
Abstract
In this research, a dispersive solid phase extraction procedure based on changing the solubility of octadecylamine with pH was proposed to determine Ag+ ions in different water samples. For this purpose, first, the pH of sample solution containing the analyte was adjusted to 10.5. Then desired volume of the octadecylamine dissolved in acidic solution was injected into the solution. Because of the low solubility of octadecylamine in alkaline solution, a cloudy state was formed. The produced octadecylamine particles acted as a complexing agent for Ag+ ions and adsorbent for the formed complex. The obtained cloudy solution was centrifuged and the sedimented particles were removed and dissolved in a diluted nitric acid solution. It was injected into a flame atomic absorption spectrometry to determine the extracted amounts of the analyte. The effect of important parameters such as the amount of octadecylamine, volume of nitric acid, and centrifugation and vortexing conditions on the extraction efficiency of the procedure was studied and optimized. In optimal conditions, the developed method showed a linear range of 0.50–200 µg L−1. The limits of detection and quantification were 0.18 and 0.50 µg L−1, respectively. Extraction recovery was 93.6%. The relative standard deviations were less than 4%. The effectiveness of the method was investigated by determination of Ag+ ions in water and wastewater samples.
Graphical abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In the current era, many efforts are made to develop the methods for detecting metal ions from biological and chemical point of view, and among the various types of metal ions, silver ion is considered for identification due to its effective roles in the biology and environment and also as a valuable metal for the production of products such as mirror, photographic film [1], coin [2], electronic components and paper jewelry [3]. Because of antibacterial properties of silver ion, it is used in antibacterial clothes and water purification [4]. It is utilized in traditional Indian and Chinese medicines because of its antimicrobial properties to eliminate contaminations in food hygiene [5]. The WHO announced that about 0.1 mg L−1 of silver(I) ions does not have adverse effects on human life, and this level in drinking water does not harm human health [6, 7]. Long time exposure to silver or its high concentrations can result in significant health problems [8]. Therefore, analysis of trace amounts of Ag(I) is significant in many fields [9].
To determine Ag+ ions, different methods such as spectrophotometry [10, 11], nanosensor [12], inductively coupled plasma (ICP)-mass spectrometry [13], liquid chromatography with electrochemical detector [14], stripping voltammetry [15], dynamic light scattering and UV spectroscopy [16], ICP-optical emission spectrometry [17], and flame atomic absorption spectroscopy (FAAS) [9, 18, 19] were used. Due to the problems of small amount of analytes in real samples and matrix effect, sample preconcentration step plays an effective role in analyses [20,21,22]. Up to now, various methods like solid phase extraction (SPE) [23,24,25], dispersive liquid–liquid microextraction [26,27,28,29], homogeneous liquid–liquid extraction [30], solid phase microextraction [31], dispersive solid phase extraction (DSPE) [32], and liquid–liquid extraction [33] have been used for extraction of Ag+ ions.
In a common DSPE, an adsorbent is usually dispersed into solution by sonication or vortexing [34,35,36]. This method does not need sorbent packing like conventional SPE and also has high extraction recoveries (ERs) [37]. DSPE has been interested by researchers and increasingly is used in different matrices by employing various adsorbents [34, 36, 38]. In-situ formation of adsorbent has attracted considerable attention in recent years; in which the adsorbent is formed from a homogeneous solution. Thus an unlimited contact area between the analytes and adsorbent is achieved during adsorption step [39]. The method is efficient and easy to use. However, the main concern of the usage of an adsorbent in the analysis of heavy metals is the presence of electrostatic interactions between metal ions and adsorbent. Electrostatic interaction occurs between the positively charged heavy metals and the negatively charged carbonous adsorbents, especially with the presence of functional groups [40,41,42,43]. Because most carbon surfaces are variably charged, prevalence of electrostatic interaction in heavy metal adsorption is dependent on solution pH [44, 45]. The charged interface between carbons and the solution depends strongly on ionization of surface groups. Due to the formation of stable complexes between silver and nitrogen-donor ligands such as amines [46, 47], octadecylamine (ODA) can act as a suitable sorbent in the sensing Ag+ in the aqueous medium. Also ODA is a solid amine which can be used as a sorbent. Also it is soluble in acidic solution.
