
Abstract
Haematopoietic stem cells (HSCs) are a rare cell population found in the bone marrow of adult mammals and are responsible for maintaining the entire haematopoietic system. Definitive HSCs are produced from mesoderm during embryonic development, from embryonic day 10 in the mouse. HSCs seed the foetal liver before migrating to the bone marrow around the time of birth. In the adult, HSCs are largely quiescent but have the ability to divide to self-renew and expand, or to proliferate and differentiate into any mature haematopoietic cell type. Both the specification of HSCs during development and their cellular choices once formed are tightly controlled at the level of transcription. Numerous transcriptional regulators of HSC specification, expansion, homeostasis and differentiation have been identified, primarily from analysis of mouse gene knockout experiments and transplantation assays. These include transcription factors, epigenetic modifiers and signalling pathway effectors. This chapter reviews the current knowledge of these HSC transcriptional regulators, predominantly focusing on the transcriptional regulation of mouse HSCs, although transcriptional regulation of human HSCs is also mentioned where relevant. Due to the breadth and maturity of this field, we have prioritised recently identified examples of HSC transcriptional regulators. We go on to highlight additional layers of control that regulate expression and activity of HSC transcriptional regulators and discuss how chromosomal translocations that result in fusion proteins of these HSC transcriptional regulators commonly drive leukaemias through transcriptional dysregulation.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The haematopoietic system performs a number of critical functions for mammalian physiology including transport of oxygen and nutrients, as well as immune protection. Blood cells have a rapid turnover and the entire haematopoietic system is maintained by haematopoietic stem cells (HSCs), a rare cell type normally found in the bone marrow of adult mammals.
1.1 Functional Definition of an HSC
The gold standard functional definition of an HSC comes from transplantation assays. In the mouse for example, a single HSC has the ability to reconstitute the entire haematopoietic system when injected intravenously into a sublethally irradiated recipient mouse (irradiation destroys the haematopoietic system), and stably maintain the haematopoietic system for the life of the recipient 1. This so-called long-term reconstitution ability defines the key characteristics of HSCs: (1) the ability to home to and colonise the bone marrow in adult mammals, (2) the ability to expand and self-renew to form and maintain a stable population size for the lifetime of the organism, and (3) the capacity to differentiate into any mature haematopoietic cell type (multipotency). Long-term self-renewal and expansion can additionally be determined by serial or competitive transplantation assays, while multipotency can also be analysed using in vitro colony forming assays.
Large efforts have been made to prospectively isolate pure HSC populations using a range of cell surface marker combinations (both their presence and absence), cellular characteristics such as their ability to efflux certain dyes, and molecular signature such as gene expression patterns. Other combinations of markers have been identified that mark the various haematopoietic progenitor and mature cell populations, alongside functional colony forming assays and morphological identification.
1.2 Key Experimental Approaches
A key approach used to characterise transcriptional regulators of mouse HSCs has been gene targeting in embryonic stem (ES) cells followed by the generation of knock-out mice, which can then be used to assay the consequences of gene deletion on haematopoiesis during embryonic development, the adult HSC pool and differentiation potential. Conditional gene targeting protocols, such as those using the Cre-Lox system, allow genes to be deleted later during development or in an adult cell population. Dosage effects can be analysed using heterozygous (+/null) mice, retrovirally inserted shRNAs or overexpression vectors. Recently, ES cell differentiation to embryoid bodies (EBs) has been validated as an in vitro model of developmental haematopoiesis (reviewed elsewhere 2), and has allowed analysis of some of these critical developmental transcriptional regulators in haematopoiesis.
Techniques such as phylogenetic footprinting, DNase I hypersensitive (DHS) assays, chromatin immunoprecipitation (ChIP) assays and mutagenesis have been used to identify cis-regulatory elements within critical gene loci and determine upstream transcriptional regulators. Importantly, the tissue and developmental time specific activity of regulatory regions identified using the above techniques can be validated using powerful in vivo assays including transient (F0) transgenic mouse embryo assays and comprehensive analysis of haematopoietic parameters in the bone marrow of established transgenic mouse lines. The advent of next generation DNA sequencing coupled to ChIP (ChIP-seq) has allowed identification of genome-wide binding sites of specific transcription factor within a given cell population and identifies putative regulatory sites and downstream targets. A current limitation of this technique is the large number of cells required, typically several million.
2 Transcriptional Regulation of HSC Formation
2.1 Biology of Mammalian Developmental Haematopoiesis
The haematopoietic system is derived from the mesoderm lineage in the developing embryo in a process called developmental haematopoiesis. Developmental haematopoiesis occurs at several distinct spatiotemporal locations in the developing embryo and can be broadly divided into two stages: (1) embryonic haematopoiesis and (2) definitive haematopoiesis 3. Embryonic haematopoiesis occurs from E7.5 in the yolk sac, initially producing primitive erythroid cells, and later multilineage progenitors 4– 7. However, these cell types do not fulfill the criteria of a true HSC as they are unable to reconstitute the entire haematopoietic system of an irradiated mouse.
True HSCs are only produced during the second wave, definitive haematopoiesis, which occurs from approximately E10 in the developing mouse embryo, when the first cells are generated that have the ability to both self-renew and reconstitute the entire haematopoietic system 6 7. Definitive HSCs are believed to bud off from the ventral wall of the dorsal aorta in a part of the embryo labelled the aorta-gonad-mesonephros (AGM) region 7. Additional contribution to the pool of definitive HSCs from extraembryonic tissue is currently unresolved (reviewed in 3).
2.2 Specification of HSCs
Two models of haematopoietic specification, the haemangioblast and haemogenic endothelium models (reviewed in 3) have recently been reconciled by Lancrin et al. who proposed a linear pathway of haematopoietic specification from mesoderm, through a tri-potent haemangioblast cell type (with the capacity of forming haematopoietic, endothelial and smooth muscle cells) to a bi-potent haemogenic endothelium (HE) cell type (with the capacity to commit to either haematopoietic or endothelial cell types), from which definitive blood cells can be derived 8 (Fig. 11.1). A number of transcriptional regulators have been identified as playing a crucial role in the specification of HSCs during development, and can be fitted into the pathway described above.

Model of definitive HSC specification from mesoderm during embryonic development. Definitive HSCs are derived from Flk-1+ mesoderm, which are specified through a tri-potent haemangioblast stage (Etv2-dependent), and bi-potent haemogenic endothelial stage (Scl-dependent). Haemogenic endothelium lineage specification to haematopoietic or endothelial cell types is dependent on the expression of antagonistic transcription factors Runx1 and HoxA3
2.3 Formation of the Haemangioblast from Mesoderm
The E-twenty-six specific (Ets) factor Etv2 (ER71) has recently been identified as a key transcriptional regulator of the mesoderm to haemangioblast transition 9. Etv2 is essential for development of both endothelial and haematopoietic lineages at an early stage; mesodermal precursors of haemangioblasts are generated in Etv2 null embryos and during ES cell differentiation, but further specification is blocked. Etv2 is expressed early in developing mesoderm, and marks a subset of the Flk-1+ mesodermal population with enhanced haematopoietic and endothelial potential 9. Etv2 expression is downregulated by E9.5 and silenced by E10.5 in endothelial cells, suggesting it only plays a role in the early steps of mesoderm specification towards endothelial and haematopoietic cell fates 9. Lee et al. have previously identified a potential regulatory cascade acting upstream of Etv2 including Notch, BMP and Wnt signalling 10. Liu et al. recently suggested that Etv2 plays a role in specifying a haematopoietic rather than cardiogenic fate of Flk-1+ mesoderm through regulating Wnt signalling 11.
2.4 Commitment of the Haemangioblast to Haemogenic Endothelium
Lancrin et al. demonstrated that the transition between haemangioblast and haemogenic endothelium was dependent on expression of the basic helix-loop-helix (bHLH) transcription factor Scl (Tal-1), when analysed using ES cell in vitro differentiation assays 8. Scl is first expressed at the haemangioblast stage, and its expression is maintained through haemogenic endothelial and HSC stages 8 12. Expression of Scl is regulated by several developmental tissue-specific enhancers, including three important for haematopoiesis. The -4 Scl enhancer was found to drive expression to endothelium and foetal haematopoetic progenitors, mediated by Ets factor binding (including Fli-1 and Elf-1) 13. The +19 enhancer is active in endothelial and haematopoietic cells, and critically depends on an Ets/Ets/Gata motif that binds Ets factors Fli-1 and Elf-1, and Gata2 14. These two enhancers appear to have overlapping roles in HSC specification, with the +19 enhancer being sufficient to drive Scl expression and blood formation in Scl −/− embryos, but not necessary as its deletion does not result in loss of haematopoiesis 13. The third enhancer is the +40 region, which drives Scl expression in embryonic and definitive haematopoietic cells. The +40 enhancer may be particularly important to sustain rather than initiate Scl expression as its activity is regulated by Scl protein, thereby forming an autoregulatory loop 15 16.
2.5 Specification of HSCs from Haemogenic Endothelium
Several critical factors have been identified as transcriptional regulators of definitive HSC specification including Runx1, Mll1, TFIIS, Gata2, Notch1, Meis1, Erg, c-Myb and c-Myc (see references below). The role of c-Myb and c-Myc in transcriptional regulation of stem cells is reviewed in Chaps. 15 and 19.
The core binding factor Runx1 (AML1) and its binding partner, CBF, are both required for definitive haematopoiesis 17– 19. Using conditional knockout mice models, Chen et al. recently identified the HE to definitive HSC transition as dependent on Runx1 20. Nottingham et al. identified an important Runx1 enhancer, the +23, which regulates Runx1 expression during HSC emergence, through binding of Gata, Ets and Scl factors 21.
The Trithrorax-related Mll1, a histone H3 lysine 3 (H3K4) methyltransferase, is required for definitive haematopoiesis from analysis of Mll1 null mouse embryos and chimera contribution 22. However, a second Mll1 knockout mouse model created by McMahon et al. survived up to E16.5 and contained a limited number of foetal HSCs 23. The reason for this discrepancy is unclear. Mll1 forms a large multi-protein complex with many proteins, which is thought to activate and maintain transcription and epigenetic memory (reviewed in 24). Although Mll1 contains a CXXC-type zinc finger DNA binding domain, its recruitment to DNA is not fully understood and the recent identification of the possible involvement of non-coding RNAs (ncRNAs) suggesting at least in part non-classical modes of recruitment to target regions 25. The transcription elongation factor S-II (TFIIS), which is known to interact and synergistically function with the Mll1-interacting PAF1 complex 26, is also required for definitive haematopoiesis 27. Recently, a physical interaction between the C-terminal SET domain of Mll1 and the Runt domain of Runx1 has been identified, responsible for recruitment of Mll1 to, and the regulation of, the Runx1 target gene Spi-1/PU.1 28. Recruitment of Mll1 by Runx1 may in part explain the apparent functional overlap of these two transcriptional regulators in haematopoiesis.
The zinc finger transcription factor Gata2 is essential for definitive haematopoiesis. Gata2 is expressed prior to HSC emergence and thought to mark haematopoietic-specified cells 29. However, a reduction of Gata2 expression or activity appears necessary for haematopoietic commitment 30. Once again, the Ets/Ets/Gata motif and E-box motifs were found in a Gata2 enhancer region (the -3 enhancer) 31 32. Gata2 appears to have an overlapping role with Runx1 in definitive haematopoiesis, as Gata2 +/− Runx1 +/− mice are not viable and display haematopoietic defects at midgestation, while single heterozygous mice are viable with only a minor haematopoietic phenotype 33.
The Ets transcription factor Erg was recently shown to be critical for early maintenance, but not specification, of definitive HSCs as deletion results in rapid loss of HSCs 34. Erg is thought to achieve this by acting as an upstream regulator of Scl, Gata2 and Runx1 14 21 31.
Notch proteins are major constituents of a highly conserved signalling pathway. Notch proteins are membrane bound receptors, which when bound by their ligand Jagged, proteolytically cleave their intracellular domain, the so-called Notch-IC domain, which translocates to the nucleus where it participates in the formation of multiprotein DNA-binding complexes to regulate transcription 35. Notch1 (but not Notch2-4) is required for generating definitive HSCs 36. Further analysis using ES cell differentiation models and the generation of chimeric mouse embryos demonstrated that Notch1-deficient ES cells are capable of producing definitive haematopoietic progenitors, but fail to establish long-term definitive HSCs 37. Runx1 appears to be a key target of Notch signalling during definitive haematopoiesis 38 39.
