
Abstract
Background/Objectives:
The relationship between obesity and chronic disease risk is well-established; the underlying biological mechanisms driving this risk increase may include obesity-related epigenetic modifications. To explore this hypothesis, we conducted a genome-wide analysis of DNA methylation and body mass index (BMI) using data from a subset of women in the Sister Study.
Subjects/Methods:
The Sister Study is a cohort of 50 884 US women who had a sister with breast cancer but were free of breast cancer themselves at enrollment. Study participants completed examinations which included measurements of height and weight, and provided blood samples. Blood DNA methylation data generated with the Illumina Infinium HumanMethylation27 BeadChip array covering 27,589 CpG sites was available for 871 women from a prior study of breast cancer and DNA methylation. To identify differentially methylated CpG sites associated with BMI, we analyzed this methylation data using robust linear regression with adjustment for age and case status. For those CpGs passing the false discovery rate significance level, we examined the association in a replication set comprised of a non-overlapping group of 187 women from the Sister Study who had DNA methylation data generated using the Infinium HumanMethylation450 BeadChip array. Analysis of this expanded 450 K array identified additional BMI-associated sites which were investigated with targeted pyrosequencing.
Results:
Four CpG sites reached genome-wide significance (false discovery rate (FDR) q<0.05) in the discovery set and associations for all four were significant at strict Bonferroni correction in the replication set. An additional 23 sites passed FDR in the replication set and five were replicated by pyrosequencing in the discovery set. Several of the genes identified including ANGPT4, RORC, SOCS3, FSD2, XYLT1, ABCG1, STK39, ASB2 and CRHR2 have been linked to obesity and obesity-related chronic diseases.
Conclusions:
Our findings support the hypothesis that obesity-related epigenetic differences are detectable in blood and may be related to risk of chronic disease.
Similar content being viewed by others

Introduction
Obesity is a well-established risk factor for chronic diseases including cancer, metabolic disease and Type 2 diabetes. High body mass index (BMI) has some degree of heritability, but is also influenced by lifestyle and environment.1, 2, 3, 4 High BMI and excess adiposity likely contribute to disease risk through a variety of biological pathways, which may include epigenetic processes such as alterations in DNA methylation patterns.5, 6
There is a growing body of evidence supporting links between DNA methylation and obesity. A recently published study of adipose tissue collected from a small number of monozygotic twins found evidence of differential methylation between lean and overweight/obese twins at multiple genes.7 Other studies have identified differentially methylated CpGs in adipose tissue between lean and obese individuals, including genes that were also differentially expressed in the adipose tissue.8, 9, 10 Because adipose tissue is difficult to obtain in large population studies, the few existing large epigenome-wide association studies have used blood DNA to identify potential methylation differences related to BMI. These studies have identified more than 40 CpG sites showing an association between methylation levels and BMI in mixed cohorts of men and women;10, 11 obesity-related differences in DNA methylation may help identify key biological pathways involved in disease pathogenesis. Using a nationwide cohort study, we have previously reported the association between obesity and risk of breast cancer.12 Here, we extend that work using a subset of the subjects to examine the association between BMI and blood DNA methylation.
Materials and methods
Study population
Study participants were drawn from the Sister Study cohort, a prospective volunteer cohort of women from the United States and Puerto Rico. From 2003 to 2009, women were recruited into the Sister Study if they had a sister with breast cancer but had not been diagnosed with breast cancer themselves. They complete annual, biennial and triennial updates on cancer and various other lifestyle, exposure and health factors. Participants have a home visit from a trained examiner at study baseline which includes blood collection and measurements of height and weight. The study was approved by the Institutional Review Board of the National Institute of Environmental Health Sciences, NIH and the Copernicus Group Institutional Review Board. Inclusion in this analysis is covered by the informed consent form all included cohort participants agreed to and signed. Because of ethical restrictions (participant confidentiality), data are available upon request. Permission to access Sister Study data may be obtained at www.sisterstudystars.org or by contacting the authors.