The purpose of this work was analysis of Ag+ ions in aqueous samples by DSPE and FAAS. The adsorbent particles are formed in-situ and simultaneously extracted the analyte. First, octadecylamine is dissolved in a diluted HCl solution and then injected into the solution adjusted at an alkaline pH. Because of the low solubility of ODA at high pHs, ODA is precipitated as fine particles and dispersed in the solution. Because of the presence of amino group in the ODA structure, it can also form complex with Ag+ ions. Therefore, the extraction step is performed without adding further complexing agent. In the following, the particles are collected, dissolved in a diluted HNO3 solution, and used in the determination step. Therefore the proposed procedure needs to no desorption step, which is time-consuming and requires sonication or vortexing. Easy operation, short extraction time, high extraction efficiency, and inexpensiveness can be the major advantages of the proposed procedure.
Experimental
Chemicals and solutions
The stock solution of 100 mg L−1 of Ag+ ion in deionized water was prepared using silver nitrate (≥ 99.0%, Merck, Darmstadt, Germany). In each case, the needed solutions were prepared by diluting the stock solution till 10 µg L−1 with deionized water and was used as working solutions. ODA (98%) was purchased from Sigma-Aldrich. Hydrochloric acid (37%, w/w), nitric acid (37%, w/w), and triethylamine (99%) were bought from Merck.
Real samples
Different water samples consisting of well water (Miandoab, Iran), dam water (Alaviyan, Maragheh, Iran), and tap water (Tabriz, Iran) were used as samples. Also, rainwater was collected during the spring of 2022 from the University of Tabriz zone. Tap water was collected from our laboratory. Two industrial wastewaters were also collected from Songun copper mine (Varzghan, Iran) and petrochemical unit (Tabriz, Iran). Before analysis, the wastewater samples were centrifuged at a speed of 7000 rpm for 6 min to remove the possible particles.
Instrumentation
The detection of analyte was done with an FAAS 6300 (Shimadzu, Kyoto, Japan), and to eliminate background effects, it was equipped with a deuterium lamp. A combination of air and acetylene was used for combustion (15 and 2.3 L min−1, respectively). The used light source was a silver hollow cathode lamp (resonance line, 328.1 nm and current, 15 mA). All pH measurements were made using a Metrohm pH meter (Herisau, Switzerland) model 654 equipped with a combined pH electrode.
Extraction procedure
First, for preparation of ODA solution, 100 mg of ODA was added to a mixture of 4 mL concentrated hydrochloric acid solution (12 M) and 36 mL deionized water. To dissolve ODA, the mixture was stirred at 300 rpm at 80 ºC for 40 min. Then for preparation of buffer solution, triethylamine solution (1.0 M) was mixed with hydrochloric acid solution (0.1 M) in the volume ratio of 1:1. In the following, 1 mL of this solution was added to 5 mL of sample solution or spiked deionized water (10 µg L−1 Ag+) was mixed with 1 mL of the prepared buffer solution in a test tube. Then, 1 mL of the prepared ODA solution was injected into it. The formed cloudy solution was vortexed for 2 min. Then the tube was centrifuged at 7000 rpm for 7 min. The supernatant was removed and the sedimented particles were dissolved in 100 µL of nitric acid solution (5%, v/v) and injected into FAAS.
Enrichment factor (EF) and extraction recovery (ER)
The ratio of analyte concentration in the final phase (Cfin) to its initial concentration in the sample (C0) is defined as EF:
Cfin can be obtained from calibration graph. The percentage of total analyte amount (n0) that is extracted into the final phase (nfin) is defined as ER:
where Vfin and V0 are volumes of the final phase and sample solution, respectively.
Results and discussion
Various parameters such as the volume of ODA solution, type and amount of desorption solvent, time and speed of centrifugation, and vortexing can affect the extraction efficiency of the procedure. Therefore, their effect should be evaluated and optimized.