Meis1, a member of TALE subfamily of homeobox proteins, is a Hox protein cofactor that modulates their DNA binding affinity and specificity. Several Meis1-deficient mouse models have been created and show similar phenotypes; mouse embryos die by E14.5 with haemorrhaging and liver hypoplasia due to defective developmental haematopoiesis 40 41. Definitive haematopoiesis is compromised but is not completely ablated, and Meis1-deficient foetal livers at E12.5 have reduced HSC populations, which lack reconstitution ability 41. Meis1 is expressed in definitive haematopoietic clusters in the AGM, which are reduced in number and size, and have reduced Runx1 expression in Meis1-deficient embryos 40.
Recently, a negative regulator of HE specification to HSC has been identified, the homeobox transcription factor HoxA3 42. HoxA3 is a positive regulator of HE specification to endothelial lineage, and with Runx1 plays a key role in this lineage decision process. Runx1 acts to induce a haematopoietic transcription factor cascade to promote HSC formation, while inhibiting essential endothelial lineage genes. HoxA3 acts to maintain these endothelial lineage genes within the HE, and represses the haematopoietic cascade, which appears to at least in part be achieved through direct repression of Runx1 42.
2.6 Migration, Expansion and Maintenance of Foetal HSCs
From approximately E12.5 of mouse embryonic development, definitive HSCs generated in the AGM region migrate to and colonise the foetal liver, the site of foetal haematopoiesis 43. Around the time of birth, HSCs move to the bone marrow niche for the rest of the life of the mammal 43. It is estimated that at E11.5 there is one HSC produced in the AGM 44. Expansion of these early HSCs is therefore critical to form a large enough population to maintain haematopoiesis for the life of the organism. This propensity to expand the HSC population, rather than maintain pool size is a key difference between foetal and adult HSCs, although adult HSCs retain this potential as demonstrated by transplantation assays.
Sox17 is a Sry-related high mobility group box transcription factor and within the haematopoietic system, is expressed in foetal and neonatal, but not adult HSCs 45. Sox17 deficiency severely impairs foetal haematopoiesis, and Sox17-null foetal and neonatal HSCs lose all reconstitution ability implicating Sox17 in generation or maintenance of definitive HSCs 45. Loss of Sox17 expression correlates with acquisition of adult HSC characteristics; slow cell cycling and adult surface marker phenotype 45. A number of other transcriptional regulators of both foetal and adult HSCs have been identified, but are discussed in Sect. 11.4.
3 Transcriptional Regulation of HSC Homeostasis
HSCs have the capacity to proliferate and self-renew to maintain their population for the lifetime of the organism, and balance this with differentiation into the committed haematopoietic cell types to replenish physiological turnover or injury. Additionally, to prevent population expansion to a physiologically dangerous size, programmed cell death (apoptosis) must also be regulated. In the adult, HSCs constitute an exceedingly rare cell population estimated at 1 in 104 to 1 in 105 bone marrow cells. Adult HSCs are believed to be predominantly quiescent, with recent estimates in the mouse suggesting one cell division every 145 days and may reversibly switch between this state and self-renewal during homeostasis and repair 46. Further modelling suggested the existence of two kinetically distinct subpopulations of HSCs, one cycling every 149–193 days, and the other cycling every 28–32 days 47.
3.1 Concepts of HSC Fate Decisions
It is generally assumed that HSC fate choices are associated with cell division, as HSC differentiation without division would likely lead to HSC exhaustion 48. These fate decisions would therefore be a result of the type of cell division; symmetrical division to produce either two HSCs or two progenitor cells, or asymmetric cell division into one HSC and one progenitor (Fig. 11.2a). These cell division options would allow HSC pool size to be regulated (e.g. expansion after transplantation) and respond to environmental stress 48.

Models of HSC fate choices. (a) HSCs may divide symmetrically into two HSCs or two progenitors, or asymmetrically into one HSC and one progenitor. (b) The two types of model of HSC fate determination. Selective/stochastic models predict HSC fate choice is random and signalling molecules (e.g. cytokines) act to promote survival and proliferation or apoptosis of the fated progenitors. Instructive/deterministic models predict signalling molecules directly determine HSC fate decisions
Cell intrinsic (e.g. transcription factor protein concentrations and distribution in daughter cells) and extrinsic (e.g. cytokines and cell-cell signalling) cues determine lineage restriction. Two types of models have been proposed to explain HSC lineage commitment (48 49 summarised in Fig. 11.2b): (1) Instructive or deterministic models predict HSCs to respond to external stimuli, which directly guide lineage decisions during differentiation. (2) Selective or permissive models predict lineage choice is predominately random, which may be due to stochastic gene expression, and that external stimuli act to regulate survival and proliferation of these randomly produced progenitors and mature cells. Evidence for both models has been reported (see 48 49 and references within, and 50 51). It is important to mention that these two models are not mutually exclusive, and it seems likely extrinsic events can be both instructive and selective 48 49.
Cell intrinsic processes, in particular transcription factor networks, are central to defining the developmental stage and lineage potential, and determine the response of an external signal. External signals must act within these intrinsic parameters to instruct and/or select cell fate. Indeed, simply the regulation of cell surface receptor expression immediately determines the ability of a cell to respond to a particular extracellular ligand. Numerous intrinsic positive and negative transcriptional regulators of HSC homeostasis have been identified, which control self-renewal, proliferation, quiescence and apoptosis, and include transcription factors, chromatin and DNA modifying enzymes, and signalling pathways, and are described below.
3.2 Transcription Factor Networks Active in Haematopoietic Cells
ChIP-seq experiments to define genome-wide occupancy of key transcription factors in HSCs have been limited by the relatively large cell numbers required for this technique, and the scarcity of HSCs. However, using cell line models, such as the mouse haematopoietic stem/progenitor cell line HPC7, has allowed analysis of transcription factor binding in early haematopoietic cells. So far, ChIP-seq of ten haematopoietic transcription factors has been published using this cell line, identifying combinatorial transcriptional regulation of key genes and putative cis-regulatory sites 33. Combining such ChIP experiments to define transcription factor occupancy with knowledge of cis-regulatory elements within gene loci has allowed modelling of the interconnections within active transcription factor networks in haematopoietic cells (Fig. 11.3; reviewed in 52). Due to the availability of mature haematopoietic cell types for ChIP-seq experiments, the regulatory networks in the later stages of haematopoiesis are more advanced. An alternate method has been used by Novershtern et al., who used gene expression analysis (which require lower cell numbers) of pure haematopoietic populations combined with known cis-regulatory interactions to identify tightly interconnecting networks that control HSC and mature haematopoietic cell state 53.

A model of a core transcriptional regulatory network active in haematopoietic stem/progenitor cells consisting of ten transcription factors predicted from Wilson et al. 33. Interactions identified from analysis of transcription factor enrichment from ChIP-seq experiments within gene loci at cis-regulatory elements. The transcription factor regulatory network is highly interconnected, rather than hierarchical in structure
3.3 Basic Helix-Loop-Helix Transcription Factors
Scl is highly expressed in HSCs and regulates quiescence by inhibiting the G0 to G1 transition 54. The same study also identified a dosage-dependent role for Scl in long-term HSC reconstitution potential. A paralogue of Scl, Lyl1, also regulates foetal and adult HSC reconstitution potential and lymphoid differentiation 55. Lyl1 appears to have a partially overlapping role with Scl, as Lyl1/Scl conditional double knockout HSCs have complete loss of reconstitution ability, and increased HSC and progenitor apoptosis, a more severe phenotype than loss of Scl or Lyl1 alone 56. Scl forms a multifactor complex with transcription factors Gata2 and E2A proteins, along with bridging molecules Lmo2 and Ldb1 in foetal and adult HSCs and differentiating haematopoietic cells. Formation of this complex is essential for regulation of HSC function, as loss of any component impairs HSC function (see below and 57 58). Depending on the context, the Scl complex may also include Lyl1, Gata1, Lmo4, HEB, Eto2 and Sp1 59– 62.
The E2A locus expresses two bHLH E-proteins; E47 and E12, which regulate HSC cycling and promote early progenitor maturation 63. Genetic deletion of E2A increases HSC cycling while reducing pool size, and HSCs lose long term reconstitution ability 63. Recent analysis of pure HSC populations has identified a role for the E47 isoform in regulating HSC proliferation and energetics 64; E47 −/− HSCs progressively lose self-renewal potential with concomitant hyperproliferation of progenitor populations. E2A protein activity is regulated through interactions with inhibitors Id1-3; Id1 also regulates HSC homeostasis 65 66, while Id2 and Id3 regulate haematopoietic lineage commitment 67– 69.
Besides its role in definitive haematopoiesis, c-Myc also regulates HSC homeostasis, playing a crucial role in balancing HSC self-renewal versus differentiation decisions 70, as well as HSC survival and HSC lineage commitment 71 72. HSCs also express a second Myc protein, N-myc, which with c-Myc regulates HSC function and survival 71. The role of Myc proteins in stem cells is considered in more detail in Chap. 19.
3.4 Homeobox Transcription Factors
Hox genes encode homeodomain transcription factors and are crucial for developmental patterning 73. In mammals, 39 Hox genes are co-ordinately expressed from four loci. DNA binding of Hox transcription factors is modulated by interaction with DNA binding cofactors; one of three Pbx family members and/or one of four Meis family members (both families are also homeobox proteins) 74. Several Hox genes have been implicated in HSC homeostasis, although deletion of a single Hox gene does not usually severely affect HSC homeostasis, possibly due to their functional redundancy. Within the haematopoietic system, Hox gene expression is largely confined to the HSC and progenitor compartment 75.
Ectopic expression of HoxA9, HoxA10, HoxA6 HoxB4 and HoxC5 expands HSCs in vitro 76– 80. Additionally, genetic deletion of HoxA9 or HoxB3 and HoxB4 mildly impairs HSC proliferation 81 82. HoxA9 null HSCs also have impaired differentiation and reduced reconstitution ability 82. Compound deletion of HoxA9, HoxB3 and HoxB4 caused an increase in bone marrow HSCs, but did not affect in vitro colony forming ability 83. Interestingly, the defect in reconstitution ability of compound null HSCs was no worse than single HoxA9 deficiency 83.
Pbx1 can dimerise with a subset of Hox proteins, and can also trimerise with Hox and Meis proteins 84 85. Pbx1 is required to maintain definitive HSCs in the foetal liver; Pbx1 null mice are embryonic lethal at E15-16, and have severe anaemia due to defective foetal liver haematopoiesis 86. Conditional deletion of Pbx1 from adult HSCs results in the upregulation of several cell cycle regulators with increased HSC cycling and progressive loss of HSC reconstitution ability 87.
3.5 Ets Transcription Factors
Several Ets transcription factors are known to regulate HSC homeostasis and differentiation including Erg, Fli-1, Tel/Etv6, GABP, PU.1/Spi-1 and Elf4 88– 95. Two of the most recently reported Ets factors are described below.
A role for Erg in adult HSC function was identified using a sensitised genetic screen in mice 88. Erg is required to maintain HSC pool size and reconstitution ability, and differentiation to committed progenitors 88 89. Furthermore, additional mutation of Fli-1 in Erg-deficient HSCs identified a partial functional redundancy of these two Ets factors, with the double deficiency causing a more severe phenotype 90. Yu et al. recently identified GABP to be a critical regulator of HSC homeostasis and differentiation 91. GABP heterodimerises with GABP to form the GABP complex and is essential for early embryogenesis 96. Conditional deletion of GABP in adult HSCs lead to a rapid loss of HSC self-renewal, increased apoptosis of HSCs and progenitors, and impaired differentiation 91.
3.6 Zinc Finger Transcription Factors
Zinc fingers are a protein domain that commonly interacts with DNA and are found in a wide range of transcription factors, several of which are known to regulate HSC homeostasis including Gata2, Gata3, Gfi1, Gfi1b, Klf4, Ikaros, Evi-1, Sall4, Zfx and Prdm16 97– 108. Interestingly, several of these zinc finger transcription factors also regulate ES cell self-renewal and pluripotency (Klf4, Sall4 and Zfx 100 109 110).
