
Abstract
Herbal and herbal dietary supplements have a massive share of the global market, and their use continues to grow worldwide. There is a general perception that herbal and herbal dietary supplements are effective and safe. However, herbal formulation-drug and natural product-drug interactions are an easily overlooked issue in using herbals and herbal dietary supplements. Positive herb-drug interactions can increase the effectiveness of drugs and reduce their toxic effects. Instead, negative natural product-drug interactions can cause adverse reactions. Quercetin, a natural flavonoid, is present in the form of quercetin glycosides in herbal dietary supplements and is widely consumed by the population daily. In recent years, quercetin has demonstrated excellent biological activities such as hypotensive, hypoglycemic, antioxidant stress, anti-inflammatory, antibacterial, antiviral, and immunomodulatory, and it is widely used in the treatment of various human diseases. Numerous in vivo, in vitro, and clinical studies have revealed pharmacokinetic and pharmacodynamic interactions between quercetin and drugs. By competitively binding to serum albumin, affecting cytochrome P450, glycoproteins, organic anion transporting peptides, and glucuronidase activity, quercetin can alter the absorption, distribution, metabolism, and clearance of some drugs in vivo, affecting their pharmacokinetic profile. In addition, its pharmacodynamic interactions with many drugs have produced potentiation, toxicity reduction, or overcoming resistance in treating several diseases such as infectious diseases, chronic infectious diseases, cardiovascular diseases, diabetes, and cancer. We present a comprehensive summary of the observed pharmacokinetic and pharmacodynamic interactions of quercetin with herbal drugs. Additionally, the potential mechanisms of these interactions and directions for future related research are described.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
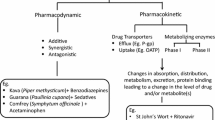
Herbal medicine has a long history of use in diverse communities worldwide. Its use continues to grow globally, capturing a massive share of the global market (Navarro et al. 2017; Zhou et al. 2021). In the perception of most people, herbs are natural and safe. However, an easily overlooked issue is that herbal formulation-drug and natural product-drug interactions may occur when herbs and pure natural products are used in combination with prescription drugs (Gerber et al. 2018). The herbal-drug interactions are divided into pharmacokinetic (altering blood levels) and pharmacodynamic interactions (acting on drug-related targets). Natural product-drug pharmacokinetic interactions refer to the effects of herbal drugs on the absorption, distribution, metabolism, and excretion of drugs in the organism (di Minno et al. 2017). Herbal drugs share the same drug-metabolizing enzymes and drug transport proteins as some clinical drugs, such as cytochrome P450 enzymes (CYP450), glucuronosyltransferases (UGT), and P-glycoproteins (Zhou et al. 2021). Herbal drugs can alter the pharmacological parameters, such as maximum concentration (Cmax), time to peak concentration (Tmax), and the area under the curve (AUC) of some clinical drugs by affecting these pharmacokinetic parameters related to the concentration of a drug in blood plasma, resulting in enhanced efficacy, even adverse effects, or reduced efficacy (Zhou et al. 2021). Herbal-drug pharmacodynamic interactions include synergistic or antagonistic pharmacological effects related to their action on associated molecular targets (Gouws and Hamman 2020). The combination of herbs and certain drugs can cause an increase or decrease in the intensity of the desired effect.
Currently, many patients in the clinic are taking herbal or herbal dietary supplements in conjunction with prescription medications (Gerber et al. 2018). This mode of combination may be at risk of causing adverse reactions due to the presence of a potential negative drug-to-drug interactions. For example, combining ginkgo with warfarin or aspirin may increase the risk of bleeding (Chen et al. 2011). Co-administration of xiyampin injection, an extract of Andrographis paniculata (Burm.f.) Nees, Acanthaceae, with ribavirin increases the risk and severity of herbal-related adverse drug reactions, especially in the elderly population (Zheng et al. 2020). Some herbal dietary supplements, such as St. John’s wort significantly increase the hepatotoxicity of certain drugs via herbal formulation-drug interactions (Zhu et al. 2018). On the bright side, some herbal-drug interactions can be of clinical benefit. For example, propolis extract in combination with doxorubicin, a chemotherapeutic agent, enhanced anti-cancer activity while reducing chemoresistance and improving the significant side effects of doxorubicin (Hossain et al. 2022). Co-administered garlic extract and metformin enhanced hypoglycemic effects and may reduce gentamicin-induced renal tubular toxicity (Gupta et al. 2017). A gel based on Aloe vera (L.) Burm.f., Xanthorrhoeaceae, increases oral absorption of the antiretroviral drug didanosine by up to 11-fold (Gerber et al. 2018). Herbal formulation-drug interactions have advantages and disadvantages and may cause adverse effects or enhance clinical efficacy benefits. Therefore, monitoring and managing possible interactions are necessary (Skalli and Bencheikh 2012).
Quercetin (1), a natural flavonoid, is widely distributed in species of fruits, vegetables, such as onions, cauliflower, cilantro, tomatoes, in fruits as nuts and apples, and beverages as tea and wine (Fig. 1) (Latos-Brozio and Masek 2019; Shabbir et al. 2021; Sobhani et al. 2021). Quercetin has biological activities such as hypotensive, hypoglycemic, antioxidant, anti-inflammatory, antibacterial, antiviral, and immunomodulatory (Chen et al. 2022). Numerous studies have shown the potential of quercetin to treat oncological, autoimmune, cardiovascular, metabolic syndrome, and viral/bacterial infectious diseases (Heinz et al. 2010; Patel et al. 2018; Reyes-Farias and Carrasco-Pozo 2019; Hosseini et al. 2021; Shen et al. 2021). Due to its safety, quercetin is widely used, which is present as glycosides in herbal dietary supplements (Andres et al. 2018). The daily intake of quercetin from vegetables and fruits included in the diet (approximately 250 mg), together with the recommended daily intake of quercetin glycosides from herbal dietary supplements (up to 1000 mg), results in a total of a daily intake of approximately 1250 mg (Andres et al. 2018). Coming from herbs, drug-to-drug interactions between quercetin and drugs is analogously present. However, to our knowledge, there is no review on the potential drug-to-drug interactions of quercetin. Therefore, after reviewing the toxicity of quercetin, attention was placed on its interactions with herbal drugs, including their pharmacokinetic and pharmacodynamics, to provide a basis for its rational dispensing in clinical practice.

The primary sources of quercetin (1)

Search Strategies
A systematic search was conducted on PubMed, Web of Science, and Google Scholar. The search keywords included “quercetin,” “herbal drug interaction,” “pharmacodynamic interaction,” “pharmacokinetic interaction,” “preclinical studies.” The filters were not used. The search lasted from inception to 2022.
Discussion
Physicochemical Properties
The pure substance of quercetin has a yellow, needle-like, or powdered appearance. Ether, methanol, ethanol, acetone, pyridine, and acetic acid are all suitable solvents for quercetin. It is soluble in ether, methanol, ethanol, acetone, pyridine, and acetic acid but not in water (Pradniwat and Chanprasert 2022). Quercetin is weakly acidic, and its pK a value was measured as 6.62 ± 0.04 (Zenkevich and Guschina 2010). There is a clear structure-instability association of specific groups in the quercetin molecule: the C3-OH group in the C ring, the catechol portion of the B ring, and the C2 = C3 bond in the C ring are also present. Due to the presence of these groups, quercetin exhibits a high degree of instability in alkaline solutions (Jurasekova et al. 2014).
Pharmacokinetics
Quercetin undergoes initial metabolism mainly in the small intestine, including glucuronidation and O-methylation. Through the hepatic portal vein, quercetin and its metabolites get to the liver, where it is further broken down, O-methylated, sulfated, and glucuronidated. Also, the bacteria in the large intestine play a role in how quercetin is broken down. For example, Clostridium orbiscindens plays an essential role in the C ring fission of quercetin. The portal vein brings metabolites of quercetin made by the gut bacteria to the liver (Wang et al. 2016). In human plasma, the important metabolites of quercetin are quercetin-3-glucuronide, quercetin-3-sulfate, and isorhamnetin-3-glucosidic acid (Patel et al. 2018). Quercetin and its metabolites are distributed in several organs, including the lungs, kidneys, heart, and liver, with the highest levels of quercetin in the lungs (Wang et al. 2016). Quercetin and its conjugates containing glucuronide groups, methyl or sulfate groups are mainly distributed in the blood and ultimately excreted in the urine (Almeida et al. 2018).
