
Abstract
In the present work, a new microextraction technique, namely in situ-produced CO2-assisted dispersive liquid–liquid microextraction was introduced for the extraction and preconcentration of cobalt, nickel, and copper from aqueous samples followed by graphite furnace atomic absorption spectrometry detection. The proposed method relies on the CO2 gas produced due to a chemical reaction as the disperser agent instead of the disperser solvent used in the conventional dispersive liquid–liquid microextraction. Initially, a solid mixture of tartaric acid and sodium bicarbonate was placed in the bottom of a dry conical glass test tube. Then µL level of 1,1,2,2-tetrachloroethane as the extraction solvent was added into the tube. An aqueous solution of the analytes containing sodium diethyldithiocarbamate (as chelating agent) was transferred into the tube. The reaction between tartaric acid and sodium bicarbonate was immediately occurred, and the produced CO2 led to dispersion of the extraction solvent as tiny droplets into the sample which resulted in extraction of the analytes into the organic solvent. The cloudy solution was centrifuged, and the sedimented phase was analyzed by the instrumental analytical method. Under the optimum conditions, the calibration curves were linear in the ranges of 20–300, 20–200, and 15–250 ng L−1 for Co2+, Ni2+, and Cu2+, respectively. Repeatability of the proposed method, expressed as relative standard deviation, ranged from 2.3 to 4.6 and 4.5–5.6% for intra- and inter-day (n = 6, C = 50 ng L−1) precisions, respectively. Moreover, the detection limits and enrichment factors of the selected analytes were obtained in the ranges of 6.2–12 and 139–150 ng L−1, respectively. The accuracy of the developed procedure was checked by analyzing NRCC-SLRS4 Riverine water as a certified reference material. Finally, the proposed method was successfully applied for the simultaneous analysis of the selected analytes in environmental water and fruit juice samples. The relative recoveries obtained for the analytes in the spiked samples were within in the range of 84–107%.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Heavy metal ions are present in the environment at relatively low concentrations. They are widely used in various industries, while at the same time they are important pollutants of the environment due to their toxic effect on human health. Cobalt, nickel, and copper are typical metal ions present in the environmental samples and have important roles in many physiological functions. However, high concentrations of these metal ions may be toxic and lead to side effects. The ingestion of relatively large doses of cobalt may cause toxic effects [1]. It may give rise to several health problems such as paralysis, diarrhea, low blood pressure, lung irritation, and bone defects. The toxic effects of nickel are well known, and it is also considered as one of the most common causes of allergic contact dermatitis and respiratory system diseases [2, 3]. High amounts of copper in human body can cause stomach and intestinal illnesses such as nausea, vomiting, diarrhea, and stomach cramps [4]. It is noteworthy to mention here that the metals contents in water can provide essential information on the level of contamination in their surrounding environment. Different analytical techniques have been used for determination of heavy metals in different samples. Prior to measurement of low concentrations of heavy metals in a complex matrix, performing separation and preconcentration techniques are inevitable in order to eliminate or minimize matrix effects which lead to lower detection limits and improved sensitivity of detection techniques. Liquid–liquid extraction [5] and solid-phase extraction [6] are the most conventional techniques suffering from several shortcomings such as use of large amounts of toxic organic solvents, and being expensive, time-consuming, and environmentally unfriendly. Modern trends in analytical chemistry are toward the development of new methods which offers such advantages as being fast, cheap, and performing in miniaturized scale leading to reduction in solvent and material usage. Cloud point extraction [7], solid-phase microextraction [8], matrix solid-phase dispersion [9], stir-bar sorptive extraction [10], and liquid-phase microextraction (LPME) methods [11] such as single-drop microextraction [12] and hollow-fiber LPME [13, 14] have been developed to overcome drawbacks of the classical extraction methods. The microextraction techniques are non-exhaustive extractions, and therefore they are time-dependent. The minimal extraction solvent or sorbent applied in a single attempt is not sufficient to completely extract the analytes. However, this weakness is also the competitive advantage of microextraction techniques, where the minimal solvent or sorbent employed enhances the analyte enrichment. Assadi et al. [15] developed a novel liquid-phase microextraction technique in 2006 termed dispersive liquid–liquid microextraction (DLLME) which is based on a ternary component solvent system and applies dispersive concept to overcome the long extraction time problem encountered in the microextraction techniques. In this extraction method, very large surface area between the fine droplets of an extraction solvent and an aqueous sample is achieved, and the corresponding fast mass transfer kinetic results in the rapid establishment of equilibrium. DLLME has been widely used for the extraction and preconcentration of heavy metals [16,17,18,19]. DLLME offers outstanding advantages such as easy operation, rapidity, low sample volume, low cost, and high EFs. In conventional DLLME, an extraction solvent is dispersed into an aqueous sample solution with the aid of a disperser solvent. The presence of relatively high volume of the disperser solvent (usually 1–2 mL for a 5-mL aqueous sample) makes the aqueous phase relatively nonpolar and results in an increased solubility of the target lipophilic analytes into the aqueous sample solution leading to relatively low extraction efficiency. Therefore, some alternatives to the conventional DLLME such as up-and-down shaker-assisted DLLME [20], ultrasonic-assisted DLLME [21], pressure-assisted dispersive microextraction [22], air-assisted liquid–liquid microextraction [23], vortex-assisted DLLME [24, 25], effervescence assisted dispersive liquid–liquid microextraction [26, 27], and salt-assisted liquid–liquid microextraction [28, 29] have been proposed.
