
Abstract
Our previous studies have reported that a putative trypsin-like serine protease, PRSS37, is exclusively expressed in testicular germ cells during late spermatogenesis and essential for sperm migration from the uterus into the oviduct and sperm-egg recognition via mediating the interaction between PDILT and ADAM3. In the present study, the global proteome profiles of wild-type (wt) and Prss37−/− mice in testis and sperm were compared employing data independent acquisition (DIA) technology. Overall, 2506 and 459 differentially expressed proteins (DEPs) were identified in Prss37-null testis and sperm, respectively, when compared to control groups. Bioinformatic analyses revealed that most of DEPs were related to energy metabolism. Of note, the DEPs associated with pathways for the catabolism such as glucose via glycolysis, fatty acids via β-oxidation, and amino acids via oxidative deamination were significantly down-regulated. Meanwhile, the DEPs involved in the tricarboxylic acid cycle (TCA cycle) and oxidative phosphorylation (OXPHOS) were remarkably decreased. The DIA data were further confirmed by a markedly reduction of intermediate metabolites (citrate and fumarate) in TCA cycle and terminal metabolite (ATP) in OXPHOS system after disruption of PRSS37. These outcomes not only provide a more comprehensive understanding of the male fertility of energy metabolism modulated by PRSS37 but also furnish a dynamic proteomic resource for further reproductive biology studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In mammals, spermiogenesis and sperm maturation are completed in the testis and epididymis, respectively, and freshly ejaculated sperm must gain their fertilizing ability after spending some time in the female genital tract, a process called capacitation, and finally sperm-egg fusion [1, 2]. Distinctly, the interruption of any link will result in an inability to fertilize properly. During the past two decades, a growing body of genes have been identified to be participated in the occurrence of these key processes [3, 4]. Especially sperm maturation during epididymal transit, it is well-known that not only posttranslational modification (PTM) of proteins synthesized in spermatogenesis but the incorporation of new factors from the epididymal epithelium are required as well [5]. It is estimated that more than 2300 expressed genes are germ cell specific within the male reproductive tract, among which many proteases such as cysteine proteases, aspartic acid proteases, serine proteases, metalloproteases and ubiquitin proteasome are known to be essential for male fertility [6,7,8,9].
Serine proteases constitute nearly one-third of all proteases [10], and a subfamily of serine proteases, trypsin-like serine proteases, can be distinguished from others due to the presence of a nucleophilic catalytic serine residue at the active site, as a result, it stands out [11]. The role of this active site is to selectively hydrolyze peptide bonds in C-terminal to basic amino acid residues for the purposes of PTMs of polypeptides [12]. It has been displayed that several trypsin-like serine proteases are expressed predominantly and specifically in testis and play crucial roles during spermatogenesis, sperm maturation, sperm migration and sperm-egg binding [13]. Antibody blocked assays indicated that PRSS41 is required for the progression of each stage of mitosis from spermatogonia to primary spermatocytes [14], while both PRSS43 and PRSS42 are essential for the progression of meiosis, among which PRSS43 is necessary for the primary spermatocytes to differentiate into secondary spermatocytes, and PRSS42 is crucial for the transformation from secondary spermatocytes to spermatids [15]. Moreover, a mutant mouse model of another serine protease, PRSS21, exhibited a subfertile phenotype owing to decreased sperm motility, morphological malformation, and susceptibility to decapitation as they transit through the epididymis. Because the proteolytic capability and membrane localization of PRSS21, it is hypothesized that this protease is participated in proteolytic cleavage of substrates indispensable for the sperm maturation process [16]. Nevertheless, our previous studies and more recent works by two other groups have reported that mice deficient for the serine protease PRSS37, PRSS55, PRSS59 (also known as TRYX5) or TMPRSS12 did not affect the spermatogenesis and sperm maturation, but gene-deletion sperm failed to migrate from the uterus into the oviduct and displayed a limited capability of sperm-egg recognition, and was accompanied by the disappearance of mature ADAM3 on sperm surface, leading to a sterile phenotype [5, 17,18,19]. Intriguingly, all of them share similar male infertility phenotypes as those shown by ADAM3 deficiency [20, 21]. Therefore, it is reasonable to speculate that above mentioned serine proteases (PRSS37, PRSS55, PRSS59 and TMPRSS12) are involved in the PTMs of ADAM3, and that the correct positioning of mature ADAM3 on sperm surface is essential for sperm migration through utero-tubal junction (UTJ) and sperm-egg interaction. Particularly, the potential molecular mechanism of PRSS37 in regulating ADAM3 maturation was further revealed by identification of PRSS37 interactome [22]. We have illustrated that PRSS37 specifically modulates the interaction of PDILT and ADAM3, which is a prerequisite for the disulfide-bond formation (a form of PTMs) and folding of ADAM3 [23], and finally it facilitates the transport of ADAM3 from the endoplasmic reticulum (ER) to the germ cell membrane surface [22]. In silico analysis predicted that PRSS59 was presented in the Golgi apparatus, indicating that this protein might participate in the formation of disulfide-bond, which can contribute to protein folding and stability. As a result, PRSS59 may involve in the PTMs for ADAM3, and loss of PRSS59 may bring ADAM3 precursor into an unstable state owing to lacking some PTMs [19]. Serine proteases PRSS55 acts as a glycosylphosphatidylinositol (GPI)-anchored membrane protein [18] whereas TMPRSS12 acts as a transmembrane protein [5], which are both specifically confined to the surface of germ cells, suggesting that they are primarily associated with the shearing of ADAM3 from an uncleaved precursor form to a functional mature form rather than folding, stability and translocation.
In addition to regulating ADAM3 protein maturation, TMPRSS12 also reported to be impacted in sperm motility to participate in UTJ migration [5], and PRSS55 played a significant role in sperm ATP synthesis, which contributes to male mouse fertility [24]. In the present study, we constructed and compared dynamic network profiles of testicular germ cells (TGCs) and sperm in wild-type (wt) and Prss37−/− mice using a data independent acquisition (DIA)-based quantitative proteomics strategy, and explored the signaling pathways involving proteins associated with energy metabolism of male reproduction. This study provides a systematic approach to thorough comprehension the potential molecular mechanisms of male fertility, and these findings also shed new lights into signaling modulated systems of relative proteins in mammals, and offer the important data for further studies on biology of reproduction.
Materials and methods
Mice
The C57BL/6 mouse strains were used in this study. Prss37 knockout (KO) mice have been described previously [17]. The wt and Prss37 KO mice were all housed under the specific pathogen-free environments at a constant room temperature of 22–24 °C with a 12-h (hrs) light/dark cycle, with free access to a diet of regular chow and water. All research protocols involving animal experiments were approved by the Institutional Animal Care and Use Committee of Shanghai Research Center for Model Organism.
Microarray analysis
Total RNAs were extracted from three testes of wt and Prss37 KO mice (6–8 weeks old), purified using RNeasy mini kit (Qiagen, GmBH, Germany) following the manufacturer’s instructions, and checked for an RNA integrity number to inspect RNA integration by an Agilent Bioanalyzer 2100 (Agilent technologies, Santa Clara, CA, USA). The sample labelling, hybridization, staining and scanning procedures were carried out at the National Engineering Research Center of Shanghai employing the whole mouse genome oligo microarray (4 × 44 k, Agilent). The data were normalized by quantile algorithm, Gene-Spring software GX 12.6.1 (Agilent). The differentially expressed genes (DEGs) were determined based on Student’s t-test (P < 0.05) and fold change (FC > 2). All DEGs were clustered by Hierarchical Clustering method. GO terms for each DEGs were obtained by using KOBAS 3.0 (http://kobas.cbi.pku.edu.cn). The calculated P value goes through Bonferroni correction, taking corrected P value < 0.05 as a threshold, and the GO terms fulfilling this condition are defined as significantly enriched GO terms.
qRT-PCR
Total RNAs were isolated from testes of wt and Prss37−/− mice (6–8 weeks old) employing a Trizol Plus RNA Purification Kit (Invitrogen, Carlsbad, CA, USA) and were reverse transcribed to cDNAs using 5 × PrimeScript RT Master Mix (Takara, Dalian, China) following the manufacturer’s instructions. cDNAs were amplified by using specific sets of primers as listed in Table S1 for qRT-PCR, which performed with a Mastercycler ep realplex thermal cycler (Eppendorf, Hamburg, Germany) via using a SYBR Premix Ex Taq Kit (Vazyme, Nanjing, China). The resolution of the product of interest from nonspecific product amplification was achieved by melt curve analysis. Relative gene expression was normalized to β-actin using the 2−ΔCt method [25]. Three biological replications (Each biological replication consists of three technical replications) were used to measure mRNA levels by qRT-PCR.
Preparation of testicular single cell suspension and percoll density gradient centrifugation
Bilateral testes from adult age-matched mice with different genotypes were dissected and placed in a 10 cm dish containing 2 ml GBSS buffer (1.5 mM CaCl2, 5 mM KCl, 0.22 mM KH2PO4, 1 mM MgCl2.6H2O, 0.285 mM MgSO4.7H2O, 137 mM NaCl, 2.7 mM NaHCO3, 0.85 mM Na2HPO4 and 5.5 mM glucose). The white membrane on the testis was removed, and the spermatogenic tubule was dissociated and transferred to 10 ml preheated enzymatic solution I, followed by incubation for 15–20 min at 37 °C water bath with oscillates multiple times. Then, the 10 ml spermatogenic tubule sample was added to the top of the 40 ml 5% percoll solution for free sedimentation for 5–10 min, and leydig cells were located in the 45 ml solution on the top. Discarding the upper liquids (about 45 ml leydig cells), transferred the spermatogenic tubule sample (about 5 ml) from the bottom to the top of 10 ml 5% percoll solution, settled freely for 5–10 min, discarded the supernatant, poured in 10 ml enzymatic solution II and incubated in 37 °C water bath for 15–20 min with several oscillations. Next the testicular single cell suspension was dispersed by gentle blowing with a 1 ml pipette for about 30 times, added 2 ml fetal bovine serum (FBS) to terminate digestion and filtered into a 50 ml centrifuge tube with a 70 μm cell filter. Finally, the spermatogenic cells could be harvested by centrifugation at 1000 rpm for 5 min at 4 °C, and then resuspended into the 5 ml enzymatic solution III. Testicular single cell suspension has thus been obtained. The descriptions for enzymatic solution I, II, III were shown in Table S2.
The testicular single cell suspension was centrifuged at 500 × g for 10 min at 4 °C, discarded the supernatant, resuspended in 2 ml 21% percoll solution, and slowly added to the top of the 12 ml 26%-52% percoll gradient solution, and next centrifuged at 10,000 rpm for 15 min at 4 °C without interruption. After centrifugation, the cells were divided into three layers from top to bottom, namely including layer 1 (about 1 ml), layer 2 (about 3 ml) and layer 3 (about 10 ml). The above three layers of cells were respectively transferred to a 50 ml centrifuge tube, added GBSS solution to 20 ml, centrifuged at 500 × g for 10 min at 4 °C, discarded the percoll supernatant and subsequent resuspended layer 1, layer 2 and layer 3 cells with 1 ml, 3 ml and 500 μl enzymatic solution III, respectively. The acquired cells were then employed for flow cytometry, propidium iodide staining, immunofluorescence, immunoblotting and mass spectrometry analyses. The formulas for different concentrations of percoll solutions were also illustrated in Table S2.