DNA methylation array analysis
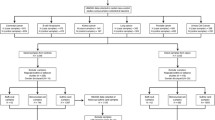
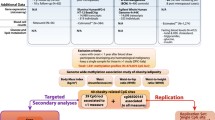
We examined the relationship between methylation and BMI using two DNA methylation datasets available within the Sister Study. The ‘discovery set’ was comprised of 871 white, non-Hispanic women with methylation array data available on 27 589 CpG (cytosine-phosphate-guanine) sites from a nested case–cohort study of breast cancer. A second, smaller ‘replication set’ was comprised of 187 white, non-Hispanic women with methylation array data on 485 512 CpG sites from a nested case-control study of diethylstilbestrol (DES) exposure. Study populations and details of methods for methylation measurements have been previously described.12, 13, 14
Statistical analysis
To examine the association between BMI and DNA methylation in the discovery and replication sets, we used robust linear regression modeling. To correct for multiple testing the FDR was set at q<0.05 as previously described;12, 14 specific CpGs were examined in the replication set for the same direction of association and an unadjusted P<0.05. Data pre-processing, normalization methods and quality control measures are detailed in Supplementary Material of the previously published papers.12, 14 Briefly, methylation intensity values were background-corrected using the Robust Multichip Average method15 and quantile-normalized across arrays. Methylation array plates included laboratory controls with known methylation levels to assess precision of measurement, and duplicate samples to assess reproducibility of results within the assay. Each methylation array included probes to assess bisulfite conversion efficiency and negative control probes to measure background fluorescent intensity. Samples with poor bisulfite conversion efficiency (<3800) or having >5% of probes with unreliable measures (detection P>0.05) were excluded (N=3 probes in the 27k discovery set and N=491 probes in the 450k replication set). The methylation outcome was estimated using beta values, which are calculated using fluorescence intensities for unmethylated (U) and methylated (M) alleles as M/(M+U+100). Beta values range from 0 (completely unmethylated) to 1 (100% methylated). In the 450 K discovery set, we also excluded CpG probes with single-nucleotide polymorphisms present at target sites or that mapped to multiple genomic regions (N=48 158), Illumina-designed single-nucleotide polymorphism probes (N=65), and CpG sites on the X and Y chromosomes (N=10 257) (Illumina, San Diego, CA, USA). In the 27 K discovery data set we examined 27 575 probes and in the 450 K replication data set we examined 426 606 probes. Singular value decomposition analysis of the raw data set revealed that the top principal components derived from the methylation β-value matrix were highly correlated with plate, bisulfite conversion intensities and age. We adjusted for these factors together with breast cancer status in all linear regression association analyses. All association tests were also adjusted for the proportions of different types of white blood cells estimated using a method described by Houseman et al.16, 17
Pyrosequencing validation
Although the sample size in the replication set was much smaller (N=187), the 450 K array used for this set provides methylation data at ~320 000 additional CpG sites not covered by the 27 K array used in the discovery set. We selected five of the top BMI-associated CpGs identified from 450 K analysis of the replication set for independent validation by pyrosequencing in samples from the discovery set. Pyrosequencing assays for five CpGs cg17501210 (RPS6KA2), cg06500161 (ABCG1), cg07728579 (FSD2), cg11775828 (STK39) and cg13134297 (CRHR2) were designed using Pyromark Assay Design version 2.0.2.15 (Qiagen, Valencia, CA, USA). Primer sequences are detailed in Supplementary Table 1. Reaction mixtures (25 μl) containing 100 ng of bisulfite-converted DNA, 5 pmol of each primer (forward and reverse) PCR buffer (Invitrogen, Carlsbad, CA, USA), 3 mm MgCl2, 1 mm dNTP and 0.8 units of taq polymerase (Invitrogen), were heated to 95 °C for 15 min, followed by 45 PCR cycles (95 °C for 20 s, 55 °C for 20 s and 72 °C for 20 s) with a final extension at 72 °C for 5 min. After PCR, the biotin-labeled PCR product was hybridized to streptavidin-coated sepharose beads (GE Healthcare, Madison, WI, USA) and denatured in 0.2 m sodium hydroxide to provide a single-stranded sequencing template. Pyrosequencing primers (0.3 μmol l−1) were annealed to the single-stranded template and the pyrosequencing was carried out using PyroMark Q96 MD System (Qiagen) according to the manufacturer’s instructions. The percentage methylation was quantified using the Pyro Q-CpG Software (Qiagen). Associations between methylation levels and BMI were examined using linear regression analysis of the methylation percentages and BMI with adjustment for age and breast cancer status.
