
Abstract
Background
Obesity is an established risk factor for several common chronic diseases such as breast and colorectal cancer, metabolic and cardiovascular diseases; however, the biological basis for these relationships is not fully understood. To explore the association of obesity with these conditions, we investigated peripheral blood leucocyte (PBL) DNA methylation markers for adiposity and their contribution to risk of incident breast and colorectal cancer and myocardial infarction.
Methods
DNA methylation profiles (Illumina Infinium® HumanMethylation450 BeadChip) from 1941 individuals from four population-based European cohorts were analysed in relation to body mass index, waist circumference, waist-hip and waist-height ratio within a meta-analytical framework. In a subset of these individuals, data on genome-wide gene expression level, biomarkers of glucose and lipid metabolism were also available. Validation of methylation markers associated with all adiposity measures was performed in 358 individuals. Finally, we investigated the association of obesity-related methylation marks with breast, colorectal cancer and myocardial infarction within relevant subsets of the discovery population.
Results
We identified 40 CpG loci with methylation levels associated with at least one adiposity measure. Of these, one CpG locus (cg06500161) in ABCG1 was associated with all four adiposity measures (P = 9.07×10−8 to 3.27×10−18) and lower transcriptional activity of the full-length isoform of ABCG1 (P = 6.00×10−7), higher triglyceride levels (P = 5.37×10−9) and higher triglycerides-to-HDL cholesterol ratio (P = 1.03×10−10). Of the 40 informative and obesity-related CpG loci, two (in IL2RB and FGF18) were significantly associated with colorectal cancer (inversely, P < 1.6×10−3) and one intergenic locus on chromosome 1 was inversely associated with myocardial infarction (P < 1.25×10−3), independently of obesity and established risk factors.
Conclusion
Our results suggest that epigenetic changes, in particular altered DNA methylation patterns, may be an intermediate biomarker at the intersection of obesity and obesity-related diseases, and could offer clues as to underlying biological mechanisms.
Similar content being viewed by others

Introduction
Obesity is a growing global public health concern with an estimated 1.9 billion adults classified as overweight or obese worldwide [1]. Obesity is a risk factor for a variety of chronic diseases including type 2 diabetes, cardiovascular disease (CVD) and a number of malignancies such as postmenopausal breast, colorectal, liver, kidney and endometrial cancer [2, 3]. Obesity-associated metabolic abnormalities such as hyperinsulinemia, hyperglycaemia and dyslipidaemia have also been associated with CVD and a number of malignancies [4]. However, the contribution of obesity and its physiological consequences to the development of these diseases remains poorly understood.
The aetiology of obesity is multifactorial, with both genetic and environmental determinants. Numerous genetic variants linked to obesity have been discovered by large meta-analyses of genome-wide association studies [5,6,7]; however, taken together, these variants explain only a modest fraction of the phenotypic variation, and most of them do not appear to directly modulate phenotypes potentially driving individual risk profiles for obesity-related diseases [8]. It has been hypothesised that adiposity influences DNA methylation and the identification of genetic loci that are epigenetically modified in obesity could help further understanding on the development of obesity and its pathophysiologic sequelae. Recently, several epigenome-wide association studies have been published that have identified several hundred CpG sites that are statistically significantly associated with body mass index (BMI) [9] and with metabolic disturbances and subsequent risk of type 2 diabetes [10, 11]. To our knowledge, no studies have investigated the association of adiposity-related CpG loci and subsequent risk of other obesity-related diseases such as myocardial infarction, breast and colorectal cancer.
In this analysis, we obtained genome-wide DNA methylation levels in peripheral blood leucocytes (PBLs) from over 1900 individuals from four prospective cohorts in Italy, the Netherlands, Norway and Sweden. Using a meta-analytical framework, we tested the association of DNA methylation levels at each CpG locus with four measures of adiposity and investigated their association with transcriptomic profiles and subsequent development of myocardial infarction, breast and colorectal cancer.
Methods
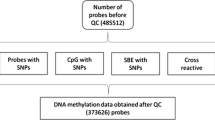
Features of the study population and the overall analytical strategy are summarised in Fig. 1.

Schematic representation of the study population and statistical analyses performed
Discovery set
The study included participants from four large population-based cohorts: the Italian and Dutch components of the European Prospective Investigation into Cancer and Nutrition (EPIC-Italy, N = 47,749 [12] and EPIC-Netherlands, N = 33,066 [13]), the Norwegian Women and Cancer Study (NOWAC, N = 53,363) [14], and the Västerbotten Intervention Program (VIP) part of the Northern Sweden Health and Disease Study (NSHDS, N = 101,790) [15].
A total of 2560 genome-wide DNA methylation profiles from PBLs were available as part of:
-
(i)
Three separate prospective case-control studies nested within EPIC-Italy on breast cancer (N = 332), colorectal cancer (N = 338), and myocardial infarction (N = 552);
-
(ii)
Two separate prospective nested case-control from EPIC-Italy and NSHDS on breast cancer and B-cell malignancies (EPIC-Italy and NSHDS components of the EnviroGenoMarkers project, N = 241 and 400, respectively).
-
(iii)
A longitudinal study in healthy women nested within EPIC-Netherlands (N = 148 pairs of baseline and follow-up samples, of which N = 66 at 5 years and N = 82 at 10 years from baseline).
-
(iv)
A prospective case-control study nested within NOWAC on breast cancer (N = 384).