Analysis of Gata2 heterozygous mouse embryos identified a role for Gata2 in expansion of definitive HSCs in the AGM and their proliferation after foetal colonisation 111. However, Gata2 is generally thought to restrict cell cycle entry in adult HSCs (reviewed elsewhere 102). Additionally, GATA2 also regulates human HSC quiescence, with enforced expression increasing G0 residency 112. More recently, a second Gata factor, Gata3, has also been identified as a regulator of HSC maintenance, with Gata3 null mice having a smaller HSC population 101.
Gfi1 is a transcriptional repressor that promotes HSC quiescence, and maintains HSC self-renewal and reconstitution potential 103 104. Additionally, Gfi1 appears to be a direct target of p53 in HSCs, a key cell cycle regulator. The paralogue Gfi1b is also responsible for maintaining HSC quiescence, although Gfi1b −/− HSCs retain self-renewal capacity 105. Gfi1 and Gfi1b appear to have partially overlapping functions as Gfi1/Gfi1b double deletion result in complete loss of HSCs 105.
Ikaros and related family of transcription factors were initially identified as regulators of lymphoid lineages (reviewed in 113). However, Ikaros is also expressed in HSCs, although different isoforms to those expressed in lymphoid progenitors 114, and plays a role in HSC activity. Ikaros mutant mice have reduced numbers of HSCs and progenitors, and have reduced reconstitution ability 115. More recently, a role for Ikaros in lymphoid lineage priming of HSCs has been identified 106.
Evi-1 contains a SET/PR-domain with a total of ten zinc fingers 116. Deletions of Evi-1 results in embryonic lethality at E10-16.5 (depending on the mouse model), and the development and expansion of definitive HSCs is severely impaired 107 108, and reviewed in 116. Conditional deletion of Evi-1 from adult HSCs causes a shift from quiescence to cell cycling, reduction of the HSC pool size and loss of reconstitution ability 108. Evi-1 expression has also been used as a marker of long term haematopoietic reconstitution potential 117. Interestingly, dosage of Evi-1 appears important as Evi-1 heterozygosity causes partial loss of HSC self-renewal while overexpression enhances HSC self-renewal at the expense of differentiation 107. However, Evi-1 is dispensable for lineage commitment 107.
A second SET/PR-domain protein, Prdm16, is also important for HSC homeostasis, and specifically expressed in HSCs and early progenitors in the adult haematopoietic system 118. Overexpression of Prdm16 has previously been found to expand HSCs in vitro, and also causes myeloproliferative disease in vivo after transplantation 119. A transposon mutagenesis screen identified a role for Prdm16 in regulation of adult stem cell reactive oxygen species (ROS) levels, apoptosis and cell cycle, and its loss lead to HSC depletion 120. Aguilo et al. identified a role of Prdm16 in HSCs using Prdm16 null mice embryos 118; foetal HSC and progenitors were reduced in number, had mild defects in vitro colony forming ability, severely impaired reconstitution ability, and increased apoptosis.
3.7 Myb Proteins
C-Myb plays an important role in HSC self-renewal and adult haematopoiesis; its conditional deletion causing a defect in HSC proliferation, increased differentiation, and loss of reconstitution ability 121. A genome-wide mutagenesis screen identified the ability of p300 to interact with the transactivation domain of c-Myb to be necessary for proper HSC proliferation and differentiation 122. The role of c-Myb in stem cells is discussed in further detail in Chap. 15.
The cyclin-D binding myb-like transcription factor 1 (Dmtf1) has been implicated in regulating HSC quiescence 123. Dmtf1 null mice are viable, but have increased blood counts, and Dmtf1 null HSCs have increased proliferation, self renewal and long term reconstitution ability 123.
3.8 Core Binding Factors
A number of conflicting papers have been published about the role of Runx1 in HSCs 124– 128. The most recent from Cai et al., has sought to resolve the experimental discrepancies by highlighting that Runx1 regulates the expression of several key markers commonly used to identify HSCs by flow cytometry, and report that conditional deletion of Runx1 only moderately decreases the number of HSCs, while increasing those of early progenitors 129. Loss of Runx1 also causes slight increases in HSC quiescence and reduces apoptosis, and combined suggest Runx1 promotes HSC proliferation and differentiation. However, the three major Runx1 isoforms (Runx1a, b and c) appear to have at least partially distinct functions in the haematopoietic system 130– 132. Ectopic expression of a short isoform of Runx1, Runx1a, expands HSCs in vitro (which retain in vivo reconstitution ability), while ectopic expression of Runx1b promotes differentiation 130 132. No functional difference between the two long isoforms, Runx1b and Runx1c, has been identified 131. Regulation of Runx1 in haematopoietic cells is considered in more detail in Sect. 11.6.
By comparison, HSCs appear to be more sensitive to CBF from hypomorph experiments; 15–30 % of WT CBF levels promote HSC and progenitor expansion as well as mature cell differentiation 133, and suggest a role for CBF in HSC quiescence. Interestingly, Miller et al. suggest a Runx1-independent role for CBF in foetal haematopoiesis in differentiation of haematopoietic progenitors, which is not due to a defect in bone marrow niche 134. CBF can also heterodimerise with the two paralogues of Runx1, Runx2 and Runx3, to form protein complexes that can bind to the same consensus DNA sequence (reviewed in 135). Partial overlap in function of Runx1– 3 in regulating HSCs would help explain the difference in Runx1 and CBF phenotypes, although are yet to be identified. Defective bone marrow haematopoiesis in Runx2 null mice has been identified, but is thought to be a result of altered HSC niche due to defective osteoblast differentiation 136 137.
3.9 Cell Cycle Regulators
Unsurprisingly, cell cycle regulators have also been identified as regulating HSC homeostasis. Two of these involved in transcriptional regulation are retinoblastoma (pRB) and p53 families 138– 141. pRb, with family members p107 and p130, inhibit cell cycle entry by repressing E2F target gene expression, and have an overlapping function in regulating HSC quiescence and self-renewal 138. Conditional deletion of all three pRB proteins causes increased HSC proliferation, expansion of HSC numbers, loss of reconstitution activity and a lethal myloproliferative phenotype 138. However, an extrinsic role for pRB in regulating HSC has also been identified 142. The functionally similar Necdin, also regulates HSC quiescence state, and interactions with p53 141 143.
A third cell cycle regulator has also been found to regulate HSCs; NF-Y, a trimeric transcription factor complex composed of NF-Ya, NF-Yb and NF-Yc 144. NF-Y is an important developmental regulator, with genetic deletion of NF-Ya causing embryonic lethality around E8.5 145. NF-Ya overexpression promotes HSC self-renewal 146 while conditional deletion of NF-Ya causes defective cell cycle G2/M progression and increased apoptosis, resulting in death 147.
3.10 Immediate Early Response Transcription Factors
Two immediate early response transcription factors, JunB and Egr1, have been implicated in regulating HSCs. Inactivation of JunB in HSCs results in increased proliferation and differentiation 148, but does not affect reconstitution ability. Additionally, JunB inactivation decreases HSC response to Notch and TGF signalling through loss of Hes1 expression 148. JunB is also a target of TGF signalling 149. Egr1 regulates HSC quiescence as well as retention within the HSC niche 150. Interestingly, Egr1 knockout HSCs expand and mobilise without losing reconstitution ability 150.
3.11 Epigenetic Regulation
A key mechanism by which transcription factors are thought to regulate eukaryotic gene expression is through their recruitment of epigenetic modifying enzymes, which catalyse histone or DNA modifications. Epigenetic modifications affect chromatin structure, recruit secondary factors and regulate transcription. Several epigenetic modifiers have been identified to play an important role in HSC homeostasis, summarised below.
Three histone lysine acetyltransferases and transcriptional co-activators are essential for HSC self-renewal; CBP, p300 and MOZ 151– 153. The H3K79 methyltransferase Dot1l is also crucial for maintaining HSC function 154 155.
Both Mll1, and its cofactor Menin, are required for HSC self-renewal 156. As described earlier, two mouse Mll1 knockout models display differing severity in phenotype, although both agree that Mll1 is necessary to maintain adult HSC self-renewal 23 157. Using a conditional gene knockout model, Gan et al. identified a role for Mll1 in post-natal but not foetal HSC maintenance 158. The distantly related Mll family member, Mll5, is also involved in regulating HSC self-renewal and haematopoietic differentiation 159– 161.
Multiple polycomb group (PcG) proteins, which epigenetically regulate transcriptional repression, have been found to regulate HSCs (review in 162). PcG proteins form two discrete complexes, polycomb repressive complex 1 and 2 (PRC1 and PRC2), which have distinct enzymatic activity (H2AK119 monoubiquitination and H3K27 trimethylation activities respectively) and discrete functions in HSCs 163. Various gene knockout models suggest PRC2 limits HSC self-renewal 162– 167. Ezh2, a core component of PRC2 is also required for maintenance of foetal HSCs 168.
Genetic deletion of PRC1 core components generally results in the loss of HSC self-renewal 169– 173. Bmi-1 is a particularly important core component, with overexpression promoting mouse and human HSC self-renewal and ex vivo expansion 170 174. A paralogue of Bmi-1, Mel-18, which replaces Bmi-1 to form a PRC1-like complex, promotes HSC proliferation and differentiation 175 176. The balance between Bmi-1 and Mel-18 expression may regulate HSC fate. A functional crosstalk between Bmi-1 and Mll1/HoxA9 has also been identified in establishing HSCs 177.
DNA methylation, generally 5-cytosine methylation (mC) in a CpG dinucleotide context, is a key epigenetic mark and is thought to inhibit transcription. DNA methyltransferases Dmnt3a and Dmnt3b (involved in de novo DNA methylation), and Dnmt1 (involved in maintaining DNA methylation patterns) have been implicated in regulating HSC self-renewal 178– 180. Additionally, Tet2, a methylcytosine dioxygenase that converts mC to 5-hydroxymethyl-cytosine (hmC), is required for HSC homeostasis 181– 183. Chromatin remodelling complexes such as the Mi-2 containing NuRD complex are also important for maintaining HSC quiescence and self-renewal 184.
3.12 Signalling Pathways
Several signalling pathways have been identified to regulate HSC self-renewal and differentiation through regulating transcription. These appear to play important, although not usually essential roles in HSC homeostasis. Functional redundancy of signalling molecules within these pathways, as well as overlap and integration of different signalling pathways help explain the often conflicting phenotypes after in vitro activation, in vivo genetic deletion, depletion, inhibition, constitutive activation or overexpression of the mediators of these pathways.
The downstream signalling transcriptional regulators Notch-IC (Notch signalling) and -catenin (Wnt signalling) have a fairly established role in HSC self-renewal and expansion (reviewed in 35 185– 187). Signalling through Smad transcription factors (TGF and BMP signalling) and Gli1-3 (Hedgehog signalling) are also thought to regulate HSC self-renewal (reviewed elsewhere 187 188). In contrast, retinoic acid receptor (retinoic acid signalling) stimulates HSC and progenitor cell differentiation 189.
Activation of the receptor tyrosine kinases c-kit (SCF receptor), c-mpl (thrombpoietin receptor) and Tie-2 (angiopoietin receptor) also regulate HSC maintenance, although through multiple signalling pathways including JAK-STAT, phosphoinositide-3 kinase (PI3K), and MAPK. JAK-STAT signalling activation but PI3K signalling inhibition appears important to maintain HSC self-renewal (reviewed in 187 190– 192).
3.13 Oxidative Stress
Regulation of oxidative stress is critical for HSC homeostasis. FoxO transcription factors are regulated by PI3K signalling, and also play a critical role in HSC resistance to oxidative stress 193 194. FoxO1/3/4 null HSCs have increased ROS levels, increased cell cycling and apoptosis, are reduced in number and defective in their reconstitution activity. Interestingly, anti-oxidative treatment alleviates the FoxO-deficient phenotype 194. Even single deletion of FoxO3a results in elevated ROS in HSCs, which impairs HSC function 195. As mentioned above, Prdm16 is also involving in regulation of adult stem cell ROS levels 120. Additionally, proper regulation of the hypoxia-inducible factor 1 alpha (HIF-1) is essential for HSC quiescence and reconstitution ability 196.