Results
BMI in the Sister Study DNA methylation datasets
Basic characteristics of Sister Study participants with methylation array data are summarized in Table 1. As the discovery set includes a substantial number of women who later developed breast cancer, characteristics are stratified by breast cancer case status. The mean BMI in both the discovery subset (27.1 kg m−2, s.d. 5.7 kg m−12) and replication subset (26.7 kg m−2, s.d. 5.6 kg m−2) was slightly above the CDC’s ‘healthy BMI’ upper boundary. In both the discovery and replication set, at least 50% of participants fell into the overweight (BMI 25.0–29.9) or obese (BMI 30+) categories. The replication set had a larger proportion of women in the healthy BMI category (49%) than the discovery set (42%). Seven percent of the discovery set and 6% of the replication set reported they were current smokers, and slightly more women in the discovery set had at least one pregnancy (87%) compared with the replication set (81%).
Analysis of BMI and DNA Methylation in discovery set
In an analysis limited to women in the discovery set who had not developed breast cancer (N=571), BMI was associated with methylation at two CpG sites located in the genes LGALS3BP (q-value=0.02) and ANGPT4 (q-value=0.05) (Table 2). When the analysis was expanded to include women who developed breast cancer during study follow-up with adjustment for case status (N=871), the same two sites remained associated (cg14870271 and cg03218374) and two additional associated CpG sites were identified in the genes RORC and SOCS3 (cg18149207 at q-value=0.02 and cg27637521 at q-value=0.02). All four of these CpG sites are present on the Infinium450K array used in the replication study.
Analysis of BMI and DNA methylation in replication set
The association between BMI and DNA methylation at these four CpGs was examined in the replication sample (N=187); P-values for all four associations passed a strict Bonferroni correction for multiple testing (P<0.0125) as shown in Table 2. The CpG sites located in LGALS3BP and RORC showed an ~0.1% increase in methylation with each increasing unit (1 kg m−2) of BMI, and the site in ANGPT4 showed 0.2% increase in methylation per 1 unit of BMI. These differences would correspond to approximately 1% higher methylation at these sites between individuals at normal weight (BMI=20) verse obese individuals (BMI=30). The CpG located in SOCS3 showed an association in the opposite direction, with 0.05% decrease in methylation with each increasing unit of BMI.
A 450 K array-wide analysis of DNA methylation and BMI in the replication set women revealed associations (q<0.05) for 23 CpG sites (Table 3). Three of these 23 sites were covered on the 27 K array; one of the three, cg21282997 located in the gene IL18RAP, showed an association with BMI at p=0.02 in the discovery set. The remaining 20 CpG sites were not covered on the 27 K array; from these 20 we selected five of the top CpG sites for pyrosequencing analysis to determine methylation status in participants from the discovery set (Table 3).
Results of pyrosequencing validation
Pyrosequencing analysis of 871 DNA samples from the discovery set validated the associations between BMI and methylation at the five selected CpG sites (Table 4). Point estimates for the associations between BMI and methylation at all five CpGs tested (cg17501210; cg06500161; cg07728579; cg11775828; and cg13134297) generated using pyrosequencing passed a Bonferroni-corrected P-value threshold of 0.01 and both the magnitude and direction of effect estimates were similar between array and pyrosequencing data. Pyrosequencing also provided information on CpGs flanking the sites of interest for cg17501210 (RPS6KA2) and cg06500161 (ABCG1), and these flanking CpGs had findings similar to that of the target CpG (Table 4).