Despite its numerous beneficial biological properties, the low bioavailability of quercetin limits its use as a clinical therapeutic agent. It is mainly due to the water insolubility of quercetin, which makes it difficult for most of it to pass through the mucus layer of the gastrointestinal tract. On the other hand, the remaining quercetin absorbed by the blood and intestinal tract is rapidly metabolized (Li et al. 2021). Quercetin glycoside-based dietary supplements are the main dietary preparations for oral supplementation with quercetin (Li et al. 2021). Therefore, improving the absorption and oral bioavailability of quercetin sapogenins is a critical factor in the efficacy of quercetin.
Toxicity
Specific Toxicity
The early specific toxicological studies of quercetin focused on its potential mutagenicity, carcinogenicity, and teratogenicity. In vitro studies of quercetin investigated its mutagenic effect to some bacteria, fungi, and somatic cells. However, the in vivo results were not consistent with previous in vitro experiments, probably due to the chemically unstable nature of the DNA adducts formed by quercetin oxidation products (Andres et al. 2018). Gene mutations are the primary way in which activation of proto-oncogenes occurs. Many in vivo experiments have also explored the carcinogenicity of quercetin. An early in vivo experiment did not demonstrate any significant carcinogenic effect of dietary levels of quercetin up to 5% in F344 rats (Ito et al. 1989). In contrast, the incidence of renal tubular cell adenomas and the severity of nephropathy increased in male F344 rats fed with a diet containing high-purity quercetin for an extended time (National Toxicology Program 1992). In another experiment, quercetin was genotoxic, causing mainly benign tumors in the male renal tubular epithelium but not elsewhere, which may be partly due to a combination of non-genotoxic and genotoxic events (Dunnick and Hailey 1992).
In 1999, the International Agency for Research on Cancer (IARC) recommended the classification of quercetin as a non-carcinogenic compound (Batiha et al. 2020). In contrast, in recent years, the anti-cancer activity of quercetin has been discovered by researchers and is emerging as a promising natural compound in the therapy of various cancers such as prostate, breast, ovarian, colorectal, esophageal, and osteosarcoma (Farooqi et al. 2018; Ezzati et al. 2020; Vafadar et al. 2020; Tang et al. 2020; Maleki et al. 2021). In addition, quercetin can cross the blood-fetal barrier and may affect pregnancy. In several in vivo studies of quercetin in pregnant mouse models, no adverse effects of quercetin on fetal development and blood status were found. On the contrary, quercetin also had a protective effect on the liver enzymatic antioxidant defense system, renal histological changes, and memory capacity in mouse fetuses (Ożarowski et al. 2018).
General Toxicity
While exploring the specific toxicity of quercetin, researchers have conducted numerous studies on its general toxicity. An early in vivo trial first demonstrated the low acute and chronic toxicity of quercetin and quercetin glycosides (Ambrose et al. 1952). In another in vivo experiment, Ruiz et al. (2006) found no adverse effects or death in Swiss mice from 30 to 3000 mg/kg/day of quercetin intake (for 28 days). Quercetin as a functional food does not affect the normal development of mice (Barrenetxe et al. 2006). In vitro, quercetin is a potent inhibitor of NF-kappa B/AP-1 activation. However, the results of an in vivo experiment suggest that quercetin unexpectedly exacerbated the induction of AP-1 in mice with adriamycin nephropathy and standard groups, possibly associated with adverse renal effects (Rangan et al. 2002). It may be related to the dose-dependent nature of quercetin. At low doses of 10 mg (kg/BW), quercetin protects renal function. In comparison, high doses of 100 mg (kg/BW) induce renal injury, leading to increased abnormalities in adriamycin nephropathy renal function (Heeba and Mahmoud 2016). The results of many later studies have shown the protective effect of quercetin against acute and chronic renal injury caused by heavy metals or drugs (Faddah et al. 2012; Jain et al. 2013; Morales et al. 2006; Abdel-Raheem et al. 2009; Yuksel et al. 2016). A meta-analysis of animal studies included 20 studies with 378 animals. The meta-analysis showed significant improvements in the renal index, urinary protein, uric acid, urinary albumin, and serum creatinine levels after quercetin administration. However, no significant association between quercetin and creatinine clearance was observed (Feng et al. 2022).
In in vivo experiments, quercetin also showed a protective effect against the hepatotoxicity of some drugs and toxicants. Quercetin (10 mg/kg) prevents carbon tetrachloride (CCl4)-induced elevation of serum enzymes and prevents CCl4-induced prolongation of pentobarbital sleep (Janbaz et al. 2004). In addition, quercetin softened Cd-induced oxidative stress by significantly ameliorating oxidative stress and increasing MT and eNOS expression, but quercetin did not prevent Cd-induced liver damage (Vicente-Sánchez et al. 2008). Quercetin inhibits DEN-induced DNA damage and apoptosis in rats and ameliorates the hepatotoxicity of diethyl nitrosamine in rats (Gupta et al. 2010). In addition, quercetin also has a protective effect against hepatotoxicity caused by ciprofloxacin, pyrrolizidine alkaloids, Ralston polysaccharide, isoniazid, rifampin, and chloroquine (Ji et al. 2014; Wang et al. 2015). In conclusion, quercetin has some potential for treating the drug-related liver injury. Regarding the above evidence, quercetin is generally non-toxic and safe. However, it also has drug-to-drug interactions, which may affect the efficacy and toxicity of the drug (Auxtero et al. 2021). Therefore, in the next section, the potential drug-to-drug interactions of quercetin will be highlighted.
Drug-to-Drug Pharmacokinetic Interactions
Binding to Serum Albumin
Human serum albumin (HSA) is an abundant carrier protein in the human body responsible for the transport, ligand binding, distribution, and metabolism of exogenous drugs (Rabbani and Ahn 2019). Competitive binding of quercetin and rutin to fluphenazine (FPZ), a piperazine derivative of phenothiazines, induces conformational changes in human serum albumin (HSA) and increases the binding distance of fluphenazine (Jing et al. 2017). Compared to gliclazide, quercetin has higher binding power to HAS, so quercetin can displace gliclazide from HSA and increase the unbound portion of gliclazide, leading to its more significant hypoglycemic effect (Kameníková et al. 2017). Similarly, quercetin can compete with neratinib (NRB) to bind HSA and replace neratinib (NRB) at its binding site (Wani et al. 2021). Quercetin competes with erlotinib (ERL) to bind to bovine serum albumin (BSA), increasing the free drug fraction in the system, which may add to the increased incidence of adverse events associated with ERL use (Wani et al. 2022). In addition, the binding constant of BSA-poziotinib showed a more significant decrease in the presence of quercetin (Zargar et al. 2020). The above information obtained from such in vitro experiments can help to optimize dose selection when combining drugs (Table S1).