4 Transcriptional Regulation of HSC Differentiation
4.1 Cellular Hierarchy of Mammalian Adult Haematopoiesis
HSCs have the ability to differentiate into at least ten different specialised mature cell types with a diverse range of functions, morphologies, lifetimes and proliferative abilities, and their relative proportions are dependent on extracellular and external influences. Adult HSCs differentiate through progressively more lineage-committed stages (progenitor cells) to form mature, terminally differentiated haematopoietic cells. This lineage commitment is represented as a haematopoietic hierarchy or tree (Fig. 11.4). Numerous cell surface markers and functional assays have been used to identify and define these intermediate progenitors and mature cell types, although the complete definition of in vivo potential of the many different progenitor populations is still ongoing. This detailed understanding of HSC differentiation pathways has greatly facilitated the identification and dissection of the role of transcriptional regulators of this process. Almost all transcriptional regulators of HSC homeostasis also regulate later lineage commitment decisions. Numerous other transcriptional regulators of this process have also been identified, predominantly controlling relatively late lineage commitment decisions. We refer to a number of recent reviews of the transcriptional regulation of these later lineage commitment decisions for further detail 198– 203.

The haematopoietic lineage tree illustrates HSC differentiation potential. HSCs differentiate through progressively more committed progenitors into at least ten mature blood cell types with diverse functions (which can be divided into myeloid and lymphoid cell types). The haematopoietic tree shows stable cell populations, which have been defined by surface marker expression, although the exact branching points and potential of progenitors are still a matter of debate (see for example 197 for further details). LT-HSC long-term haematopoietic stem cell, ST-HSC short-term haematopoietic stem cell, MPP multipotent progenitor, CLP common lymphoid progenitor, CMP common myeloid progenitor, GMP granulocyte monocyte progenitor, MEP myeloerythroid progenitor, MK megakaryocyte, RBC red blood cell
4.2 HSC Differentiation and Lineage Specification
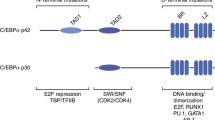
HSC differentiation is closely linked to proliferation, and many of the transcriptional regulators of HSC homeostasis also play a role in differentiation, and have been mentioned above. Few transcriptional regulators of the initial steps of HSC differentiation and commitment have so far been identified that do not also regulate HSC homeostasis. Here we briefly describe two example transcriptional regulators of HSC differentiation: C/EBP and Hmgb3.
The CCAAT-enhancer binding protein alpha (C/EBP) is required for development of granulocytes 204, but also functions in HSCs to promote differentiation. C/EBP null HSCs have increased repopulating ability and self-renewal, and also display a block of early myeloid differentiation 205. Additionally, C/EBP determines cell fate of multipotent progenitors, inducing myeloid differentiation while inhibiting erythroid differentiation 206.
The high mobility group binding protein B3 (Hmgb3) is a sequence-independent chromatin binding protein. Loss of Hmgb3 does not affect HSC numbers, self-renewal or reconstitution ability, but does result in reduced CLP and CMP numbers 207 208. Of note, even though in vitro differentiation of Hmgb3-deficient CLP and CMP are unaffected, loss of Hmgb3 appears to bias HSCs to self-renewal rather than differentiation into progenitors 207.
5 Regulation of HSC Transcriptional Regulator Expression
Much of our understanding of the regulation of HSCs is at the transcriptional level, and transcriptional regulation of haematopoietic transcription factors by cis-regulatory elements has allowed modelling of transcription factor networks. However, additional regulatory mechanisms overlay and interconnect with these networks, including alternative promoter usage and splicing, post-transcriptional and translational control mechanisms, and post-translational regulation of protein activity and degradation. To highlight this additional complexity, we discuss the regulatory mechanisms known to control expression and activity of a single transcription factor, Runx1, within the haematopoietic system.
5.1 Transcriptional and Co-transcriptional Regulation
Runx1 is expressed from two promoters, a distal P1 and proximal P2, which play nonredudant roles in definitive haematopoiesis, with the P2 being critically required 209. Different transcription factor binding at the Runx1 promoters confers specificity of promoter activity, and explain differential promoter activity during developmental haematopoiesis 131. Haematopoietic expression is also regulated by the activity of the Runx1 +23 enhancer 21. As mentioned above, three major isoforms of Runx1 appear to have partially distinct functions 130. However, over 12 differentially spliced Runx1 cDNAs have so far been identified, which may play additional roles in the haematopoietic system 209 210.
5.2 Post-transcriptional and Translational Regulation
MicroRNAs (miRNAs) are a class of small ncRNA that play a critical role in regulating gene expression (see Chap. 18 for further details). Ben-Ami et al. identified five miRNAs with the ability to bind the Runx1 3UTR and inhibit expression 211. Alternative splicing determines the length of the 3UTR, and therefore the ability of these miRNAs to bind and interfere with translation of Runx1. Ben-Ami et al. went onto describe a feedback loop active during megakaryocytic differentiation (of a myeloid cell line) between Runx1 and miR-27a 211. MiR-27 has also been identified as inhibiting Runx1 expression during granulocyte development 212.
Runx1 promoter activity determines the 5UTR transcribed and site of translational initiation. Transcripts from the distal P1 promoter are translated by a Cap-dependent mechanism, while transcripts from the proximal P2 promoter are translated from an internal ribosome entry site (IRES) 213. Regulation of these two translational start sites by different mechanisms adds an additional level of control to Runx1 expression. Interestingly, several studies suggest miRNAs do not inhibit translation from IRES 214 215, and could represent a further mechanism by which expression of alternative isoforms is differentially regulated.
5.3 Post-translational Modification by Phosphorylation, Acetylation and Methylation
Post-translational modification of proteins by phosphorylation, acetylation and methylation are common mechanisms to regulate protein activity through modulating tertiary structure and protein-protein or protein-DNA interactions. Runx1 is phosphorylated by cyclin-dependent kinases (CDKs) in a cell cycle-specific manner, which regulates Runx1 activity, protein-protein interactions, stability and degradation 216– 218. Runx1-DNA binding stability is also regulated by transcriptional co-activator p300- and MOZ-mediated lysine acetylation 219 220. Runx1 methylation has also been reported to alter its activity and transcriptional co-activator interactions 221 222. Post-translational modification also appears important for the ability of transcriptional regulator fusion proteins to drive leukaemias; lysine acetylation of RUNX1-ETO is necessary for its ability to mediate leukaemogenesis 223.
5.4 Regulation of Runx1 Activity by Smad6
Besides protein-protein interactions with transcriptional co-activators and co-repressors that regulate Runx1 activity, Runx1 is also regulated by interaction with Smad6, a downstream regulator of the BMP and TGF signalling pathways. Smad6 regulates Runx1 (as well as Runx2) activity by acting as an adaptor, mediating ubiquitination of Runx1 by Smurf2 (an E3 ubiquitin ligase), which results in proteosomal degradation 224 225. A novel self-regulatory mechanism has recently been identified by Knezevic et al., whereby Runx1 controls its own expression during definitive haematopoiesis through regulation of Smad6 expression, an inhibitor of Runx1 activity 226. Three key Runx1 expression regulators Scl, Gata2 and Fli-1 also regulate Smad6 expression, in combination with Runx1 226. Runx1 activity therefore determines Smad6 expression, which in turn regulates Runx1 activity, and acts to maintain steady Runx1 activity during this process 226.
6 Transcriptional Regulation in Leukaemogenesis
6.1 Mutation, Translocation and Aberrant Expression of Haematopoietic Transcriptional Regulators
Haematological malignancies are a heterogenous group of diseases, genotypically and phenotypically, and include leukaemias and lymphomas. Chromosomal translocations that produce gene fusions are particularly common in haematological malignancies, with over 264 different gene fusions identified so far 227. Mutation, translocation, or aberrant expression of many transcriptional regulators discussed above is associated with haematological malignancies, in particular leukaemias (reviewed elsewhere 197 227 228). A large number were in fact originally identified from cytogenetic analysis of chromosomal abnormalities in leukaemias. The molecular pathogenesis of translocations of the haematopoietic transcriptional regulator, MLL1, one the best understood examples, is discussed below.
6.2 MLL1 Translocations and Fusion Proteins
Chromosomal translocations involving MLL1 account for approximately 10 % of all leukaemias and cause a variety of phenotypes (from which MLL1 gets its name; Mixed Lineage Leukaemia 1). Over 60 different in-frame gene fusion partners of MLL1 have been identified as well as MLL1 partial duplication events 229 230. However, over 90 % of cases are accounted for by gene fusion with AF4, AF9, ELL, ENL, AF6 or AF10 231. Expression from the MLL1 promoter after translocation produces an MLL fusion protein consisting of the N-terminus of MLL1 and the C-terminus of the fusion partner, which does not contain H3K4 methyltransferase activity 230. An increasing understanding of the molecular mechanisms by which MLL fusion proteins initiate and maintain leukaemias has helped developed targeted therapies.
MLL fusion proteins appear to “hijack” normal transcriptional regulators to mediate leukaemogenesis. Continued expression of the MLL fusion protein is required to maintain leukaemic growth 232, and MLL-AF9 also requires expression of wild-type MLL1 to initiate and maintain leukaemia 233. The MLL1 cofactor Menin is also required for maintenance of MLL1 leukaemias 234, and Menin-MLL inhibitors have recently been found to ablate leukaemogenic activity of MLL fusion proteins 235. Additionally, a PcG protein Cbx8 (and PRC1 component) has recently been found to be necessary for initiation and maintenance of MLL-AF9 leukaemias, suggesting cooperation between PcG and MLL fusion proteins in leukaemogenesis 236.
Four of the most common fusion partners of MLL1 (AF4, AF9, ENL and ELL), along with the transcriptional coactivator pTEFb, the polymerase associated factor 1 complex, the H3K79 methyltransferase DOT1L, and the BET family protein BRD4 are thought to form large molecular complexes with MLL fusion proteins at target genes 237– 241. These data, combined with reports that H3K79 methylation profiles define multiple MLL fusion protein leukaemias 242, led to the design of a DOT1L inhibitor, which was recently reported to selectively kill MLL1 leukaemias 243. BET inhibitors prevent BET proteins (including BRD4) from binding to acetylated histones. BET inhibitors are thought to destabilise MLL fusion protein complexes at target genes, and have also recently been reported to be an effective treatment of MLL1 leukaemias, inducing downregulation of MYC, cell cycle arrest and apoptosis 244.
The reports summarised above highlight the notion of how a molecular understanding of the transcriptional dysregulation that occurs in leukaemias can facilitate the design of effective targeted therapies. Furthermore, they suggest MLL fusion proteins may mediate leukaemogenesis through a common molecular mechanism involving inappropriate recruitment of transcriptional elongation promoting factors to MLL target genes. MLL fusion proteins are thought to target a subset of wild-type MLL1 targets, their aberrant expression promoting cellular proliferation and survival 245. Perhaps unsurprisingly, several key MLL fusion protein targets are transcriptional regulators of HSC self-renewal: Hox genes (in particular HOXA9, HOXA10), MEIS1, EVI-1, MYC and MYB, which contribute to MLL1 leukaemogenesis 244 246– 249.
However, a comparison of two ChIP-seq data sets of genome-wide MLL fusion protein occupancy (MLL-AF4 and MLL-AF9) identified few common gene targets 250. This suggests that although MLL fusion proteins may act by a common mechanism to dysregulate transcription of target genes, many of these target genes are likely to be unique to the particular MLL1 leukaemia, and may depend on cell of origin, MLL fusion partner, and/or additional mutations present. This may help to explain the heterogeneity in cellular phenotype and pathology of MLL1 leukaemias.
7 Conclusions
Over the last 30 years, transcription factors have been identified as key regulators of every stage of normal and malignant haematopoiesis. However, most work to date has involved focusing on the role of single transcription factors within this system. However, it is becoming increasingly clear that transcription factors act within large regulatory networks, often functionally and physically interacting. Further work is needed to synthesise all this information, as well as integrating the additional layers of regulation acting on these transcription factors, into a wider, coherent network model.
Besides serving as a model of mammalian development, the overall aim of such research is its application to clinical problems, such as production or expansion of HSCs for bone marrow transplantation and mature blood cell types for transfusion medicine, as well as rational design of treatments of HSC-associated diseases, such as leukaemias. As mentioned in Sect. 11.7, several small molecule inhibitors have recently been identified as potential revolutionary treatments of MLL1 leukaemias. However, our understanding of the leukaemogenic mechanisms of many other fusion proteins is less well advanced. Recent cancer genome sequencing projects are discovering ever more transcriptional regulators as candidate leukaemic oncogenes and/or tumour suppressors 251 252. However, further work is required to confirm their role and determine their function in driving leukaemia, as well as in normal haematopoiesis.