Replication of findings from published studies of BMI and genome-wide DNA methylation
Recent epigenome-wide association studies have used the Infinium HumanMethylation450 BeadChip to investigate BMI and peripheral blood DNA methylation data. We examined whether differentially methylated CpGs reported in those studies were differentially methylated in our replication set data (Supplementary Table 2). A study from the Cardiogenics Consortium identified five CpGs associated with BMI in their discovery cohort of men and women and replicated three of these associations.18 Our data provide additional support for their observed associations in a women-only cohort with BMI at two CpG sites: one in KLF13 (cg07814318 at P⩽0.0003) and one in KIAA0664/CLUH (cg09664445 at P⩽0.04). A second study utilizing the Atherosclerosis Risk in Communities cohort and multiple replication cohorts identified 76 CpGs that were differentially associated with BMI and replicated 37 sites.10 Eighteen of these associations were recently replicated in an Arab population,19 and we found that 27 of these associations replicated in our data set, including the site cg06500161 in ABCG1 identified in our discovery set. We provide the first replication for associations with BMI at the CpG sites cg09554443 in CD247, cg04986899 in XYLT1, cg05242915 on chromosome 19 and cg03562528 in ASB2.
Discussion
Using 27 K, 450 K, and pyrosequencing data on 1066 women from the Sister Study cohort, we identified and replicated eight previously unreported associations between BMI and CpG site DNA methylation and confirm a previously reported and replicated association at cg06500161 in ABCG1. Our results also provide support for association at 27 previously reported additional CpG sites and provide the first replication for associations with BMI at four of these sites.
We identified and confirmed methylation differences associated with BMI at CpG sites in the genes LGALS3BP, ANGPT4, RORC, SOCS3, RSPK6A2, FSD2, ABCG1, STK39 and CRHR2. Several of the genes with differentially methylated sites in our study have been linked to obesity and obesity-related chronic diseases. The decreased methylation with increasing BMI observed at cg27637521 is of particular interest as it is 25 bp from the transcriptional start site of SOCS3 (suppressor of cytokine signaling 3). The two flanking CpGs in SOCS3 (cg10508317 and cg10279487) also show decreased methylation with increasing BMI (P⩽0.003). Animal models indicate that SOCS3 expression inhibits leptin signaling and is likely involved in the decreased leptin sensitivity observed in obese individuals.20, 21, 22, 23, 24 SOCS3 is a negative regulator of cytokine signaling; expression is induced by various cytokines, including IL-6, IL-10 and interferon (IFN)-γ.25, 26, 27 Genetic polymorphisms in and near SOCS3 are associated with obesity in human population studies.28, 29, 30 In cohort studies of Indian Asians and Europeans, decreased DNA methylation in blood at another CpG (cg18181703) in the coding region of SOCS3 has been linked to risk of Type 2 (T2) diabetes.31
We also identified a differentially methylated region in the gene CRHR2 (corticotrophin-releasing hormone receptor 2); the CpG site we identified (cg13134297) is in the first intron of this gene. As with SOCS3, examination of flanking CpGs on the 450 K array (cg23068772 and cg22007110) shows associations with BMI (P⩽0.002) with the same directionality. CRHR2 is involved in corticotrophin-releasing hormone signaling and likely has a role in coordinating endocrine and autonomic responses to stress.32 STK39 (Serine Threonine Kinase 39) also plays a role in cellular responses to stress; we identified and confirmed increasing methylation with increasing BMI at this gene at cg11775828. Flanking CpGs on the 450 K array (cg21899461 and cg03331300) were also associated with BMI at P<0.05. Polymorphisms in this gene have been linked to risk of hypertension.33, 34, 35 Epigenetic silencing of STK39 through DNA hypermethylation in B-cell lymphoma appears to promote cancer progression by inhibiting apoptosis in cells with DNA damage.36