Cytochrome P450 Activity
Quercetin can also influence the process of drug transformation by mediating effects on the activity of drug-metabolizing enzymes. Among them is cytochrome P450 (CYP450), the primary enzyme involved in drug metabolism. Using caffeine metabolite ratios as indicators of drug enzyme activity, Chen et al. (2009) found that quercetin intake (500 mg for 13 days) inhibited CYP1A2 activity and enhanced CYP2A6 activity (Chen et al. 2009). In addition, an in vitro study showed that quercetin strongly inhibited CYP2D6 (Elbarbry et al. 2018). There is increasing evidence that quercetin interacts pharmacokinetically with some drugs by inhibiting CYP450. By mediating the inhibition of CYP2E1 enzyme activity in pairs, quercetin can alter the pharmacokinetics of chlorzoxazone. Moreover, since quercetin can reduce the ethanol-induced increase in CYP2E1 enzyme activity, it may be a potential natural compound for the treatment of alcohol-induced liver injury (Bedada and Neerati 2018). Long-term administration of quercetin (500 mg/day for 3 weeks) does not inhibit CYP2C8 activity, and therefore, HDI is unlikely to occur with drugs metabolized by CYP2C8, such as rosiglitazone (Kim et al. 2005). By inhibiting the CYP1A pathway, quercetin improves the plasma clearance of melatonin (Jana and Rastogi 2017). In addition, Nguyen et al. (2015) found that intake of quercetin (1500 mg/day for 1 week) reduced systemic exposure to oral midazolam by increasing CYP3A-mediated metabolism. Quercetin doubly inhibited CYP3A4 and P-GP to promote intestinal absorption and reduce the first-pass metabolism of triamcinolone, increasing triamcinolone bioavailability (Shin et al. 2006). Similarly, Challa et al. (2013) found that dual inhibition of CYP3A4 and P-gp by quercetin in vitro significantly increased intestinal absorption and decreased efflux of valsartan. Warfarin, a common anticoagulant, increased mRNA and protein expression of porcine primary hepatocytes CYP3A and did not affect CYP1A2. However, interestingly, the co-administration of warfarin with quercetin decreased CYP1A2 and CYP3A activity (Burkina et al. 2022), suggesting the interaction of both compounds by inhibition of CYP450 activity.
P-Glycoprotein Activity
P-Glycoprotein (P-gp) is an efflux transport protein that affects the pharmacokinetics of various drugs (Elmeliegy et al. 2020). As mentioned previously, quercetin can reduce triclosan and valsartan efflux by inhibiting P-gp (Shin et al. 2006; Challa et al. 2013). In an in vitro experiment, Lan et al. (2008) found that isorhamnetin, when combined with quercetin, significantly increased the cellular permeability of both, possibly related to P-gp. Digoxin, a cardiac glycoside with a narrow therapeutic range and high drug-to-drug interactions, is a substrate of P-gp. In an in vivo experiment, Wang et al. (2004) found that the combined application (50 mg/kg) significantly increased the maximum blood concentration (Cmax) and the mean area under the blood concentration–time curve (AUC0-t) of digoxin. Within 30 min, two cases of sudden death in pigs occurred due to digoxin toxicity by the poisoning of P-gp and Na–K transporters, suggesting that combined digoxin or plant species containing digoxin (Digitalis species) and quercetin or quercetin-containing herbs and dietary supplements should be avoided (Wang et al. 2004). Another study exploring quercetin effect on P-gp in broiler chickens showed a dose-dependent effect of quercetin on P-gp expression in chicken Caco-2 cells and tissues. By interacting with the exogenous chicken receptor (CXR), quercetin may induce P-gp expression in tissues, which limits the absorption of enrofloxacin in the intestinal tract (Bhutto et al. 2018). Based on this finding, quercetin was used in combination with enrofloxacin and its similar antibiotics with dose adjustment to ensure adequate blood levels of enrofloxacin.
Anion Transporting Polypeptides
Organic anion transporting polypeptides (OATP) are present in the liver, small intestine, and blood–brain barrier and mediate drug transport at these sites (Kalliokoski and Niemi 2009). Modifying OATP family carrier proteins by quercetin is another mechanism that causes drug-to-drug interactions (Mandery et al. 2010). A crossover clinical study unexpectedly found a 23.9 to 60.6% reduction in talinolol AUC0–48 h in 5 subjects and a 29.2–78.7% reduction in Cmax in 7 subjects after co-administration with quercetin. The experimental results are inconsistent with those expected from the benefits of quercetin in inhibiting intestinal P-gp-mediated drug efflux. The mechanism behind this may involve overlapping modifications of efflux and uptake transport of OATP family carrier proteins and site-dependent interactions (Nguyen et al. 2015). In addition, in vitro, quercetin inhibited the transport of some OATP1B1-mediated drugs, such as estrone 3-sulfate and pravastatin. Furthermore, the inhibitory effect of quercetin on the pharmacokinetics of pravastatin was demonstrated in healthy Chinese Han male volunteers (Wu et al. 2012).
Glucuronidase Inhibition
Glucuronidase (UGT) is involved in the clearance of many classes of drugs such as non-steroidal anti-inflammatory drugs (NSAIDs), analgesics, antivirals, and benzodiazepines by catalyzing the “glucuronidation” reaction (Rowland et al. 2013). In vitro, similar to myricetin, quercetin, with potent inhibition of most human UGTs, has the potential risk of causing drug-to-drug interactions (Li et al. 2022). By using inducers to assess drug-metabolizing enzymes in precise sections of the human intestine, van de Kerkhof et al. (2008) found that quercetin induced elevated UGT1A6 mRNA expression, particularly in colon tissue, by 6.7-fold. In addition, in vitro studies by Zhang et al. (2021) revealed that quercetin has an inhibitory effect on human UDP-glucuronosyltransferase 1A (UGT1A) isoforms. Quercetin inhibits UGT1A isomers, with residual enzyme activities of UGT1A6 (23.1%), UGT1A1 (8.4%), UGT1A3 (30.8%), and UGT1A9 (11.7%) (Zhang et al. 2021). Several other in vitro studies explored the potential of drug-to-drug interactions of quercetin by affecting the glucuronidase activity. Acetaminophen, an antipyretic and analgesic drug, causes liver damage in overdose. Cao et al. (2017) found low inhibition of acetaminophen glucuronidation (not calculated) by quercetin, suggesting that quercetin is unlikely to ameliorate acetaminophen-associated hepatotoxicity. Furthermore, in vitro, quercetin in red wine inhibited UGT2B17 by 72% and reduced testosterone glucuronidation responses. It seems feasible for quercetin to increase testosterone blood levels by reducing urinary excretion; however, further in vivo studies and human studies are needed to verify this effect (Jenkinson et al. 2012). The combinations of quercetin-containing drugs that lead to HDIPK and the targets affected by them (Fig. 2).

Drug-to-drug pharmacokinetic interactions of quercetin with neoplastic, endocrine, psychiatric, and cardiovascular drugs and the associated targets behind them. Abbreviations: ERL, erlotinib; ES3, estrone 3-sulfate; FPZ, fluphenazine; GL, gliclazide; ISR, isorhamnetin; MDZ, midazolam; QR, quercetin; TAM, trichlorophenol amine; VAL, valsartan; WF, warfarin. HSA, human serum albumin; CYP450, cytochrome P-450; P-gp, P-glycoprotein; OATP, anion transporting polypeptides
Drug-to-Drug Pharmacodynamic Interactions
Quercetin and Antibiotics
Quercetin has some antibacterial activity (Nguyen and Bhattacharya 2022). Several studies have explored its pharmacodynamic interactions with antibiotic-like drugs. In vitro, Pal and Tripathi (2019) found that quercetin enhanced the antibacterial efficacy of meropenem against carbapenem-resistant Pseudomonas aeruginosa and Acinetobacter baumannii by enhancing the disruption of cell wall integrity and altering cell morphology. The antibacterial efficacy of low-dose quercetin (amoxycillin + quercitin: 5 μl of 5 mg/ml + 5 μl of 10 mg/ml; amoxycillin: 10 μl of 5 mg/ml) in combination with antibiotics such as amoxicillin and ciprofloxacin was similar to that of high-dose antibiotics. Moreover, this combination of drugs helps to reduce antibiotic resistance in mastitis treatment (Srividya et al. 2018). In addition, quercetin antagonized the toxic effects of some antibiotics. Metronidazole causes neurotoxicity by reducing the proportion of antioxidants in brain tissue, inducing nitric oxide synthesis, and promoting apoptosis. Quercetin antagonized the neuronal toxicity induced by metronidazole (oral) (Chaturvedi et al. 2020). Following antibiotic administration, quercetin counteracts intestinal flora dysbiosis (Shi et al. 2020).