Mouse models have provided powerful tools to investigate the transcriptional regulation of HSCs, and in many ways account for our greater understanding of mouse HSCs over human HSCs. However, an over-reliance on mouse experiments must be avoided if research is to be successfully translated into clinical application. Although the roles of many transcriptional regulators of HSCs are likely conserved, differences in the basic biology of mice and humans (such as life expectancy) as well as those specific to HSCs will limit translation of knowledge. For example, HoxB4 is a potent regulator of mouse HSC expansion 78, but has very limited ability to expand human HSCs 253. Additionally, current isolation protocols for human HSCs provide less pure cell populations than mouse HSCs. Further characterisation and dissection of human HSCs will therefore be important in the future.
In summary, transcriptional regulation of HSCs is a mature area of research that is continuing at an exciting pace, and one which holds real promise for further clinical application in the near future.
References
Krause DS, Theise ND, Collector MI, Henegariu O et al (2001) Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell 105(3):369–377
Keller G (2005) Embryonic stem cell differentiation: emergence of a new era in biology and medicine. Genes Dev 19(10):1129–1155
Medvinsky A, Rybtsov S, Taoudi S (2011) Embryonic origin of the adult hematopoietic system: advances and questions. Development 138(6):1017–1031
Silver L, Palis J (1997) Initiation of murine embryonic erythropoiesis: a spatial analysis. Blood 89(4):1154–1164
Medvinsky AL, Samoylina NL, Muller AM, Dzierzak EA (1993) An early pre-liver intraembryonic source of CFU-S in the developing mouse. Nature 364(6432):64–67
Muller AM, Medvinsky A, Strouboulis J, Grosveld F et al (1994) Development of hematopoietic stem cell activity in the mouse embryo. Immunity 1(4):291–301
Medvinsky A, Dzierzak E (1996) Definitive hematopoiesis is autonomously initiated by the AGM region. Cell 86(6):897–906
Lancrin C, Sroczynska P, Stephenson C, Allen T et al (2009) The haemangioblast generates haematopoietic cells through a haemogenic endothelium stage. Nature 457(7231):892–895
Kataoka H, Hayashi M, Nakagawa R, Tanaka Y et al (2011) Etv2/ER71 induces vascular mesoderm from Flk1 + PDGFR{alpha} + primitive mesoderm. Blood 118:6975–6986
Lee D, Park C, Lee H, Lugus JJ et al (2008) ER71 acts downstream of BMP, notch, and Wnt signaling in blood and vessel progenitor specification. Cell Stem Cell 2(5):497–507
Liu F, Kang I, Park C, Chang LW et al (2012) ER71 specifies Flk-1+ hemangiogenic mesoderm by inhibiting cardiac mesoderm and Wnt signaling. Blood 119(14):3295–3305
Kallianpur AR, Jordan JE, Brandt SJ (1994) The SCL/TAL-1 gene is expressed in progenitors of both the hematopoietic and vascular systems during embryogenesis. Blood 83(5):1200–1208
Gottgens B, Broccardo C, Sanchez MJ, Deveaux S et al (2004) The scl +18/19 stem cell enhancer is not required for hematopoiesis: identification of a 5 bifunctional hematopoietic-endothelial enhancer bound by Fli-1 and Elf-1. Mol Cell Biol 24(5):1870–1883
Gottgens B, Nastos A, Kinston S, Piltz S et al (2002) Establishing the transcriptional programme for blood: the SCL stem cell enhancer is regulated by a multiprotein complex containing Ets and GATA factors. EMBO J 21(12):3039–3050
Ogilvy S, Ferreira R, Piltz SG, Bowen JM et al (2007) The SCL +40 enhancer targets the midbrain together with primitive and definitive hematopoiesis and is regulated by SCL and GATA proteins. Mol Cell Biol 27(20):7206–7219
Delabesse E, Ogilvy S, Chapman MA, Piltz SG et al (2005) Transcriptional regulation of the SCL locus: identification of an enhancer that targets the primitive erythroid lineage in vivo. Mol Cell Biol 25(12):5215–5225
Okuda T, van Deursen J, Hiebert SW, Grosveld G et al (1996) AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 84(2):321–330
Wang Q, Stacy T, Miller JD, Lewis AF et al (1996) The CBF subunit is essential for CBF2 (AML1) function in vivo. Cell 87(4):697–708
Sasaki K, Yagi H, Bronson RT, Tominaga K et al (1996) Absence of fetal liver hematopoiesis in mice deficient in transcriptional coactivator core binding factor beta. Proc Natl Acad Sci U S A 93(22):12359–12363
Chen MJ, Yokomizo T, Zeigler BM, Dzierzak E et al (2009) Runx1 is required for the endothelial to haematopoietic cell transition but not thereafter. Nature 457(7231):887–891
Nottingham WT, Jarratt A, Burgess M, Speck CL et al (2007) Runx1-mediated hematopoietic stem-cell emergence is controlled by a gata/Ets/SCL-regulated enhancer. Blood 110(13):4188–4197
Ernst P, Fisher JK, Avery W, Wade S et al (2004) Definitive hematopoiesis requires the mixed-lineage leukemia gene. Dev Cell 6(3):437–443
McMahon KA, Hiew SYL, Hadjur S, Veiga-Fernandes H et al (2007) Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell Stem Cell 1(3):338–345
Schuettengruber B, Martinez AM, Iovino N, Cavalli G (2011) Trithorax group proteins: switching genes on and keeping them active. Nat Rev Mol Cell Biol 12(12):799–814
Bertani S, Sauer S, Bolotin E, Sauer F (2011) The noncoding RNA mistral activates Hoxa6 and Hoxa7 expression and stem cell differentiation by recruiting MLL1 to chromatin. Mol Cell 43(6):1040–1046
Kim J, Guermah M, Roeder RG (2010) The human PAF1 complex acts in chromatin transcription elongation both independently and cooperatively with SII/TFIIS. Cell 140(4):491–503
Ito T, Arimitsu N, Takeuchi M, Kawamura N et al (2006) Transcription elongation factor S-II is required for definitive hematopoiesis. Mol Cell Biol 26(8):3194–3203
Huang G, Zhao X, Wang L, Elf S et al (2011) The ability of MLL to bind RUNX1 and methylate H3K4 at PU.1 regulatory regions is impaired by MDS/AML-associated RUNX1/AML1 mutations. Blood 118(25):6544–6552
Minegishi N, Ohta J, Yamagiwa H, Suzuki N et al (1999) The mouse GATA-2 gene is expressed in the para-aortic splanchnopleura and aorta-gonads and mesonephros region. Blood 93(12):4196–4207
Minegishi N, Suzuki N, Yokomizo T, Pan X et al (2003) Expression and domain-specific function of GATA-2 during differentiation of the hematopoietic precursor cells in midgestation mouse embryos. Blood 102(3):896–905
Pimanda JE, Ottersbach K, Knezevic K, Kinston S et al (2007) Gata2, Fli1, and Scl form a recursively wired gene-regulatory circuit during early hematopoietic development. Proc Natl Acad Sci U S A 104(45):17692–17697
Kobayashi-Osaki M, Ohneda O, Suzuki N, Minegishi N et al (2005) GATA motifs regulate early hematopoietic lineage-specific expression of the Gata2 gene. Mol Cell Biol 25(16):7005–7020
Wilson NK, Foster SD, Wang X, Knezevic K et al (2010) Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell 7(4):532–544
Taoudi S, Bee T, Hilton A, Knezevic K et al (2011) ERG dependence distinguishes developmental control of hematopoietic stem cell maintenance from hematopoietic specification. Genes Dev 25(3):251–262
Pajcini KV, Speck NA, Pear WS (2011) Notch signaling in mammalian hematopoietic stem cells. Leukemia 25(10):1525–1532
Kumano K, Chiba S, Kunisato A, Sata M et al (2003) Notch1 but not Notch2 is essential for generating hematopoietic stem cells from endothelial cells. Immunity 18(5):699–711
Hadland BK, Huppert SS, Kanungo J, Xue Y et al (2004) A requirement for Notch1 distinguishes 2 phases of definitive hematopoiesis during development. Blood 104(10):3097–3105
Burns CE, Traver D, Mayhall E, Shepard JL et al (2005) Hematopoietic stem cell fate is established by the notch-runx pathway. Genes Dev 19(19):2331–2342
Nakagawa M, Ichikawa M, Kumano K, Goyama S et al (2006) AML1/Runx1 rescues Notch1-null mutation-induced deficiency of para-aortic splanchnopleural hematopoiesis. Blood 108(10):3329–3334
Azcoitia V, Aracil M, Martínez-A C, Torres M (2005) The homeodomain protein Meis1 is essential for definitive hematopoiesis and vascular patterning in the mouse embryo. Dev Biol 280(2):307–320
Hisa T, Spence SE, Rachel RA, Fujita M et al (2004) Hematopoietic, angiogenic and eye defects in Meis1 mutant animals. EMBO J 23(2):450–459
Iacovino M, Chong D, Szatmari I, Hartweck L et al (2011) HoxA3 is an apical regulator of haemogenic endothelium. Nat Cell Biol 13(1):72–U165
Dzierzak E, Speck NA (2008) Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nat Immunol 9(2):129–136
Kumaravelu P, Hook L, Morrison AM, Ure J et al (2002) Quantitative developmental anatomy of definitive haematopoietic stem cells/long-term repopulating units (HSC/RUs): role of the aorta-gonad-mesonephros (AGM) region and the yolk sac in colonisation of the mouse embryonic liver. Development 129(21):4891–4899
Kim I, Saunders TL, Morrison SJ (2007) Sox17 dependence distinguishes the transcriptional regulation of fetal from adult hematopoietic stem cells. Cell 130(3):470–483
Wilson A, Laurenti E, Oser G, van der Wath RC et al (2008) Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135(6):1118–1129
van der Wath RC, Wilson A, Laurenti E, Trumpp A et al (2009) Estimating dormant and active hematopoietic stem cell kinetics through extensive modeling of bromodeoxyuridine label-retaining cell dynamics. PLoS One 4(9):e6972
Morrison SJ, Kimble J (2006) Asymmetric and symmetric stem-cell divisions in development and cancer. Nature 441(7097):1068–1074
Mansson R, Zandi S, Bryder D, Sigvardsson M (2009) The road to commitment: lineage restriction events in hematopoiesis. In: Wickrema A, Kee B (eds) Molecular basis of hematopoiesis. Springer, New York, pp 23–46
Stoffel R, Ziegler S, Ghilardi N, Ledermann B et al (1999) Permissive role of thrombopoietin and granulocyte colony-stimulating factor receptors in hematopoietic cell fate decisions in vivo. Proc Natl Acad Sci U S A 96(2):698–702
Rieger MA, Hoppe PS, Smejkal BM, Eitelhuber AC et al (2009) Hematopoietic cytokines can instruct lineage choice. Science 325(5937):217–218
Pimanda JE, Gottgens B (2010) Gene regulatory networks governing haematopoietic stem cell development and identity. Int J Dev Biol 54(6–7):1201–1211
Novershtern N, Subramanian A, Lawton LN, Mak RH et al (2011) Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 144(2):296–309
Lacombe J, Herblot S, Rojas-Sutterlin S, Haman A et al (2010) Scl regulates the quiescence and the long-term competence of hematopoietic stem cells. Blood 115(4):792–803
Capron C, Lécluse Y, Kaushik AL, Foudi A et al (2006) The SCL relative LYL-1 is required for fetal and adult hematopoietic stem cell function and B-cell differentiation. Blood 107(12):4678–4686
Souroullas GP, Salmon JM, Sablitzky F, Curtis DJ et al (2009) Adult hematopoietic stem and progenitor cells require either Lyl1 or Scl for survival. Cell Stem Cell 4(2):180–186
Li L, Jothi R, Cui K, Lee JY et al (2011) Nuclear adaptor Ldb1 regulates a transcriptional program essential for the maintenance of hematopoietic stem cells. Nat Immunol 12(2):129–136