NOWAC study was approved by the Regional Committee for Medical and Health Research Ethics and the Norwegian Data Inspectorate. The EnviroGenomarkers project and its associated studies (EPIC-Italy and NHSDS) and experimental protocols were approved by the Regional Ethical Review Board of the Umeå Division of Medical Research, for NHSDS, and the Florence Health Unit Local Ethical Committee, for EPIC Italy, and by local review board for EPIC-NL. Other studies involving EPIC-Italy samples were approved by the HuGeF Ethics, Committee. All participants gave written informed consent.
For the EPIC participants who contributed to more than one case-control study, multiple DNA methylation profiles were available. We only retained the single profile that exhibited the best technical quality (based on control probes). Participants from prospective nested case-control studies who developed the relevant condition less than 1 year after blood draw (to guard against reverse causation), or who developed any kind of haematological malignancy at any time after enrolment, were excluded, as were those with no anthropometric data. Characteristics of the resulting 1941 participants (588 men and 1353 women) are reported in Supplementary Table 1. Cancer cases were ascertained by linkage to national cancer registries, and myocardial infarction cases were confirmed by a cardiologist based on clinical records. Across all studies, completeness of follow-up was close to 100%, and cancer cases were histologically confirmed. Healthy controls were matched to cases by gender, time since blood sampling, and year of birth in order to control for effects of blood storage time and ageing.
Replication set
In order to validate the adiposity−methylation associations identified in the discovery set, we defined a replication sample (N = 384) from EPIC-Italy (independent of those individuals tested in the discovery sample). Samples exclusion criterion included: (i) prevalent dyslipidaemia or diabetes (self-reported); (ii) diagnosis of any non-haematological malignancy less than 5 years after blood draw; (iii) diagnosis of any haematological malignancy at any time after enrolment. Adiposity was assessed using a combination of BMI and waist circumference. To maximise contrast in obesity metrics and explore possible departure from a linear dose−response curve linking obesity and methylation levels, at high levels of obesity, we oversampled obese participants and randomly selected from the obese group 288 (75%) participants. The remaining 96 (25%) participants were sampled from the reference, normal-weight group. Samples that did not pass quality control procedures, as well as participants with incomplete anthropometric information, were excluded from further analyses, leaving a total of 358 participants (127 men and 231 women) whose characteristics are reported in Supplementary Table 2.
Adiposity parameters
Four adiposity measures were considered: BMI (kg/m2), waist circumference (cm), waist to hip ratio (WHR), and waist to height ratio (WHtR). Anthropometric measurements concurrent with blood sampling were obtained by a trained nurse at enrolment for all EPIC and NSHDS participants and were self-reported for NOWAC. Waist circumference and related measures were only available for EPIC participants.
Biochemical and biological parameters
Fasting blood levels of circulating total and HDL cholesterol, triglycerides, glucose, and insulin were available in a subset of non-diabetic EPIC-Italy participants (independent of the samples used in the replication set) who declared not to be taking lipid-lowering medications (N = 412), and for whom DNA methylation profiles were also available. Differential blood counts were available for a subset of EPIC-Netherlands participants at baseline (N = 56).
Laboratory analyses and data pre-processing
DNA methylation analyses
Genome-wide DNA methylation profiles were obtained using the Illumina Infinium® HumanMethylation450 (HM450) BeadChip assaying 470,870 autosomal CpG sites across the genome. Genomic DNA extracted from PBLs was bisulphite-converted using the Zymo Research EZ-96 DNA Methylation-Gold™ Kit, and hybridised to HM450 BeadChips according to the manufacturer’s protocol. Microarrays were subsequently scanned using the Illumina HiScanSQ system, and raw intensity data were exported from Illumina GenomeStudio (version 2011.1). Control probes included in the microarray were used to assess bisulphite conversion efficiency and to exclude lower-quality samples from further analyses (probes with detection P-values greater than 0.05 were excluded). Data pre-processing was performed using in-house software written for the R statistical computing environment. In particular, for each sample and each probe, measurements were set to missing if obtained by averaging intensities over less than three beads, or if averaged intensities were below detection thresholds estimated from negative control probes. Background subtraction and dye bias correction (for probes using the Infinium II design) were also performed. DNA methylation levels at each locus were assumed to be proportional to the ratio of intensities arising from methylated cytosine residues over total intensities. Details of the DNA-methylation pre-processing procedures are given in the Appendix. DNA methylation levels in the replication sample were assessed using bisulphite pyrosequencing. Primers were designed using QIAGEN PyroMark Assay Design (version 2.0). Preliminary PCR reactions were performed using the following cycling protocol: 95 °C for 10 min, followed by 45 cycles of denaturation at 95 °C for 30 s, annealing at 58 °C for 1 min, and extension at 72 °C for 1 min; a final extension was performed at 72 °C for 10 min. All samples were analysed two or three times (following bisulphite conversion) to assess and account for technical variability.
Gene expression analyses
Blood samples from the EnviroGenoMarkers and NOWAC studies additionally underwent genome-wide gene expression profiling using the Agilent Whole Human Genome 4 × 44 K Microarray [16], and the Illumina HumanWG-6 and HumanHT-12 BeadChip (NOWAC), respectively, as previously reported [17, 18]. Samples that failed quality control procedures were excluded from further analyses, leaving a total of 672 genome-wide gene expression profiles (N = 353 in EnviroGenoMarkers, and N = 319 in NOWAC) with associated DNA methylation data. For both studies, pre-processing steps included within- and between-array normalisation, followed by removal of un-annotated and non-specific probes, leaving a total of 14,698 transcripts for EnviroGenoMarkers, and 14,980 transcripts for NOWAC.