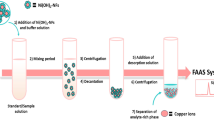
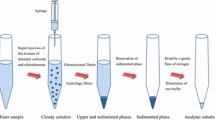
The goal of the present study was to develop a simple and rapid microextraction method that can extract the selected heavy metals. In this method, the disperser solvent was replaced by CO2 gas produced in situ. In the proposed method, despite conventional DLLME methods, the use of mL volume of an organic disperser solvent was avoided, and the dispersion state was done by CO2 as a disperser resulted from the reaction between tartaric acid and sodium bicarbonate. It provided an efficient dispersion of the extraction solvent into the aqueous phase by creating numerous microdrops of the extractant, and led to good extraction efficiency. Then, the obtained cloudy solution was centrifuged and the extraction solvent containing the analytes was settled down at the bottom of a conical test tube. To investigate the efficiency of the method, 10 µL of the settled phase was removed and injected into graphite furnace atomic absorption spectrometry (GFAAS). The influence of different operational parameters on the extraction performance of the target analytes was systematically investigated and optimized.
Materials and methods
Reagents and solutions
A mixture stock solution of Co(II), Cu(II), and Ni(II) (10 mg L−1 of each) was prepared from analytical reagent grade Co(NO3)2·6H2O, Cu(NO3)2·6H2O, and Ni(NO3)2·6H2O (all from Merck, Darmstadt, Germany) by dissolving appropriate amount of each salt in deionized water (Ghazi Company, Tabriz, Iran). A working standard solution (50 ng L−1 of each) was prepared daily by diluting the stock solution with deionized water. Also a mixture standard solution with a concentration of 0.1 mg L−1 of each analyte was prepared and injected into GFAAS each day (three times) for quality control, and the obtained signals were used to calculate EFs and extraction recoveries (ERs) of the analytes. Sodium diethyldithiocarbamate (SDDTC), a chelating agent, was purchased from Fluka (Buchs, Switzerland). Tartaric acid, sodium bicarbonate, carbon tetrachloride, chloroform, 1,2-dibromoethane (1,2-DBE), sodium hydroxide, and sodium chloride were also from Merck. 1,1,2-Trichloroethane (1,1,2-TCE) and 1,1,2,2-tetrachloroethane (1,1,2,2-TCE) were from Janssen Chimica (Beerse, Belgium).
Real samples
Well water sample was collected from suburb of Tabriz (East Azarbaijan province, Iran). River water was collected from Mehranrood River (Tabriz, Iran). Grape and apple juice samples were supplied from local supermarkets (in Tabriz, Iran). All of the real samples were directly subjected to the extraction method without any pretreatment.
Instrumentation
The measurements were performed with a Shimadzu 6300 atomic absorption spectrometer (Kyoto, Japan) equipped with a heated graphite tube atomizer. The instrument settings and optimized furnace programs for determination of the analytes are listed in Table 1. Cobalt, nickel, and copper hollow-cathode lamps (Hamamatsu Photonics, Shizuoka, Japan) were used as radiation sources. An ASC 6100 autosampler (Shimadzu, Kyoto, Japan) was used to deliver standard solutions and samples from the cup to the graphite tube. A Hettich centrifuge, model ROTOFIX 32A (Kirchlengern, (Germany), was used for accelerating phase separation.