The differentially methylated CpGs we identified in the genes RORC and FSD2 may also be promising targets for future studies of obesity and development of cardiovascular and metabolic diseases. RORC (RAR-Related Orphan Receptor C) regulates production of the inflammatory cytokine IL-17 by T helper-17 (Th17) cells; Th17 cell activation and cytokine production appears to play a critical role in the pathogenesis of diabetes.37, 38, 39, 40, 41 Additionally, genetic variation in RORC has been linked to body fat composition levels in cattle.42 Single-nucleotide polymorphisms in FSD2 (Fibronectin Type III and SPRY Domain Containing 2) have been linked to the presence and volume of carotid plaque in a Caribbean Hispanic population.43
We provided the first replication of associations with BMI at the CpGs cg09554443 in CD247, cg04986899 in XYLT1, cg05242915 on chromosome 19, and cg03562528 in ASB2. CD247 (T-cell surface glycoprotein CD3 zeta chain) plays an important role in antigen recognition in the immune response and shows altered expression patterns in peripheral blood in individuals with Type 2 Diabetes.44 XYLT1 (Xylosyltransferase 1) is necessary for biosynthesis of glycosaminoglycan chains. Genetic variations in this gene can increase risk of abdominal aortic aneurysm.45 ASB2 (Ankyrin repeat and SOCS box protein 2) is differentially methylated in atherosclerotic aorta tissue compared with healthy aortic tissue.46
The observed association between adult BMI and methylation at cg03218374 in the Angiopoetin-4 (ANGPT4) gene is consistent with an existing study that links methylation at this same CpG in cord blood to infant birth weight,47 indicating that this association between body size and methylation is consistent in both infants and adults. This CpG is located in the promoter region of ANGPT4, 18 bp upstream of the transcription start site. ANGPT4 plays in important role in vascular angiogenesis, and may help to mediate lipid breakdown.48 We have also confirmed the association between BMI and methylation at ABCG1 (cg06500161) observed in previous studies.10, 11 Methylation of multiple CpG sites in ABCG1 is linked to individual variability in blood lipid levels.49 In our pyrosequencing validation analysis of RPS6KA2 and ABCG1, our primers covered additional CpG sites (two sites in RPS6KA2 and 1 in ABCG1) that were not covered on the Infinium450K array. These additional CpG sites exhibited the same associations with BMI as the adjacent sites on the array (Table 4). Both sites in ABCG1, 1 bp apart, and all three sites in RPS6KA2 (spanning a 9 bp region) show increasing methylation with increasing BMI.
We have identified and validated associations between BMI and methylation at nine CpG sites using a combination of array-based and pyrosequencing methylation measures, and provided further confirmation for an additional 27 associations reported in previous studies. A potential limitation of our study is the inclusion of DES-exposed women in our replication set, which might potentially affect our results if DES also affects methylation patterns. However, previous analysis of these participants’ DES exposure status and the DNA methylation array data showed no detectable associations between DES exposure and DNA methylation.14 Although this study’s cross-sectional design limits our ability to determine whether these methylation differences are caused by obesity, it has the advantage of large number of participants with both DNA methylation data and examiner-measured height and weight.
References
Bouchard C, Tremblay A . Genetic influences on the response of body fat and fat distribution to positive and negative energy balances in human identical twins. J Nutr 1997; 127: 943S–947S.
Albuquerque D, Stice E, Rodriguez-Lopez R, Manco L, Nobrega C . Current review of genetics of human obesity: from molecular mechanisms to an evolutionary perspective. Mol Genet Genomics 2015; 290: 1191–1221.
Hemani G, Yang J, Vinkhuyzen A, Powell JE, Willemsen G, Hottenga JJ et al. Inference of the genetic architecture underlying BMI and height with the use of 20,240 sibling pairs. Am J Hum Genet 2013; 93: 865–875.
Arabshahi S, Ibiebele TI, Hughes MC, Lahmann PH, Williams GM, van der Pols JC . Dietary patterns and weight change: 15-year longitudinal study in Australian adults. Eur J Nutr 2016. e-pub ahead of print 26 February 2016.
Martinez JA, Milagro FI, Claycombe KJ, Schalinske KL . Epigenetics in adipose tissue, obesity, weight loss, and diabetes. Adv Nutr 2014; 5: 71–81.