Yamada Y, Warren AJ, Dobson C, Forster A et al (1998) The T cell leukemia LIM protein Lmo2 is necessary for adult mouse hematopoiesis. Proc Natl Acad Sci U S A 95(7):3890–3895
Soler E, Andrieu-Soler C, de Boer E, Bryne JC et al (2010) The genome-wide dynamics of the binding of Ldb1 complexes during erythroid differentiation. Genes Dev 24(3):277–289
Goardon N, Lambert JA, Rodriguez P, Nissaire P et al (2006) ETO2 coordinates cellular proliferation and differentiation during erythropoiesis. EMBO J 25(2):357–366
Fujiwara T, Lee HY, Sanalkumar R, Bresnick EH (2010) Building multifunctionality into a complex containing master regulators of hematopoiesis. Proc Natl Acad Sci U S A 107(47):20429–20434
Song SH, Hou CH, Dean A (2007) A positive role for NLI/Ldb1 in long-range beta-globin locus control region function. Mol Cell 28(5):810–822
Semerad CL, Mercer EM, Inlay MA, Weissman IL et al (2009) E2A proteins maintain the hematopoietic stem cell pool and promote the maturation of myelolymphoid and myeloerythroid progenitors. Proc Natl Acad Sci U S A 106(6):1930–1935
Yang Q, Kardava L, St. Leger A, Martincic K et al (2008) E47 controls the developmental integrity and cell cycle quiescence of multipotential hematopoietic progenitors. J Immunol 181(9):5885–5894
Jankovic V, Ciarrocchi A, Boccuni P, DeBlasio T et al (2007) Id1 restrains myeloid commitment, maintaining the self-renewal capacity of hematopoietic stem cells. Proc Natl Acad Sci U S A 104(4):1260–1265
Perry SS, Zhao Y, Nie L, Cochrane SW et al (2007) Id1, but not Id3, directs long-term repopulating hematopoietic stem-cell maintenance. Blood 110(7):2351–2360
Ji M, Li H, Suh HC, Klarmann KD et al (2008) Id2 intrinsically regulates lymphoid and erythroid development via interaction with different target proteins. Blood 112(4):1068–1077
Deed RW, Jasiok M, Norton JD (1998) Lymphoid-specific expression of the Id3 gene in hematopoietic cells—selective antagonism of E2A basic helix-loop-helix protein associated with Id3-induced differentiation of erythroleukemia cells. J Biol Chem 273(14):8278–8286
Miyazaki M, Rivera RR, Miyazaki K, Lin YC et al (2011) The opposing roles of the transcription factor E2A and its antagonist Id3 that orchestrate and enforce the naive fate of T cells. Nat Immunol 12(10):992–103
Wilson A, Murphy MJ, Oskarsson T, Kaloulis K et al (2004) c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev 18(22):2747–2763
Laurenti E, Varnum-Finney B, Wilson A, Ferrero I et al (2008) Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell 3(6):611–624
Baena E, Ortiz M, Martínez-A C, de Alborán IM (2007) c-Myc is essential for hematopoietic stem cell differentiation and regulates Lin(−)Sca-1(+)c-Kit(−) cell generation through p21. Exp Hematol 35(9):1333–1343
Pearson JC, Lemons D, McGinnis W (2005) Modulating Hox gene functions during animal body patterning. Nat Rev Genet 6(12):893–904
Moens CB, Selleri L (2006) Hox cofactors in vertebrate development. Dev Biol 291(2):193–206
Argiropoulos B, Humphries RK (2007) Hox genes in hematopoiesis and leukemogenesis. Oncogene 26(47):6766–6776
Thorsteinsdottir U, Mamo A, Kroon E, Jerome L et al (2002) Overexpression of the myeloid leukemia-associated Hoxa9 gene in bone marrow cells induces stem cell expansion. Blood 99(1):121–129
Magnusson M, Brun ACM, Miyake N, Larsson J et al (2007) HOXA10 is a critical regulator for hematopoietic stem cells and erythroid/megakaryocyte development. Blood 109(9):3687–3696
Antonchuk J, Sauvageau G, Humphries RK (2002) HOXB4-induced expansion of adult hematopoietic stem cells ex vivo. Cell 109(1):39–45
Auvray C, Delahaye A, Pflumio F, Haddad R et al (2012) HOXC4 homeoprotein efficiently expands human hematopoietic stem cells and triggers similar molecular alterations as HOXB4. Haematologica 97(2):168–178
Fischbach NA, Rozenfeld S, Shen W, Fong S et al (2005) HOXB6 overexpression in murine bone marrow immortalizes a myelomonocytic precursor in vitro and causes hematopoietic stem cell expansion and acute myeloid leukemia in vivo. Blood 105(4):1456–1466
Bjornsson JM, Larsson N, Brun ACM, Magnusson M et al (2003) Reduced proliferative capacity of hematopoietic stem cells deficient in Hoxb3 and Hoxb4. Mol Cell Biol 23(11):3872–3883
Lawrence HJ, Christensen J, Fong S, Hu YL et al (2005) Loss of expression of the hoxa-9 homeobox gene impairs the proliferation and repopulating ability of hematopoietic stem cells. Blood 106(12):3988–3994
Magnusson M, Brun ACM, Lawrence HJ, Karlsson S (2007) Hoxa9/hoxb3/hoxb4 compound null mice display severe hematopoietic defects. Exp Hematol 35(9):1421–1428
Chang CP, Jacobs Y, Nakamura T, Jenkins NA et al (1997) Meis proteins are major in vivo DNA binding partners for wild-type but not chimeric Pbx proteins. Mol Cell Biol 17(10):5679–5687
Mann RS, Lelli KM, Joshi R (2009) Hox specificity: unique roles for cofactors and collaborators. Curr Top Dev Biol 88:63–101
DiMartino JF (2001) The Hox cofactor and proto-oncogene Pbx1 is required for maintenance of definitive hematopoiesis in the fetal liver. Blood 98(3):618–626
Ficara F, Murphy MJ, Lin M, Cleary ML (2008) Pbx1 regulates self-renewal of long-term hematopoietic stem cells by maintaining their quiescence. Cell Stem Cell 2(5):484–496
Loughran SJ, Kruse EA, Hacking DF, de Graaf CA et al (2008) The transcription factor Erg is essential for definitive hematopoiesis and the function of adult hematopoietic stem cells. Nat Immunol 9(7):810–819
Ng AP, Loughran SJ, Metcalf D, Hyland CD et al (2011) Erg is required for self-renewal of hematopoietic stem cells during stress hematopoiesis in mice. Blood 118(9):2454–2461
Kruse EA, Loughran SJ, Baldwin TM, Josefsson EC et al (2009) Dual requirement for the ETS transcription factors Fli-1 and Erg in hematopoietic stem cells and the megakaryocyte lineage. Proc Natl Acad Sci U S A 106(33):13814–13819
Yu S, Cui K, Jothi R, Zhao D-M et al (2011) GABP controls a critical transcription regulatory module that is essential for maintenance and differentiation of hematopoietic stem/progenitor cells. Blood 117(7):2166–2178
Iwasaki H, Somoza C, Shigematsu H, Duprez EA et al (2005) Distinctive and indispensable roles of PU.1 in maintenance of hematopoietic stem cells and their differentiation. Blood 106(5):1590–1600
Hock H, Meade E, Medeiros S, Schindler JW et al (2004) Tel/Etv6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes Dev 18(19):2336–2341
Lacorazza HD, Yamada T, Liu Y, Miyata Y et al (2006) The transcription factor MEF/ELF4 regulates the quiescence of primitive hematopoietic cells. Cancer Cell 9(3):175–187
Wang LC, Swat W, Fujiwara Y, Davidson L et al (1998) The TEL/ETV6 gene is required specifically for hematopoiesis in the bone marrow. Genes Dev 12(15):2392–2402
Ristevski S, O’Leary DA, Thornell AP, Owen MJ et al (2004) The ETS transcription factor GABPalpha is essential for early embryogenesis. Mol Cell Biol 24(13):5844–5849
Alder JK, Georgantas RW, Yu X, Civin CI (2004) KLF4 as a mediator of quiescence in hematopoietic stem/progenitor cells. Blood 104(11, Part 2):123B–123B
Yang J, Aguila JR, Alipio Z, Lai R et al (2011) Enhanced self-renewal of hematopoietic stem/progenitor cells mediated by the stem cell gene Sall4. J Hematol Oncol 4(1):38–38
Aguila JR, Liao W, Yang J, Avila C et al (2011) SALL4 is a robust stimulator for the expansion of hematopoietic stem cells. Blood 118(3):576–585
Galan-Caridad JM, Harel S, Arenzana TL, Hou ZE et al (2007) Zfx controls the self-renewal of embryonic and hematopoietic stem cells. Cell 129(2):345–357
Ku CJ, Hosoya T, Maillard I, Engel JD (2012) GATA-3 regulates hematopoietic stem cell maintenance and cell cycle entry. Blood 119(10):2242–2251
Rodrigues NP, Tipping AJ, Wang Z, Enver T (2012) GATA-2 mediated regulation of normal hematopoietic stem/progenitor cell function, myelodysplasia and myeloid leukemia. Int J Biochem Cell Biol 44(3):457–460
Zeng H, Yücel R, Kosan C, Klein-Hitpass L et al (2004) Transcription factor Gfi1 regulates self-renewal and engraftment of hematopoietic stem cells. EMBO J 23(20):4116–4125
Hock H, Hamblen MJ, Rooke HM, Schindler JW et al (2004) Gfi-1 restricts proliferation and preserves functional integrity of haematopoietic stem cells. Nature 431(7011):1002–1007
Khandanpour C, Sharif-Askari E, Vassen L, Gaudreau M-C et al (2010) Evidence that growth factor independence 1b regulates dormancy and peripheral blood mobilization of hematopoietic stem cells. Blood 116(24):5149–5161
Ng SY-M, Yoshida T, Zhang J, Georgopoulos K (2009) Genome-wide lineage-specific transcriptional networks underscore ikaros-dependent lymphoid priming in hematopoietic stem cells. Immunity 30(4):493–507
Goyama S, Yamamoto G, Shimabe M, Sato T et al (2008) Evi-1 is a critical regulator for hematopoietic stem cells and transformed leukemic cells. Cell Stem Cell 3(2):207–220
Zhang Y, Stehling-Sun S, Lezon-Geyda K, Juneja SC et al (2011) PR-domain-containing Mds1-Evi1 is critical for long-term hematopoietic stem cell function. Blood 118(14):3853–3861
Jiang J, Chan YS, Loh YH, Cai J et al (2008) A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat Cell Biol 10(3):353–360
Zhang J, Tam WL, Tong GQ, Wu Q et al (2006) Sall4 modulates embryonic stem cell pluripotency and early embryonic development by the transcriptional regulation of Pou5f1. Nat Cell Biol 8(10):1114–1123
Ling K-W, Ottersbach K, van Hamburg JP, Oziemlak A et al (2004) GATA-2 plays two functionally distinct roles during the ontogeny of hematopoietic stem cells. J Exp Med 200(7):871–882
Tipping AJ, Pina C, Castor A, Hong D et al (2009) High GATA-2 expression inhibits human hematopoietic stem and progenitor cell function by effects on cell cycle. Blood 113(12):2661–2672
John LB, Ward AC (2011) The ikaros gene family: transcriptional regulators of hematopoiesis and immunity. Mol Immunol 48(9–10):1272–1278
Klug CA (1998) Hematopoietic stem cells and lymphoid progenitors express different ikaros isoforms, and ikaros is localized to heterochromatin in immature lymphocytes. Proc Natl Acad Sci 95(2):657–662
Nichogiannopoulou A (1999) Defects in hemopoietic stem cell activity in ikaros mutant mice. J Exp Med 190(9):1201–1214
Kumano K, Kurokawa M (2010) The role of Runx1/AML1 and Evi-1 in the regulation of hematopoietic stem cells. J Cell Physiol 222(2):282–285
Kataoka K, Sato T, Yoshimi A, Goyama S et al (2011) Evi1 is essential for hematopoietic stem cell self-renewal, and its expression marks hematopoietic cells with long-term multilineage repopulating activity. J Exp Med 208(12):2403–2416, jem.20110447-jem.20110447-
Aguilo F, Avagyan S, Labar A, Sevilla A et al (2011) Prdm16 is a physiologic regulator of hematopoietic stem cells. Blood 117(19):5057–5066
Deneault E, Cellot S, Faubert A, Laverdure JP et al (2009) A functional screen to identify novel effectors of hematopoietic stem cell activity. Cell 137(2):369–379
Chuikov S, Levi BP, Smith ML, Morrison SJ (2010) Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat Cell Biol 12(10):999–1006