Statistical analyses
In the discovery set, separate analyses were conducted for each study and each adiposity measure. DNA methylation levels at each assayed CpG locus were modelled as the dependent variable in a generalised linear model accommodating beta-distributed responses [19], and for paired samples from EPIC-Netherlands, in a linear mixed model. All models were adjusted for microarray and position on the microarray, sex (if applicable), age at blood draw, and case-control status (if applicable). For each adiposity measure, estimated effect sizes (on the logit scale) and associated standard errors were obtained. Results across studies were combined in a meta-analytic framework using fixed effects models with inverse-variance weighting. For each CpG locus the model estimated, effect sizes, standard errors and corresponding P values as well as the I2 heterogeneity statistic [20]. Associations were declared statistically significant based on their meta-analytic P value with a 5% Bonferroni-corrected significance threshold α = 0.05/470,870 ≈ 1.06×10−7. Sensitivity analyses included stratification by sex, adopting a 5% significance threshold after Bonferroni correction for the number of informative CpG loci identified in the meta-analysis for each adiposity measure. DNA methylation measurements in the replication sample consisted of 2−3 technical replicates per participant and all were analysed together. In order to capture the possible technical variation within each participant, we used a linear mixed model with a participant random effect and adjusted for the same technical covariates as described above. CpG loci identified by the meta-analysis were only retained if they were not reported in the literature as cross-hybridising probes [21].
Informative CpG loci from the meta-analysis were further characterised by relating DNA methylation levels to transcriptional activity of the closest gene in EnviroGenoMarkers and NOWAC, using a log2-linear model adjusted for technical confounders, cohort and sex (EnviroGenoMarkers data only), age, case-control status, and BMI. As previously reported, linear models and linear mixed models (including a random intercept dependent on the main technical confounders) were used for the NOWAC and EnviroGenoMarkers data, respectively [16, 22]. Associations were declared statistically significant at a nominal 5% significance threshold.
Fasting levels of circulating total and HDL cholesterol, triglycerides, glucose, and insulin, as well as two further indicators of cardiometabolic risk (total-to-HDL cholesterol ratio and triglycerides-to-HDL cholesterol ratio) [23], and two indicators of β-cell function and insulin resistance (HOMA-%Β and HOMA-IR [24]), were measured in the EPICOR study (N = 412, see Fig. 1). These were analysed in relation to DNA methylation levels at informative CpG loci using log-linear models adjusted for sex, age, case-control status (separately for cancers and myocardial infarction), BMI and WHR.
We also investigated the potential for bias induced by differential blood cell counts adjusting for measured counts that were available in 56 EPIC-Netherlands study participants. These analyses were performed for our informative CpG loci using linear models adjusted for age, and were corrected for multiple comparisons using a 5% Bonferroni corrected significance level based on the total number of informative CpG loci identified in the meta-analysis. In addition, we used the ‘double bootstrap’ approach by Houseman et al. [25] and DNA methylation profiles in purified leukocytes made available by Reinus et al. [26] to infer associations between adiposity measures and blood differentials estimated from DNA methylation data. Adiposity-related CpG loci were then analysed with respect to incident post-menopausal breast cancer (from EPIC-Italy, EnviroGenoMarkers, and NOWAC studies N = 283 cases/282 controls, see Fig. 1), colorectal cancer (from EPIC-Italy CRC study N = 171 cases/132 controls, see Fig. 1), and myocardial infarction (from EPICOR study N = 131 cases/195 controls, see Fig. 1) in a logistic regression model for case-control status adjusted for age, sex (except post-menopausal breast cancer), BMI and WHR, as well as disease-specific confounders (Supplementary Table 3).
Data and code availability
Most of the microarray data are already publicly available (accession numbers available upon request to the corresponding author). The remaining data could be accessed upon request to the originating cohorts. Access will be conditional to adherence to local ethical and security policy. R-codes used for the analyses presented in the paper are available upon request.
Results
Epigenome-wide association analysis of adiposity
The meta-analysis of studies in the discovery set identified a list of 40 unique informative CpG loci associated with one or more adiposity measure (P < 1.06×10−7, Table 1). In particular, after correction for multiple testing, 26 CpG loci were statistically significantly associated with BMI (Table 1A, P values ranging between 9.89×10−8 and 2.76×10−18), of which 8 are directly associated to BMI and 18 were characterised by low-to-moderate heterogeneity across studies (I2 < 50%). Analyses for waist circumference and derived measures identified fewer associations: 12 for waist circumference (including 6 direct associations, Table 1B), 9 for WHR (including only one direct association, Table 1C), and 12 for WHtR (including three direct associations, Table 1D). All 12 CpG loci associated with WHtR exhibited low-to-moderate heterogeneity across studies (I2 < 50%). Methylation levels at a single CpG locus (cg06500161, ABCG1) were significantly (and positively) associated with all four adiposity measures (P values ranging between 3.07×10−11 and 2.76×10−18). Associations with DNA methylation levels at cg06500161 (ABCG1) were confirmed in the replication experiment (P values 5.61×10−10, 8.17×10−11, 7.68×10−9, and 4.71×10−11 for BMI, waist circumference, WHR and WHtR, respectively). DNA methylation measurements obtained using bisulphite pyrosequencing were additionally used to examine the shape of the dose−response curve of DNA methylation levels with each of the four adiposity measures; this showed linear direct relationships (Supplementary Figure 1). Additional stratification by sex did not yield any substantial differences in the results (Supplementary Table 4). However, associations appeared stronger in women than in men. Among men, 15 of the CpG loci associated with BMI did not reach statistical significance; however, seven (nominally statistically significant) showed effect size estimates similar to those obtained in the main analysis. This might at least partially be attributed to the smaller number of men included in our study (N = 588 men, vs. N = 1353 women).