Studying the effect of important parameters
Optimizing the volume of ODA solution
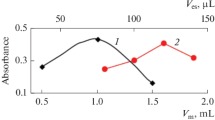
In this method, the adsorbent (ODA) is produced in-situ. It also acts as a complexing agent. Therefore, the volume of ODA solution (ODA amount) can affect the ER of Ag+ ions. To investigate the effect of this parameter, different volumes of ODA solution in the range of 0.50–2.00 mL were injected into the working solutions and extraction efficiency of the method was evaluated. According to Fig. 1, up to 1.00 mL the ER increases and then it decreases. More sorbent is formed by increasing the volume of ODA solution, and therefore, the active sites to adsorb the analyte increase, and it leads to high ER. But at higher volumes of ODA, the ER gradually decreases. It may be due to agglomeration of the formed sorbent particles. Also by increasing volume of the ODA solution, pH of the final solution decreased due to dissolution of ODA in HCl solution. Therefore formation of complex between Ag+ and amino group of ODA was disturbed. As a result, 1.00 mL of the ODA solution was finalized for the further experiments.

Study of the effect of the volume of ODA solution on ER of Ag+. Extraction conditions: sample, 5 mL standard solution containing 10 µg L−1 of Ag+; pH = 10.4 ± 0.2; centrifugation conditions, 5 min, and 5000 rpm; and desorption solvent (volume); HNO3 (300 µL)
Optimization of vortexing conditions
To more enhance the contact area between the analyte solution and the produced sorbent, agitation of the mixture was performed by vortexing. Vortex is a widely used method in laboratories to mix samples inside tubes with a small volume and has a rotational movement and an orbital state on its axis. Its optimization in term of time can be important. Vortexing time has a direct effect on the efficiency of the procedure since it changes the numbers of contact between the analyte and sorbent. Therefore, the time period from 0 to 6 min was studied. The results in Fig. 2 showed that vortex has less effect. Therefore, it verifies that the main part of the analyte adsorption was done during the adsorbent formation, and vortexing of the solution and the passage of time have less effects on the extraction efficiency of the method. According to these experiments, vortexing time was adjusted at 2 min.

Study of the effect of vortex time on ER of Ag+. Extraction conditions: volume of ODA solution, 1.00 mL; and the other conditions were the same as those used in Fig. 1
Study of conditions of centrifugation
Centrifugation is an important step for rapid collection of the formed ODA particles. Therefore, its optimization in terms of time and speed can be important, which were investigated in two stages. The results (presented in Electronic Supplementary Material, ESM) proved that this parameter had no significant effect on the ER. Therefore the centrifuge was performed at 7000 rpm for 5 min.
Recovery of Ag+ in the ODA particles
The collected particles of ODA (containing analyte) must be placed in contact with an elution solvent to desorb of Ag+. For this purpose, the efficiency of methanol, acetonitrile [48], and nitric acid solution (5%, v/v) was studied. Therefore, after collecting the particles of ODA, the aqueous solution was removed. Then 0.3 mL of each mentioned solvent was mixed with the particles and vortexed for 2 min. In the case of nitric acid solution (5%, w/v), the particles were dissolved. According to the results in Fig. 3, nitric acid solution (5%, w/v) was selected as the best solvent to release the analyte into the solution. According to these results, it can be concluded that the most of the analyte was simultaneously extracted with re-formation of the solid particles of ODA (most of the extracted analyte was in the inside of the particles of ODA, not on their surface). All analyte was transferred into the nitric acid solution by dissolving ODA particles. Whereas only the analytes adsorbed onto the adsorbent surface were desorbed by methanol and acetonitrile. This is the reason for obtaining low analytical signals in the cases of methanol and acetonitrile compared to nitric acid solution. Therefore nitric acid solution (5%, w/v) was selected for the rest of the experiments.

Study of the type of desorption solvent. Extraction conditions: vortexing time, 2 min; centrifuge rate, 7000 rpm; and the other conditions were the same as those used in Fig. 2
Studying the volume of nitric acid solution (5%, w/v)
For FAAS determinations, a volume of 1–2 mL of solution is usually needed. To analyze the samples with low volumes dilution is needed. However, by dilution of the sample, concentration of analyte may reach less than the detection limit of the apparatus. To solve this problem, a home-made micro-sample introduction system was constructed [49] to determine the analytes by FAAS in a microliter level without dilution. In this study, to investigate the effect of the volume of nitric acid solution on the ER of the analyte, dissolving of the collected particles of ODA was performed in the presence of low volumes of the nitric acid solution to access high EFs. For this purpose, 80, 100, 120, 160, 210, 260, 300, 360, and 410 µL of the nitric acid solution was investigated, According to the results in Fig. 4, 100 µL of nitric acid was selected as the suitable volume to dissolve the adsorbent with high analytical signals and low detection limit.

