sagot C, Boissel N, Cayuela JM, Tigaud I, de Botton S et al. Favorable prognostic significance of CEBPA mutations in patients with de novo acute myeloid leukemia: a study from the Acute Leukemia French Association (ALFA). Blood 2002; 100: 2717–2723.
Gombart AF, Hofmann WK, Kawano S, Takeuchi S, Krug U, Kwok SH et al. Mutations in the gene encoding the transcription factor CCAAT/enhancer binding protein alpha in myelodysplastic syndromes and acute myeloid leukemias. Blood 2002; 99: 1332–1340.
Landschulz WH, Johnson PF, McKnight SL . The DNA binding domain of the rat liver nuclear protein C/EBP is bipartite. Science 1989; 243: 1681–1688.
Dufour A, Schneider F, Metzeler KH, Hoster E, Schneider S, Zellmeier E et al. Acute myeloid leukemia with biallelic CEBPA gene mutations and normal karyotype represents a distinct genetic entity associated with a favorable clinical outcome. J Clin Oncol 2010; 28: 570–577.
Pabst T, Eyholzer M, Fos J, Mueller BU . Heterogeneity within AML with CEBPA mutations; only CEBPA double mutations, but not single CEBPA mutations are associated with favourable prognosis. Br J Cancer 2009; 100: 1343–1346.
Rapin N, Bagger FO, Jendholm J, Mora-Jensen H, Krogh A, Kohlmann A et al. Comparing cancer vs normal gene expression profiles identifies new disease entities and common transcriptional programs in AML patients. Blood 2014; 123: 894–904.
Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009; 114: 937–951.
Wouters BJ, Lowenberg B, Erpelinck-Verschueren CA, van Putten WL, Valk PJ, Delwel R . Double CEBPA mutations, but not single CEBPA mutations, define a subgroup of acute myeloid leukemia with a distinctive gene expression profile that is uniquely associated with a favorable outcome. Blood 2009; 113: 3088–3091.
Smith ML, Cavenagh JD, Lister TA, Fitzgibbon J . Mutation of CEBPA in familial acute myeloid leukemia. N Engl J Med 2004; 351: 2403–2407.
Green CL, Tawana K, Hills RK, Bodor C, Fitzgibbon J, Inglott S et al. GATA2 mutations in sporadic and familial acute myeloid leukaemia patients with CEBPA mutations. Br J Haematol 2013; 161: 701–705.
Fasan A, Alpermann T, Haferlach C, Grossmann V, Roller A, Kohlmann A et al. Frequency and prognostic impact of CEBPA proximal, distal and core promoter methylation in normal karyotype AML: a study on 623 cases. PLoS One 2013; 8: e54365.
Zhang H, Alberich-Jorda M, Amabile G, Yang H, Staber PB, Di Ruscio A et al. Sox4 is a key oncogenic target in C/EBPalpha mutant acute myeloid leukemia. Cancer Cell 2013; 24: 575–588.
Pabst T, Mueller BU, Harakawa N, Schoch C, Haferlach T, Behre G et al. AML1-ETO downregulates the granulocytic differentiation factor C/EBPalpha in t(8;21) myeloid leukemia. Nat Med 2001; 7: 444–451.
Ptasinska A, Assi SA, Martinez-Soria N, Imperato MR, Piper J, Cauchy P et al. Identification of a dynamic core transcriptional network in t(8;21) AML that regulates differentiation block and self-renewal. Cell Rep 2014; 8: 1974–1988.
Helbling D, Mueller BU, Timchenko NA, Hagemeijer A, Jotterand M, Meyer-Monard S et al. The leukemic fusion gene AML1-MDS1-EVI1 suppresses CEBPA in acute myeloid leukemia by activation of Calreticulin. Proc Natl Acad Sci USA 2004; 101: 13312–13317.
Helbling D, Mueller BU, Timchenko NA, Schardt J, Eyer M, Betts DR et al. CBFB-SMMHC is correlated with increased calreticulin expression and suppresses the granulocytic differentiation factor CEBPA in AML with inv(16). Blood 2005; 106: 1369–1375.
Perrotti D, Cesi V, Trotta R, Guerzoni C, Santilli G, Campbell K et al. BCR-ABL suppresses C/EBPalpha expression through inhibitory action of hnRNP E2. Nat Genet 2002; 30: 48–58.