Using measured blood differentials available in 56 study participants from EPIC-Netherlands, sensitivity analyses showed evidence of potential confounding induced by leucocyte high turnover rates and inter-individual tissue heterogeneity [25, 27] for only one of our 40 informative CpG loci, cg27117792 (intergenic). We additionally estimated blood cell composition using the ‘double bootstrap’ approach by Houseman et al. [25] in EPIC-Italy and NOWAC. We did not identify any associations between adiposity measures and blood differentials estimated from DNA methylation data (all P values > 0.05, see Supplementary Table 5). Furthermore, using results from the genome-wide meta-analyses from the GIANT consortium [5,6,7] we did not identify any SNP, with minor allele frequencies above 1% in the European population [28], and located within 500 kb windows centred at each informative CpG locus, associated with BMI or WHR (lowest reported P values 6.36×10−5 and 5.42×10−6 for BMI and WHR, respectively). We believe these data argue against potential genetic confounding. In addition, using genotype data available for 552 EPIC-Italy participants from the EPICOR study, we investigated associations between DNA methylation levels and 38 assayed SNPs that were also reported in the most recent GIANT meta-analysis, separately and combined in a (partial) genetic risk score. As illustrated in Supplementary Figure 2, we did not identify any significant associations, which also supports the lack of (non-local) genetic confounding.
Associations with transcriptional activity
Among the 40 informative CpG loci, six exhibited DNA methylation levels that were consistently associated with transcriptional activity in PBLs for the nearest gene in both EnviroGenoMarkers and NOWAC data (Table 2). With the exception of DNA methylation levels at cg00574958, which were inversely associated with transcription levels of CPT1A in EnviroGenoMarkers, but positively in NOWAC, all such associations showed consistent directions in both studies. In particular, DNA methylation levels at cg11024682 were inversely associated with transcriptional activity of SREBF1, and DNA methylation levels at both pairs of informative CpG loci found in close proximity (cg27243685 and cg06500161 on ABCG1, and cg16246545 and cg14476101 on PHGDH) were consistently inversely associated with transcriptional activity of the corresponding genes. In addition, transcription levels of ABCG1 were independently inversely associated with BMI (P = 7.38×10−7) in the NOWAC data; an association in the same direction was also found in the EnviroGenoMarkers data (P = 1.18×10−2).
Associations with biomarkers of lipid and glucose metabolism
Statistically significant associations between methylation levels at the 40 informative CpG loci and biomarkers of lipid and glucose metabolism are reported in Table 3. Associations were investigated without and with adjustment for BMI and WHR. While effect size estimates remained stable after this additional adjustment, some associations lost statistical significance. Adjusting for the effect of BMI and WHR, fasting blood levels of HDL cholesterol were associated with DNA methylation levels at cg0650016 (ABCG1) (the other CpG locus on ABCG1, cg27243685 was found associated in the model not adjusted for BMI and WHR), and cg09831562 (SOX2OT), though we did not identify any association with total cholesterol levels. Triglyceride levels were associated with DNA methylation levels at five informative CpG loci, cg00574958 (CPT1A), both CpG loci in ABCG1 (cg06500161, cg27243685), cg17901584 (DHCR24), and cg11024682 (SREBF1). A significant association was found between total-to-HDL cholesterol ratio and DNA methylation levels at cg06500161 (ABCG1), and between triglycerides-to-HDL cholesterol ratio and five informative CpG loci: cg00574958 (CPT1A), cg27243685 and cg06500161 (both on ABCG1), cg17901584 (DHCR24), and cg11024682 (SREBF1, only for the model not adjusted for BMI and WHR). Finally, DNA methylation levels at cg06500161 (ABCG1) were associated with fasting blood glucose levels; no significant associations were found with insulin or HOMA index.
Associations of adiposity-related CpG loci with breast cancer, colorectal cancer and myocardial infarction
Among the 40 informative CpG loci, we identified one that was also associated with colorectal cancer (cg21307484 in IL2RB, Table 4B, Model 2). To investigate the potential for an independent effect of these markers, we ran similar models adjusted for BMI and WHR (Model 1). While the strength of the association with cg21307484 was slightly weakened, one additional association with colorectal cancer emerged involving methylation levels at cg11661512 in FGF18 (P = 1.6×10−3). We also identified one CpG locus associated with myocardial infarction (cg12593793, Table 4C, Model 2). This locus was found to be inversely associated with smoking (P = 2.93×10−6 for current-to-never smoker comparison), even after adjustment for BMI and WHR (Model 1).
Discussion
We conducted a series of genome-wide association studies of DNA methylation levels for four adiposity measures in over 1900 individuals from large European population-based cohorts. Forty CpG loci were associated with at least one adiposity measure using a fixed effects meta-analysis. This model assumes that the obesity−methylation associations are similar across studies, which was supported by the modest heterogeneity estimates we obtained. We related DNA methylation levels to transcriptional activity in PBLs of the nearest gene and identified and replicated associations between higher DNA methylation levels at five CpG loci and downregulation of ABCG1, PHGDH, and SREBF1 gene expression. In addition, we found transcription levels of ABCG1 to be inversely associated with BMI. Further, of the 40 adiposity-associated CpG loci, two were statistically significantly associated with subsequent colorectal cancer risk while one was related to myocardial infarction.