Flow cytometry analysis DNA types and propidium iodide (PI) nuclear staining
100 μl of layer 1, layer 2 and layer 3 cells were respectively placed at the bottom of a 15 ml centrifuge tube. The cells were oscillated on the vortex while added 7 ml precooled 75% alcohol, and then fixed at 4 °C overnight. On the next day, the cells were centrifuged at 2,000 rpm for 10 min at 4 °C, discarded supernatant, washed with 7 ml 1 × PBS for three times. After the third cleaning, left about 500 μl 1 × PBS, added 0.5 μl PI (50 μg/μl) and 0.5 μl RNase A (100 μg/μl) and put it on ice for more than 30 min. Then, the DNA types were analyzed by flow cytometry (BD FACS Celesta, Becton, Dickinson and Company, USA) and the nuclear morphology was observed in a fluorescence microscope (Nikon Eclipse 80i, Nikon, Tokyo, Japan).
Immunofluorescence
Three layers of the testicular cells from wt mice were obtained employing percoll density gradient centrifugation as described above. Cell smears were acquired via spotting different layers of cells in the testis onto glass slides, which were then air dried for 20 min and fixed in 4% paraformaldehyde (PFA) for 15 min. The samples were washed with 1 × PBS, blocked in 10% normal goat serum and incubated with primary antibody anti-PRSS37 (Item No. HPA020541, Sigma-Aldrich, 1:500 dilution) overnight at 4 °C, followed by incubation for 1 h at room temperature with Alexa Fluor-conjugated goat-anti-rabbit IgG secondary antibody (Invitrogen), then co-labeled with Alexa Fluor 488-conjugated PNA (Invitrogen) and DAPI (Invitrogen). Glass slides were next mounted with fluorescence mounting medium (DAKO, Glostrup, Denmark), coverslipped and examined under a fluorescence microscope (Nikon Eclipse 80i, Nikon, Tokyo, Japan).
Immunoblotting
Proteins from three layers of the testicular cells of adult age-matched mice with different genotypes were extracted in RIPA lysis buffer supplemented with protease and phosphatase inhibitor cocktails (Roche) for 30 min on ice. After centrifugation at 16,000 × g for 10 min, the supernatant was harvested and the protein concentration was determined using the Bradford method. Proteins were then separated by SDS-PAGE under reducing conditions and transferred onto nitrocellulose membranes. After blocking, blots were incubated with primary antibodies overnight at 4 °C. Antibodies used were a rabbit polyclonal antibody raised against PRSS37 (code HPA020541, Sigma-Aldrich, 1:1000 dilution) and a rabbit monoclonal antibody against GAPDH (code 5174, Cell Signaling Technology, 1:1000 dilution). To visualize specific protein bands, the secondary antibodies conjugated with IRdye800CW (LI-COR, Lincoln, USA) were employed and membranes were scanned by Odyssey Infrared Imager (LI-COR). GAPDH was used as a protein loading control. This experiment was repeated four times and the bands of a representative experiment are shown.
Preparation of mature sperm from caudal epididymis
The mature sperm samples were collected as described previously [17, 18] with minor modifications. Briefly, wt and Prss37−/− male mice (8-weeks old) were sacrificed by cervical dislocation and sperm cells from the cauda epididymis were isolated and dispersed in 500 μl of TYH medium. After incubation for 30 min at 37 °C with 5% CO2 in air, the sperm were acquired by centrifugation at 2,000 × g for 10 min at room temperature to discard supernatants. Next, the samples were washed with 500 μl of 1 × PBS for three times in order to avoid blood cell contamination. Finally, sperm precipitates were quickly frozen in liquid nitrogen and then stored at -80 °C until machine testing and ATP detection.
DIA analysis
Testicular layer 3 cells and sperm from wt and Prss37-deficient mice were treated with the same method for DIA analysis. Each bioassay was replicated three times. The all samples were denatured by 2% SDS buffer containing 50 mM dithiothreitol (DTT) for 20 min at room temperature and boiled at 100℃ for 5 min. The proteins were alkylated for 1 h at room temperature in the dark by addition of a final concentration of 200 mM iodoacetamide (IAA). Subsequently, the five times volume of pre-cooled acetone was added, and the proteins were precipitated overnight at -20℃. The precipitates were resolved and digested by sequencing grade modified trypsin (Promega) at a protein-to-enzyme of 50:1 at 37℃ overnight. The resulting peptides were collected by centrifugation at 14,000 × g for 20 min at 20 °C, and then treated with 1% trifluoroacetic acid (TFA), and purified using the C18 Ziptips, eluted with 0.1% TFA in 50% ~ 70% acetonitrile. The eluted peptides were lyophilized by a SpeedVac (Thermo Savant), and then resuspended in 1% formic acid and 5% acetonitrile. The iRT peptides (Biognosys, Schlieren, Switzerland) were added into the samples prior to analysis according to manufacturer’s instructions.
The pooled digests were further fractionated using high pH reversed phase chromatography for building each spectral library. Briefly, the pooled digests were lyophilized employing a SpeedVac (Thermo Savant), and resuspended in 5% acetonitrile in 0.05 M ammonium formate, and fractionated using high pH reversed phase separation on a Dionex UHPLC (Thermo Scientific, Waltham, Massachusetts, USA) with a 2.1 × 150 mm Ethylene Bridged Hybrid (EBH) C18 column (3 μm) at 40 °C with 0.2 ml/min flow and a 60 min acetonitrile gradient in 5 mM ammonium formate (pH 10). Fractions were collected at 1 min intervals and pooled at various intervals to 12 fractions. Then the samples were dried and resuspended in 1% formic acid and 5% acetonitrile.
The data dependent acquisition (DDA) is used to generate spectral library. DDA was performed on an Orbitrap Fusion LUMOS mass spectrometer (Thermo Fisher Scientific) connected to an Easy-nLC 1200 through an Easy Spray (Thermo Fisher Scientific). The peptide mixture was loaded onto a self-packed analytical PicoFrit column with integrated spray tip (New Objective, Woburn, MA, USA) packed with ReproSil-Pur 120A C18-AQ 1.9 μm (Dr. Maisch GmbH, Ammerbuch, Germany) and separated within a 2 h linear gradient from 95% solvent A (0.1% formic acid, 2% acetonitrile) to 28% solvent B (0.1% formic acid, 80% acetonitrile) at a flow rate of 250 nL/min at 50 °C. The mass spectrometer were operated in positive ion mode and employed in the data-dependent mode within the specialized cycle time (3 s) to automatically switch between MS and MS/MS. One full MS scan from 350 to 1500 m/z was acquired at high resolution of 120,000; MS/MS scans were performed at a resolution of 30,000 with an isolation window of 4 Da. Dynamic exclusion was set to 30 s.
All the raw data were analysed using the Pulsar search engine (only available in Spectronaut Pulsar). The DDA raw data were searched against the mouse Uniprot fasta database within the default parameters. In brief, the digestion enzyme was allowed for specific trypsin, max missed cleavages was set 2, carbamidomethyl of cysteine was used as a fixed modification, and oxidation of methionine was used as variable modifications. iRTs were calculated derived from median iRTs across all DDA runs. Fragment ions for the targeted data analysis were selected from 300–1800 m/z, minimal relative intensity was set to > 5%, and fragment ion number > 3. False discovery rate (FDR) was set to 1% for protein and peptide spectrum matches. Protein inference was performed employing the ID Picker algorithm integrated within the spectronaut software.
To best resolve the DIA data based on the spectral library of the DDA data, the DIA MS/MS acquisition should perform with the same LC–MS systems and LC linear gradient method as DDA. For MS/MS acquisition, DIA was set 50 variable isolation windows according to the FWTH (Full width at half maximum), the specific window lists were constructed based on the respective DDA data of the pooled sample. The full scan was set at a resolution of 1,200,000 over range of 350 to 1500 m/z and DIA scans was set at a resolution of 30,000; collision energy was set 30%; AGC target was set 1e6; maximal injection time was set 54 ms.
The DIA raw results were analyzed in Spectronaut X (Biognosys, Schlieren, Switzerland). The default settings were used for targeted analysis of DIA data. Retention time prediction type was set to dynamic iRT. Correction factor was for window 1. Interference correction on MS2 level was enabled as well. Systematic variance was normalized by local normalization strategy. The FDR was estimated with the mProphet approach and set to 1% at peptide precursor level and at 1% at protein level. The protein intensity was summed by the intensity of their respective peptides which was calculated by the peak areas of their respective fragment ions of MS2. All results were filtered by a Q value cutoff of 0.01 (corresponds to a FDR of 1%).
Bioinformatic analyses
To screen out differentially expressed proteins (DEPs), the threshold of fold change difference between two groups was set at 1.5 (up or down) as well as the corrected P-value was less than 0.05. Gene Ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment were used as previously described to explore the roles of the DEPs [26]. Briefly, GO annotation was performed to discover the gene regulatory networks based on hierarchical categories according to the molecular function, biological process, and cellular component terms of the DEPs (http://www.geneontology.org). The calculated P value was subjected to a Bonferroni correction via using a corrected P value ≤ 0.05 as a threshold. GO terms fulfilling this condition were defined as significantly enriched GO terms in DEPs. In addition, KEGG pathway analysis was further employed to investigate the significant pathways enriched by the DEPs (http://www.genome.jp/kegg/). KEGG pathways with a P value ≤ 0.05 were significantly enriched pathways with DEPs.
Multiple reaction monitoring (MRM)-based quantitative detection of targeted energy metabolism in sperm cells
The eight sperm samples were collected from cauda epididymidis of four wt and four Prss37−/− mice as mentioned above. The cells were homogenized with 200 μl precooling ultrapure water, and then added into 800 μl precooling methanol/acetonitrile solution (1:1, v/v) and vortex mixed. After 20 min ice-water bath ultrasonic extraction, the mixture was incubated at -20 °C for 60 min to precipitate protein, and then was centrifuged at 14,000 × g for 20 min at 4 °C. The supernatant was transferred into a new EP tube and vacuum dried. Before the detection, 100 μl acetonitrile/H2O solution (1:1, v/v) was added to the dried extract to dissolve completely, and centrifuged at 14,000 × g for 20 min at 4 °C. The supernatant was then used for the LC–MS/MS analysis.