Studying the volume of HNO3 solution (5%, w/v). Extraction conditions were the same as those used in Fig. 3
Ionic strength effect
According to the results in Fig. S3 (in ESM) the extraction efficiency of the procedure in the presence of salt decreased, and therefore, the next experiments were performed without adding salt.
Study of interferences
To evaluate the potential interference of different ions in the analysis of Ag+, different concentrations of various anions and cations were added to working solution. When the added ion caused ± 5% change in the ER of Ag+, it was considered as an interfering ion. For this purpose, different tests were designed and the results are collected in Table 1. According to these results, the desired cations and anions do not have significant effect on analysis of Ag+ by the developed method.
Analytical data
The analytical characteristics of the optimized method, such as repeatability, linear range (LR), limit of quantification (LOQ), limit of detection (LOD), ER, and enrichment factor (EF) were evaluated. The LR of the calibration curve was 0.50–200 μg L−1. The LOD (3 Sb/m, where m is slope of the calibration curve and Sb is standard deviation of blank measurements, n = 7) was 0.18 μg L−1. The LOQ (10 Sb/m) was obtained 0.50 μg L−1. The precision of the proposed method was evaluated in the solutions containing 10 μg L−1 of Ag+ (intra-day, n = 6) and in 4 consecutive days (inter-day). The results of the relative standard deviations (RSDs) were in the range of 2.7–3.9%,. The ER and EF were 93.6% and 46.8, respectively. The results are summarized in Table 2.
Real samples analysis and method validation
Using the proposed method, concentration of Ag+ was determined in real samples such as well water, Songun copper mine (Varzaqan county, East Azarbaijan, Iran) wastewater, tap water, petrochemical unit (Tabriz, Iran) wastewater, dam water, and rain water. To study the matrix effect of, the real samples were spiked at the concentrations of 20 and 50 μg L−1 of Ag+. The found concentrations along with the calculated recoveries are collected in Table 3. As shown, the recoveries of the analyte are good and the values are in the range of 82.5–94.8%, which confirm the accuracy of the developed method. Furthermore, the accuracy was also verified by determining concentration of Ag+ in a certified reference material (CRM), CWW-TM-D waste-water, with certified value of 0.25 ± 0.01 mg L−1. Because of high concentration of Ag+ in the CRM, it was diluted 10 times before analysis. The concentration of Ag+ obtained by the proposed method (0.238 ± 0.011 mg L−1) was compared with the certified value (0.250 ± 0.012 mg L−1) by t-test. It was confirmed that there was a good agreement (tobtained = 1.73˂tcritical = 4.3) between the obtained concentration and the approved value. Therefore, the proposed method is reliable for analysis of Ag+ concentration in various wastewater and water samples.
Comparison of the proposed extraction procedure with other approaches
The figures of merit of the developed procedure were compared with other published extraction methods in the determination of Ag+ in various types of real samples. Examining the results of the comparison (Table 4) shows that the proposed procedure is comparable with and even better than the previously published procedure in most parameters The prominent advantages of the developed method are wider LR, low LOD (except Ref. [30]), acceptable RSD, simplicity, and using no organic solvent.
Conclusions
In this study, ODA was used as adsorbent and complexing agent to extract Ag+ ions from aqueous solutions. This extraction method was suggested for the first time based on the dependence of ODA solubility on pH. The results showed that the most amounts of Ag+ ions were extracted in the stage of formation of ODA. Therefore, it can be concluded that the most of extracted analyte were in the inside of the particles of ODA, not on the surface of sorbent. Because of this, the diluted solution of nitric acid, which is able to dissolve the particles, had better results than methanol and acetonitrile, because in the case of the mentioned solvents, only the desorption of the extracted analytes from the surface of the sorbent was occurred. The use of nitric acid solution led to the easy coupling the method with FAAS. Verification of the method in the determination amount of Ag+ ions was done by analyzing a CRM and several water and wastewater samples. The approach provided satisfactory results including broad LR of the calibration curve, low LOD and LOQ, and high ER.