Of the 40 obesity-related CpG sites we report, 21 were also found to be associated with BMI at genome-wide significance level in a recent meta-analysis including more than 10,000 individuals [10]. Using a Bonferroni correction for 40 tests, we found that 26 of our obesity-related CpG sites, of which 18 were related to BMI in our data, replicated in the BMI meta-analysis. The 26 replicated CpG sites included both cg21307484, and cg12593793, which we found to be associated with colorectal cancer and myocardial infarction, respectively. Reciprocally, of the 187 BMI-related CpG that meta-analysis reported, 41 replicated in our data, and 2 were found to be associated with breast cancer, 2 with colorectal cancer, and 5 (including cg12593793) with myocardial infarction.
Of the 40 obesity-related CpG loci, only one (cg06500161, ABCG1) showed associations with all four adiposity measures [29]. DNA methylation levels at this locus were also associated with fasting triglyceride levels, HDL cholesterol, total-to-HDL cholesterol ratio, and triglycerides-to-HDL cholesterol ratio. These results are consistent with recent findings on the role of ABCG1 in cellular cholesterol efflux and reverse cholesterol transport [30] through removal of excess cholesterol and promotion of the maturation of nascent HDL particles to larger, cholesterol-rich lipoproteins [31]. ABCG1 is highly expressed in cholesterol-loaded macrophages, where it guards against lipid overloading [32], and targeted disruption of Abcg1 in mice results in substantial lipid accumulation in macrophages and multiple tissues [33]. ABCG1 also plays a role in lipoprotein lipase regulation as demonstrated in knockdown experiments from cultured human macrophages [34], and may thus result in less efficient uptake of circulating triglycerides. Our results are in agreement with the ‘conjoint trait’ hypothesis of a combined low HDL cholesterol/high triglycerides phenotype that is regulated pleiotropically [35]. We also found an association between DNA methylation levels at one CpG locus on CPT1A and fasting triglyceride levels, consistent with recent reports in the literature [36,37,38] and with previous results in animal models [39]. DNA methylation levels at one CpG locus on DHCR24 were associated with HDL cholesterol and triglycerides-to-HDL cholesterol ratio. Expression of this gene is known to be under epigenetic control [40], and its products are thought to mediate the anti-inflammatory effect of HDL cholesterol in endothelial cells [41, 42]; they also appear to regulate cellular response to oncogenic and oxidative stress [43].
Among the 40 informative CpG loci, two loci in IL2RB and FGF18 and one CpG locus in an intergenic region of chromosome 1 were associated with colorectal cancer and myocardial infarction development, respectively, after controlling for anthropometric parameters and established risk factors. None of the informative CpG loci were associated with post-menopausal breast cancer following Bonferroni correction, though we note that the number of post-menopausal breast cancer cases included in the study was relatively small.
Differential DNA methylation levels in the IL2RB and FGF18 genes were associated with risk of colorectal cancer, which suggests that changes in the activity of these genes may be contributing to the development of this malignancy. The IL2RB gene encodes the interleukin(IL)-2 receptor-β which binds IL-2, a cytokine which plays a major role in T-cell differentiation and proliferation. Overexpression of lymphocyte IL2RB has been linked to risk of colorectal cancer in small clinical case-control studies and was shown to predict risk when combined with six additional genes [44, 45]. Given that obesity is accompanied by alterations in T-cell populations, inflammation and immune dysregulation [46], our results provide a potential link between obesity, T-cell proliferative pathways and colorectal cancer development that warrants further study. The FGF18 gene, which encodes the fibroblast growth factor 18, has also been previously linked to colorectal tumorigenesis [47]. FGF is a downstream target of the Wnt-signalling and β-catenin pathways which are central to cell growth and proliferation, and frequently mutated in colorectal cancer [48]. Alterations in the Wnt/β-catenin pathway have been associated with obesity-related disorders such as insulin resistance and type 2 diabetes [49, 50] and FGF18 interacts with PI3K/mTOR and growth factor signalling pathways. Our findings may provide new evidence that obesity is associated with specific changes in the Wnt/β-catenin signalling pathways, that could, in-turn, drive colorectal cancer development. Methylation levels at cg12593793 were found to be inversely associated with risk of myocardial infarction, independent of obesity and established risk factors, including smoking status. We may speculate that both smoking and adiposity influence myocardial infarction risk through a mechanism that alters methylation status at this locus.
In support of the biological basis of these relationships, some of the obesity-related CpG loci we report have been identified in previous studies, not only of adiposity measures, but also of lipid and glycaemic traits. In particular, cg06500161 (ABCG1) was found to be linked to incident type 2 diabetes in a recent study by Chambers et al. [11]; CpG loci on CPT1A and PHGDH both with adiposity measures [11] and the metabolic syndrome [51]; and HIF3A, CPT1A and ABCG1 with BMI and waist circumference [52]. Nevertheless, we note that the recently identified associations between BMI and DNA methylation at three CpG loci on HIF3A [9] did not reach genome-wide statistical significance in our meta-analysis. Inconsistencies between the prior data and the current analysis may partially reflect different modelling strategies: we opted for a more conventional parameterisation using DNA methylation levels as dependent variable, with adjustment for technical (microarray and position on the microarray) and other confounders.