Quercetin and Anti-inflammatory Drugs
Quercetin has activity against several human chronic inflammatory diseases (Kashyap et al. 2019). Haleagrahara et al. (2018) found that combined treatment with quercetin and methotrexate significantly reduced the levels of inflammatory mediators in collagen-induced arthritis mice. Quercetin can be used as an adjuvant to improve the inadequate response to anti-rheumatic monotherapy (Haleagrahara et al. 2018). However, in arthritic gout-pain model rats, quercetin antagonism reduced the diclofenac anti-inflammatory effect. Therefore, this combination is not recommended for pain relief in arthritic gout (Ventura-Martínez et al. 2021). In addition, quercetin has the potential to treat non-steroidal anti-inflammatory drug-induced gastrointestinal disease and related complications, given that it overcomes the exacerbating effect of ranitidine on diclofenac-induced small intestinal toxicity (Singh et al. 2017). In addition, in a clinical study, Hickson et al. (2019) found that quercetin and senolytics combined to reduce the senescent cell burden in humans by reducing essential senescence-associated secretory phenotype factors, including the interleukins IL-1α, IL-6, and matrix metalloproteinases (MMPs) 9 and 12, in patients with diabetic nephropathy.
Quercetin and Cardiovascular Drugs
Quercetin also has a cardiovascular protective effect. Both quercetin and tamsulosin have vasodilatory effects and their combinations are more effective than the compounds alone. The combination of quercetin, quercetin-3-glucosinolate, or 4′-O-methyl-quercetin with tamsulosin reduced the sensitivity of phenylephrine, a potent alpha agonist that constricts small arteries and veins, by approximately 200-fold, 35-fold, and 150-fold, respectively (Vrolijk et al. 2015). Compared to curcumin, the combination of quercetin and curcumin had a more significant protective effect against ischemia–reperfusion injury-induced myocardial toxicity in rats (Chakraborty et al. 2018). Both captopril and quercetin had cardioprotective effects. The combination of adriamycin and quercetin showed diminished cardioprotection in rats with adriamycin-induced cardiotoxicity compared with the drug alone. It suggests a possible pharmacodynamic antagonism between them (Thangavel and Chenniappan 2021).
Quercetin and Hypoglycemic Agents
Quercetin has hypoglycemic biological activity. Many studies have explored the hypoglycemic synergism of quercetin with hypoglycemic drugs. When saxagliptin, a potent and specific DPP-4 inhibitor, is combined with quercetin, its hypoglycemic potency is enhanced (Sowjanya et al. 2017). Similarly, quercetin increased the hypoglycemic effect on streptozotocin-induced diabetic rats when combined with glimepiride, a sulfonylurea hypoglycemic agent, compared to the drug alone (Samala et al. 2015). The antioxidant and chelating properties of quercetin may be related to their protective effects against diabetes. The combined combination of the two lowered blood glucose and increased urinary glucose in tetraoxypyrimidine-induced diabetes in rats. It suggests that inhibition of renal glucose reabsorption may play a hypoglycemic role (Lukačínová et al. 2008). Furthermore, in vitro, a docking simulation experiment showed that quercetin-3-oleoyl is a novel G-protein-coupled receptor 40 agonist (Carullo et al. 2019). Quercetin possesses excellent hypoglycemic potency, and studies on its pharmacodynamic interactions in combination with other hypoglycemic agents should be widely conducted to provide new combination strategies for diabetes treatment.
Quercetin and Anti-cancer Drugs
Quercetin has a combined anti-cancer effect of promoting cancer cell apoptosis, preventing cancer metastasis, and improving cancer metabolism (Reyes-Farias and Carrasco-Pozo 2019). Currently, many studies have explored the additional enhanced anti-cancer potency of quercetin in combination with anti-cancer drugs (Table S2 and Fig. 3). Lipid polymer hybrid nanoparticles (LPNs) of mycophenolic acid and quercetin showed higher and higher cellular uptake rates and combined cellular toxicity compared to individual congeners in MCF-7 cells (low metastatic breast cancer cells) (Patel et al. 2020). In addition, quercetin was able to attenuate doxorubicin-induced vascular toxicity. However, there may be a potential anti-cancer antagonistic pharmacodynamic interaction between them, which may be due to the potent antioxidant activity of quercetin, which eliminates the production of doxorubicin-associated reactive oxygen species, resulting in weaker tumor cell killing activity (Henidi et al. 2020). In another animal study, Liu et al. (2020) explored the effect of co-administration of natural compounds and chemotherapeutic agents on multidrug resistance in breast tumors. Pretreatment with mixed micelles of quercetin not only downregulated P-glycoprotein expression to reduce doxorubicin efflux and increased doxorubicin sequencing in MDA-MB-231 (high-metastatic breast cancer cells)/MDR1 cells but also initiated a mitochondria-dependent apoptotic pathway to accelerate doxorubicin-induced apoptosis. Quercetin reduces cytokinin kinase 6 (CDK6) expression in MCF-7 cells and can potentially be an emerging lead against breast and non-small cell lung cancer as a CDK6 inhibitor (Yousuf et al. 2020). Furthermore, in vivo, clinical trials are needed to validate the pharmacokinetic interactions of quercetin as a relevant CDK6 inhibit. Researchers have also explored the potentiation options of quercetin in other tumor treatments. Quercetin may synergistically enhance the anti-cervical cancer effects of cisplatin by enhancing apoptosis and inhibiting cancer cell proliferation, migration, and invasion (Xu et al. 2021). In addition, considering the low bioavailability of quercetin, some novel drug delivery modalities have been applied in antitumor therapy (Alizadeh and Ebrahimzadeh 2022). Quercetin and gefitinib simultaneously loaded with polyvinylpyrrolidone (PVP)-GO-PVP exerted higher cytotoxicity against PA-1 ovarian cancer cells (Tiwari et al. 2019). Nanoparticles loaded with quercetin and paclitaxel synergistically combat non-small cell lung cancer and improve chemotherapeutic efficacy against A549/paclitaxel-resistant cells (Wang et al. 2021).

Quercetin combines with antineoplastic drugs (chemotherapy) for the treatment of cancer
Perspectives and Future Directions
Currently, the pharmacokinetic interactions associated with quercetin are mainly focused on synergistic effects with drugs, including enhanced efficacy and reduced resistance. These synergies have been shown to enhance the efficacy of quercetin in bacterial infections, chronic inflammatory diseases, cardiovascular diseases, diabetes mellitus, and oncology-related drugs. However, there is a lack of clinical studies evaluating adverse effects, especially related to quercetin drug-to-drug interactions. These studies will further provide a basis and reference for the rational combination of quercetin in the clinical setting. In addition, considering the shortcomings of quercetin low bioavailability, additional drug delivery methods to improve quercetin bioavailability need to be utilized in future studies.
Conclusion
In this review, the potential natural product-drug interactions of quercetin and their pharmacokinetic and pharmacodynamic mechanisms with various drugs were summarized. The pharmacokinetic interactions of quercetin with clinical drugs were mainly related to the regulation of serum albumin, cytochrome P450, and P-glycoprotein by quercetin. In addition, organic anion transport peptides and glucuronidases are potential targets in the pharmacokinetic interactions induced by quercetin. However, most studies on quercetin-related pharmacokinetic interactions have been conducted in vitro with cellular models while a few of them with animals. The consistency of in vitro and in vivo studies needs to be further validated. Moreover, there are still many gaps in the research for translating these preclinical drug-to-drug interactions for the clinical translation of quercetin.
Data Availability
Not applicable.