Perrotti D, Calabretta B . Post-transcriptional mechanisms in BCR/ABL leukemogenesis: role of shuttling RNA-binding proteins. Oncogene 2002; 21: 8577–8583.
Mueller BU, Pabst T . C/EBPalpha and the pathophysiology of acute myeloid leukemia. Curr Opin Hematol 2006; 13: 7–14.
Keeshan K, He Y, Wouters BJ, Shestova O, Xu L, Sai H et al. Tribbles homolog 2 inactivates C/EBPalpha and causes acute myelogenous leukemia. Cancer Cell 2006; 10: 401–411.
Wouters BJ, Jorda MA, Keeshan K, Louwers I, Erpelinck-Verschueren CA, Tielemans D et al. Distinct gene expression profiles of acute myeloid/T-lymphoid leukemia with silenced CEBPA and mutations in NOTCH1. Blood 2007; 110: 3706–3714.
Dedhia PH, Keeshan K, Uljon S, Xu L, Vega ME, Shestova O et al. Differential ability of Tribbles family members to promote degradation of C/EBPalpha and induce acute myelogenous leukemia. Blood 2010; 116: 1321–1328.
Nerlov C . C/EBPalpha mutations in acute myeloid leukaemias. Nat Rev Cancer 2004; 4: 394–400.
Zhang P, Iwasaki-Arai J, Iwasaki H, Fenyus ML, Dayaram T, Owens BM et al. Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBPalpha. Immunity 2004; 21: 853–863.
Wagner K, Zhang P, Rosenbauer F, Drescher B, Kobayashi S, Radomska HS et al. Absence of the transcription factor CCAAT enhancer binding protein alpha results in loss of myeloid identity in bcr/abl-induced malignancy. Proc Natl Acad Sci USA 2006; 103: 6338–6343.
Ohlsson E, Hasemann MS, Willer A, Lauridsen FK, Rapin N, Jendholm J et al. Initiation of MLL-rearranged AML is dependent on C/EBPalpha. J Exp Med 2014; 211: 5–13.
Ye M, Zhang H, Yang H, Koche R, Staber PB, Cusan M et al. Hematopoietic differentiation is required for initiation of acute myeloid leukemia. Cell Stem Cell 2015; 17: 611–623.
Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J et al. C/EBPbeta is required for ‘emergency’ granulopoiesis. Nat Immunol 2006; 7: 732–739.
Jones LC, Lin ML, Chen SS, Krug U, Hofmann WK, Lee S et al. Expression of C/EBPbeta from the C/ebpalpha gene locus is sufficient for normal hematopoiesis in vivo. Blood 2002; 99: 2032–2036.
Collins C, Wang J, Miao H, Bronstein J, Nawer H, Xu T et al. C/EBPalpha is an essential collaborator in Hoxa9/Meis1-mediated leukemogenesis. Proc Natl Acad Sci USA 2014; 111: 9899–9904.
Madsen MS, Siersbaek R, Boergesen M, Nielsen R, Mandrup S . Peroxisome proliferator-activated receptor gamma and C/EBPalpha synergistically activate key metabolic adipocyte genes by assisted loading. Mol Cell Biol 2014; 34: 939–954.
Pedersen TA, Kowenz-Leutz E, Leutz A, Nerlov C . Cooperation between C/EBPalpha TBP/TFIIB and SWI/SNF recruiting domains is required for adipocyte differentiation. Genes Dev 2001; 15: 3208–3216.
Porse BT, Bryder D, Theilgaard-Monch K, Hasemann MS, Anderson K, Damgaard I et al. Loss of C/EBP alpha cell cycle control increases myeloid progenitor proliferation and transforms the neutrophil granulocyte lineage. J Exp Med 2005; 202: 85–96.
Kirstetter P, Schuster MB, Bereshchenko O, Moore S, Dvinge H, Kurz E et al. Modeling of C/EBPalpha mutant acute myeloid leukemia reveals a common expression signature of committed myeloid leukemia-initiating cells. Cancer Cell 2008; 13: 299–310.
Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 2006; 442: 818–822.
Reya T, Morrison SJ, Clarke MF, Weissman IL . Stem cells, cancer, and cancer stem cells. Nature 2001; 414: 105–111.
Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature 2014; 506: 328–333.