LC–MS/MS was conducted with an Agilent 1290 Infinity chromatography system and AB SCIEX 5500 QTRAP mass spectrometer. In all, 10 mM ammonium acetate solution and acetonitrile were employed as mobile phase A and B, respectively. The samples were placed in 4 °C automatic sampler with column temperature of 45 °C, and the flow rate was set 300 μL/min and the injection volume was 2 μl. The gradient of mobile phase B was as follows: 0–18 min at 90–40% acetonitrile; 18–18.1 min at 40–90% acetonitrile and 18.1–23 min at 90%.
Moreover, in the sample queue, a QC sample was set at a certain number of experimental samples at an interval for testing and evaluating the stability and repeatability of the system. The sample queue was set up with a standard mixture of energy metabolites for correction of chromatographic retention time. The 5500 QTRAP system was operated with a negative ion mode under the following conditions: source temperature, 450 °C; ion source gas 1, 45; ion source gas 2, 45; curtain gas, 30. ion sapary voltage floating (ISVF), -4500 V. Analyses were determined by electrospray ionization employing multiple reaction monitoring. Peak chromatographic area and retention time were analyzed with Multiquant software. The standard substance of energy metabolites was used to calculate the retention time and identify metabolites.
Besides, the clustering analysis of identified metabolites was carried by Shanghai Applied Protein Technology Co. Ltd. A fold change of metabolite greater than 1 indicates an up-regulation whereas less than 1 indicates a down-regulation. The statistical analysis value of metabolites in comparison group P value < 0.05 was the metabolites with significant difference.
Adenosine triphosphate (ATP) measurement
The ATP levels of testis and sperm in wt and Prss37-null mice were detected using Enhanced ATP Assay Kit (Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s instruction with minor modifications. The liver tissues derived from two genotypes of mice were employed as the negative controls. At least three biological replications were performed to measure ATP levels.
Statistical analysis
All data are presented as mean ± standard error (SEM) (n ≥ 3). A two-tailed unpaired Student’s t-test was employed to determine statistical significance between two groups. P value < 0.05 was considered statistically significant.
Results
PRSS37 does not primarily act through transcriptional regulation
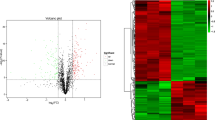
Although our previous studies have shown that PRSS37 is supposed to modulate the interaction between PDILT and ADAM3 to involve in male fertility [17, 22], the detailed molecular mechanisms remain unclear. Considering that PRSS37 is also distributed in nucleus in addition to membrane and cytoplasmic [22], and is predicted to play a key function in transcriptional regulation. We therefore employed oligonucleotide microarrays with full-genome coverage to profile transcriptional differences between adult testes isolated from wt and Prss37-null mice. Hierarchical clustering of the data from three biological replications merely identified 31 differentially expressed genes (DEGs) (Fig. 1A and Table S3), among which 12 DEGs were up-regulated and 19 DEGs were down-regulated in Prss37-deficient testes when compared to wt tissues (Fig. 1B), and the six genes were also selected for qRT-PCR validation (Fig. 1C). Nevertheless, GO analysis of these DEGs displayed that the top 10 most significantly enriched items had nothing to do with male reproductive feature (Fig. S1), and more importantly, a literature review of these genes showed that they were not specifically expressed in testis and/or sperm, and were not associated with PRSS37 function. It implied that PRSS37 acts primarily through non-transcriptional mechanism.

Microarray analysis of transcriptional differences between wt and Prss37-null testis. (A) Total RNAs extracted from the testes of three pairs of wt and Prss37−/− mice were used for gene expression analysis using whole mouse genome 4 × 44 k microarray. A total of 31 genes were identified to be differentially expressed significantly (P < 0.05 and fold change > 2) as shown in heat map. (B) The number of genes up-regulated and down-regulated in Prss37-deficient mice relative to the control were indicated. (C) Comparison of the expression profiles of the selected genes as determined by microarray analysis and qRT-PCR
PRSS37 is largely enriched in layer 3 cells of wt testis
Because PRSS37 appears to have little effect on the transcription regulation, the question then arose as to whether PRSS37 modulated the protein translation and influenced the protein expression? Prior to the differential proteomic analysis, we attempted to separate a group of cells with high expression of PRSS37 in the testis. The testicular single cell suspensions in wt and Prss37-mutant mice were obtained by a combination of collagenase and trypsin digestion, and layer 1, layer 2 and layer 3 cells were further isolated by percoll density gradient centrifugation. Flow cytometry DNA ploidy analysis and nuclear propidium iodide (PI) staining revealed that there was no significant difference of each layer cell composition between wt and Prss37-null mice (Fig. 2A & 2B). Based on the observation of cell morphology, we found that layer 1 was chiefly cell debris, layer 2 was mainly round spermatozoa, and layer 3 was primarily elongating spermatid cells (Fig. 2B). Intriguingly, the expression of PRSS37 was principally enriched in layer 3 cells compared to layer 1 and layer 2 cells as shown by immunofluorescence and immunoblot analyses, respectively (Fig. 2C & 2D & 2E).

Percoll density gradient centrifugation of testicular single cell suspension. (A) Flow cytometry analysis of DNA ploid types of wt and Prss37-null mice in total (T), layer 1 (L1), layer 2 (L2) and layer 3 (L3) cells. (B) Propidium iodide (PI) nuclear staining (red) of T, L1, L2 and L3 cells between wt and Prss37-null mice. (C) Immunofluorescence staining of T, L1, L2 and L3 cells with antibody against PRSS37 (red). Nuclei and acrosome were respectively visualized by DAPI (blue) and PNA (green) staining. (D) Immunoblotting analysis of PRSS37 levels in T, L1, L2 and L3 cells. GAPDH was shown as a loading control. (E) Quantitative analysis of relative intensities of proteins. Error bars indicate the standard error (SE) of the mean data from four biological repetitions. *P-value < 0.05, **P-value < 0.01, ***P-value < 0.001
Changes in protein expression profiles between wt and Prss37−/− testes
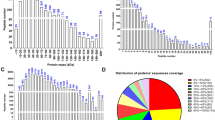
In view of the highest expression of PRSS37 in layer 3 cells, we then compared the global network profiles of wt and Prss37-null layer 3 cells employing a DIA-based quantitative proteomics strategy. A total of 6796 proteins were identified (Fig. 3A), and to determine which of these proteins were expressed significantly and differentially between two samples, we required a fold-change (FC > 1.5) and corrected P-value (P < 0.05), which resulted in a set of 2506 differentially expressed proteins (DEPs). Compared with the controls, 1321 DEPs were up-regulated while 1185 DEPs were down-regulated in Prss37-deficient samples, and the number of up-regulated was almost as many as those of down-regulated of DEPs (Fig. 3A & 3B). Hierarchical clustering displayed that these DEPs well distinguished the two groups, indicating them to be potential downstream signaling molecules of PRSS37 (Fig. 3C). The GO and KEGG analyses were respectively performed to classify the functions and pathways of the DEPs between two samples. The DEPs were annotated to three main GO functional categories, of which 10 most significantly enriched items (corrected P-value < 0.05) were selected from biological process (BP), cell component (CC) and molecular function (MF), respectively. Of note, the entries in BP including oxidation–reduction process (241 DEPs, GO:0,055,114), small molecule metabolic process (359 DEPs, GO:0,044,281), single-organism metabolic process (719 DEPs, GO:0,044,710), catabolic process (369 DEPs, GO:0,009,056), and single-organism catabolic process (221 DEPs, GO:0,044,712) et al. were all involved in energy metabolism (Fig. 3D). Meanwhile, the DEPs were mapped to the reference canonical pathways in the KEGG, and the top 10 pathways with the greatest significantly (corrected P-value < 0.05) included the metabolic pathways (247 DEPs, mmu01100), carbon metabolism (43 DEPs, mmu01200), valine, leucine and isoleucine degradation (28 DEPs, mmu00280), fatty acid metabolism (25 DEPs, mmu01212), and propanoate metabolism (19 DEPs, mmu04146) et al. (Fig. 3E).

DIA quantitative proteomic comparison analysis of three pairs of wt and Prss37-deficient testis layer 3 cells. (A) Volcano plot depicting the proteomic differences. The differentially expressed proteins (DEPs) that exhibited a fold change > 1.5 in abundance and had a P value < 0.05 were shown in green (down-regulated) or red (up-regulated). (B) Statistics on the numbers of down-regulated and up-regulated DEPs. (C) The heat map from three independent proteomic analyses of DEPs from wt and Prss37-mutant testis layer 3 cells. (D) Gene Ontology (GO) classification of DEPs. The top 10 terms (corrected P-value < 0.05) were respectively selected in three main categories: biological process (blue), cellular component (red), and molecular function (yellow). Y-axis showed each GO term, X-axis represented the percent and number of DEPs in each main category. (E) Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment of DEPs. The top 10 pathways (corrected P-value < 0.05) were displayed. X-axis indicated each KEGG pathway, Y-axis represented the percent and number of DEPs in each main pathway
Changes in protein expression profiles between wt and Prss37−/− sperm
Because the main function of PRSS37 was to act on mature sperm, namely sperm from mice deficient for PRSS37 failed to migrate from the uterus into the oviduct and impaired sperm-egg binding [17], therefore, we further investigated the global proteome profiles between wt and Prss37-null sperm using the same DIA method. In all, 3093 proteins were quantified and 459 DEPs were identified significantly differentially expressed between two groups (Fig. 4A), of which 52 DEPs were up-regulated whereas 407 DEPs were down-regulated in Prss37-mutant groups (Fig. 4B). The number of down-regulated DEPs was obviously much larger than the number of up-regulated DEPs. The heat map illustrated that the reproducibility of three independent experiments was well (Fig. 4C). The DEPs were annotated to three main GO functional categories, of which 10 most significantly enriched items (corrected P-value < 0.05) were selected from BP, CC and MF, respectively. Coincidently, all items in BP such as the single-organism metabolic process (176 DEPs, GO:0,044,710), small molecule metabolic process (97 DEPs, GO:0,044,281), oxidation–reduction process (65 DEPs, GO:0,055,114), carboxylic acid metabolic process (59 DEPs, GO:0,019,752), and oxoacid metabolic process (61 DEPs, GO:0,043,436) et al. were associated with the energy metabolism (Fig. 4D), which similar to BP functional categories of layer 3 cells of testis. Moreover, the DEPs were also mapped to the reference canonical pathways in the KEGG, and the top 10 pathways with the greatest significantly (corrected P-value < 0.05) included metabolic pathways (78 DEPs, mmu01100), valine, leucine and isoleucine degradation (14 DEPs, mmu00280), propanoate metabolism (7 DEPs, mmu00640), carbon metabolism (12 DEPs, mmu01200), and arginine and proline metabolism (8 DEPs, mmu00330) et al. (Fig. 4E).