References
Abdel-Raheem IT, Abdel-Ghany AA, Mohamed GA (2009) Protective effect of quercetin against gentamicin-induced nephrotoxicity in rats. Biol Pharm Bull 32:61–67. https://doi.org/10.1248/bpb.32.61
Almeida AF, Borge GIA, Piskula M, Tudose A, Tudoreanu L, Valentová K, Williamson G, Santos CN (2018) Bioavailability of quercetin in humans with a focus on interindividual variation: variability in quercetin bioavailability. Compr Rev Food Sci Food Saf 17:714–731. https://doi.org/10.1111/1541-4337.12342
Alizadeh SR, Ebrahimzadeh MA (2022) Quercetin derivatives: drug design, development, and biological activities, a review. Eur J Med Chem 229:114068. https://doi.org/10.1016/J.EJMECH.2021.114068
Ambrose AM, Robbins DJ, Deeds F (1952) Comparative toxicites of quercetin and quercitrin. J Am Pharm Assoc Am Pharm Assoc 41:119–122. https://doi.org/10.1002/jps.3030410303
Andres S, Pevny S, Ziegenhagen R, Bakhiya N, Schäfer B, Hirsch-Ernst KI, Lampen A (2018) Safety aspects of the use of quercetin as a dietary supplement. Mol Nutr Food Res 62:1700447. https://doi.org/10.1002/mnfr.201700447
Auxtero MD, Chalante S, Abade MR, Jorge R, Fernandes AI (2021) Potential herb-drug interactions in the management of age-related cognitive dysfunction. Pharmaceutics 13:124. https://doi.org/10.3390/pharmaceutics13010124
Barrenetxe J, Aranguren P, Grijalba A, Martínez-Peñuela JM, Marzo F, Urdaneta E (2006) Effect of dietary quercetin and sphingomyelin on intestinal nutrient absorption and animal growth. Br J Nutr 95:455–461. https://doi.org/10.1079/bjn20051651
Batiha GE, Beshbishy AM, Ikram M, Mulla ZS, El-Hack ME, Taha AE, Algammal AM, Elewa YH (2020) The pharmacological activity, biochemical properties, harmacokinetics of the major natural polyphenolic flavonoid: quercetin. Foods 9:374. https://doi.org/10.3390/FOODS9030374
Bedada SK, Neerati P (2018) The effect of quercetin on the pharmacokinetics of chlorzoxazone, a CYP2E1 substrate, in healthy subjects. Eur J Clin Pharmacol 74:91–97. https://doi.org/10.1007/s00228-017-2345-9
Bhutto ZA, He F, Zloh M, Yang J, Huang J, Guo T, Wang L (2018) Use of quercetin in animal feed: effects on the P-gp expression and pharmacokinetics of orally administrated enrofloxacin in chicken. Sci Rep 8. https://doi.org/10.1038/s41598-018-22354-1
Burkina V, Zamaratskaia G, Rasmussen MK (2022) Curcumin and quercetin modify warfarin-induced regulation of porcine CYP1A2 and CYP3A expression and activity in vitro. Xenobiotica 52:435–441. https://doi.org/10.1080/00498254.2022.2089932
Cao L, Kwara A, Greenblatt DJ (2017) Metabolic interactions between acetaminophen (paracetamol) and two flavonoids, luteolin and quercetin, through in-vitro inhibition studies. J Pharm Pharmacol 69:1762–1772. https://doi.org/10.1111/jphp.12812
Carullo G, Perri M, Manetti F, Aiello F, Caroleo MC, Cione E (2019) Quercetin-3-oleoyl derivatives as new GPR40 agonists: molecular docking studies and functional evaluation. Bioorg Med Chem Lett 29:1761–1764. https://doi.org/10.1016/j.bmcl.2019.05.018
Chakraborty M, Ahmed MG, Bhattacharjee A (2018) Effect of quercetin on myocardial potency of curcumin against ischemia reperfusion induced myocardial toxicity. Synergy 7:25–29. https://doi.org/10.1016/j.synres.2018.09.001
Challa VR, Babu PR, Challa SR, Johnson B, Maheswari C (2013) Pharmacokinetic interaction study between quercetin and valsartan in rats and in vitro models. Drug Dev Ind Pharm 39:865–872. https://doi.org/10.3109/03639045.2012.693502
Chaturvedi S, Malik MY, Rashid M, Singh, Sandeep, Tiwari V, Gupta P, Shukla S, Singh, Sarika, Wahajuddin M (2020) Mechanistic exploration of quercetin against metronidazole induced neurotoxicity in rats: Possible role of nitric oxide isoforms and inflammatory cytokines. Neurotoxicology 79:1–10. https://doi.org/10.1016/j.neuro.2020.03.002
Chen J, Li G, Sun C, Peng F, Yu L, Chen Y, Tan Y, Cao X, Tang Y, Xie X, Peng C (2022) Chemistry, pharmacokinetics, pharmacological activities, and toxicity of Quercitrin. Phytother Res 36:1545–1575. https://doi.org/10.1002/ptr.7397
Chen X-W, S. Serag E, B. Sneed K, Liang J, Chew H, Pan S-Y, Zhou S-F (2011) Clinical herbal interactions with conventional drugs: from molecules to maladies. Curr Med Chem 18:4836–4850. https://doi.org/10.2174/092986711797535317
Chen Y, Xiao P, Ou-Yang D-S, Fan L, Guo D, Wang Y-N, Han Y, Tu J-H, Zhou G, Huang Y-F, Zhou H-H (2009) Simultaneous action of the flavonoid quercetin on cytochrome P450 (CYP) 1A2, CYP2A6, N-acetyltransferase and xanthine oxidase activity in healthy volunteers. Clin Exp Pharmacol Physiol 36:828–833. https://doi.org/10.1111/j.1440-1681.2009.05158.x
Di Minno A, Frigerio B, Spadarella G, Ravani A, Sansaro D, Amato M, Kitzmiller JP, Pepi M, Tremoli E, Baldassarre D (2017) Old and new oral anticoagulants: food, herbal medicines and drug interactions. Blood Rev 31:193–203. https://doi.org/10.1016/j.blre.2017.02.001
Dunnick JK, Hailey JR (1992) Toxicity and carcinogenicity studies of quercetin, a natural component of foods. Fundam Appl Toxicol 19:423–431. https://doi.org/10.1016/0272-0590(92)90181-g
Elbarbry F, Ung A, Abdelkawy K (2018) Studying the inhibitory effect of quercetin and thymoquinone on human cytochrome P450 enzyme activities. Pharmacogn Mag 13:S895–S899. https://doi.org/10.4103/0973-1296.224342
Elmeliegy M, Vourvahis M, Guo C, Wang DD (2020) Effect of P-glycoprotein (P-gp) inducers on exposure of P-gp substrates: review of clinical drug-drug interaction studies. Clin Pharmacokinet 59:699–714. https://doi.org/10.1007/s40262-020-00867-1
Ezzati M, Yousefi B, Velaei K, Safa A (2020) A review on anti-cancer properties of quercetin in breast cancer. Life Sci 248:117463. https://doi.org/10.1016/j.lfs.2020.117463
Faddah LM, Abdel Baky NA, Al-Rasheed NM, Fatani AJ, Atteya M (2012) Role of quercetin and arginine in ameliorating nano zinc oxide-induced nephrotoxicity in rats. BMC Complement Altern Med 12:60. https://doi.org/10.1186/1472-6882-12-60
Farooqi AA, Jabeen S, Attar R, Yaylim I, Xu B (2018) Quercetin-mediated regulation of signal transduction cascades and microRNAs: natural weapon against cancer. J Cell Biochem 119:9664–9674. https://doi.org/10.1002/jcb.27488
Feng X, Bu F, Huang L, Xu W, Wang W, Wu Q (2022) Preclinical evidence of the effect of quercetin on diabetic nephropathy: a meta-analysis of animal studies. Eur J Pharmacol 921:174868. https://doi.org/10.1016/j.ejphar.2022.174868
Gerber W, Steyn JD, Kotzé AF, Hamman JH (2018) Beneficial pharmacokinetic drug interactions: a tool to improve the bioavailability of poorly permeable drugs. Pharmaceutics 10:106. https://doi.org/10.3390/pharmaceutics10030106