We identified several CpG loci that are associated with adiposity, metabolic traits, and which appear to be associated with gene expression. Among these, two CpG loci in IL2RB and FGF18, and one intergenic CpG locus on chromosome 1 were associated with subsequent risk of colorectal cancer and myocardial infarction, respectively. These associations survived adjustment for adiposity measures and known risk factors, suggesting a potential role for DNA methylation as intermediate biomarker in the complex interplay between adiposity, metabolic health and risk of obesity-related diseases. However, because of the overlap between the methylation-obesity discovery and methylation-disease analyses, we cannot fully discard potential residual confounding in our BMI-adjusted analyses. In addition, as previously reported in a larger family-based study [9], and because our data do not arise from a structured population, our sample size renders any attempts to assess causality, for example using instrumental variable approaches, statistically underpowered and our analyses of the genetic data available in a fraction of our study population preclude the use of established BMI-related (combinations of) genetic variants as potential instruments in an attempt to infer causality.
Despite these limitations, we believe that our observational study data provide evidence for potential mechanisms involving DNA methylation alterations that may partly explain the association of obesity with obesity-related chronic diseases.
In conclusion, we have identified a number of CpG sites that are associated with adiposity and metabolic traits and which appear to have functional effects on gene expression. Among these, one was consistently associated to all four adiposity measures: cg06500161, ABCG1. In addition, two CpG sites in IL2RB and FGF18 and one intergenic CpG locus on chromosome 1 were associated, independently of obesity and known risk factors, with subsequent risk of CRC and MI, respectively. This suggests a potential pleiotropic role for these epigenetic markers, and substantiates the involvement of DNA methylation in the complex interplay between adiposity, metabolic health, and risk of obesity-related diseases.
References
Stevens GA, Singh GM, Lu Y, Danaei G, Lin JK, Finucane MM et al. National, regional, and global trends in adult overweight and obesity prevalences. Popul Health Metr. 2012;10:16. https://doi.org/10.1186/1478-7954-10-22
Arnold M, Pandeya N, Byrnes G, Renehan AG, Stevens GA, Ezzati M, et al. Global burden of cancer attributable to high body-mass index in 2012: a population-based study. Lancet Oncol. 2015;16:36–46.
Bhaskaran K, Douglas I, Forbes H, dos-Santos-Silva I, Leon DA, Smeeth L. Body-mass index and risk of 22 specific cancers: a population-based cohort study of 5.24 million UK adults. Lancet. 2014;384:755–65.
Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–71.
Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–48.
Heid IM, Jackson AU, Randall JC, Winkler TW, Qi L, Steinthorsdottir V, et al. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet. 2010;42:949–60.
Yang J, Loos RJF, Powell JE, Medland SE, Speliotes EK, Chasman DI, et al. FTO genotype is associated with phenotypic variability of body mass index. Nature. 2012;490:267–72.
Scott RA, Lagou V, Welch RP, Wheeler E, Montasser ME, Luan Ja, et al. Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet. 2012;44:991–1005.
Dick KJ, Nelson CP, Tsaprouni L, Sandling JK, Aissi D, Wahl S, et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet. 2014;383:1990–8.
Wahl S, Drong A, Lehne B, Loh M, Scott WR, Kunze S, et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2017;541:81–86.
Chambers JC, Loh M, Lehne B, Drong A, Kriebel J, Motta V, et al. Epigenome-wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: a nested case-control study. Lancet Diabetes Endocrinol. 2015;3:526–34.
Palli D, Berrino F, Vineis P, Tumino R, Panico S, Masala G, et al. A molecular epidemiology project on diet and cancer: the EPIC-Italy Prospective Study. Design and baseline characteristics of participants. Tumori. 2003;89:586–93.
Beulens JWJ, Monninkhof EM, Verschuren WMM, van der Schouw YT, Smit J, Ocke MC, et al. Cohort profile: the EPIC-NL study. Int J Epidemiol. 2010;39:1170–8.
Lund E, Kumle M, Braaten T, Hjartaker A, Bakken K, Eggen E, et al. External validity in a population-based national prospective study—the Norwegian Women and Cancer Study (NOWAC). Cancer Causes Control. 2003;14:1001–8.
Hallmans G, Agren A, Johansson G, Johansson A, Stegmayr B, Jansson J-H, et al. Cardiovascular disease and diabetes in the Northern Sweden Health and Disease Study Cohort—evaluation of risk factors and their interactions. Scand J Public Health. 2003;61:18–24.
Chadeau-Hyam M, Vermeulen RCH, DGAJ Hebels, Castagne R, Campanella G, Portengen L, et al. Prediagnostic transcriptomic markers of chronic lymphocytic leukemia reveal perturbations 10 years before diagnosis. Ann Oncol. 2014;25:1065–72.
Dumeaux V, Borresen-Dale A-L, Frantzen J-O, Kumle M, Kristensen VN, Lund E. Gene expression analyses in breast cancer epidemiology: the Norwegian Women and Cancer postgenome cohort study. Breast Cancer Res. 2008;10:R13 https://doi.org/10.1186/bcr1859
DGAJ Hebels, Georgiadis P, Keun HC, Athersuch TJ, Vineis P, Vermeulen R, et al. Performance in omics analyses of blood samples in long-term storage: opportunities for the exploitation of existing biobanks in environmental health research. Environ Health Perspect. 2013;121:480–7.