DIA quantitative proteomic comparison analysis of three pairs of wt and Prss37−/− sperm. (A) Volcano plot depicting the proteomic differences. The differentially expressed proteins (DEPs) that exhibited a fold change > 1.5 in abundance and had a P value < 0.05 were displayed in green (down-regulated) or red (up-regulated). (B) Statistics on the numbers of down-regulated and up-regulated DEPs. (C) The heat map from three independent proteomic analyses of DEPs from wt and Prss37-mutant sperm. (D) Gene Ontology (GO) classification of DEPs. The top 10 terms (corrected P-value < 0.05) were respectively selected in three main categories: biological process (blue), cellular component (red), and molecular function (yellow). Y-axis showed each GO term, X-axis represented the percent and number of DEPs in each main category. (E) Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment of DEPs. The top 10 pathways (corrected P-value < 0.05) were shown. X-axis denoted each KEGG pathway, Y-axis represented the percent and number of DEPs in each main pathway
Functional analysis of DEPs in testis and sperm based on the DIA data
Based on the GO annotation and KEGG enrichment data in the testis and sperm, we found that DEPs related to energy metabolism were primarily participated in glycometabolism, fatty acid metabolism, amino acid metabolism, tricarboxylic acid cycle (TCA cycle), electron transport chain (ETC) and oxidative phosphorylation (OXPHOS). Interestingly, all these processes or pathways were compromised after perturbing of PRSS37. Specifically, ADPGK, PGM2, GPI, ALDOC, TPI1, PGAM1, PKM, PYGM, PRKCSH, MANBA, H6PD, PGLX and TALDO1 were down-regulated in testis, ENO3, GANAB and MOGS were down-regulated in sperm, and GIB1 and GIB1L2 were concurrently down-regulated in testis and sperm, which inhibited the glycometabolism including glycolysis, glycogen degradation and pentose phosphate pathway (Table 1). ACSS1, ACSS2, ACSS3, ACSF2, ACSBG1, FABP3, FABP5, DBI, ACADS, ACADL, ACADVL, ACAD11, ACAD12, HADH, HSD17B10, ACAA2 and ACAT1 were reduced in testis, ACSL4 and FABP9 were reduced in sperm, and ACADM, ACADSB and ECHS1 were synchronously reduced in testis and sperm, which suppressed the fatty acid metabolism including activation, transmembrane transport and β-oxidation (Table 2). SFXN5, ALDH4A1, GLUD1 and ACAD8 were decreased in testis, AMT, GLDC, SHMT2, MPST and BCKDK were decreased in sperm, and IVD, MCCC1, MCCC2, AUH, GPT2, PRODH, ALDH18A1 and ALDH6A1 were simultaneously decreased in testis and sperm, which repressed the amino acid metabolism resulting in a reduction of the end products of acetyl-CoA, α-ketoglutaric acid, succinyl-CoA, fumaric acid and oxaloacetic acid (Table 3). Moreover, the key enzymes involved in TCA cycle such as IDH2, OGDHL, SUCLG2, SDHB, ME1 were weakened in testis and/or sperm, which hindered the further oxidation of finally substrates of glycometabolism, fatty acid metabolism and amino acid metabolism (Table 4). Obviously, PDK1, PDK2, MPC2, PDHA2, PDHB, PDHX and DLAT were changed and this is the result of the breakdown of the TCA cycle (Table 4). Meanwhile, the components of Complexes I-IV of respiratory chain were diminished, which impeded the OXPHOS of NADH and FADH2 originated from the catabolic pathways of sugar, fatty acids, amino acids, and TCA cycle (Table 5 & Table 6). Consistent with these, 16 members of SLC25A subfamily and 3 members of SFXN family (Table S4), as well as CHCHD3, IMMT, MIGA2, FIS1, MTFP1, MGARP, RHOT2, LACTB2, STOML2, TXN2 and OXA1L et al. (Table S5) were declined, which possible impaired the mitochondrial metabolites transport and crippled the mitochondrial morphology and function. These data suggested that PRSS37 acts a significant role in energy metabolism, possibly through regulating the expression of these DEPs.
Validation of DIA data by comparison metabolite levels between wt and Prss37−/− mice
The down-regulation of DEPs involved in energy metabolism in Prss37−/− mice inevitably led to a reduction of intermediate metabolites in TCA cycle and OXPHOS system. Thus, we next applied the MRM ion ratio strategy to identify differences in metabolite abundance in wt and Prss37-null sperm. As expected, we found that the abundances of 21 identified metabolites were changed in response to PRSS37 deletion. Seventeen metabolites were down-regulated (< onefold) and only 4 metabolites were up-regulated (> onefold) in Prss37−/− samples. Of note, fumarate, citrate, GMP and cyclic AMP were significantly decreased while lactate and GDP were markedly increased (Fig. 5A). Meanwhile, we further detected the final metabolite ATP levels in testis and sperm between wt and Prss37-mutant mice. The data showed that ATPs were remarkably reduced in Prss37-deficient testis and sperm, and obviously there was no apparent difference in an unrelated tissue of liver compared to the wt control groups (Fig. 5B). It demonstrated that PRSS37 indeed paly a crucial role in energy metabolism, which possibly contributes to the male fertility such as sperm migration through UTJ and sperm-egg recognition. These results indirectly validate that our proteomic data in testis and sperm are authentic and dependable, which could further be used to identity and explore the potential molecular regulatory mechanisms of downstream signaling of PRSS37.

Comparison metabolite levels between wt and Prss37−/− mice. (A) Metabolic profile of regulated metabolites in four pairs of wt and Prss37-null sperm. The heat map from four independent MRM-based targeted metabolomics analyses of sperm from the WT and KO mice. Only significantly difference (P < 0.05) metabolites are shown in red. (B) Comparison of ATP level between three wt and three Prss37-deficient mice in the testis, sperm and liver. The liver was employed as the negative controls in here. *P-value < 0.05, **P-value < 0.01
Discussion
Our previous studies have shown that PRSS37 was associated inextricably with the sperm migration from the uterus into the oviduct and sperm-egg recognition [17, 22]. Intriguingly, PRSS55 also displayed the similar male sterility phenotypes of PRSS37 after disturbance [18], and recent study reported that PRSS55 plays a significant role in ATP production to maintain the male mouse fertility [24]. But little is known about whether loss of PRSS37 is involved in energy metabolism imbalance. We attamed to use an oligonucleotide microarray technology to identify the potential downstream signaling pathway of PRSS37, and the results implied that PRSS37 appears to have little regulatory effect on the transcription level. This may be determined by the nature of PRSS37 itself, as an important member of the serine protease family, which is more likely to be involved in protein level regulation. Therefore, we further compared the differences of protein expression between wt and Prss37−/− mice in testis layer 3 cells and sperm employing a DIA-based quantitative proteomics strategy. In all, 2506 and 459 DEPs were identified in Prss37−/− testis and sperm, respectively, when compared to control groups. GO and KEGG analyses suggested that most of these DEPs were enriched in energy metabolism associated biological processes and pathways. The DIA data were also confirmed by a decline in key intermediate metabolites such as fumarate and citrate of TCA cycle and end product ATP levels of all metabolic pathways. These results indicated that PRSS37, which may be similar to PRSS55, acts a crucial role in the pathway of energy metabolism in mice as well. In the present study, the DEPs involved in glycometabolism, fatty acid metabolism, amino acid metabolism, TCA cycle, electron transport chain and oxidative phosphorylation, as well as transport of mitochondrial metabolites and mitochondrial morphology and function were discussed in detail as they were most likely related to PRSS37 functions in mice (Fig. 6).

Schematic illustration of PRSS37 regulating energy metabolism. PRSS37 facilitates the synthesis of ATP by positively modulating the glycometabolism, fatty acid metabolism, amino acid metabolism, tricarboxylic acid cycle (TCA cycle) and oxidative phosphorylation (OXPHOS) to participate in male fertility
The DEPs involved in glycometabolism
Generally, glycolysis is a key metabolic pathway that is employed by all cells for the oxidation of glucose to generate energy in the form of ATP and intermediates for use in other metabolic pathways including gluconeogenesis [27]. Besides glucose, other hexose sugars such as fructose and galactose end up in the glycolytic pathway for catabolism as well [28]. Glycolysis consists of a series of 10 step reactions occurring in the cytoplasm where one molecule of glucose is oxidized to produce two molecules of pyruvate [29]. ADPGK catalyzes the phosphorylation of glucose to glucose 6-phosphate (G6P) using the phosphate from the hydrolysis of ADP, this irreversible step serves to trap the glucose molecule within the cells [30], meanwhile, PGM2 also catalyzes the interconversion of glucose-1-phosphate (G1P) and G6P to supplement the levels of the latter for the beginning of glycolysis [31]; G6P is isomerized to fructose-6-phosphate (F6P) catalyzed by GPI [32,33,34]; PFKM catalyzes the phosphorylation of F6P to fructose 1,6-bisphosphate (FBP) by ATP, and this is an irreversible, rate limiting, regulatory step [35]; Next, cleavage of FBP leads to the formation of glyceraldehyde-3-phosphate (GAP) and dihydroxyacetone phosphate (DHAP) by ALDOC [36]; While TPI1 is an extremely efficient metabolic enzyme that catalyzes the interconversion between DHAP and GAP. This isomerization causes producing of two molecules of GAP [37]; Oxidation of GAP is subsequently catalyzed by GAPDHS and results in the synthesis of 1, 3-bisphosphoglycerate (1,3-BPG), this is the first oxidation reduction step in glycolysis where NAD+ is reduced to NADH, while the aldehyde group of GAP is oxidized to a carboxyl group coupled to the attachment of a phosphate group [38]; Formation of 3-phosphoglycerate (3-PG) from 1,3-BPG is a first ATP generating step in glycolysis. The phosphate group attached during the formation of 1,3-BPG in the previous step is employed to phosphorylates ADP with the help of PGK2, this substrate level phosphorylation therefore generates 2 ATPs [39]; A reversible isomerization reaction of 3-PG to 2-phosphoglycerate (2-PG) is then carried out through PGAM1 [40]; 2-PG is converted to phosphoenolpyruvate, which contains the high energy enol phosphate, via ENO3 [41]; The final step is the enzymatic conversion of phosphoenolpyruvate to pyruvate by PKM. Substrate level phosphorylation occurs in this irreversible step to generate 2 molecules of ATP [42]. The whole glycolysis process has been completed (Fig. 6). Herein, we offer the first evidence to demonstrate that disruption of PRSS37 led to the down-expression of ADPGK, PGM2, GPI, ALDOC, TPI1, PGAM1, ENO3, and PKM in testis or sperm (Table 1), suggesting that PRSS37 participated in glycolysis process through cross-talk with these key enzymes. Intriguingly, loss of the activities of these significant enzymes caused by lacking of PRSS37 in mice is also likely compensated by increasing the expression of other major enzymes such as PFKM, GAPDHS, and PGK2 (Table 1), at least partially. It should be noted that PRSS37 and GAPDHS were identified as having potential interactions as well [22].