Gouws C, Hamman JH (2020) What are the dangers of drug interactions with herbal medicines? Expert Opin Drug Metab Toxicol 16:165–167. https://doi.org/10.1080/17425255.2020.1733969
Gupta C, Vikram A, Tripathi DN, Ramarao P, Jena GB (2010) Antioxidant and antimutagenic effect of quercetin against DEN induced hepatotoxicity in rat: effect of quercetin against DEN induced hepatotoxicity. Phytother Res 24:119–128. https://doi.org/10.1002/ptr.2883
Gupta RC, Chang D, Nammi S, Bensoussan A, Bilinski K, Roufogalis BD (2017) Interactions between antidiabetic drugs and herbs: an overview of mechanisms of action and clinical implications. Diabetol Metab Syndr 9:59. https://doi.org/10.1186/s13098-017-0254-9
Haleagrahara N, Hodgson K, Miranda-Hernandez S, Hughes S, Kulur AB, Ketheesan N (2018) Flavonoid quercetin-methotrexate combination inhibits inflammatory mediators and matrix metalloproteinase expression, providing protection to joints in collagen-induced arthritis. Inflammopharmacology 26:1219–1232. https://doi.org/10.1007/s10787-018-0464-2
Heeba GH, Mahmoud ME (2016) Dual effects of quercetin in doxorubicin-induced nephrotoxicity in rats and its modulation of the cytotoxic activity of doxorubicin on human carcinoma cells: effects of Quercetin on Dox-Induced Nephrotoxicity. Environ Toxicol 31:624–636. https://doi.org/10.1002/tox.22075
Heinz SA, Henson DA, Austin MD, Jin F, Nieman DC (2010) Quercetin supplementation and upper respiratory tract infection: a randomized community clinical trial. Pharmacol Res 62:237–242. https://doi.org/10.1016/j.phrs.2010.05.001
Henidi HA, Al-Abbasi FA, El-Moselhy MA, El-Bassossy HM, Al-Abd AM (2020) Despite blocking doxorubicin-induced vascular damage, quercetin ameliorates its antibreast cancer activity. Oxid Med Cell Longev 2020:8157640. https://doi.org/10.1155/2020/8157640
Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, Herrmann SM, Jensen MD, Jia Q, Jordan KL, Kellogg TA, Khosla S, Koerber DM, Lagnado AB, Lawson DK, LeBrasseur NK, Lerman LO, McDonald KM, McKenzie TJ, Passos JF, Pignolo RJ, Pirtskhalava T, Saadiq IM, Schaefer KK, Textor SC, Victorelli SG, Volkman TL, Xue A, Wentworth MA, Wissler Gerdes EO, Zhu Y, Tchkonia T, Kirkland JL (2019) Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 47:446–456. https://doi.org/10.1016/j.ebiom.2019.08.069
Hossain S, Yousaf M, Liu Y, Chang D, Zhou X (2022) An overview of the evidence and mechanism of drug-herb interactions between propolis and pharmaceutical drugs. Front Pharmacol 13:876183. https://doi.org/10.3389/fphar.2022.876183
Hosseini A, Razavi BM, Banach M, Hosseinzadeh H (2021) Quercetin and metabolic syndrome: a review. Phytother Res 35:5352–5364. https://doi.org/10.1002/ptr.7144
Ito N, Hagiwara A, Tamano S, Kagawa M, Shibata M, Kurata Y, Fukushima S (1989) Lack of carcinogenicity of quercetin in F344/DuCrj rats. Jpn J Cancer Res 80:317–325. https://doi.org/10.1111/j.1349-7006.1989.tb02313.x
Jain S, Jain AK, Pohekar M, Thanki K (2013) Novel self-emulsifying formulation of quercetin for improved in vivo antioxidant potential: implications for drug-induced cardiotoxicity and nephrotoxicity. Free Radic Biol Med 65:117–130. https://doi.org/10.1016/j.freeradbiomed.2013.05.041
Jana S, Rastogi H (2017) Effects of caffeic acid and quercetin on in vitro permeability, metabolism and in vivo pharmacokinetics of melatonin in rats: potential for herb-drug interaction. Eur J Drug Metab Pharmacokinet 42:781–791. https://doi.org/10.1007/s13318-016-0393-7
Janbaz KH, Saeed SA, Gilani AH (2004) Studies on the protective effects of caffeic acid and quercetin on chemical-induced hepatotoxicity in rodents. Phytomedicine 11:424–430. https://doi.org/10.1016/j.phymed.2003.05.002
Jenkinson C, Petroczi A, Naughton DP (2012) Red wine and component flavonoids inhibit UGT2B17 in vitro. Nutr J 11:67. https://doi.org/10.1186/1475-2891-11-67
Ji L, Ma Y, Wang Z, Cai Z, Pang C, Zhengtao W (2014) Quercetin prevents pyrrolizidine alkaloid clivorine-induced liver injury in mice by elevating body defense capacity. PLoS ONE 9:e98970. https://doi.org/10.1371/journal.pone.0098970
Jing J-J, Liu B, Wang X, Wang X, He L-L, Guo X-Y, Xu M-L, Li Q-Y, Gao B, Dong B-Y (2017) Binding of fluphenazine with human serum albumin in the presence of rutin and quercetin: an evaluation of food-drug interaction by spectroscopic techniques. Luminescence 32:1056–1065. https://doi.org/10.1002/bio.3291
Jurasekova Z, Domingo C, Garcia-Ramos JV, Sanchez-Cortes S (2014) Effect of pH on the chemical modification of quercetin and structurally related flavonoids characterized by optical (UV-visible and Raman) spectroscopy. Phys Chem Chem Phys 16:12802–12811. https://doi.org/10.1039/c4cp00864b
Kalliokoski A, Niemi M (2009) Impact of OATP transporters on pharmacokinetics: OATP transporters and pharmacokinetics. Br J Pharmacol 158:693–705. https://doi.org/10.1111/j.1476-5381.2009.00430.x
Kameníková M, Furtmüller PG, Klacsová M, Lopez-Guzman A, Toca-Herrera JL, Vitkovská A, Devínsky F, Mučaji P, Nagy M (2017) Influence of quercetin on the interaction of gliclazide with human serum albumin – spectroscopic and docking approaches. Luminescence 32:1203–1211. https://doi.org/10.1002/bio.3312
Kashyap D, Garg VK, Tuli HS, Yerer MB, Sak K, Sharma AK, Kumar M, Aggarwal V, Sandhu SS (2019) Fisetin and quercetin: promising flavonoids with chemopreventive potential. Biomolecules 9:174. https://doi.org/10.3390/biom9050174
Kim K-A, Park P-W, Kim H-K, Ha J-M, Park J-Y (2005) Effect of quercetin on the pharmacokinetics of rosiglitazone, a CYP2C8 substrate, in healthy subjects. J Clin Pharmacol 45:941–946. https://doi.org/10.1177/0091270005278407
Lan K, He J-L, Tian Y, Tan F, Jiang X-H, Wang L, Ye L-M (2008) Intra-herb pharmacokinetics interaction between quercetin and isorhamentin. Acta Pharmacol Sin 29:1376–1382. https://doi.org/10.1111/j.1745-7254.2008.00884.x
Latos-Brozio M, Masek A (2019) Structure-activity relationships analysis of monomeric and polymeric polyphenols (quercetin, rutin and catechin) obtained by various polymerization methods. Chem Biodivers 16:e1900426. https://doi.org/10.1002/cbdv.201900426
Li H, Li M, Fu J, Ao H, Wang W, Wang X (2021) Enhancement of oral bioavailability of quercetin by metabolic inhibitory nanosuspensions compared to conventional nanosuspensions. Drug Deliv 28:1226–1236. https://doi.org/10.1080/10717544.2021.1927244