Lieu YK, Reddy EP (2009) Conditional c-myb knockout in adult hematopoietic stem cells leads to loss of self-renewal due to impaired proliferation and accelerated differentiation. Proc Natl Acad Sci U S A 106(51):21689–21694
Sandberg ML, Sutton SE, Pletcher MT, Wiltshire T et al (2005) c-Myb and p300 regulate hematopoietic stem cell proliferation and differentiation. Dev Cell 8(2):153–166
Kobayashi M, Srour EF (2011) Regulation of murine hematopoietic stem cell quiescence by Dmtf1. Blood 118(25):6562–6571
Growney JD, Shigematsu H, Li Z, Lee BH et al (2005) Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood 106(2):494–504
Ichikawa M, Asai T, Saito T, Seo S et al (2004) AML-1 is required for megakaryocytic maturation and lymphocytic differentiation, but not for maintenance of hematopoietic stem cells in adult hematopoiesis. Nat Med 10(3):299–304
Ichikawa M, Goyama S, Asai T, Kawazu M et al (2008) AML1/Runx1 negatively regulates quiescent hematopoietic stem cells in adult hematopoiesis. J Immunol 180(7):4402–4408
Motoda L, Osato M, Yamashita N, Jacob B et al (2007) Runx1 protects hematopoietic stem/progenitor cells from oncogenic insult. Stem Cells 25(12):2976–2986
Jacob B, Osato M, Yamashita N, Wang CQ et al (2010) Stem cell exhaustion due to Runx1 deficiency is prevented by Evi5 activation in leukemogenesis. Blood 115(8):1610–1620
Cai X, Gaudet JJ, Mangan JK, Chen MJ et al (2011) Runx1 loss minimally impacts long-term hematopoietic stem cells. PLoS One 6(12):e28430–e28430
Tsuzuki S, Hong DL, Gupta R, Matsuo K et al (2007) Isoform-specific potentiation of stem and progenitor cell engraftment by AML1/RUNX1. PLoS Med 4(5):880–896
Challen GA, Goodell MA (2010) Runx1 isoforms show differential expression patterns during hematopoietic development but have similar functional effects in adult hematopoietic stem cells. Exp Hematol 38(5):403–416
Tsuzuki S, Seto M (2012) Expansion of functionally defined mouse hematopoietic stem and progenitor cells by a short isoform of RUNX1/AML1. Blood 119(3):727–735
Talebian L, Li Z, Guo YL, Gaudet J et al (2007) T-lymphoid, megakaryocyte, and granulocyte development are sensitive to decreases in CBF beta dosage. Blood 109(1):11–21
Miller J, Horner A, Stacy T, Lowrey C et al (2002) The core-binding factor beta subunit is required for bone formation and hematopoietic maturation. Nat Genet 32(4):645–649
Link KA, Chou FS, Mulloy JC (2010) Core binding factor at the crossroads: determining the fate of the HSC. J Cell Physiol 222(1):50–56
Deguchi K, Yagi H, Inada M, Yoshizaki K et al (1999) Excessive extramedullary hematopoiesis in Cbfa1-deficient mice with a congenital lack of bone marrow. Biochem Biophys Res Commun 255(2):352–359
Komori T, Yagi H, Nomura S, Yamaguchi A et al (1997) Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 89(5):755–764
Viatour P, Somervaille TC, Venkatasubrahmanyam S, Kogan S et al (2008) Hematopoietic stem cell quiescence is maintained by compound contributions of the retinoblastoma gene family. Cell Stem Cell 3(4):416–428
Asai T, Liu Y, Bae N, Nimer SD (2011) The p53 tumor suppressor protein regulates hematopoietic stem cell fate. J Cell Physiol 226(9):2215–2221
Liu Y, Elf SE, Asai T, Miyata Y et al (2009) The p53 tumor suppressor protein is a critical regulator of hematopoietic stem cell behavior. Cell Cycle 8(19):3120–3124
Liu Y, Elf SE, Miyata Y, Sashida G et al (2009) p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell 4(1):37–48
Walkley CR, Shea JM, Sims NA, Purton LE et al (2007) Rb regulates interactions between hematopoietic stem cells and their bone marrow microenvironment. Cell 129(6):1081–1095
Kubota Y, Osawa M, Jakt LM, Yoshikawa K et al (2009) Necdin restricts proliferation of hematopoietic stem cells during hematopoietic regeneration. Blood 114(20):4383–4392
Mantovani R (1999) The molecular biology of the CCAAT-binding factor NF-Y. Gene 239(1):15–27
Bhattacharya A, Deng JM, Zhang Z, Behringer R et al (2003) The B subunit of the CCAAT box binding transcription factor complex (CBF/NF-Y) is essential for early mouse development and cell proliferation. Cancer Res 63(23):8167–8172
Zhu J, Zhang Y, Joe GJ, Pompetti R et al (2005) NF-Ya activates multiple hematopoietic stem cell (HSC) regulatory genes and promotes HSC self-renewal. Proc Natl Acad Sci U S A 102(33):11728–11733
Bungartz G, Land H, Scadden DT, Emerson SG (2012) NF-Y is necessary for hematopoietic stem cell proliferation and survival. Blood 119(6):1380–1389
Santaguida M, Schepers K, King B, Sabnis AJ et al (2009) JunB protects against myeloid malignancies by limiting hematopoietic stem cell proliferation and differentiation without affecting self-renewal. Cancer Cell 15(4):341–352
Verrecchia F, Tacheau C, Schorpp-Kistner M, Angel P et al (2001) Induction of the AP-1 members c-Jun and JunB by TGF-beta/smad suppresses early smad-driven gene activation. Oncogene 20(18):2205–2211
Min IM, Pietramaggiori G, Kim FS, Passegué E et al (2008) The transcription factor EGR1 controls both the proliferation and localization of hematopoietic stem cells. Cell Stem Cell 2(4):380–391
Rebel VI, Kung AL, Tanner EA, Yang H et al (2002) Distinct roles for CREB-binding protein and p300 in hematopoietic stem cell self-renewal. Proc Natl Acad Sci U S A 99(23):14789–14794
Katsumoto T, Aikawa Y, Iwama A, Ueda S et al (2006) MOZ is essential for maintenance of hematopoietic stem cells. Genes Dev 20(10):1321–1330
Chan WI, Hannah RL, Dawson MA, Pridans C et al (2011) The transcriptional coactivator Cbp regulates self-renewal and differentiation in adult hematopoietic stem cells. Mol Cell Biol 31(24):5046–5060
Nguyen AT, He J, Taranova O, Zhang Y (2011) Essential role of DOT1L in maintaining normal adult hematopoiesis. Cell Res 21(9):1370–1373
Jo SY, Granowicz EM, Maillard I, Thomas D et al (2011) Requirement for Dot1l in murine postnatal hematopoiesis and leukemogenesis by MLL translocation. Blood 117(18):4759–4768
Maillard I, Hess JL (2009) The role of menin in hematopoiesis. Adv Exp Med Biol 668:51–57
Jude CD, Climer L, Xu D, Artinger E et al (2007) Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell 1(3):324–337
Gan T, Jude CD, Zaffuto K, Ernst P (2010) Developmentally induced Mll1 loss reveals defects in postnatal haematopoiesis. Leukemia 24(10):1732–1741
Heuser M, Yap DB, Leung M, de Algara TR et al (2009) Loss of MLL5 results in pleiotropic hematopoietic defects, reduced neutrophil immune function, and extreme sensitivity to DNA demethylation. Blood 113(7):1432–1443
Madan V, Madan B, Brykczynska U, Zilbermann F et al (2009) Impaired function of primitive hematopoietic cells in mice lacking the mixed-lineage-leukemia homolog MLL5. Blood 113(7):1444–1454
Zhang Y, Wong J, Klinger M, Tran MT et al (2009) MLL5 contributes to hematopoietic stem cell fitness and homeostasis. Blood 113(7):1455–1463
Konuma T, Oguro H, Iwama A (2010) Role of the polycomb group proteins in hematopoietic stem cells. Dev Growth Differ 52(6):505–516
Majewski IJ, Ritchie ME, Phipson B, Corbin J et al (2010) Opposing roles of polycomb repressive complexes in hematopoietic stem and progenitor cells. Blood 116(5):731–739
Iwama A, Oguro H, Negishi M, Kato Y et al (2005) Epigenetic regulation of hematopoietic stem cell self-renewal by polycomb group genes. Int J Hematol 81(4):294–300
Lessard J, Schumacher A, Thorsteinsdottir U, van Lohuizen M et al (1999) Functional antagonism of the polycomb-group genes eed and Bmi1 in hemopoietic cell proliferation. Genes Dev 13(20):2691–2703
Majewski IJ, Blewitt ME, de Graaf CA, McManus EJ et al (2008) Polycomb repressive complex 2 (PRC2) restricts hematopoietic stem cell activity. PLoS Biol 6(4):e93
Su IH, Basavaraj A, Krutchinsky AN, Hobert O et al (2003) Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol 4(2):124–131
Mochizuki-Kashio M, Mishima Y, Miyagi S, Negishi M et al (2011) Dependency on the polycomb gene Ezh2 distinguishes fetal from adult hematopoietic stem cells. Blood 118(25):6553–6561
Calés C, Román-Trufero M, Pavón L, Serrano I et al (2008) Inactivation of the polycomb group protein Ring1B unveils an antiproliferative role in hematopoietic cell expansion and cooperation with tumorigenesis associated with Ink4a deletion. Mol Cell Biol 28(3):1018–1028
Iwama A, Oguro H, Negishi M, Kato Y et al (2004) Enhanced self-renewal of hematopoietic stem cells mediated by the polycomb gene product Bmi-1. Immunity 21(6):843–851
Kim JY, Sawada A, Tokimasa S, Endo H et al (2004) Defective long-term repopulating ability in hematopoietic stem cells lacking the polycomb-group gene rae28. Eur J Haematol 73(2):75–84
Lessard J, Sauvageau G (2003) Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 423(6937):255–260
Ohta H (2002) Polycomb group gene rae28 is required for sustaining activity of hematopoietic stem cells. J Exp Med 195(6):759–770
Rizo A, Dontje B, Vellenga E, de Haan G et al (2008) Long-term maintenance of human hematopoietic stem/progenitor cells by expression of BMI1. Blood 111(5):2621–2630
Elderkin S, Maertens GN, Endoh M, Mallery DL et al (2007) A phosphorylated form of Mel-18 targets the Ring1B histone H2A ubiquitin ligase to chromatin. Mol Cell 28(1):107–120
Kajiume T, Ninomiya Y, Ishihara H, Kanno R et al (2004) Polycomb group gene mel-18 modulates the self-renewal activity and cell cycle status of hematopoietic stem cells. Exp Hematol 32(6):571–578
Smith L-L, Yeung J, Zeisig BB, Popov N et al (2011) Functional crosstalk between Bmi1 and MLL/Hoxa9 axis in establishment of normal hematopoietic and leukemic stem cells. Cell Stem Cell 8(6):649–662
Challen GA, Sun D, Jeong M, Luo M et al (2011) Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet 44(1):23–31
Tadokoro Y, Ema H, Okano M, Li E et al (2007) De novo DNA methyltransferase is essential for self-renewal, but not for differentiation, in hematopoietic stem cells. J Exp Med 204(4):715–722
Trowbridge JJ, Snow JW, Kim J, Orkin SH (2009) DNA methyltransferase 1 is essential for and uniquely regulates hematopoietic stem and progenitor cells. Cell Stem Cell 5(4):442–449
Ko M, Bandukwala HS, An J, Lamperti ED et al (2011) Ten-eleven-translocation 2 (TET2) negatively regulates homeostasis and differentiation of hematopoietic stem cells in mice. Proc Natl Acad Sci 108(35):14566–14571
Li Z, Cai X, Cai C, Wang J et al (2011) Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 118(17):4509–4518
Moran-Crusio K, Reavie L, Shih A, Abdel-Wahab O et al (2011) Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20(1):11–24
Yoshida T, Hazan I, Zhang J, Ng SY et al (2008) The role of the chromatin remodeler Mi-2beta in hematopoietic stem cell self-renewal and multilineage differentiation. Genes Dev 22(9):1174–1189
Staal FJT, Clevers HC (2005) WNT signalling and haematopoiesis: a WNT-WNT situation. Nat Rev Immunol 5(1):21–30
Staal FJT, Luis TC (2010) Wnt signaling in hematopoiesis: crucial factors for self-renewal, proliferation, and cell fate decisions. J Cell Biochem 109(5):844–849
Blank U, Karlsson G, Karlsson S (2008) Signaling pathways governing stem-cell fate. Blood 111(2):492–503