Moreover, the glycan degradation and pentose phosphate pathway (PPP) were also destroyed in Prss37-deficient mice. The process by which glycogen, stored in the cytoplasm, is broken down into glucose for use in glycolysis when the body needs energy, and PYGM, as a rate-limiting enzyme acts in glycogen degradation [43]. PRKCSH and GANAB, the regulatory and catalytic subunits of glucosidase II, respectively, cleave sequentially the two innermost α-1,3-linked glucose residues from the Glc2Man9GlcNAc2 oligosaccharide precursor [44, 45], whereas MOGS can cleave the distal α-1,2-linked glucose residue from the Glc3Man9GlcNAc2 oligosaccharide precursor [46]. MANBA cleaves the single β-linked mannose residue from the non-reducing end of all N-linked glycoprotein oligosaccharides [47], and GIB1 or GIB1L2 protein cleaves the β-linked terminal galactosyl residues from gangliosides, glycoproteins and glycosaminoglycans [48]. According to our DIA proteomics data, the expression of all enzymes involved in glycogen or glycan degradation were down-regulated after deletion of PRSS37, among which PYGM, PRKCSH and MANBA were specifically decreased in testis, GANAB and MOGS were specifically reduced in sperm, and GIB1 and GIB1L2 were concurrently declined in testis and sperm (Table 1). It implies that the primary substrate (glucose) or intermediate metabolites (mannose, fructose and galactose et.al) of glycolysis process is not replenished in time in testis or sperm. Additionally, PPP, an alternative to glycolysis process and a major source of reducing power and metabolic intermediates for biosynthetic processes. Typically, the reaction of PPP is generally divided into two stages, oxidative phase and non-oxidative phase (Fig. 6). H6PD and PGLX are participated in the oxidative phase that synthesize ribulose 5-phosphate from glucose 6-phosphate [49,50,51,52,53], while TALDO1 is involved in the non-oxidative phase that synthesizes glyceraldehyde 3-phosphate and fructose 6-phosphate from ribose 5-phosphate and xylulose 5-phosphate [54]. In this study, the expression of H6PD, PGLX and TALDO1 were down-regulated in Prss37−/− testis (Table 1). Taken together, the glycometabolism such as glycolysis, glycan degradation and PPP were paralyzed when PRSS37 was inhibited in testis and sperm.
The DEPs associated with fatty acid metabolism
Fatty acid β-oxidation is an another major pathway of energy metabolism and occurs primarily in mitochondria, and it can provide a large amount ATP required for the testis and sperm as well [55, 56]. Four sets of enzymes are participated in the cyclic process of fatty acid β- oxidation in mitochondrial matrix (Fig. 6). Since these enzymes act on CoA, a prelude to fatty acid β-oxidation is the conversion of fatty acids to acyl-CoA by acyl-CoA synthases, which is called as fatty acid activation. The acyl-CoA then enters a four-step cyclic process in which they are converted to one molecule of acetyl-CoA by acyl-CoA dehydrogenases, 2,3-enoyl-CoA hydratases, L-3-hydroxyacyl-CoA dehydrogenases and 3-ketoacyl-CoA thiolases [57], and is accompanied by the generation of FADH2 and NADH at first step and at third step, respectively [58]. In contrast to short chain acyl-CoA (chain length less than C6) and medium chain acyl-CoA (chain length C6 to C12), which are able to directly enter the mitochondrial matrix, long chain acyl-CoA (chain length C14 to C18) fail to directly across the mitochondrial membrane. It needs a special transport mechanism with help of carnitine palmitoyl transferase to enter the mitochondrial matrix, and then enter the cyclic four-step fatty acid β-oxidation processes [57]. On the basis of DIA results of testis and sperm, we have identified a number of DEPs participated in fatty acid metabolism including activation, translocation and β-oxidation (Table 2). ACSS1, ACSS2 and ACSS3 catalyze the synthesis of acyl-CoA from short chain fatty acid [59,60,61], ACSF2 has some preference toward medium chain substrates to synthesis of acyl-CoA forms [62], ACSBG1 and ACSL4 catalyze the conversion of fatty acids such as long chain and very long-chain fatty acids to their active form acyl-CoA forms [63, 64]. Moreover, FABP3, FABP5 and FABP9 [65, 66] and DBI [67] are thought to play a crucial role in the intracellular transport of long-chain fatty acids and their acyl-CoA esters, and CPT1A catalyzes the transfer of the acyl group of long chain acyl-CoAs conjugates onto carnitine, a key step for the mitochondrial matrix uptake of long chain acyl-CoAs [68]. Furthermore, ACADS, ACADM, ACADL, ACADVL, ACADSB, ACAD11, and ACAD12 are the most important members of the acyl-CoA dehydrogenases family that catalyze the first step of β-oxidation [69]. ECHS1 as an enoyl-CoA thioesters [70], HADH and HSD17B10 as the hydroxyacyl-CoA dehydrogenases [71, 72], they respectively function in the second step and third step of β-oxidation. Whereas ACAA2 and ACAT1 are two of the enzymes that catalyzes the last step of β-oxidation, which catalyzes the thiolytic cleavage of acyl-CoA into acetyl-CoA and an acyl-CoA shortened by two carbons [73]. Coincidently, all of these factors discussed above were declined in testis and/or sperm (Table 2), suggesting that the fatty acid metabolism, to some extent, is blocked after loss of PRSS37 protein.
The DEPs related to amino acid metabolism
As we have already seen in times of interruption of glycometabolism and fatty acid metabolism, amino acid metabolism can be vital to maintain ATP levels and provide alternative carbon sources. The carbon skeleton of amino acids is decomposed by multi-enzyme systems. Although the oxidation pathways of amino acids are varied, they are transported into mitochondria by SFXN5 and then all concentrated into five products (acetyl-CoA, α-ketoglutaric acid, succinyl-CoA, fumaric acid and oxaloacetic acid) that enter the TCA cycle and ultimately oxidize to CO2 and H2O [74] (Fig. 6). Typically, GPT2 [75], GLDC [76], AMT [76], SHMT2 [77] and MPST [78] are took part in the pathway of alanine, glycine, serine and cysteine degradation from pyruvate to acetyl-CoA. Meanwhile, FAH [79] and GSTZ1 [80] are involved directly in the pathway of phenylalanine degradation, and IVD [81], MCCC1 [82], MCCC2 [82] and AUH [83] are respectively participated in the step 1, 2 and 3 of the pathway of leucine degradation, GCDH [84] and GOT2 [85] are associated with the pathway of lysine and tryptophan degradation, respectively. The degradations of phenylalanine, leucine, lysine and tryptophan were from acetoacetyl-CoA to acetyl-CoA. The intermediate metabolites of phenylalanine can also enter TCA cycle through fumaric acid [74]. Moreover, PRODH and ALDH4A1 are involved in the pathway of proline degradation [86], GLUD1 [87] and ALDH18A1 [88] are participated in the pathway of glutamate degradation, which finally can form α-ketoglutaric acid. Additionally, ACAD8 [89], ALDH6A1 [90] and BCKDK [91] are related to the valine degradation to form succinyl-CoA. Intriguingly, all these enzymes associated with amino acid metabolism including SFXN5 were impaired by disturbance PRSS37 function (Table 3). At the same time, PRSS37 possible regulate the expression of these proteins such as BCKDK through closely interaction with them [22].
The DEPs participated in TCA cycle
The TCA cycle is the final common oxidative pathway for carbohydrates, fatty acids and amino acids. It is the most important metabolic pathway connecting almost all the individual metabolic pathways for the energy supply to the tissues [92]. Particularly, glucose-derived pyruvate in glycolysis can be transported to mitochondria matrix by MPC2 and then converted to acetyl-CoA by pyruvate dehydrogenase, and finally acetyl-CoA enters the TCA cycle and is accompanied by the production of GTP, NADH and FADH2 [93]. Fatty acid-derived acetyl-CoA enters the TCA cycle directly and the end products of amino acid metabolism also confluence into TCA cycle in the form of the intermetabolites. The TCA cycle is formed from a set of 8 enzyme reactions as follows: 1) acetyl-CoA and oxaloacetate are converted to citrate by citrate synthetase; 2) citrate is transformed into cis-aconitate and further converted to isocitrate by aconitase; 3) isocitrate can subsequently be oxidatively decarboxylated to α-ketoglutarate by NAD-dependent isocitrate dehydrogenase, generating CO2 and NADH; 4) a second oxidative decarboxylation step is performed by a α-ketoglutarate dehydrogenation complex. In this reaction, α-ketoglutarate is converted into succinyl-CoA by α-ketoglutarate dehydrogenase, which producing CO2 and NADH; 5) substrate level phosphorylation, namely succinyl-CoA and GDP are transformed into succinate and GTP by succinyl-CoA thiokinase; 6) succinate and FAD are converted to fumaric acid and FADH2 through succinate dehydrogenase; 7) fumaric acid is catalyzed by fumarase and malate is formed; 8) malate is dehydrogenated to produce oxaloacetate by malate dehydrogenase. NAD is converted to NADH and oxaloacetate formed in this reaction reacts with acetyl-CoA to form citrate in order to start another round of the TCA cycle [92] (Fig. 6). Here, the pivotal enzymes of energy generation steps including IDH2 in third step, OGDHL, DBT, BCKDHA and BCKDHB in fourth step, SUCLG2 in fifth step, SDHB and SDHAF2 in sixth step and ME1 in eighth step were all down-regulated (Table 4). Meanwhile, PDK1 and PDK2, which act a significant role in regulation of glucose and fatty acid metabolism by phosphorylation of the pyruvate dehydrogenase subunit PDHA2, were reduced in Prss37−/− testis, thereby preventing the formation of acetyl-CoA from pyruvate and suppresses metabolite flux in TCA cycle. As a result, PDHA2, and even including PDHB, PDHX, and DLAT were accumulated and up-regulated in testis. When glycolysis process and TCA cycle were paralyzed, MPC2 that mediates the uptake of pyruvate into mitochondria was accumulated in testis as well (Table 4). These results indicated that PRSS37 likely participates in TCA cycle pathway to regulate energy metabolism cooperating with IDH2, OGDHL, SUCLG2, SDHB, ME1, PDK, PDHA2, MPC2 and et al. Alternatively, two key intermediate metabolites such as citrate and fumarate in TCA cycle were also highly deregulated in Prss37−/− sperm (Fig. 5A), which consistent with our DIA proteomics data.