Li X, Wang C, Chen J, Hu X, Zhang H, Li Z, Lan B, Zhang W, Su Y, Zhang C (2022) Potential interactions among myricetin and dietary flavonols through the inhibition of human UDP-glucuronosyltransferase in vitro. Toxicol Lett 358:40–47. https://doi.org/10.1016/j.toxlet.2022.01.007
Liu S, Li R, Qian J, Sun J, Li G, Shen J, Xie Y (2020) Combination therapy of doxorubicin and quercetin on multidrug-resistant breast cancer and their sequential delivery by reduction-sensitive hyaluronic acid-based conjugate/d-α-tocopheryl poly(ethylene glycol) 1000 succinate mixed micelles. Mol Pharm 17:1415–1427. https://doi.org/10.1021/acs.molpharmaceut.0c00138
Lukačínová A, Mojžiš J, Beňačka R, Keller J, Maguth T, Kurila P, Vaško L, Rácz O, Ništiar F (2008) Preventive effects of flavonoids on alloxan-induced diabetes mellitus in rats. Acta Vet Brno 77:175–182. https://doi.org/10.2754/avb200877020175
Maleki DP, Sadoughi F, Asemi Z, Yousefi B (2021) Anti-cancer properties of quercetin in osteosarcoma. Cancer Cell Int 21:349. https://doi.org/10.1186/s12935-021-02067-8
Maleki Dana P, Sadoughi F, Asemi Z, Yousefi B (2021) Anti-cancer properties of quercetin in osteosarcoma. Cancer Cell Int 21:349. https://doi.org/10.1186/s12935-021-02067-8
Mandery K, Bujok K, Schmidt I, Keiser M, Siegmund W, Balk B, König J, Fromm MF, Glaeser H (2010) Influence of the flavonoids apigenin, kaempferol, and quercetin on the function of organic anion transporting polypeptides 1A2 and 2B1. Biochem Pharmacol 80:1746–1753. https://doi.org/10.1016/j.bcp.2010.08.008
Morales AI, Vicente-Sánchez C, Sandoval JMS, Egido J, Mayoral P, Arévalo MA, Fernández-Tagarro M, López-Novoa JM, Pérez-Barriocanal F (2006) Protective effect of quercetin on experimental chronic cadmium nephrotoxicity in rats is based on its antioxidant properties. Food Chem Toxicol 44:2092–2100. https://doi.org/10.1016/j.fct.2006.07.012
National Toxicology Program (1992) Toxicology and carcinogenesis studies of quercetin (CAS no. 117–39-5) in F344 rats (feed studies). Natl Toxicol Program Tech Rep Ser 409:1–171
Navarro VJ, Khan I, Björnsson E, Seeff LB, Serrano J, Hoofnagle JH (2017) Liver injury from herbal and dietary supplements. Hepatology 65:363–373. https://doi.org/10.1002/hep.28813
Nguyen MA, Staubach P, Wolffram S, Langguth P (2015) The influence of single-dose and short-term administration of quercetin on the pharmacokinetics of midazolam in humans. J Pharm Sci 104:3199–3207. https://doi.org/10.1002/jps.24500
Nguyen TLA, Bhattacharya D (2022) Antimicrobial activity of quercetin: an approach to its mechanistic principle. Molecules 27:2494. https://doi.org/10.3390/molecules27082494
Ożarowski M, Mikołajczak PŁ, Kujawski R, Wielgus K, Klejewski A, Wolski H, Seremak-Mrozikiewicz (2018) Pharmacological effect of quercetin in hypertension and its potential application in pregnancy-induced hypertension: review of in vitro, in vivo, and clinical studies. Evid Based Complement Alternat Med 2018:7421489. https://doi.org/10.1155/2018/7421489
Pal A, Tripathi A (2019) Quercetin potentiates meropenem activity among pathogenic carbapenem-resistant Pseudomonas aeruginosa and Acinetobacter baumannii. J Appl Microbiol 127:1038–1047. https://doi.org/10.1111/jam.14388
Patel G, Thakur NS, Kushwah V, Patil MD, Nile SH, Jain S, Kai G, Banerjee UC (2020) Mycophenolate co-administration with quercetin via lipid-polymer hybrid nanoparticles for enhanced breast cancer management. Nanomedicine 24:102147. https://doi.org/10.1016/j.nano.2019.102147
Patel RV, Mistry BM, Shinde SK, Syed R, Singh V, Shin H-S (2018) Therapeutic potential of quercetin as a cardiovascular agent. Eur J Med Chem 155:889–904. https://doi.org/10.1016/j.ejmech.2018.06.053
Pradniwat P, Chanprasert S (2022) Properties, pharmacological activities and toxicities of quercetin. Thai J Toxicol 37:1–19
Rabbani G, Ahn SN (2019) Structure, enzymatic activities, glycation and therapeutic potential of human serum albumin: a natural cargo. Int J Biol Macromol 123:979–990. https://doi.org/10.1016/J.IJBIOMAC.2018.11.053
Rangan GK, Wang Y, Harris DCH (2002) Dietary quercetin augments activator protein-1 and does not reduce nuclear factor-kappa B in the renal cortex of rats with established chronic glomerular disease. Nephron 90:313–319. https://doi.org/10.1159/000049067
Reyes-Farias M, Carrasco-Pozo C (2019) The anti-cancer effect of quercetin: molecular implications in cancer metabolism. Int J Mol Sci 20:3177. https://doi.org/10.3390/ijms20133177
Rowland A, Miners JO, Mackenzie PI (2013) The UDP-glucuronosyltransferases: their role in drug metabolism and detoxification. Int J Biochem Cell Biol 45:1121–1132. https://doi.org/10.1016/j.biocel.2013.02.019
Ruiz MJ, Fernández M, Estela JM, Asensi MÁ, Mañes J, Picó Y (2006) Short-term oral toxicity of quercetin and pterostibene in Swiss mice. Toxicol Lett 164:S275–S276. https://doi.org/10.1016/j.toxlet.2006.07.232
Samala S, Research CV, Of -J (2015) Altered pharmacokinetics and pharmacodynamics of glimepiride by the concomitant use of quercetin in diabetic rats: PK/PD modeling
Shabbir U, Rubab M, Daliri EB-M, Chelliah R, Javed A, Oh D-H (2021) Curcumin, quercetin, catechins and metabolic diseases: the role of gut Microbiota. Nutrients 13:206. https://doi.org/10.3390/nu13010206
Shen P, Lin W, Deng X, Ba X, Han L, Chen Z, Qin K, Huang Y, Tu S (2021) Potential implications of quercetin in autoimmune diseases. Front Immunol 12:689044. https://doi.org/10.3389/fimmu.2021.689044
Shi T, Bian X, Yao Z et al (2020) Quercetin improves gut dysbiosis in antibiotic-treated mice. Food Funct 11:8003–8013. https://doi.org/10.1039/d0fo01439g
Shin S-C, Choi J-S, Li X (2006) Enhanced bioavailability of tamoxifen after oral administration of tamoxifen with quercetin in rats. Int J Pharm 313:144–149. https://doi.org/10.1016/j.ijpharm.2006.01.028
Singh DP, Borse SP, Nivsarkar M (2017) Overcoming the exacerbating effects of ranitidine on NSAID-induced small intestinal toxicity with quercetin: providing a complete GI solution. Chem Biol Interact 272:53–64. https://doi.org/10.1016/j.cbi.2017.04.006
Skalli S, Bencheikh RS (2012) Safety monitoring of herb-drug interactions: a component of pharmacovigilance. Drug Saf 35:785–791. https://doi.org/10.1007/bf03261975
Sobhani M, Farzaei MH, Kiani S, Khodarahmi R (2021) Immunomodulatory; anti-inflammatory/antioxidant effects of polyphenols: a comparative review on the parental compounds and their metabolites. Food Rev Int 37:759–811. https://doi.org/10.1080/87559129.2020.1717523
Srividya G, Deepthi B, Lakshminarasaiah S, Srinivasarao G (2018) Ex vivo studies on pharmacodynamic interaction of phytochemicals with antibiotics against clinical isolates from mastitic milk samples.