Garcia-Jimenez C, Gutierrez-Salmeron M, Chocarro-Calvo A, Garcia-Martinez JM, Castano A, De la Vieja A . From obesity to diabetes and cancer: epidemiological links and role of therapies. Br J Cancer 2016; 114: 716–722.
Pietilainen KH, Ismail K, Jarvinen E, Heinonen S, Tummers M, Bollepalli S et al. DNA methylation and gene expression patterns in adipose tissue differ significantly within young adult monozygotic BMI-discordant twin pairs. Int J Obes (Lond) 2015; 40: 654–661.
Arner P, Sinha I, Thorell A, Ryden M, Dahlman-Wright K, Dahlman I . The epigenetic signature of subcutaneous fat cells is linked to altered expression of genes implicated in lipid metabolism in obese women. Clin Epigenet 2015; 7: 93.
Gillberg L, Perfilyev A, Brons C, Thomasen M, Grunnet LG, Volkov P et al. Adipose tissue transcriptomics and epigenomics in low birthweight men and controls: role of high-fat overfeeding. Diabetologia 2016; 59: 799–812.
Demerath EW, Guan W, Grove ML, Aslibekyan S, Mendelson M, Zhou YH et al. Epigenome-wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum Mol Genet 2015; 24: 4464–4479.
Aslibekyan S, Demerath EW, Mendelson M, Zhi D, Guan W, Liang L et al. Epigenome-wide study identifies novel methylation loci associated with body mass index and waist circumference. Obesity (Silver Spring, Md) 2015; 23: 1493–1501.
Xu Z, Bolick SC, DeRoo LA, Weinberg CR, Sandler DP, Taylor JA . Epigenome-wide association study of breast cancer using prospectively collected sister study samples. J Natl Cancer Inst 2013; 105: 694–700.
Harlid S, Xu Z, Panduri V, Sandler DP, Taylor JA . CpG sites associated with cigarette smoking: analysis of epigenome-wide data from the sister study. Environ Health Perspect 2014; 122: 673–678.
Harlid S, Xu Z, Panduri V, D'Aloisio AA, DeRoo LA, Sandler DP et al. In utero exposure to diethylstilbestrol and blood DNA methylation in women ages 40-59 years from the sister study. PLoS One 2015; 10: e0118757.
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics (Oxford, England) 2003; 4: 249–264.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics 2012; 13: 86.
Accomando WP, Wiencke JK, Houseman EA, Butler RA, Zheng S, Nelson HH et al. Decreased NK cells in patients with head and neck cancer determined in archival DNA. Clin Cancer Res 2012; 18: 6147–6154.
Dick KJ, Nelson CP, Tsaprouni L, Sandling JK, Aissi D, Wahl S et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet 2014; 383: 1990–1998.
Al Muftah WA, Al-Shafai M, Zaghlool SB, Visconti A, Tsai P-C, Kumar P et al. Epigenetic associations of type 2 diabetes and BMI in an Arab population. Clin Epigenet 2016; 8: 13.
Bjørbæk C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS . Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell 1998; 1: 619–625.
Pedroso JAB, Buonfiglio DC, Cardinali LI, Furigo IC, Ramos-Lobo AM, Tirapegui J et al. Inactivation of SOCS3 in leptin receptor-expressing cells protects mice from diet-induced insulin resistance but does not prevent obesity. Mol Metab 2014; 3: 608–618.
Howard JK, Flier JS . Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol Metab 2006; 17: 365–371.
Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H et al. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med 2004; 10: 739–743.
Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjoørbæk C, Flier JS . Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med 2004; 10: 734–738.
Alexander WS, Hilton DJ . The role of suppressors of cytokine signaling (SOCS) proteins in regulation of the immune response. Annu Rev Immunol 2004; 22: 503–529.
Cohney SJ, Sanden D, Cacalano NA, Yoshimura A, Mui A, Migone TS et al. SOCS-3 is tyrosine phosphorylated in response to interleukin-2 and suppresses STAT5 phosphorylation and lymphocyte proliferation. Mol Cell Biol 1999; 19: 4980–4988.