Data availability
Not applicable.
References
A.S. Kholmogorova, M.L. Chernysh, L.K. Neudachina, I.S. Puzyrev, React. Funct. Polym. 152, 104596 (2020)
M.T. Doménech-Carbó, F.D. Turo, N. Montoya, F. Catalli, A. Doménech-Carbó, C.D. Vito, Sci. Rep. 8, 10676 (2018)
E.Y. Frag, N.A. El-ZaherSally, E.A. Elashery, Microchem. J. 155, 104750 (2020)
N. Duran, P.D. Marcato, G.I.H. De Souza, O.L. Alves, E. Esposito, J. Biomed. Nanotech. 3, 203–208 (2007)
S.P. Deshmukh, S.M. Patil, S.B. Mullani, S.D. Delekar, Mater. Sci. Eng. 97, 954–965 (2019)
P.D. Howe, S. Dobson, Silver and silver compounds: environmental aspects, In: Concise International Chemical Assessment Document, 44, World Health Organi-zation, Geneva, 2002.
L. Kocurova, I.S. Balogh, L. Nagy, F. Billes, A. Simon, V. Andruch, Microchem. J. 99, 514 (2011)
Eisler R. Silver hazards to fish, wildlife and invertebrates: a synoptic review, US Department of the Interior, National Biological Service, Washington, DC, 1996p. 44.
X. Yang, Zh. Ji, X. Yang, G. Li, X. Liao, Saudi J. Biol. Sci. 24, 589–594 (2017)
M. Sikder, J.R. Lead, G.T. Chandler, M. Baalousha, Sci. Total. Environ. 618, 597–607 (2018)
R.K. Shah, Orien. J. Chem. 32, 499–507 (2016)
R. Cheng, L. Li, S. Ou, Y. Bu, L. Dai, Y. Xue, Roy. Soc. Ch. 6, 36218–36222 (2016)
Y. Yang, L. Luo, H.P. Li, Q. Wang, Z.G. Yang, C.L. Long, Talanta 161, 342–349 (2016)
R. Mikelova, J. Baloun, J. Petrlova, V. Adam, L. Havel, J. Petrek, A. Horna, R. Kizek, Bioelectrochem. 70, 508–518 (2007)
H. Zejli, J.L. Hidalgo-Hidalgo de Cisneros, I. Naranjo-Rodriguez, K.R. Temsamani, Talanta 71, 1594–1598 (2007)
E. Tomaszewska, K. Soliwoda, K. Kadziola, B. Tkacz-Szczesna, G. Celichowski, M. Cichomski, W. Szmaja, J. Grobelny, Microchem. J. 2013, 60 (2013)
P. Anekthirakun, A. Imyim, Microchem. J. 145, 470–475 (2019)
H. Tavallali, H. Malekzadeh, F. Dadvar, M. Tabandeh, M. Haghshenas, Arab. J. Chem. 10, S2090–S2097 (2017)
T. Daşbaş, Ş Sacmacı, S. Şahan, Ş Kartal, A. Ülgen, Talanta 103, 1 (2013)
F. Pena-Pereira, I. Lavilla, C. Bendicho, Anal. Chim. Acta 669, 1 (2010)
F. Pena-Pereira, I. Lavilla, C. Bendicho, Spectrochim. Acta B 64, 1–15 (2009)
M.A. Farajzadeh, S.M. Sorouraddin, M.R. Afshar Mogaddam, Microchim. Acta 181, 829–851 (2014)
H. Abdolmohammad-Zadeh, Z. Javan, Microchim. Acta 182, 1447–1456 (2015)
E. Tahmasebi, Y. Yamini, Microchim. Acta 181, 543–551 (2014)
T. Cetin, S. Tokalıoglu, A. Ulgen, S. Sahan, I. Ozenturk, C. Soykan, Talanta 105, 340–346 (2013)
Sh. Chen, Y. Sun, J. Chao, L. Cheng, Y. Chen, J. Liu, J. Environ. Sci. 41, 211–217 (2016)
S. Jafarvand, A. Bidari, P. Hemmatkhah, M.R. Milani Hosseini, Y. Assadi, Anal. Lett. 42, 2214–2231 (2009)
T. Daşbaşı, Ş Saçmacı, A. Ülgen, Ş Kartal, J. Ind. Eng. Chem. 28, 316–321 (2015)