The DEPs took part in respiratory chain and oxidative phosphorylation
The NADH and FADH2 produced by the TCA cycle, β-oxidation and even glycolysis (generated in the cytoplasm and then entered in the mitochondria) further enter the respiratory chain for oxidative phosphorylation to generate ATP [94]. The electron transport chain consists of five protein complexes integrated into the inner mitochondrial membrane, and the mitochondrial matrix supplies NADH and FADH2 to the electron transport chain, each of which donates a pair of electrons to the electron transport chain through Complexes I (NADH-Q oxidoreductase) and Complex II (succinate dehydrogenase), respectively. The electrons from either Complex I or Complex II are donated to ubiquinone which is reduced to ubiquinol [95]. Ubiquinol is oxidized by Complex III (cytochrome c reductase) allowing one electron at a time to continue the journey through cytochrome c, and the cytochrome c further transports electrons to Complex IV (cytochrome c oxidase) where O2 acts as a terminal electron acceptor and is reduced to H2O. In response to electron transport, a total of ten protons are pumped from the mitochondrial matrix into intermembrane space, where they accumulate to generate an electrochemical proton gradient known as the mitochondrial membrane potential (ΔΨ). ΔΨ next combined with proton concentration (pH) generates a protonmotive force (Δp), which is an essential component in the process of energy storage during oxidative phosphorylation since it links electron transport chains (Complexes I-IV) to the activity of Complex V (ATP synthase), where protons re-enter the matrix to dissipate the proton gradient and this is coupled to the production of ATP from ADP [96] (Fig. 6). Of note, all DEPs associated Complexes I-V were decreased after perturbing of PRSS37 (Table 5 & Table 6). Specifically, NDUFB11, NDUFA4, NDUFB9, NDUFB5, NDUFA13, NDUFB10, NDUFA3, NDUFB4, NDUFB7, NDUFS4, NDUFA1, NDUFA10, NDUFA5, NDUFB1, NDUFB8, NDUFC2, NDUFA2, NDUFAB1 and NDUFA7 are the key accessory subunits of the Complex I, and NDUFV2, NDUFS3, NDUFV3, NDUFS8 and NDUFS7 are the core subunits of the Complex I (Table 5). HSCB, SDHB and SDHAF2 are the components of the Complex II, while UQCRH, UQCRQ and UQCR10 are the components of the Complex III (Table 5). The members of COX family such as COX6B1, COX7A2L, COX5B, COX7A2, COX7C and COX6C are necessary for the assembly of the Complex IV (Table 5). Besides, 18 ATP synthases related to the assembly of the Complex V were also weakened (Table 6). Among these factors, NDUFB8, NDUFC2, NDUFA2, SDHAF2, COX7A2, ATP5O, ATP5J and ATP5J2, as well as other associated factors ETFB and POR were declined simultaneously in testis and sperm (Table 5 & Table 6). Distinctly, these data supported that PRSS37 participated in respiratory chain and oxidative phosphorylation via regulating these DEPs.
The DEPs involved in the transport of mitochondrial metabolites and mitochondria morphology and function
Combining the DEPs related to facilitating the entry of metabolites into the mitochondria (Table S4) as well as the DEPs participated in the mitochondria morphology and function (Table S5), we detected that most of proteins were down-regulated in response to deletion of PRSS37. The solute carrier superfamily consists of a group of proteins especially solute carrier 25A subclass responsible for transporting different metabolites across the mitochondrial membrane [97]. These molecules are reported to be essential for efficient synthesis of ATP [98]. Typically, SLC25A16 and SLC25A20 respectively mediates the transport of CoA and acylcarnitines across the mitochondrial inner membrane from the cytosol to the mitochondrial matrix, which required for fatty acid beta oxidation pathway [99, 100]. SLC25A29 and SLC25A18 are involved in amino acid including glutamate and lysine translocation to mitochondrial for oxidation and decomposition [101]. SLC25A1 is a citrate carrier, which promotes efflux of tricarboxylic citrate to the cytoplasm in exchange for dicarboxylic cytosolic malate [102], SLC25A10 is necessary for translocation of malonate, malate and succinate in exchange for phosphate, sulfate, sulfite and thiosulfate [103], and SLC25A11 catalyzes the transport of 2-oxoglutarate across the mitochondrial inner membrane in an electroneutral exchange for malate or other dicarboxylic acids [104]. The occurrence of all these metabolite uptake events is critical to the TCA cycle. Concurrently, SLC25A4 as an ADP/ATP antiporter that modulates import of ADP into the mitochondrial matrix for ATP synthesis, and export of ATP out to fuel the cells [105]. Apparently, obstructing the transport of metabolites undoubtedly results in impaired metabolic pathways such as β-oxidation, amino acid metabolism, TCA cycle and oxidative phosphorylation.
In consistent with, the mitochondria morphology and function were also damaged. Concretely, CHCHD3 and IMMT, two components of the MICOS complex, a large protein complex of the mitochondrial inner membrane that plays crucial roles in the maintenance of crista junctions, inner membrane architecture, and formation of contact sites to the outer membrane [106]. MIGA2 acts as a crucial regulator of mitochondrial fusion [107], whereas FIS1 and MTFP1 are essential for the mitochondrial division [108]. Both MGARP and RHOT2 are involved in control of anterograde transport of mitochondria and their subcellular distribution [109]. Moreover, LACTB2 is required for normal mitochondrial function and cell viability [110], STOML2 probably regulates the biogenesis and the activity of mitochondria [111], and TRAP1 is participated in maintaining mitochondrial function and polarization [112]. Additionally, TXN2 is important for the control of mitochondrial reactive oxygen species homeostasis [113], and OXA1L is required for the activity and assembly of cytochrome oxidase, which is a prerequisite for the correct biogenesis of ATP synthase and complex I in mitochondria [114]. All the proteins discussed above as well as other mitochondrial function related proteins including TOMM6, TIMM13, MICU1, C1QBP, GRPEL1, MTHFD1L, MTCH1, MIPEP and MTX2 were markedly decreased in testis and/or sperm after elimination of PRSS37 (Table S5), demonstrating that site where energy metabolism occurs is also collapsed.
The associations between energy metabolism and male fertility
Typically, in addition to a reduction in some metabolic substrates, the ATP levels were significantly decreased by derange of PRSS37 (Fig. 5B), confirming that all these energy metabolism pathways as shown in DIA data were disorganized in the testis and sperm, although the further verification of key enzymes in each metabolic pathway will be pursued. There is an increasing evidences that energy metabolism is associated inextricably with the male fertility [115, 116]. Facilitating spermatogenesis, sustaining sperm motility, and activating protein modifications such as phosphorylation and glycosylation could be the reasons why testicular germ cells and sperm require exceptionally more ATP than other cells [115]. Apart from this, the functions of ATP in germ cell proliferation, mitotic regulation, and the elimination of germ cells by apoptosis are now well recognized. Eventually, mutations in mitochondrial genome have been reported in male infertility, particularly in sluggish sperm [116]. Following our previous studies, lack of PRSS37 did not affect spermatogenesis [17] although energy metabolism was arrested in the testis. Nevertheless, we are not sure whether PRSS37 is involved in fertilization protein modifications via modulating energy metabolism in the testicular germ cells. Herein, PRSS37 appears to be more involved in regulating sperm motility by promoting ATP synthesis thereby participating in male fertility including sperm migration from the uterus into the oviduct and sperm-egg binding. Most recently, PRSS55 has been identified to be played an essential role in energy metabolism to regulate sperm migration and sperm-egg interaction [24]. As a result, in the present study, we demonstrated a previously unexpected role for PRSS37, which similar to PRSS55, in modulating energy metabolism and maintaining ATP levels to take part in male reproduction. Alternatively, more details of the mechanism of action of PRSS37 remain to be determined.
Conclusion
This study identified 2506 DEPs (testis) and 459 DEPs (sperm) from the wt and Prss37−/− mice. Among these proteins, ADPGK, PGM2, GPI, ALDOC, TPI1, PGAM1, ENO3, PKM, PRKCSH, GANAB, MOGS, MANBA, GIB1, GIB1L2, H6PD, PGLX and TALDO1 were down-regulated, which would hinder the glycometabolism signaling pathway especially the glycolysis process; ACSSs, FABPs, ACADs, ECHS1, HADH, HSD17B10, ACAA2 and ACAT1 were reduced, which would suppress the fatty acid metabolism especially β-oxidation; GPT2, GLDC, AMT, SHMT2, MPST, IVD, MCCC1, MCCC2 and AUH were lowered, which would inhibit the amino acid metabolism. Moreover, IDH2, OGDHL, DBT, BCKDHA, BCKDHB, SUCLG2, SDHB, SDHAF2 and ME1 were impaired, which would impede the TCA cycle, and NDUFs, HSCB, UQCRs, COXs, and ATPs were declined, which would block the electron transport chain for oxidative phosphorylation. Additionally, the DEPs associated with mitochondrial metabolites transport and mitochondria morphology and function were also compromised by loss of PRSS37 in testis or sperm. These results indicated that PRSS37 is involved in the regulation of energy metabolism to participate in male mouse fertility. The identification and understanding of the regulatory mechanisms of male fertility will pave the way toward novel approaches for both contraception and treatment of unexplained male infertility (UMI).
Data availability
Original data of microarray are available in the Gene Expression Omnibus (GEO) at the National Center for Biotechnology Information (NCBI) website (Accession Number GSE183009). The mass spectrometry proteomics data of testis and sperm have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) through the iProX partner repository [117] with the dataset identifier PXD028133 and PXD028135, respectively.
Abbreviations
- wt:
-
Wild-type
- DIA:
-
Data independent acquisition
- DEPs:
-
Differentially expressed proteins
- TCA:
-
Tricarboxylic acid
- OXPHOS:
-
Oxidative phosphorylation
- PTM:
-
Posttranslational modification
- UTJ:
-
Utero tubal junction
- ER:
-
Endoplasmic reticulum
- TGCs:
-
Testicular germ cells
- GPI:
-
Glycosylphosphatidylinositol
- KO:
-
Knockout
- hrs:
-
Hours
- DEGs:
-
Differentially expressed genes
- FC:
-
Fold change
- IAA:
-
Iodoacetamide
- TFA:
-
Trifluoroacetic acid
- EBH:
-
Ethylene Bridged Hybrid
- ISVF:
-
Ion sapary voltage floating
- FBS:
-
Fetal bovine serum
- PI:
-
Propidium iodide
- PFA:
-
Paraformaldehyde
- DDA:
-
Data dependent acquisition
- DTT:
-
Dithiothreitol
- MS:
-
Mass spectrometer
- FDR:
-
False discovery rate
- MRM:
-
Multiple reaction monitoring
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- ATP:
-
Adenosine triphosphate
- SEM:
-
Mean ± standard error
- ETC:
-
Electron transport chain
- DHAP:
-
Dihydroxyacetone phosphate
- PPP:
-
Pentose phosphate pathway
- GAP:
-
Glyceraldehyde-3-phosphate
- 1,3-BPG:
-
1, 3-Bisphosphoglycerate
- G6P:
-
Glucose 6-phosphate
- G1P:
-
Glucose 1-phosphate
- FBP:
-
Fructose 1,6-bisphosphate
- F6P:
-
Fructose 6-phosphate
- 3-PG:
-
3-Phosphoglycerate
- 2-PG:
-
2-Phosphoglycerate
- NCBI:
-
National Center for Biotechnology Information
- GEO:
-
Gene Expression Omnibus
- UMI:
-
Unexplained male infertility
References
Chang MC. Fertilizing capacity of spermatozoa deposited into the fallopian tubes. Nature. 1951;168(4277):697–8.