Procedure
Initially, 30 mg tartaric acid and 120 mg sodium bicarbonate were mixed and placed into a dry 10-mL glass tube with a conical bottom. Then 43 µL of the extraction solvent, 1,1,2,2-TCE, was added into the mixture. Five milliliters of the standard solution (50 ng L−1 of each analyte) or real sample (pH 4–7) containing SDDTC (2.5 × 10−3 mol L−1) as the chelating agent was transferred into the tube. Upon presence of the aqueous solution, the reaction between tartaric acid and sodium bicarbonate was triggered and the produced CO2 led to dispersion of the extraction solvent as tiny droplets into the aqueous solution and a cloudy solution was formed. After centrifugation for 5 min at 5000 rpm, the fine droplets of organic phase containing the extracted analyte–SDDT complexes were sedimented at the bottom of the tube. Volume of the sedimented phase (33 ± 1 μL) was measured using a 50-μL microsyringe (zero dead volume, Hamilton, Switzerland). In order to investigate the extracted amount of the analytes, three 10 μL aliquots of the settled phase were removed and separately injected into GFAAS.
Calculation of EF and ER
EF is defined as the ratio between the analyte concentration in the sedimented phase (C sed) and the initial concentration of the analyte (C 0) in the sample:
C sed is obtained from a calibration graph. ER is defined as the percentage of the total analyte amount (n0) which is extracted to the sedimented phase (n sed).
where V sed and V aq are the volumes of sedimented phase and aqueous solution, respectively.
Results and discussion
In order to investigate the effect of experimental conditions on the extraction efficiency of the proposed method, the effect of varying different parameters were optimized. It should be mentioned that optimization of the procedure was carried out using 5 mL deionized water spiked with 50 ng L−1 of each heavy metal.
Optimization of furnace temperature program
Drying, ashing, and atomization temperatures have important effects on the determination of cations with GFAAS. So, the effect of these temperatures on the analytical signals was investigated, and the results are shown in Fig. S1 in Electronic Supplementary Material (ESM). The optimum conditions are listed in Table 1.
Optimization of the amount of tartaric acid and sodium bicarbonate
In order to study the effect of these parameters, first, the extraction recovery of the analytes was investigated in the presence of different weight ratios of tartaric acid and sodium bicarbonate (mg:mg; 2:18, 3:17, 4:16, 5:15, 6:14, and 7:13) with the total weight kept constant (200 mg). The results depicted in Fig. S2 in ESM showed that the ratio 4:16 (tartaric acid and sodium bicarbonate) provides the most effective condition resulting in the highest extraction efficiency for all analytes. The total weight of the mixture is another important factor that can affect the extraction efficiency. By taking into account the optimized ratio of tartaric acid and sodium bicarbonate in the previous step, the total weight of the mixture was varied in the range of 100–600 mg. The results (Fig. S3) showed that the ERs increase up to 150 mg and then decrease gradually. Therefore, 30 mg of tartaric acid and 120 mg of sodium bicarbonate were selected as the optimum amounts for the subsequent experiments.
Study the effect of SDDTC to the analyte ratios
SDDTC is a suitable chelating reagent that can react with many metallic ions to form stable complexes. It was selected as a nonspecific chelating agent in order to achieve multielement analysis so that simultaneous determination of Co(II), Ni(II), and Cu(II) using the proposed technique could become possible. The influence of the ratio of SDDTC to the analytes (w/w) on the ERs of Co(II), Ni(II), and Cu(II) was evaluated in the range of 3.4 × 106–13.6 × 106 (w/w). The results shown in Fig. S4 reveal that the ERs are increased by increasing the ratio up to 8.5 × 106 and thereafter remained nearly constant. Therefore, the ratio of 8.5 × 106 was chosen as the optimum ratio in the extraction of the selected heavy metal ions.