Ferrari SLP, Cribari-Neto F. Beta regression for modelling rates and proportions. J Appl Stat. 2004;31:799–815.
Higgins JPT, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21:1539–58.
Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, et al. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013;8:203–9.
Dumeaux V, Olsen KS, Nuel G, Paulssen RH, Borresen-Dale AL, Lund E Deciphering normal blood gene expression variation—the NOWAC Postgenome Study. PLoS Genet. 2010;6:e1000873
Kannel WB, Vasan RS, Keyes MJ, Sullivan LM, Robins SJ. Usefulness of the triglyceride-high-density lipoprotein versus the cholesterol-high-density lipoprotein ratio for predicting insulin resistance and cardiometabolic risk (from the Framingham offspring cohort). Am J Cardiol. 2008;101:497–501.
Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–9.
Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012;13:86.
Reinius LE, Acevedo N, Joerink M, Pershagen G, Dahlén S-E, Greco D, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS ONE. 2012;7:e41361.
Michels KB, Binder AM, Dedeurwaerder S, Epstein CB, Greally JM, Gut I, et al. Recommendations for the design and analysis of epigenome-wide association studies. Nat Methods. 2013;10:949–55.
Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65.
Pfeifferm L, Wahl S, Pilling LC, Reischl E, Sandling JK, Kunze S et al. DNA methylation of lipid-related genes affects blood lipid levels. Circ Cardiovasc Genet. 2015;8:334–342
Li G, Gu H-M, Zhang D-W. ATP-binding cassette transporters and cholesterol translocation. IUBMB Life. 2013;65:505–12.
Vaughan AM, Oram JF. ABCA1 and ABCG1 or ABCG4 act sequentially to remove cellular cholesterol and generate cholesterol-rich HDL. J Lipid Res. 2006;47:2433–43.
Klucken J, Buchler C, Orso E, Kaminski WE, Porsch-Ozcurumez M, Liebisch C, et al. ABCG1 (ABC8), the human homolog of the Drosophila white gene, is a regulator of macrophage cholesterol and phospholipid transport. Proc Natl Acad Sci USA. 2000;97:817–22.
Kennedy MA, Barrera GC, Nakamura K, Baldan A, Tarr P, Fishbein MC, et al. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005;1:121–31.
Olivier M, Tanck MW, Out R, Villard EF, Lammers B, Bouchareychas L, et al. Human ATP-binding cassette G1 controls macrophage lipoprotein lipase bioavailability and promotes foam cell formation. Arterioscler Thromb Vasc Biol. 2012;32:2223–31.
Mahaney MC, Blangero J, Comuzzie AG, Vandeberg JL, Stern MP, Maccluer JW. Plasma HDL, cholesterol, triglycerides, and adiposity—a quantitative genetic test of the conjoint trait hypothesis in the San Antonio Family Heart study. Circulation. 1995;92:3240–8.
Frazier-Wood AC, Aslibekyan S, Absher DM, Hopkins PN, Sha J, Tsai MY, et al. Methylation at CPT1A locus is associated with lipoprotein subfraction profiles. J Lipid Res. 2014;55:1324–30.
Gagnon F, Aissi D, Carrie A, Morange P-E, Tregouet D-A. Robust validation of methylation levels association at CPT1A locus with lipid plasma levels. J Lipid Res. 2014;55:1189–91.
Tobi EW, Goeman JJ, Monajemi R, Gu H, Putter H, Zhang Y, et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun. 2014;5:5592.
Stefanovic-Racic M, Perdomo G, Mantell BS, Sipula IJ, Brown NF, O’Doherty RM. A moderate increase in carnitine palmitoyltransferase 1a activity is sufficient to substantially reduce hepatic triglyceride levels. Am J Physiol—Endocrinol Metab. 2008;294:E969–77.
Drzewinska J, Walczak-Drzewiecka A, Ratajewski M. Identification and analysis of the promoter region of the human DHCR24 gene: involvement of DNA methylation and histone acetylation. Mol Biol Rep. 2011;38:1091–101.
McGrath KCY, Li XH, Puranik R, Liong EC, Tan JTM, Dy VM, et al. Role of 3β-hydroxysteroid-Δ24 reductase in mediating antiinflammatory effects of high-density lipoproteins in endothelial cells. Arterioscler Thromb Vasc Biol. 2009;29:877–82.
Wu BJ, Chen K, Shrestha S, Ong KL, Barter PJ, Rye K-A. High-density lipoproteins inhibit vascular endothelial inflammation by increasing 3β-hydroxysteroid-Δ24 reductase expression and inducing heme oxygenase-1. Circ Res. 2013;112:278–88.
Wu C, Miloslavskaya I, Demontis S, Maestro R, Galaktionov K. Regulation of cellular response to oncogenic and oxidative stress by seladin-1. Nature. 2004;432:640–5.
Chang YT, Huang CS, Yao CT, Su SL, Terng HJ, Chou HL, et al. Gene expression profile of peripheral blood in colorectal cancer. World J Gastroenterol. 2014;20:14463–71.
Marshall KW, Mohr S, Khettabi FE, Nossova N, Chao S, Bao W, et al. A blood-based biomarker panel for stratifying current risk for colorectal cancer. Int J Cancer. 2010;126:1177–86.
Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, et al. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15:914–20.