Mora-Jensen H, Jendholm J, Rapin N, Andersen MK, Roug AS, Bagger FO et al. Cellular origin of prognostic chromosomal aberrations in AML patients. Leukemia 2015; 29: 1785–1789.
Bereshchenko O, Mancini E, Moore S, Bilbao D, Mansson R, Luc S et al. Hematopoietic stem cell expansion precedes the generation of committed myeloid leukemia-initiating cells in C/EBPalpha mutant AML. Cancer Cell 2009; 16: 390–400.
Kato N, Kitaura J, Doki N, Komeno Y, Watanabe-Okochi N, Togami K et al. Two types of C/EBPalpha mutations play distinct but collaborative roles in leukemogenesis: lessons from clinical data and BMT models. Blood 2011; 117: 221–233.
Reckzeh K, Bereshchenko O, Mead A, Rehn M, Kharazi S, Jacobsen SE et al. Molecular and cellular effects of oncogene cooperation in a genetically accurate AML mouse model. Leukemia 2012; 26: 1527–1536.
Alberich-Jorda M, Wouters B, Balastik M, Shapiro-Koss C, Zhang H, Di Ruscio A et al. C/EBPgamma deregulation results in differentiation arrest in acute myeloid leukemia. J Clin Invest 2012; 122: 4490–4504.
Togami K, Kitaura J, Uchida T, Inoue D, Nishimura K, Kawabata KC et al. A C-terminal mutant of CCAAT-enhancer-binding protein alpha (C/EBPalpha-Cm) downregulates Csf1r, a potent accelerator in the progression of acute myeloid leukemia with C/EBPalpha-Cm. Exp Hematol 2015; 43: 300–308, e301.
Grebien F, Vedadi M, Getlik M, Giambruno R, Grover A, Avellino R et al. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPalpha N-terminal leukemia. Nat Chem Biol 2015; 11: 571–578.
Laiosa CV, Stadtfeld M, Xie H, de Andres-Aguayo L, Graf T . Reprogramming of committed T cell progenitors to macrophages and dendritic cells by C/EBP alpha and PU.1 transcription factors. Immunity 2006; 25: 731–744.
Di Stefano B, Sardina JL, van Oevelen C, Collombet S, Kallin EM, Vicent GP et al. C/EBPalpha poises B cells for rapid reprogramming into induced pluripotent stem cells. Nature 2014; 506: 235–239.
Kallin EM, Rodriguez-Ubreva J, Christensen J, Cimmino L, Aifantis I, Helin K et al. Tet2 facilitates the derepression of myeloid target genes during CEBPalpha-induced transdifferentiation of pre-B cells. Mol Cell 2012; 48: 266–276.
Hanna J, Markoulaki S, Schorderet P, Carey BW, Beard C, Wernig M et al. Direct reprogramming of terminally differentiated mature B lymphocytes to pluripotency. Cell 2008; 133: 250–264.
Bagger FO, Rapin N, Theilgaard-Monch K, Kaczkowski B, Thoren LA, Jendholm J et al. HemaExplorer: a database of mRNA expression profiles in normal and malignant haematopoiesis. Nucleic Acids Res 2013; 41: D1034–D1039.
Acknowledgements
This work was supported by grants from the Danish Council for Strategic Research, the Danish Cancer Society and through a centre grant from the Novo Nordisk Foundation Section for Stem Cell Biology in Human Disease. We thank Geer Rift for help with the illustrations.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Ohlsson, E., Schuster, M., Hasemann, M. et al. The multifaceted functions of C/EBPα in normal and malignant haematopoiesis. Leukemia 30, 767–775 (2016). https://doi.org/10.1038/leu.2015.324
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/leu.2015.324
- Springer Nature Limited
This article is cited by
-
A C/ebpα isoform specific differentiation program in immortalized myelocytes
Leukemia (2023)
-
The histone demethylase KDM5C functions as a tumor suppressor in AML by repression of bivalently marked immature genes
Leukemia (2023)
-
Fbxw11 impairs the repopulation capacity of hematopoietic stem/progenitor cells
Stem Cell Research & Therapy (2022)
-
Targeting cancer cell plasticity by HDAC inhibition to reverse EBV-induced dedifferentiation in nasopharyngeal carcinoma
Signal Transduction and Targeted Therapy (2021)
-
The AML-associated K313 mutation enhances C/EBPα activity by leading to C/EBPα overexpression
Cell Death & Disease (2021)