Selection of extraction solvent
The selection of a suitable extraction solvent is of great importance for the optimization of a DLLME-based procedure. The extraction solvent has to fulfill some requirements: It has to be lighter or heavier than water, be of low solubility in water, show high extraction efficiency toward the analytes, and should be easily dispersed into aqueous phase during the dispersing step. Based on these requirements, different volumes of carbon tetrachloride, chloroform, 1,2-DBE, 1,1,2-TCE, and 1,1,2,2-TCE were (separately) tested to give 33 ± 1 μL of the sedimented organic phase. The used volumes were 56, 80, 43, 48, and 43 µL for carbon tetrachloride, chloroform, 1,2-DBE, 1,1,2-TCE, and 1,1,2,2-TCE, respectively. Figure 1 shows the effect of extraction solvent type on the extraction efficiency of the analytes. As it can be seen, 1,1,2,2-TCE has the highest extraction efficiency with low consumption of organic solvent (43 µL) among the tested solvents. So it was selected as the extraction solvent for the subsequent experiments.

Effect of extraction solvent type on the extraction efficiency of the method. Extraction conditions: sample, 5 mL deionized water containing 50 ng L−1 of Co2+, Cu2+ and Ni2+ (each cation); SDDTC concentration, 2.5 × 10−3 mol L−1; extraction solvent (volume), 1,2-DBE (43 µL); temperature, 20 °C; centrifuge rate, 5000 rpm; and centrifuge time, 5 min. The error bars represent standard deviations (n = 3)
Optimization of extraction solvent volume
The volume of 1,1,2,2-TCE is another important factor that affects the extraction efficiency. With the aim of obtaining the optimal volume of the extraction solvent, different volumes of 1,1,2,2-TCE were used. According to the results (Fig. S5) by increasing the volume of 1,1,2,2-TCE the analytical signals decreased. Increasing the 1,1,2,2-TCE volume would increase the extracted amount of analytes, whereas their concentration in the sedimented phase will be more dilute. This behavior was observed for all of the studied cations, and in all cases the highest extraction efficiency was observed at 43 µL. It is worth noting here that in cases where ˂ 43 µL of 1,1,2,2-TCE was used, the volume of the sedimented phase was < 33 µL, by which the analysis of three analytes was impossible regarding to the fact that 10 µL of the sedimented phase was required for each analysis. Hence, a volume of 43 µL was selected as the optimal value for 1,1,2,2-TCE to carry out the subsequent steps.
Study the effect of sample solution pH
Extraction of the heavy metals by the proposed method involves prior formation of a complex (the cation–SDDTC complex) with sufficient hydrophobicity which is to be extracted into the small dispersed tiny droplets of the 1,1,2,2-TCE. Therefore, the pH of the aqueous phase is one of the most important factors in the extraction of Co(II), Ni(II), and Cu(II) from aqueous solution in view of the extent of formation of their corresponding complexes. The influence of pH on the ERs of the analytes was investigated in the pH range of 2.0–12.0 by adding appropriate amounts of 1 M hydrochloric acid or sodium hydroxide solutions. According to the results in Fig. 2, at pH 2, the ERs of the analytes are low which may be attributed to the interaction of SDDTC with hydronium ions rather than with the analytes. The ERs increase with increasing pH and they reach a maximum at pHs 4–7. It is in agreement with the previous papers [30,31,32,33,34]. Decrease in ERs in alkaline pHs is probably due to the precipitation of the cations in the solution. The reasonable ERs for the target cations were obtained at pH range of 4–7, and the pH of the samples used was in this range. Therefore, the original aqueous samples were used without any pH adjustment.

Study of sample pH on the ERs of the analytes. Extraction conditions: SDDTC concentration: 2.5 mM. Other conditions are the same as used in Fig. 1. The error bars represent standard deviations (n = 3)
The effect of ionic strength
From the theoretical point of view, increasing of ionic strength could show two opposing effects on the extraction efficiency: (1) salting-out effect, which normally favors the extraction efficiency, and (2) increasing viscosity of the aqueous phase which leads to a decrease in diffusion coefficients and ERs. The effect of ionic strength of the aqueous phase on the extraction efficiency was evaluated by adding various amounts of sodium chloride into the aqueous phase in the range of 0–15% (w/v). The results (Fig. S6) indicated that salt addition has no considerable effect on the extraction recoveries in the mentioned range. Hence the subsequent studies were performed without deliberate manipulation of the ionic strength.