Shimokawa T, Furukawa Y, Sakai M, Li M, Miwa N, Lin Y-M, et al. Involvement of the FGF18 gene in colorectal carcinogenesis, as a novel downstream target of the β-catenin/T-cell factor complex. Cancer Res. 2003;63:6116–20.
Bienz M, Clevers H. Linking colorectal cancer to Wnt signaling. Cell. 2000;103:311–20.
Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38:320–3.
Welters HJ, Kulkarni RN. Wnt signaling: relevance to beta-cell biology and diabetes. Trends Endocrinol Metab. 2008;19:349–55.
Das M, Sha J, Hidalgo B, Aslibekyan S, Do AN, Zhi D, et al. Association of DNA methylation at CPT1A locus with metabolic syndrome in the genetics of lipid lowering drugs and Diet Network (GOLDN) Study. PLoS ONE. 2016;11:e0145789.
Demerath EW, Guan W, Grove ML, Aslibekyan S, Mendelson M, Zhou YH, et al. Epigenome-wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum Mol Genet. 2015;24:4464–79.
Author contributions
Samples from EPIC-Italy were provided by Vittorio Krogh, Domenico Palli, Salvatore Panico, Carlotta Sacerdote, and Rosario Tumino. Samples from EPIC-Netherlands were provided by H. Bas Bueno-de-Mesquita, Anne M. May, N. Charlotte Onland-Moret, Elio Riboli, and W. M. Monique Verschuren. Samples and data from NOWAC were provided by Eiliv Lund, Nicolle Mode, and Torkjel M. Sandanger. Samples and data from NSHDS were provided by Ingvar A. Bergdahl, Beatrice Melin, and Per Lenner. Giuseppe Matullo provided DNA methylation profiles, and Giovanni Fiorito performed management and data quality assurance for the EPICOR study. Soterios A. Kyrtopoulos provided DNA methylation profiles from the EnviroGenoMarkers project. Laboratory analyses were performed by Silvia Polidoro (EPIC-Italy and NOWAC), Simonetta Guarrera (EPIC-Italy), Panagiotis Georgiadis (DNA methylation in EnviroGenoMarkers), Theo M. C. M. de Kok and Jos C. S. Kleinjans (gene expression in EnviroGenoMarkers), and Karen A. Lillycrop and Robert Murray (EPIC-Netherlands). Measurements of blood lipids, glucose, and insulin for EPIC-Italy samples were provided by Licia Iacoviello. Bisulphite pyrosequencing of the Italian replication samples was conducted by Valentina Fiano and Morena Trevisan. Marc Chadeau-Hyam supervised the statistical analyses; Gianluca Campanella compiled the data, reviewed its quality, devised and carried out all statistical analyses. Gianluca Campanella, Marc Gunter and Silvia Polidoro drafted the manuscript. Philippe Froguel provided critical comments and contributed to the manuscript preparation. Paul Elliott, Paolo Vineis, and Marc Chadeau-Hyam coordinated the work and contributed to writing the manuscript. All authors approved the final version of this article.
Funding
EPIC-Italy was financially supported by the Italian Association for Cancer Research (AIRC). Genome-wide DNA methylation profiling and bisulphite pyrosequencing of EPIC-Italy samples was financially supported by the Human Genetics Foundation (HuGeF) and Compagnia di San Paolo. The EnviroGenoMarkers project was financially supported by the European Union (grant agreement 226756 to Soterios A. Kyrtopoulos). EPIC-Netherlands was financially supported by the Dutch Ministry of Public Health, Welfare, and Sports (VWS), by the Netherlands Cancer Registry, by LK Research Funds, by Dutch Prevention Funds, by the Netherlands Organisation for Health Research and Development (ZON), and by the World Cancer Research Fund (WCRF). Genome-wide DNA methylation profiling of EPIC-Netherlands samples was financially supported by internal Imperial College funds. Genome-wide DNA methylation and gene expression profiling of NOWAC samples was financially supported by the European Research Council for frontier research, Advanced Grant TICE—Transcriptomics in Cancer Epidemiology (number 232997, period 2009–2014). Gianluca Campanella received a Doctoral Prize studentship awarded by the Engineering and Physical Sciences Research Council (EPSRC). Paul Elliott is a National Institute for Health Research (NIHR) senior investigator and acknowledges support from the NIHR Biomedical Research Centre at Imperial College Healthcare NHS Trust and Imperial College London, and the NIHR Health Protection Unit on Health Impact of Environmental Hazards. He is supported by the Medical Research Council and Public Health England as part of joint funding for the MRC-PHE Centre for Environment and Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Campanella, G., Gunter, M.J., Polidoro, S. et al. Epigenome-wide association study of adiposity and future risk of obesity-related diseases. Int J Obes 42, 2022–2035 (2018). https://doi.org/10.1038/s41366-018-0064-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41366-018-0064-7
- Springer Nature Limited
This article is cited by
-
A prospective study of smoking-related white blood cell DNA methylation markers and risk of bladder cancer
European Journal of Epidemiology (2024)
-
DNA methylation and cardiovascular disease in humans: a systematic review and database of known CpG methylation sites
Clinical Epigenetics (2023)
-
Genetics and Epigenetics in Obesity: What Do We Know so Far?
Current Obesity Reports (2023)
-
ABCA1 and ABCG1 DNA methylation in epicardial adipose tissue of patients with coronary artery disease
BMC Cardiovascular Disorders (2021)
-
DNA methylation signatures of incident coronary heart disease: findings from epigenome-wide association studies
Clinical Epigenetics (2021)