Blank U, Karlsson S (2011) The role of smad signaling in hematopoiesis and translational hematology. Leukemia 25(9):1379–1388
Purton LE, Dworkin S, Olsen GH, Walkley CR et al (2006) RARgamma is critical for maintaining a balance between hematopoietic stem cell self-renewal and differentiation. J Exp Med 203(5):1283–1293
Kent D, Copley M, Benz C, Dykstra B et al (2008) Regulation of hematopoietic stem cells by the steel factor/KIT signaling pathway. Clin Cancer Res 14(7):1926–1930
de Graaf CA, Metcalf D (2011) Thrombopoietin and hematopoietic stem cells. Cell Cycle 10(10):1582–1589
Arai F, Hirao A, Ohmura M, Sato H et al (2004) Tie2/Angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell 118(2):149–161
Tothova Z, Gilliland DG (2007) FoxO transcription factors and stem cell homeostasis: insights from the hematopoietic system. Cell Stem Cell 1(2):140–152
Tothova Z, Kollipara R, Huntly BJ, Lee BH et al (2007) FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128(2):325–339
Miyamoto K, Araki KY, Naka K, Arai F et al (2007) Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell 1(1):101–112
Takubo K, Goda N, Yamada W, Iriuchishima H et al (2010) Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell 7(3):391–402
Orkin SH, Zon LI (2008) Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132(4):631–644
Kiritoa K, Kaushansky K (2006) Transcriptional regulation of megakaryopoiesis: thrombopoietin signaling and nuclear factors. Curr Opin Hematol 13(3):151–156
Dore LC, Crispino JD (2011) Transcription factor networks in erythroid cell and megakaryocyte development. Blood 118(2):231–239
Goldfarb AN (2007) Transcriptional control of megakaryocyte development. Oncogene 26(47):6795–6802
Kim SI, Bresnick EH (2007) Transcriptional control of erythropoiesis: emerging mechanisms and principles. Oncogene 26(47):6777–6794
Dias S, Xu W, McGregor S, Kee B (2008) Transcriptional regulation of lymphocyte development. Curr Opin Genet Dev 18(5):441–448
Friedman AD (2007) Transcriptional control of granulocyte and monocyte development. Oncogene 26(47):6816–6828
Friedman AD, Keefer JR, Kummalue T, Liu HT et al (2003) Regulation of granulocyte and monocyte differentiation by CCAAT/enhancer binding protein alpha. Blood Cells Mol Dis 31(3):338–341
Zhang P, Iwasaki-Arai J, Iwasaki H, Fenyus ML et al (2004) Enhancement of hematopoietic stem cell repopulating capacity and self-renewal in the absence of the transcription factor C/EBP alpha. Immunity 21(6):853–863
Suh HC, Gooya J, Renn K, Friedman AD et al (2006) C/EBP alpha determines hematopoietic cell fate in multipotential progenitor cells by inhibiting erythroid differentiation and inducing myeloid differentiation. Blood 107(11):4308–4316
Nemeth MJ, Kirby MR, Bodine DM (2006) Hmgb3 regulates the balance between hematopoietic stem cell self-renewal and differentiation. Proc Natl Acad Sci U S A 103(37):13783–13788
Nemeth MJ, Cline AP, Anderson SM, Garrett-Beal LJ et al (2005) Hmgb3 deficiency deregulates proliferation and differentiation of common lymphoid and myeloid progenitors. Blood 105(2):627–634
Bee T, Swiers G, Muroi S, Pozner A et al (2010) Nonredundant roles for Runx1 alternative promoters reflect their activity at discrete stages of developmental hematopoiesis. Blood 115(15):3042–3050
Levanon D, Glusman C, Bangsow T, Ben-Asher E et al (2001) Architecture and anatomy of the genomic locus encoding the human leukemia-associated transcription factor RUNX1/AML1. Gene 262(1–2):23–33
Ben-Ami O, Pencovich N, Lotem J, Levanon D et al (2009) A regulatory interplay between miR-27a and Runx1 during megakaryopoiesis. Proc Natl Acad Sci U S A 106(1):238–243
Feng J, Iwama A, Satake M, Kohu K (2009) MicroRNA-27 enhances differentiation of myeloblasts into granulocytes by post-transcriptionally downregulating Runx1. Br J Haematol 145(3):412–423
Pozner A, Goldenberg D, Negreanu V, Le SY et al (2000) Transcription-coupled translation control of AML1/RUNX1 is mediated by cap- and internal ribosome entry site-dependent mechanisms. Mol Cell Biol 20(7):2297–2307
Pillai RS, Bhattacharyya SN, Artus CG, Zoller T et al (2005) Inhibition of translational initiation by Let-7 microRNA in human cells. Science 309(5740):1573–1576
Humphreys DT, Westman BJ, Martin DIK, Preiss T (2005) MicroRNAs control translation initiation by inhibiting eukaryotic initiation factor 4E/cap and poly(a) tail function. Proc Natl Acad Sci U S A 102(47):16961–16966
Biggs JR, Peterson LF, Zhang Y, Kraft AS et al (2006) AML1/RUNX1 phosphorylation by cyclin-dependent kinases regulates the degradation of AML1/RUNX1 by the anaphase-promoting complex. Mol Cell Biol 26(20):7420–7429
Guo H, Friedman AD (2011) Phosphorylation of RUNX1 by cyclin-dependent kinase reduces direct interaction with HDAC1 and HDAC3. J Biol Chem 286(1):208–215
Zhang L, Fried FB, Guo H, Friedman AD (2008) Cyclin-dependent kinase phosphorylation of RUNX1/AML1 on 3 sites increases transactivation potency and stimulates cell proliferation. Blood 111(3):1193–1200
Yamaguchi Y, Kurokawa M, Imai Y, Izutsu K et al (2004) AML1 is functionally regulated through p300-mediated acetylation on specific lysine residues. J Biol Chem 279(15):15630–15638
Yoshida H, Kitabayashi I (2008) Chromatin regulation by AML1 complex. Int J Hematol 87(1):19–24
Zhao X, Jankovic V, Gural A, Huang G et al (2008) Methylation of RUNX1 by PRMT1 abrogates SIN3A binding and potentiates its transcriptional activity. Genes Dev 22(5):640–653
Chakraborty S, Sinha KK, Senyuk V, Nucifora G (2003) SUV39H1 interacts with AML1 and abrogates AML1 transactivity. AML1 is methylated in vivo. Oncogene 22(34):5229–5237
Wang L, Gural A, Sun XJ, Zhao XY et al (2011) The leukemogenicity of AML1-ETO is dependent on site-specific lysine acetylation. Science 333(6043):765–769
Pimanda JE, Donaldson IJ, de Bruijn MF, Kinston S et al (2007) The SCL transcriptional network and BMP signaling pathway interact to regulate RUNX1 activity. Proc Natl Acad Sci U S A 104(3):840–845
Shen R, Chen M, Wang YJ, Kaneki H et al (2006) Smad6 interacts with Runx2 and mediates smad ubiquitin regulatory factor 1-induced Runx2 degradation. J Biol Chem 281(6):3569–3576
Knezevic K, Bee T, Wilson NK, Janes ME et al (2011) A Runx1-Smad6 rheostat controls Runx1 activity during embryonic hematopoiesis. Mol Cell Biol 31(14):2817–2826
Mitelman F, Johansson B, Mertens F (2007) The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 7(4):233–245
Crans HN, Sakamoto KM (2001) Transcription factors and translocations in lymphoid and myeloid leukemia. Leukemia 15(3):313–331
Hess JL (2004) MLL: a histone methyltransferase disrupted in leukemia. Trends Mol Med 10(10):500–507
Marschalek R (2010) Mixed lineage leukemia: roles in human malignancies and potential therapy. FEBS J 277(8):1822–1831
Meyer C, Kowarz E, Hofmann J, Renneville A et al (2009) New insights to the MLL recombinome of acute leukemias. Leukemia 23(8):1490–1499
Thomas M, Gessner A, Vornlocher HP, Hadwiger P et al (2005) Targeting MLL-AF4 with short interfering RNAs inhibits clonogenicity and engraftment of t(4;11)-positive human leukemic cells. Blood 106(10):3559–3566
Thiel AT, Blessington P, Zou T, Feather D et al (2010) MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell 17(2):148–159
Yokoyama A, Somervaille TCP, Smith KS, Rozenblatt-Rosen O et al (2005) The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 123(2):207–218
Grembecka J, He S, Shi A, Purohit T et al (2012) Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol 8(3):277–284
Tan JY, Jones M, Koseki H, Nakayama M et al (2011) CBX8, a polycomb group protein, is essential for MLL-AF9-induced leukemogenesis. Cancer Cell 20(5):563–575
Yokoyama A, Lin M, Naresh A, Kitabayashi I et al (2010) A higher-order complex containing AF4 and ENL family proteins with P-TEFb facilitates oncogenic and physiologic MLL-dependent transcription. Cancer Cell 17(2):198–212
Biswas D, Milne TA, Basrur V, Kim J et al (2011) Function of leukemogenic mixed lineage leukemia 1 (MLL) fusion proteins through distinct partner protein complexes. Proc Natl Acad Sci U S A 108(38):15751–15756
Okada Y, Feng Q, Lin YH, Jiang Q et al (2005) hDOT1L links histone methylation to leukemogenesis. Cell 121(2):167–178
Milne TA, Kim J, Wang GG, Stadler SC et al (2010) Multiple interactions recruit MLL1 and MLL1 fusion proteins to the HOXA9 locus in leukemogenesis. Mol Cell 38(6):853–863
Jang MK, Mochizuki K, Zhou MS, Jeong HS et al (2005) The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell 19(4):523–534
Krivtsov AV, Feng Z, Lemieux ME, Faber J et al (2008) H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell 14(5):355–368
Daigle SR, Olhava EJ, Therkelsen CA, Majer CR et al (2011) Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell 20(1):53–65
Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G et al (2011) Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 478(7370):529–533
Wang QF, Wu G, Mi SL, He FH et al (2011) MLL fusion proteins preferentially regulate a subset of wild-type MLL target genes in the leukemic genome. Blood 117(25):6895–6905
Orlovsky K, Kalinkovich A, Rozovskaia T, Shezen E et al (2011) Down-regulation of homeobox genes MEIS1 and HOXA in MLL-rearranged acute leukemia impairs engraftment and reduces proliferation. Proc Natl Acad Sci U S A 108(19):7956–7961
Arai S, Yoshimi A, Shimabe M, Ichikawa M et al (2011) Evi-1 is a transcriptional target of mixed-lineage leukemia oncoproteins in hematopoietic stem cells. Blood 117(23):6304–6314
Zeisig BB, Milne T, Garcia-Cuellar MP, Schreiner S et al (2004) Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalization. Mol Cell Biol 24(2):617–628
Zuber J, Rappaport AR, Luo WJ, Wang E et al (2011) An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev 25(18):1628
Bernt KM, Zhu N, Sinha AU, Vempati S et al (2011) MLL-rearranged leukemia is dependent on aberrant H3K79 methylation by DOT1L. Cancer Cell 20(1):66–78
Puente XS, Pinyol M, Quesada V, Conde L et al (2011) Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 475(7354):101–105
Ding L, Ley TJ, Larson DE, Miller CA et al (2012) Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481(7382):506–510
Amsellem S, Pflumio F, Bardinet D, Izac B et al (2003) Ex vivo expansion of human hematopoietic stem cells by direct delivery of the HOXB4 homeoprotein. Nat Med 9(11):1423–1427
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Wilkinson, A.C., Göttgens, B. (2013). Transcriptional Regulation of Haematopoietic Stem Cells. In: Hime, G., Abud, H. (eds) Transcriptional and Translational Regulation of Stem Cells. Advances in Experimental Medicine and Biology, vol 786. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-6621-1_11
Download citation
DOI: https://doi.org/10.1007/978-94-007-6621-1_11
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-6620-4
Online ISBN: 978-94-007-6621-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)