Investigating the effect of temperature of the solution
Temperature is another effective parameter on the efficiency of the extraction with the proposed method. High temperatures could affect the reaction of tartaric acid and sodium bicarbonate and result in better dispersion of 1,1,2,2-TCE by the produced CO2 and the reaction between the studied cations and the complexing agent can also be affected by the temperature. Furthermore, mass transfer is an important phenomenon and plays a key role in the extraction methods. Altering the temperature can make it possible to improve the mass transfer. It is mentioned that temperature can also affect the solubility of the extraction solvent into the aqueous phase and subsequently the volume of the collected phase after performing the method. Therefore, the effect of temperature on the ERs of the analytes was evaluated in the range of 25–75 °C. Figure 3 shows that the ERs increase with increasing the temperature from 25 to 45 °C. However, when the temperature of the solution is larger than 45 °C, the ERs decrease probably due to faster escape of the produced CO2 gas from the solution at higher temperatures. Also the volume of the collected organic phase was relatively constant at the tested temperatures. It seems that high concentrations of the ions produced from dissolution of the solids (sodium bicarbonate and tartaric acid) avoided most dissolving of the organic solvent into the aqueous solution at the high temperatures. Hence, calibration graphs and other analytical characteristic of the proposed method were investigated at 45 °C.

Effect of temperature on the ERs of the cations. The experimental conditions are the same as in Fig. 2 without pH adjustment. The error bars represent standard deviations (n = 3)
Centrifugation time and rate
Centrifugation is usually used to accelerate the collection of extractant droplets at the bottom or on the top of the aqueous sample depending on the fact that which one is denser: the organic solvent or the aqueous phase. In this study, after dispersion of 1,1,2,2-TCE by the produced CO2, the equilibrium status could be achieved in a few seconds due to large contact area between tiny droplets of the extraction solvent and the sample. Therefore, centrifugation was only used to help to make the cloudy solution clear by separating tiny droplets of 1,1,2,2-TCE and making them to settle at the bottom of the conical tube. To study the effect of centrifugation rate and time on the ERs of the analytes, two series of experiments were carried out: (a) first, a constant centrifugation time (6 min) was selected, while the speed varied in the range of 3000–8000 rpm, and (b) other experiments were performed at the selected optimum centrifugation speed (5000 rpm), while the run time was varied between 2 and 10 min. According to the obtained results (Fig. S7), centrifugation rate and time of 5000 rpm and 5 min, respectively, were selected.
Effect of coexisting ions
Common coexisting ions in real samples can affect complexation and consequently extraction efficiency of the analytes. Therefore, the effect of these ions on the ERs of the selected analytes was studied. For this purpose, 5.0 mL aqueous solution containing 50 ng L−1 of each analyte and various concentrations of coexisting ions was treated according to the recommended extraction procedure. A given species was considered to interfere if it resulted in a ± 5% variation in the analytical signal. The tolerable concentration ratios of the coexisting ions to the analytes are shown in Table 2. The results show that the proposed method can be used without significant interferences from the mentioned coexisting cations and anions.
Analytical figures of merit
Under the optimized conditions, quantitative characteristics of the method, namely limit of detection (LOD), limit of quantification (LOQ), linear range (LR), correlation coefficient of the calibration curve (r 2), and precision as relative standard deviation (RSD), were evaluated in order to determine efficiency of the method in analysis of the target analytes in the aqueous samples. The results are summarized in Table 3. It can be observed that RSD values were equal to or less than 6% for intra- and inter-day precisions which indicate acceptable repeatability of the proposed method. The LODs calculated as 3s B/m (s B and m are the standard deviation of the blank, and the slope of the calibration graph, respectively) were between 6.2 and 12 ng L−1. The EFs and ERs for the selected cations were calculated and found to be in the ranges of 139–150 and 92–99%, respectively. The accuracy of the proposed method was assessed with the measurement of the analytes in NRCC-SLRS-4 Riverine water as a certified reference material. For analysis of Ni2+ and Cu2+, the certified reference material was diluted 12-fold with deionized water and then subjected to the proposed procedure. The certified and observed values are given in Table 4. The student t test was applied to compare the certified and the obtained values. The obtained results are listed in Table 4. It can be seen that a good agreement between the determined values and the certified values are obtained.