Dagvadorj J, Naiki Y, Tumurkhuu G, Shadat Mohammod Noman A, Iftakhar-E-Khuda I, Komatsu T et al. Tumor necrosis factor-α augments lipopolysaccharide-induced suppressor of cytokine signalling 3 (SOCS-3) protein expression by preventing the degradation. Immunology 2010; 129: 97–104.
Li P, Tiwari HK, Lin WY, Allison DB, Chung WK, Leibel RL et al. Genetic association analysis of 30 genes related to obesity in a European American population. Int J Obes (Lond) 2014; 38: 724–729.
Tang W, Zou JJ, Chen XF, Zheng JY, Zeng HZ, Liu ZM et al. Association of two polymorphisms within and near SOCS3 gene with obesity in three nationalities in Xinjiang province of China. Acta Pharmacol Sin 2011; 32: 1381–1386.
Talbert ME, Langefeld CD, Ziegler J, Mychaleckyj JC, Haffner SM, Norris JM et al. Polymorphisms near SOCS3 are associated with obesity and glucose homeostasis traits in Hispanic Americans from the Insulin Resistance Atherosclerosis Family Study. Hum Genet 2009; 125: 153–162.
Chambers JC, Loh M, Lehne B, Drong A, Kriebel J, Motta V et al. Epigenome-wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: a nested case-control study. Lancet Diabetes Endocrinol 2015; 3: 526–534.
Grammatopoulos DK . Insights into mechanisms of corticotropin-releasing hormone receptor signal transduction. Br J Pharmacol 2012; 166: 85–97.
Persu A, Evenepoel L, Jin Y, Mendola A, Staessen JA, Vikkula M . 6C.05: STK39 and WNK1 are potential hypertension susceptibility genes in the Belhypgen Cohort. J Hypertension 2015; 33 ():e80.
Maatta KM, Nikkari ST, Lahteela KH, Palmroos PB, Kunnas TA . A functional variant in the serine-threonine kinase coding gene is associated with hypertension: a case-control study in a Finnish population, the Tampere adult population cardiovascular risk study. J Hypertension 2013; 31: 516–520.
Li B, Yang M, Liu JW . Expression of STK39 in peripheral blood of hypertension patients and the relationship between its genetic polymorphism and blood pressure. Genet Mol Res 2015; 14: 16461–16468.
Balatoni CE, Dawson DW, Suh J, Sherman MH, Sanders G, Hong JS et al. Epigenetic silencing of Stk39 in B-cell lymphoma inhibits apoptosis from genotoxic stress. Am J Pathol 2009; 175: 1653–1661.
Endo Y, Asou HK, Matsugae N, Hirahara K, Shinoda K, Tumes DJ et al. Obesity drives Th17 cell differentiation by inducing the lipid metabolic kinase, ACC1. Cell Rep 2015; 12: 1042–1055.
Ip B, Cilfone NA, Belkina AC, DeFuria J, Jagannathan-Bogdan M, Zhu M et al. Th17 cytokines differentiate obesity from obesity-associated type 2 diabetes and promote TNFalpha production. Obesity (Silver Spring, Md) 2016; 24: 102–112.
Reinert-Hartwall L, Honkanen J, Salo HM, Nieminen JK, Luopajarvi K, Harkonen T et al. Th1/Th17 plasticity is a marker of advanced beta cell autoimmunity and impaired glucose tolerance in humans. J Immunol 2015; 194: 68–75.
Romanowski M, Domanski L, Pawlik A, Osekowska B, Dziedziejko V, Safranow K et al. Interleukin-17 gene polymorphisms in patients with post-transplant diabetes mellitus. Eur Rev Med Pharmacol Sci 2015; 19: 3152–3156.
Bellemore SM, Nikoopour E, Krougly O, Lee-Chan E, Fouser LA, Singh B . Pathogenic T helper type 17 cells contribute to type 1 diabetes independently of interleukin-22. Clin Exp Immunol 2016; 183: 380–388.