A.N. Anthemidis, K.I.G. Ioannou, Talanta 84, 1215–1220 (2011)
A.R. Ghiasvand, F. Moradi, H. Sharghi, A.R. Hasaninejad, Anal. Sci. 21, 387–390 (2005)
Y. Su, T. Ren, Y. Lin, Ch. Zheng, Adv. Sample Prep. 3, 100033 (2022)
F. Omidi, M. Behbahani, S. Jamaleddin Shahtaheri, S. Salimi, Environ. Monit. Assess. 187, 361 (2015)
O. Heitzsch, K. Gloe, H. Stephan, E. Weber, Solvent Extr. Ion Exc. 12, 475–496 (1994)
A. Chisvert, S. Cárdenas, R. Lucena, Trends Anal. Chem. 112, 226–233 (2019)
T. Khezeli, A. Daneshfar, Trends Anal. Chem. 89, 99–118 (2017)
M. Ghorbani, M. Aghamohammadhassan, M. Chamsaz, H. Akhlaghi, T. Pedramrad, Trends Anal. Chem. 118, 793–809 (2019)
M. Sajid, M. Khaled Nazal, I. Ihsanullah, Anal. Chim. Acta 1141, 246–262 (2021)
A. Chisvert, J.L. Benedé, A. Salvador, Anal. Chim. Acta 1034, 22–38 (2018)
M.A. Farajzadeh, M. Sattari Dabbagh, J. Chromatogr. A 1627, 461389 (2020)
R. Fu, Y. Liu, Z. Lou, Z. Wang, S.A. Baig, X. Xu, J. Taiwan Inst. Chem. Eng. 62, 247–258 (2016)
D. Lv, Y. Liu, J. Zhou, K. Yang, Z. Lou, S.A. Baig, X. Xu, Appl. Surf. Sci. 428, 648–658 (2018)
K. Li, J. Cao, H. Li, J. Liu, M. Lu, D. Tang, RSC Adv. 6, 92186–92196 (2016)
F. Fang, L. Kong, J. Huang, S. Wu, K. Zhang, X. Wang, B. Sun, Z. Jin, J. Wang, X.-J. Huang, J. Hazard. Mater. 270, 1–10 (2014)
X. Dong, L.Q. Ma, Y. Li, J. Hazard. Mater. 190, 909–915 (2011)
A. Mukherjee, A. Zimmerman, W. Harris, Geoderma 163, 247–255 (2011)
L. G. Sillen, A.E. Martell, The Chemical Society, London, 1964, no. 17; 1973, no. 25.
D. D. Perrin, IUPAC Chemical Data Series no. 22, Pergamon Press, Oxford, 1979.
K. Parvizzad, S.M. Sorouraddin, M.A. Farajzadeh, J. Food. Compos. 112, 104654 (2022).
S.M. Sorouraddin, M.A. Farajzadeh, A. Hassanyani, M.R. Afshar Mogaddam, RSC Adv. 6, 108603 (2016)
Gh. Khayatian, Sh. Hassanpoor, J. Incl. Phenom. Macrocycl. Chem. 73, 151–159 (2012)
H. Tavallali, S. Yazdandoust, M. Yazdandoust, Clean: Soil, Air, Water 38, 242–247 (2010)
S. Rastegarzadeh, N. Pourreza, A. Larki, J. Ind. Eng. Chem. 24, 297–301 (2015)
Funding
The cost of the research was received by Saeed Mohammad Sorouraddin from University of Tabriz.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Ethical approval
This article does not contain any studies with human or animal subjects.
Informed consent
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zendehdel, A.A., Sorouraddin, S.M. & Farajzadeh, M.A. In-situ formation of the adsorbent based on octadecylamine for the extraction of Ag+ ions from aqueous solutions and its determination by microinjection flame atomic absorption spectrometry. ANAL. SCI. 39, 1901–1908 (2023). https://doi.org/10.1007/s44211-023-00399-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s44211-023-00399-1