Comparison of the developed method with other methods
Table 5 shows figures of merit of the proposed method together with those of other methods reported for analysis of the studied cations in different matrices. As it can be seen, the LRs and LODs of the proposed method are better than the reported methods (except first method). In addition, the repeatability and reproducibility of the method is good and its RSDs are lower than or equal (second method) to those obtained by other methods. Also, the presented method greatly reduces volume of the organic solvent compared to other mentioned DLLME-based procedures (except second method) considering the dispersion state. It is noted that in the cases of the second method, ultrasonication was used instead of disperser solvent to disperse extraction solvent into aqueous sample. This needs an additional device and may decompose the analytes in sonification. These results reveal that the presented method is sensitive, simple, rapid, and repeatable and can be used for preconcentration and determination of ultra-trace amounts of Co2+, Ni2+, and Cu2+ in aqueous samples.
Real sample analysis
To evaluate applicability of the proposed method, it was used for the analysis of the studied cations in real samples including well water, river water, apple juice, and grape juice. All samples were extracted and analyzed according to the method described in “Procedure” section. To evaluate matrix effect, relative recoveries were calculated for real samples spiked at three concentration levels (25, 50 and 100 ng L−1). It was done by comparing the absorbances obtained when the extraction method was performed on real samples and deionized water (all spiked at the same concentrations).The obtained relative recoveries (Table 6) were between 84 and 107% which indicated that matrices of the real samples had tolerable effect on the result of the proposed method.
Conclusion
In this study, a new microextraction technique namely in situ-produced CO2-assisted DLLME coupled with GFAAS was used for extraction and preconcentration and determination of Co2+, Ni2+, and Cu2+ in aqueous samples at ng L−1 level. In the proposed method, much less volume of an organic solvent (at μL level) is used as the extraction solvent in the absence of disperser solvent. The CO2 produced from the reaction between tartaric acid and sodium bicarbonate was used to disperse the extraction solvent as tiny droplets into aqueous sample solution with the subsequent extraction of the analytes. The proposed method is rapid, precise, efficient, and sensitive. Finally, the proposed method was successfully applied for the determination of the selected cations in different water and food samples. The results indicate that the proposed extraction procedure is noticeable due to its outstanding advantages including less organic solvent consumption, simplicity, low cost, rapidness, high efficiency, low LODs, and environmentally friendly. Hence, it seems possible to extend this method to extract the other analytes in various samples by varying the extraction conditions.
Abbreviations
- AALLME:
-
Air-assisted liquid–liquid microextraction
- 1,2-DBE:
-
1,2-Dibromoethane
- DLLME:
-
Dispersive liquid–liquid microextraction
- ER:
-
Extraction recovery
- GFAAS:
-
Graphite furnace atomic absorption spectrometry
- LLE:
-
Liquid–liquid extraction
- LPME:
-
Liquid-phase microextraction
- LOD:
-
Limit of detection
- LOQ:
-
Limit of quantification
- MSPD:
-
Matrix solid-phase dispersion
- RSD:
-
Relative standard deviation
- SALLME:
-
Salt-assisted liquid–liquid microextraction
- SDDTC:
-
Sodium diethyldithiocarbamate
- SPE:
-
Solid-phase extraction
- SPME:
-
Solid-phase microextraction