Austin CR. Observations on the penetration of the sperm in the mammalian egg. Aust J Sci Res B. 1951;4(4):581–96.
Okabe M. Mechanisms of fertilization elucidated by gene-manipulated animals. Asian J Androl. 2015;17(4):646–52.
Ikawa M, et al. Fertilization: a sperm’s journey to and interaction with the oocyte. J Clin Invest. 2010;120(4):984–94.
Larasati T, et al. Tmprss12 is required for sperm motility and uterotubal junction migration in micedagger. Biol Reprod. 2020;103(2):254–63.
Mruk DD, Cheng CY. Sertoli-Sertoli and Sertoli-germ cell interactions and their significance in germ cell movement in the seminiferous epithelium during spermatogenesis. Endocr Rev. 2004;25(5):747–806.
Schultz N, Hamra FK, Garbers DL. A multitude of genes expressed solely in meiotic or postmeiotic spermatogenic cells offers a myriad of contraceptive targets. Proc Natl Acad Sci U S A. 2003;100(21):12201–6.
Matzuk MM, Lamb DJ. The biology of infertility: research advances and clinical challenges. Nat Med. 2008;14(11):1197–213.
Matzuk MM, Lamb DJ. Genetic dissection of mammalian fertility pathways. Nat Cell Biol. 2002;4(Suppl):s41–9.
Hedstrom L. Serine protease mechanism and specificity. Chem Rev. 2002;102(12):4501–24.
Puente XS, et al. Human and mouse proteases: a comparative genomic approach. Nat Rev Genet. 2003;4(7):544–58.
Chen C, et al. A novel serine protease predominately expressed in macrophages. Biochem J. 2003;374(Pt 1):97–107.
Holcomb RJ, et al. The testis-specific serine proteases PRSS44, PRSS46, and PRSS54 are dispensable for male mouse fertilitydagger. Biol Reprod. 2020;102(1):84–91.
Yoneda R, Kimura AP. A testis-specific serine protease, Prss41/Tessp-1, is necessary for the progression of meiosis during murine in vitro spermatogenesis. Biochem Biophys Res Commun. 2013;441(1):120–5.
Yoneda R, et al. Three testis-specific paralogous serine proteases play different roles in murine spermatogenesis and are involved in germ cell survival during meiosis. Biol Reprod. 2013;88(5):118.
Netzel-Arnett S, et al. The glycosylphosphatidylinositol-anchored serine protease PRSS21 (testisin) imparts murine epididymal sperm cell maturation and fertilizing ability. Biol Reprod. 2009;81(5):921–32.
Shen C, et al. Prss37 is required for male fertility in the mouse. Biol Reprod. 2013;88(5):123.
Shang X, et al. Serine protease PRSS55 is crucial for male mouse fertility via affecting sperm migration and sperm-egg binding. Cell Mol Life Sci. 2018;75(23):4371–84.
Zhang H, et al. Male fertility in Mus musculus requires the activity of TRYX5 in sperm migration into the oviduct. J Cell Physiol. 2020;235(9):6058–72.
Yamaguchi R, et al. Aberrant distribution of ADAM3 in sperm from both angiotensin-converting enzyme (Ace)- and calmegin (Clgn)-deficient mice. Biol Reprod. 2006;75(5):760–6.
Yamaguchi R, et al. Disruption of ADAM3 impairs the migration of sperm into oviduct in mouse. Biol Reprod. 2009;81(1):142–6.
Xiong W, et al. Dissecting the PRSS37 interactome and potential mechanisms leading to ADAM3 loss in PRSS37-null sperm. J Cell Sci. 2021;134(10):
Tokuhiro K, et al. Protein disulfide isomerase homolog PDILT is required for quality control of sperm membrane protein ADAM3 and male fertility [corrected]. Proc Natl Acad Sci U S A. 2012;109(10):3850–5.
Zhu F, et al. PRSS55 plays an important role in the structural differentiation and energy metabolism of sperm and is required for male fertility in mice. J Cell Mol Med. 2021;25(4):2040–51.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–8.
Li L, et al. Systematic Identification and Analysis of Expression Profiles of mRNAs and Incrnas in Macrophage Inflammatory Response. Shock. 2019;51(6):770–9.
Xiong Y, et al. Regulation of glycolysis and gluconeogenesis by acetylation of PKM and PEPCK. Cold Spring Harb Symp Quant Biol. 2011;76:285–9.
Jeckelmann JM, Erni B. Transporters of glucose and other carbohydrates in bacteria. Pflugers Arch. 2020;472(9):1129–53.
Naifeh, J., M. Dimri, and M. Varacallo, Biochemistry, Aerobic Glycolysis, in StatPearls. 2021: Treasure Island (FL).
Grudnik P, et al. Structural basis for ADP-dependent glucokinase inhibition by 8-bromo-substituted adenosine nucleotide. J Biol Chem. 2018;293(28):11088–99.
Beck W. Differential function of the phosphoglucomutase isozymes PGM1 and PGM2. Hum Genet. 1979;50(1):93–100.
Buehr M, McLaren A. An electrophoretically detectable modification of glucosephosphate isomerase in mouse spermatozoa. J Reprod Fertil. 1981;63(1):169–73.
Merkle S, Pretsch W. Glucose-6-phosphate isomerase deficiency associated with nonspherocytic hemolytic anemia in the mouse: an animal model for the human disease. Blood. 1993;81(1):206–13.
Pretsch W, Merkle S. Glucose phosphate isomerase enzyme-activity mutants in Mus musculus: genetical and biochemical characterization. Biochem Genet. 1990;28(1–2):97–110.
Nakamura N, Mori C, Eddy EM. Molecular complex of three testis-specific isozymes associated with the mouse sperm fibrous sheath: hexokinase 1, phosphofructokinase M, and glutathione S-transferase mu class 5. Biol Reprod. 2010;82(3):504–15.
Izraely S, et al. The melanoma brain metastatic microenvironment: aldolase C partakes in shaping the malignant phenotype of melanoma cells - a case of inter-tumor heterogeneity. Mol Oncol. 2021;15(5):1376–90.
Roland BP, et al. Missense variant in TPI1 (Arg189Gln) causes neurologic deficits through structural changes in the triosephosphate isomerase catalytic site and reduced enzyme levels in vivo. Biochim Biophys Acta Mol Basis Dis. 2019;1865(9):2257–66.
Miki K, et al. Glyceraldehyde 3-phosphate dehydrogenase-S, a sperm-specific glycolytic enzyme, is required for sperm motility and male fertility. Proc Natl Acad Sci U S A. 2004;101(47):16501–6.
Danshina PV, et al. Phosphoglycerate kinase 2 (PGK2) is essential for sperm function and male fertility in mice. Biol Reprod. 2010;82(1):136–45.
Toriyama K, et al. T cell-specific deletion of Pgam1 reveals a critical role for glycolysis in T cell responses. Commun Biol. 2020;3(1):394.
De Rosa V, et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat Immunol. 2015;16(11):1174–84.
Israelsen WJ, Vander Heiden MG. Pyruvate kinase: Function, regulation and role in cancer. Semin Cell Dev Biol. 2015;43:43–51.
Agius L. Role of glycogen phosphorylase in liver glycogen metabolism. Mol Aspects Med. 2015;46:34–45.
Caputo AT, et al. Structures of mammalian ER alpha-glucosidase II capture the binding modes of broad-spectrum iminosugar antivirals. Proc Natl Acad Sci U S A. 2016;113(32):E4630–8.
Arendt CW, Ostergaard HL. Identification of the CD45-associated 116-kDa and 80-kDa proteins as the alpha- and beta-subunits of alpha-glucosidase II. J Biol Chem. 1997;272(20):13117–25.
Peiwei Z, et al. Identification and characterization of novel mutations in MOGS in a Chinese patient with infantile spams. Neurogenetics. 2020;21(2):97–104.
Zhu M, et al. Beta-mannosidosis mice: a model for the human lysosomal storage disease. Hum Mol Genet. 2006;15(3):493–500.
Nanba E, Suzuki K. Molecular cloning of mouse acid beta-galactosidase cDNA: sequence, expression of catalytic activity and comparison with the human enzyme. Biochem Biophys Res Commun. 1990;173(1):141–8.
Beutler E, Morrison M. Localization and characteristics of hexose 6-phosphate dehydrogenase (glucose dehydrogenase). J Biol Chem. 1967;242(22):5289–93.
Clarke JL, Mason PJ. Murine hexose-6-phosphate dehydrogenase: a bifunctional enzyme with broad substrate specificity and 6-phosphogluconolactonase activity. Arch Biochem Biophys. 2003;415(2):229–34.
Lavery GG, et al. Hexose-6-phosphate dehydrogenase knock-out mice lack 11 beta-hydroxysteroid dehydrogenase type 1-mediated glucocorticoid generation. J Biol Chem. 2006;281(10):6546–51.
Lavery GG, et al. Deletion of hexose-6-phosphate dehydrogenase activates the unfolded protein response pathway and induces skeletal myopathy. J Biol Chem. 2008;283(13):8453–61.
Gao X, et al. gamma-6-Phosphogluconolactone, a Byproduct of the Oxidative Pentose Phosphate Pathway, Contributes to AMPK Activation through Inhibition of PP2A. Mol Cell. 2019;76(6):857–871 e9.
Samland AK, Sprenger GA. Transaldolase: from biochemistry to human disease. Int J Biochem Cell Biol. 2009;41(7):1482–94.
Zhu, Z., et al., Exogenous Oleic Acid and Palmitic Acid Improve Boar Sperm Motility via Enhancing Mitochondrial Beta-Oxidation for ATP Generation. Animals (Basel), 2020. 10(4).
Fukasawa M, et al. Immunohistochemical localization of mitochondrial fatty acid beta-oxidation enzymes in rat testis. J Histochem Cytochem. 2010;58(2):195–206.
Rinaldo P, Matern D, Bennett MJ. Fatty acid oxidation disorders. Annu Rev Physiol. 2002;64:477–502.
Rogers GW, et al. Assessment of fatty acid beta oxidation in cells and isolated mitochondria. Curr Protoc Toxicol. 2014. 60;25 3 1–19.
Yoshimura Y, et al. Molecular cloning of rat acss3 and characterization of mammalian propionyl-CoA synthetase in the liver mitochondrial matrix. J Biochem. 2017;161(3):279–89.
Hallows WC, Lee S, Denu JM. Sirtuins deacetylate and activate mammalian acetyl-CoA synthetases. Proc Natl Acad Sci U S A. 2006;103(27):10230–5.
Fujino T, et al. Acetyl-CoA synthetase 2, a mitochondrial matrix enzyme involved in the oxidation of acetate. J Biol Chem. 2001;276(14):11420–6.
Yu S, et al. Molecular characterization, alternative splicing and expression analysis of ACSF2 and its correlation with egg-laying performance in geese. Anim Genet. 2016;47(4):451–62.