Barendse W, Bunch RJ, Kijas JW, Thomas MB . The effect of genetic variation of the retinoic acid receptor-related orphan receptor C gene on fatness in cattle. Genetics 2007; 175: 843–853.
Dong C, Beecham A, Slifer S, Wang L, Blanton SH, Wright CB et al. Genomewide linkage and peakwide association analyses of carotid plaque in Caribbean Hispanics. Stroke 2010; 41: 2750–2756.
Eldor R, Klieger Y, Sade-Feldman M, Vaknin I, Varfolomeev I, Fuchs C et al. CD247, a novel T cell-derived diagnostic and prognostic biomarker for detecting disease progression and severity in patients with type 2 diabetes. Diabetes Care 2015; 38: 113–118.
Gotting C, Prante C, Schillinger M, Exner M, Domanovits H, Raith M et al. Xylosyltransferase I variants and their impact on abdominal aortic aneurysms. Clin Chim Acta 2008; 391: 41–45.
Zaina S, Heyn H, Carmona FJ, Varol N, Sayols S, Condom E et al. DNA methylation map of human atherosclerosis. Circ Cardiovasc Genet 2014; 7: 692–700.
Turan N, Ghalwash MF, Katari S, Coutifaris C, Obradovic Z, Sapienza C . DNA methylation differences at growth related genes correlate with birth weight: a molecular signature linked to developmental origins of adult disease? BMC Med Genomics 2012; 5: 10.
Gray NE, Lam LN, Yang K, Zhou AY, Koliwad S, Wang JC . Angiopoietin-like 4 (Angptl4) protein is a physiological mediator of intracellular lipolysis in murine adipocytes. J Biol Chem 2012; 287: 8444–8456.
Pfeiffer L, Wahl S, Pilling LC, Reischl E, Sandling JK, Kunze S et al. DNA methylation of lipid-related genes affects blood lipid levels. Circ Cardiovasc Genet 2015; 8: 334–342.
Ollikainen M, Ismail K, Gervin K, Kyllonen A, Hakkarainen A, Lundbom J et al. Genome-wide blood DNA methylation alterations at regulatory elements and heterochromatic regions in monozygotic twins discordant for obesity and liver fat. Clin Epigen 2015; 7: 39.
Acknowledgements
We thank Dr Sarah Reese and Dr Yong-Moon Park for their critical review of this manuscript. We would also like to thank the women who volunteered to participate in the Sister Study. We thank the National Institutes of Health (NIH) Center for Inherited Disease Research and the National Institute of Environmental Health Sciences (NIEHS) Molecular Genetics Core and Microarray Core for their technical support. This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (Z01 ES044005, Z01 ES044032, and Z01 ES049033) which provided funding for design and conduct of the study; collection, management, analysis and interpretation of the data; and preparation, review and approval of the manuscript.
Author contributions
All authors contributed to the design of the study, the generation, analysis, and interpretation of results, and the drafting and revision of the manuscript. All authors have reviewed and approved the final manuscript. LEW and JAT confirm they had full access to the data and final responsibility for the decision to submit for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on International Journal of Obesity website
Supplementary information
Rights and permissions
About this article

Cite this article
Wilson, L., Harlid, S., Xu, Z. et al. An epigenome-wide study of body mass index and DNA methylation in blood using participants from the Sister Study cohort. Int J Obes 41, 194–199 (2017). https://doi.org/10.1038/ijo.2016.184
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ijo.2016.184
- Springer Nature Limited
This article is cited by
-
Negative association between higher maternal pre-pregnancy body mass index and breastfeeding outcomes is not mediated by DNA methylation
Scientific Reports (2024)
-
Genetics and Epigenetics in Obesity: What Do We Know so Far?
Current Obesity Reports (2023)
-
DNA methylation at the suppressor of cytokine signaling 3 (SOCS3) gene influences height in childhood
Nature Communications (2023)
-
Recent progress in epigenetics of obesity
Diabetology & Metabolic Syndrome (2022)
-
Placental DNA methylation changes associated with maternal prepregnancy BMI and gestational weight gain
International Journal of Obesity (2020)