Tang S-M, Deng X-T, Zhou J, Li QP, Ge XX, Miao L (2020) Pharmacological basis and new insights of quercetin action in respect to its anti-cancer effects. Biomed Pharmacother 121:109604. https://doi.org/10.1016/j.biopha.2019.109604
Thangavel, S., Chenniappan, S (2021) Pharmacodynamics interaction of quercetin and captopril on doxorubicin induced myocardial toxic rats. Biointerface Res Appl Chem 12, 3002–3011. https://doi.org/10.33263/briac123.30023011
Tiwari H, Karki N, Pal M, Basak S, Verma RK, Bal R, Kandpal ND, Bisht G, Sahoo NG (2019) Functionalized graphene oxide as a nanocarrier for dual drug delivery applications: the synergistic effect of quercetin and gefitinib against ovarian cancer cells. Colloids Surf B Biointerfaces 178:452–459. https://doi.org/10.1016/j.colsurfb.2019.03.037
Vafadar A, Shabaninejad Z, Movahedpour A, Fallahi F, Taghavipour M, Ghasemi Y, Akbari M, Shafiee A, Hajighadimi S, Moradizarmehri S, Razi E, Savardashtaki A, Mirzaei H (2020) Quercetin and cancer: new insights into its therapeutic effects on ovarian cancer cells. Cell Biosci 10:32. https://doi.org/10.1186/s13578-020-00397-0
van de Kerkhof EG, de Graaf IAM, Ungell A-LB, Groothuis GMM (2008) Induction of metabolism and transport in human intestine: validation of precision-cut slices as a tool to study induction of drug metabolism in human intestine in vitro. Drug Metab Dispos 36:604–613. https://doi.org/10.1124/dmd.107.018820
Ventura-Martínez R, Déciga-Campos M, Bustamante-Marquina A, Ángeles-López GE, Aviles-Herrera J, González-Trujano ME, Navarrete-Vázquez G (2021) Quercetin decreases the antinociceptive effect of diclofenac in an arthritic gout-pain model in rats. J Pharm Pharmacol 73:1310–1318. https://doi.org/10.1093/jpp/rgab093
Vicente-Sánchez C, Egido J, Sánchez-González PD, Pérez-Barriocanal F, López-Novoa JM, Morales AI (2008) Effect of the flavonoid quercetin on cadmium-induced hepatotoxicity. Food Chem Toxicol 46:2279–2287. https://doi.org/10.1016/j.fct.2008.03.009
Vrolijk MF, Haenen GRMM, Opperhuizen A, Jansen EHJM, Schiffers PM, Bast A (2015) The supplement-drug interaction of quercetin with tamsulosin on vasorelaxation. Eur J Pharmacol 746:132–137. https://doi.org/10.1016/j.ejphar.2014.11.006
Wang J, Miao M, Zhang Y, Liu R, Li X, Cui Y, Qu L (2015) Quercetin ameliorates liver injury induced with Tripterygium glycosides by reducing oxidative stress and inflammation. Can J Physiol Pharmacol 93:427–433. https://doi.org/10.1139/cjpp-2015-0038
Wang W, Sun C, Mao L, Ma P, Liu F, Yang J, Gao Y (2016) The biological activities, chemical stability, metabolism and delivery systems of quercetin: a review. Trends Food Sci Technol 56:21–38. https://doi.org/10.1016/j.tifs.2016.07.004
Wang Y, Yu H, Wang S, Gai C, Cui X, Xu Z, Li W, Zhang W (2021) Targeted delivery of quercetin by nanoparticles based on chitosan sensitizing paclitaxel-resistant lung cancer cells to paclitaxel. Mater Sci Eng C Mater Biol Appl 119:111442. https://doi.org/10.1016/j.msec.2020.111442
Wang Y-H, Chao P-DL, Hsiu SL, Wen K-C, Hou Y-C (2004) Lethal quercetin-digoxin interaction in pigs. Life Sci 74:1191–1197. https://doi.org/10.1016/j.lfs.2003.06.044
Wani TA, Alanazi MM, Alsaif NA, Bakheit AH, Zargar S, Alsalami OM, Khan AA (2022) Interaction characterization of a tyrosine kinase inhibitor erlotinib with a model transport protein in the presence of quercetin: a drug-protein and drug-drug interaction investigation using multi-spectroscopic and computational approaches. Molecules 27:1265. https://doi.org/10.3390/molecules27041265
Wani TA, Bakheit AH, Zargar S, Alanazi ZS, Al-Majed AA (2021) Influence of antioxidant flavonoids quercetin and rutin on the in-vitro binding of neratinib to human serum albumin. Spectrochim Acta A Mol Biomol Spectrosc 246:118977. https://doi.org/10.1016/j.saa.2020.118977
Wu L-X, Guo C-X, Chen W-Q, Yu J, Qu Q, Chen Y, Tan Z-R, Wang G, Fan L, Li Q, Zhang W, Zhou H-H (2012) Inhibition of the organic anion-transporting polypeptide 1B1 by quercetin: an in vitro and in vivo assessment: effect of quercetin on the pharmacokinetics of the OATP1B1 substrate, pravastatin. Br J Clin Pharmacol 73:750–757. https://doi.org/10.1111/j.1365-2125.2011.04150.x
Xu W, Xie S, Chen X, Pan S, Qian H, Zhu X (2021) Effects of quercetin on the efficacy of various chemotherapeutic drugs in cervical cancer cells. Drug Des Devel Ther 15:577–588. https://doi.org/10.2147/DDDT.S291865
Yousuf M, Khan P, Shamsi A, Shahbaaz M, Hasan GM, Haque QMR, Christoffels A, Islam A, Hassan MI (2020) Inhibiting CDK6 activity by quercetin is an attractive strategy for cancer therapy. ACS Omega 5:27480–27491. https://doi.org/10.1021/acsomega.0c03975
Yuksel Y, Yuksel R, Yagmurca M, Haltas H, Erdamar H, Toktas M, Ozcan O (2016) Effects of quercetin on methotrexate-induced nephrotoxicity in rats. Hum Exp Toxicol 36:51–61. https://doi.org/10.1177/0960327116637414
Zargar S, Alamery S, Bakheit AH, Wani TA (2020) Poziotinib and bovine serum albumin binding characterization and influence of quercetin, rutin, naringenin and sinapic acid on their binding interaction. Spectrochim Acta A Mol Biomol Spectrosc 235:118335. https://doi.org/10.1016/j.saa.2020.118335
Zenkevich IG, Guschina SV (2010) Determination of dissociation constants of species oxidizable in aqueous solution by air oxygen on an example of quercetin. J Anal Chem 65:371–375. https://doi.org/10.1134/s1061934810040064
Zhang R, Wei Y, Yang T et al (2021) Inhibitory effects of quercetin and its major metabolite quercetin-3-O-β-D-glucoside on human UDP-glucuronosyltransferase 1A isoforms by liquid chromatography-tandem mass spectrometry. Exp Ther Med 22:842. https://doi.org/10.3892/etm.2021.10274
Zheng R, Tao L, Kwong JSW, Zhong C, Li C, Chen S, Sun Y, Zhang X, Shang H (2020) Risk factors associated with the severity of adverse drug reactions by Xiyanping injection: a propensity score-matched analysis. J Ethnopharmacol 250:112424. https://doi.org/10.1016/j.jep.2019.112424
Zhou X, Fu L, Wang P, Yang L, Zhu X, Li CG (2021) Drug-herb interactions between Scutellaria baicalensis and pharmaceutical drugs: insights from experimental studies, mechanistic actions to clinical applications. Biomed Pharmacother 138:111445. https://doi.org/10.1016/j.biopha.2021.111445
Zhu J, Seo J-E, Wang S, Ashby K, Ballard R, Yu D, Ning B, Agarwal R, Borlak J, Tong W, Chen M (2018) The development of a database for herbal and dietary supplement induced liver toxicity. Int J Mol Sci 19:2955. https://doi.org/10.3390/ijms19102955
Acknowledgements
The authors would like to thank Professor Xueping Li of Department of Geriatrics, Hospital of Chengdu University of Traditional Chinese Medicine, for critical reviewing of the manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed equally to this paper. KD and HJdetermined the topic of this study; K D, WJ, and YQ collected the data; KD, HJ, and WJ wrote the main content of the manuscript; WJ and YQ drew all the pictures; YW developed the tables; ML was responsible for all revisions of the full text. All authors reviewed the final version of this paper and agreed to publish it.
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ding, K., Jia, H., Jiang, W. et al. A Double-Edged Sword: Focusing on Potential Drug-to-Drug Interactions of Quercetin. Rev. Bras. Farmacogn. 33, 502–513 (2023). https://doi.org/10.1007/s43450-022-00347-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43450-022-00347-6