Fraisl P, et al. Murine bubblegum orthologue is a microsomal very long-chain acyl-CoA synthetase. Biochem J. 2004;377(Pt 1):85–93.
Cheng J, et al. ACSL4 suppresses glioma cells proliferation via activating ferroptosis. Oncol Rep. 2020;43(1):147–58.
Kane CD, et al. Expression, purification, and ligand-binding analysis of recombinant keratinocyte lipid-binding protein (MAL-1), an intracellular lipid-binding found overexpressed in neoplastic skin cells. Biochemistry. 1996;35(9):2894–900.
Maeda K, et al. Role of the fatty acid binding protein mal1 in obesity and insulin resistance. Diabetes. 2003;52(2):300–7.
Bravo-San Pedro JM, et al. Acyl-CoA-Binding Protein Is a Lipogenic Factor that Triggers Food Intake and Obesity. Cell Metab. 2019;30(4):754–767 e9.
Deberardinis RJ, Lum JJ, Thompson CB. Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J Biol Chem. 2006;281(49):37372–80.
Wang B, et al. New Ratios for Performance Improvement for Identifying Acyl-CoA Dehydrogenase Deficiencies in Expanded Newborn Screening: A Retrospective Study. Front Genet. 2019;10:811.
Burgin HJ, McKenzie M. Understanding the role of OXPHOS dysfunction in the pathogenesis of ECHS1 deficiency. FEBS Lett. 2020;594(4):590–610.
Hiltunen JK, et al. 17B-hydroxysteroid dehydrogenases as acyl thioester metabolizing enzymes. Mol Cell Endocrinol. 2019;489:107–18.
Nwosu ZC, et al. Liver cancer cell lines distinctly mimic the metabolic gene expression pattern of the corresponding human tumours. J Exp Clin Cancer Res. 2018;37(1):211.
Jia J, et al. Transcriptome Analysis of Choroid and Retina From Tree Shrew With Choroidal Neovascularization Reveals Key Signaling Moieties. Front Genet. 2021;12:654955.
Tabe Y, Lorenzi PL, Konopleva M. Amino acid metabolism in hematologic malignancies and the era of targeted therapy. Blood. 2019;134(13):1014–23.
Sakagishi Y. Alanine aminotransferase (ALT). Nihon Rinsho. 1995;53(5):1146–50.
Simmons RM, et al. Obesity increases hepatic glycine dehydrogenase and aminomethyltransferase expression while dietary glycine supplementation reduces white adipose tissue in Zucker diabetic fatty rats. Amino Acids. 2020;52(10):1413–23.
Kim D, et al. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature. 2015;520(7547):363–7.
Pedre B, Dick TP. 3-Mercaptopyruvate sulfurtransferase: an enzyme at the crossroads of sulfane sulfur trafficking. Biol Chem. 2021;402(3):223–37.
Kaushal K, et al. E3 Ubiquitin Ligase APC/C(Cdh1) Negatively Regulates FAH Protein Stability by Promoting Its Polyubiquitination. Int J Mol Sci. 2020;21(22).
Yang H, et al. Hypersuccinylacetonaemia and normal liver function in maleylacetoacetate isomerase deficiency. J Med Genet. 2017;54(4):241–7.
Vockley J, Ensenauer R. Isovaleric acidemia: new aspects of genetic and phenotypic heterogeneity. Am J Med Genet C Semin Med Genet. 2006;142C(2):95–103.
Yang L, et al. Identification of eight novel mutations and transcript analysis of two splicing mutations in Chinese newborns with MCC deficiency. Clin Genet. 2015;88(5):484–8.
Mack M, et al. Biochemical characterization of human 3-methylglutaconyl-CoA hydratase and its role in leucine metabolism. FEBS J. 2006;273(9):2012–22.
Bouchereau J, Schiff M. Inherited Disorders of Lysine Metabolism: A Review. J Nutr. 2020;150(Suppl 1):2556S-2560S.
Han Q, et al. Biochemical and structural characterization of mouse mitochondrial aspartate aminotransferase, a newly identified kynurenine aminotransferase-IV. Biosci Rep. 2011;31(5):323–32.
van de Ven S, et al. Long-term clinical outcome, therapy and mild mitochondrial dysfunction in hyperprolinemia. J Inherit Metab Dis. 2014;37(3):383–90.
Shang M, et al. Macrophage-derived glutamine boosts satellite cells and muscle regeneration. Nature. 2020;587(7835):626–31.
Holmes RS. Comparative and evolutionary studies of ALDH18A1 genes and proteins. Chem Biol Interact. 2017;276:2–8.
Rani N, et al. Functional annotation of putative fadE9 of Mycobacterium tuberculosis as isobutyryl-CoA dehydrogenase involved in valine catabolism. Int J Biol Macromol. 2019;122:45–57.
Zhang B, et al. The promising novel biomarkers and candidate small molecule drugs in kidney renal clear cell carcinoma: Evidence from bioinformatics analysis of high-throughput data. Mol Genet Genomic Med. 2019;7(5):e607.
Pode-Shakked N, et al. Clues and challenges in the diagnosis of intermittent maple syrup urine disease. Eur J Med Genet. 2020;63(6):103901.
Akram M. Citric acid cycle and role of its intermediates in metabolism. Cell Biochem Biophys. 2014;68(3):475–8.
Li Z, Zhang H. Reprogramming of glucose, fatty acid and amino acid metabolism for cancer progression. Cell Mol Life Sci. 2016;73(2):377–92.
Signes A, Fernandez-Vizarra E. Assembly of mammalian oxidative phosphorylation complexes I-V and supercomplexes. Essays Biochem. 2018;62(3):255–70.
Judge A, Dodd MS. Metabolism. Essays Biochem. 2020;64(4):607–47.
Nolfi-Donegan D, Braganza A, Shiva S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Biol. 2020;37:101674.
Palmieri F. The mitochondrial transporter family SLC25: identification, properties and physiopathology. Mol Aspects Med. 2013;34(2–3):465–84.
Piomboni P, et al. The role of mitochondria in energy production for human sperm motility. Int J Androl. 2012;35(2):109–24.
Fukushima T, et al. Three novel mutations in the carnitine-acylcarnitine translocase (CACT) gene in patients with CACT deficiency and in healthy individuals. J Hum Genet. 2013;58(12):788–93.
Brym P, et al. Gene promoter polymorphisms in boar spermatozoa differing in freezability. Theriogenology. 2021;166:112–23.
Porcelli V, et al. Asymmetric dimethylarginine is transported by the mitochondrial carrier SLC25A2. Amino Acids. 2016;48(2):427–36.
Palmieri F, Pierri CL. Mitochondrial metabolite transport. Essays Biochem. 2010;47:37–52.
Mizuarai S, et al. Identification of dicarboxylate carrier Slc25a10 as malate transporter in de novo fatty acid synthesis. J Biol Chem. 2005;280(37):32434–41.
Buffet A, et al. Germline Mutations in the Mitochondrial 2-Oxoglutarate/Malate Carrier SLC25A11 Gene Confer a Predisposition to Metastatic Paragangliomas. Cancer Res. 2018;78(8):1914–22.
Hoshino A, et al. The ADP/ATP translocase drives mitophagy independent of nucleotide exchange. Nature. 2019;575(7782):375–9.
Sastri M, et al. Sub-mitochondrial localization of the genetic-tagged mitochondrial intermembrane space-bridging components Mic19, Mic60 and Sam50. J Cell Sci. 2017;130(19):3248–60.
Zhang Y, et al. Mitoguardin Regulates Mitochondrial Fusion through MitoPLD and Is Required for Neuronal Homeostasis. Mol Cell. 2016;61(1):111–24.
Tondera D, et al. The mitochondrial protein MTP18 contributes to mitochondrial fission in mammalian cells. J Cell Sci. 2005;118(Pt 14):3049–59.
Bastian C, et al. Preserving Mitochondrial Structure and Motility Promotes Recovery of White Matter After Ischemia. Neuromolecular Med. 2019;21(4):484–92.
Levy S, et al. Identification of LACTB2, a metallo-beta-lactamase protein, as a human mitochondrial endoribonuclease. Nucleic Acids Res. 2016;44(4):1813–32.
Qu H, et al. STOML2 as a novel prognostic biomarker modulates cell proliferation, motility and chemo-sensitivity via IL6-Stat3 pathway in head and neck squamous cell carcinoma. Am J Transl Res. 2019;11(2):683–95.
Purushottam Dharaskar S, et al. Mitochondrial chaperone, TRAP1 modulates mitochondrial dynamics and promotes tumor metastasis. Mitochondrion. 2020;54:92–101.
Nonn L, et al. The absence of mitochondrial thioredoxin 2 causes massive apoptosis, exencephaly, and early embryonic lethality in homozygous mice. Mol Cell Biol. 2003;23(3):916–22.
Thompson K, et al. OXA1L mutations cause mitochondrial encephalopathy and a combined oxidative phosphorylation defect. EMBO Mol Med. 2018;10(11).
Miki K. Energy metabolism and sperm function. Soc Reprod Fertil Suppl. 2007;65:309–25.
Vertika S, Singh KK, Rajender S. Mitochondria, spermatogenesis, and male infertility - An update. Mitochondrion. 2020;54:26–40.
Ma J, et al. iProX: an integrated proteome resource. Nucleic Acids Res. 2019;47(D1):D1211–7.
Acknowledgements
This work was supported by the grants from the National Natural Science Foundation of China (81430028 to Z.W., 81901529 to C.S., 81671538 to H.Z., 81900799 to L.T.); the grants from the Science and Technology Commission of Shanghai Municipality (19DZ2280500 to Z.W., 18ZR1423500 to C.S., 19YF1430400 to L.T.).
Funding
Zhugang Wang, Chunling Shen.
Resources: Zhugang Wang.
Investigation: Haoyang Ge, Chaojie Li, Xiaohong Zhang.
Methodology: Lingyun Tang, Hongxing Zhang.
Supervision: Zhugang Wang, Chunling Shen.
Validation: Yan Shen, Shunyuan Lu.
Writing-original draft: Wenfeng Xiong, Chunling Shen.
Writing-review & editing: Zhugang Wang, Chunling Shen.
Author information
Authors and Affiliations
Contributions
Conceptualization: Zhugang Wang, Chunling Shen.
Data curation: Wenfeng Xiong, Chunling Shen.
Formal analysis: Wenfeng Xiong.
Corresponding authors
Ethics declarations
Disclosure statement
The authors declare no conflicts of interest.
Additional information
Summary Statement
We identified the global proteome profiles of wt and Prss37−/− mice in the testis and sperm, and found that PRSS37 specifically modulates energy metabolism related pathways to participate in male mouse fertility.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xiong, W., Ge, H., Shen, C. et al. PRSS37 deficiency leads to impaired energy metabolism in testis and sperm revealed by DIA-based quantitative proteomic analysis. Reprod. Sci. 30, 145–168 (2023). https://doi.org/10.1007/s43032-022-00918-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43032-022-00918-x