- SBSE:
-
Stir bar sorptive extraction
- 1,1,2,2-TCE:
-
1,1,2,2-Tetrachloroethane
- 1,1,2-TCE:
-
1,1,2-Trichloroethane
- UDSA–DLLME:
-
Up-and-down shaker-assisted dispersive liquid–liquid microextraction
References
Y. Wang, X. Ke, X. Zhou, J. Li, J. Ma, J. Saudi Chem. Soc. 20, S145 (2016)
C.E. Borba, R. Guirardello, E.A. Silva, M.T. Veit, C.R.G. Tavares, Biochem. Eng. J. 30, 184 (2006)
F. Fu, Q. Wang, J. Environ. Manag. 92, 407 (2011)
A.T. Paulino, F.A.S. Minasse, M.R. Guilherme, A.V. Reis, E.C. Muniz, J. Nozaki, J. Colloid Interface Sci. 301, 479 (2006)
A. Oliva, A. Molinari, F. Zuniga, P. Ponce, Microchim. Acta 140, 201 (2002)
T. Daşbaşı, Ş. Saçmacı, N. Çankaya, C. Soykan, Food Chem. 211, 68 (2016)
D. Citak, M. Tuzen, Food Chem. Toxicol. 48, 1399 (2010)
A.K. Malik, V. Kaur, N. Verma, Talanta 68, 842 (2006)
X.P. Yao, Z.J. Fu, Y.G. Zhao, L. Wang, L.Y. Fang, H.Y. Shen, Talanta 97, 124 (2012)
X. Huang, N. Qiu, D. Yuan, B. Huang, Talanta 78, 101 (2009)
M.A. Farajzadeh, S.M. Sorouraddin, M.R. Afshar Mogaddam, Microchim. Acta 181, 829 (2014)
P. Liang, R. Liu, J. Cao, Microchim. Acta 160, 135 (2008)
Y. Liu, Y. Wang, Y. Hu, L. Ni, J. Han, T. Chen, H. Chen, Y. Liu, J. Iran. Chem. Soc. 12, 371 (2015)
J. Abulhassani, J.L. Manzoori, M. Amjadi, J. Hazard. Mater. 176, 481 (2010)
M. Rezaee, Y. Assadi, M.R. Milani Hosseini, E. Aghaee, F. Ahmadi, S. Berijani, J. Chromatogr. A 1116, 1 (2006)
Y. Yamini, M. Rezaee, A. Khanchi, M. Faraji, A. Saleh, J. Chromatogr. A 1217, 2358 (2010)
H. Sereshti, V. Khojeh, S. Samadi, Talanta 83, 885 (2011)
M. Mirzaei, M. Behzadi, N.M. Abadi, A. Beizaei, J. Hazard. Mater. 186, 1739 (2011)
M. Shamsipur, N. Fattahi, M. Sadeghi, M. Pirsaheb, J. Iran. Chem. Soc. 11, 249 (2014)
S.P. Chu, W.C. Tseng, P.H. Kong, C.K. Huang, J.H. Chen, P.S. Chen, S.D. Huang, Food Chem. 185, 377 (2015)
N. Wei, X.E. Zhao, S. Zhu, Y. He, L. Zheng, G. Chen, J. You, S. Liu, Z. Liu, Talanta 161, 253 (2016)
Naeemullah, M. Tuzen, T.G. Kazi, D. Citak, M. Soylak, J. Anal. At. Spectrom. 28, 1441 (2013)
M.A. Farajzadeh, M.R. Afshar Mogaddam, Anal. Chim. Acta 728, 31 (2012)
M. Soylak, E. Kiranartligiller, Arab. J. Sci. Eng. 42, 175 (2017)
J. Xue, X. Chen, W. Jiang, F. Liu, H. Li, J. Chromatogr. B 975, 9 (2015)
G.L. Aragonés, R. Lucena, S. Cárdenas, M. Valcárcel, Anal. Chim. Acta 807, 61 (2014)
K. Medinskaia, C. Vakh, D. Aseeva, V. Andruch, L. Moskvin, A. Bulatov, Anal. Chim. Acta 902, 129 (2016)
M. Gupta, A. Jain, K.K. Verma, Talanta 80, 526 (2009)
H. Ma, Y. Li, H. Zhang, S.M. Shah, J. Chen, J. Chromatogr. A 1358, 14 (2014)
I. Rapp, Ch. Schlosser, D. Rusiecka, M. Gledhill, E.P. Achterberg, Anal. Chim. Acta 976, 1 (2017)
L. Hejazi, D.E. Mohammadi, Y. Yamini, R.G. Brereton, Talanta 62, 185 (2004)
S.M. Sorouraddin, M.A. Farajzadeh, T. Okhravi, Talanta 175, 359 (2017)
M. Niinae, T. Suzuki, Y. Inoue, H. Saito, J. Shibata, J. Min. Mater. Process. Inst. Jpn. 130, 16 (2014)
V.T. Nguyen, J.C. Lee, J. Jeong, B.S. Kim, B.D. Pandey, Met. Mater. Int. 20, 357 (2014)
P. Liang, J. Yu, E. Yang, Y. Mo, Food Anal. Methods 7, 1506 (2014)
H. Jiang, Y. Qin, B. Hu, Talanta 74, 1160 (2008)
Acknowledgements
The authors would like to thank the Research Office at the University of Tabriz for financial support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sorouraddin, S.M., Farajzadeh, M.A. & Ghorbani, M. In situ-produced CO2-assisted dispersive liquid–liquid microextraction for extraction and preconcentration of cobalt, nickel, and copper ions from aqueous samples followed by graphite furnace atomic absorption spectrometry determination. J IRAN CHEM SOC 15, 201–209 (2018). https://doi.org/10.1007/s13738-017-1224-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13738-017-1224-8