
Abstract
Congenital disorders of glycosylation (CDGs) are a genetically heterogeneous group of disorders caused by the defects in the synthesis and processing of glycoproteins. CDG is caused by mannosyl-oligosaccharide glucosidase (MOGS) deficiency, and is an extremely rare type, and only six patients have been reported. Here, we report a patient from China with facial dysmorphism, infantile spams, developmental delay, low vison, and abnormal liver function and low immunoglobulin. Brain MRI showed hypoplasia of the corpus callosum and slightly wide sulci at bilateral frontal parietal lobes. Compound heterozygous mutations of (c.1694G>A: R565Q and c.1619G>A: R540H) in exon 4 of MOGS gene (NM_006302.2) were identified by whole exome sequencing. Further investigation showed that the gene expression of MOGS in patients’ peripheral blood was decreased. We observed that two mutations were associated with lower protein expression of MOGS, cell growth, and cell cycle in transiently transfected Hela cells. We also noticed that cell cycle–related proteins, β-catenin, cyclin D1, and C-myc, were decreased in mutant cells. In conclusion, our study suggested whole exome sequencing, and genes associated with CDGs should be analyzed in patients with infantile spams and multiple system involvement, and mutant MOGS–impaired cell cycle progression. Our work broadens the mutation spectrum of MOGS gene.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Congenital disorders of glycosylation (CDGs) are a group of metabolic disorders that result from abnormal glycosylation of proteins or lipids [1]. Glycosylation is the principal form of post-translational modification of proteins and lipids, and half of all cellular proteins are glycosylated and glycosylation occurs in every cell in every organ; thus, CDGs usually is a multisystem disease and the central nervous system is mainly involved, manifesting developmental delay, seizures, intellectual disability, hypotonia, and microcephal [2, 3], but other organs can also be affected, including the liver, heart, eyes, skeleton, and immune system [4,5,6].
Glycosylation can be classified into N-glycosylation and O-glycosylation, depending on whether the glycans are linked to the amino group of asparagine (N-glycosylation) or the hydroxyl group of serine or threonine (O-glycosylation) [7]. Traditionally, CDGs were divided into CDG-I and CDG-II, according to the affected step in the synthesis and/or of the processing of N-glycans [8]. Recently, novel nomenclature, the gene name followed by “-CDG”, has been used.
Until now, more than 125 types of CDG have been reported [9, 10]. MOGS-CDG (MIM:606056) is caused by mannosyl-oligosaccharide glucosidase (MOGS) deficiency, and is an extremely rare CDG type, and to our knowledge, there is only 6 cases from 4 families reported [4, 10,11,12,13]. MOGS gene is located on chromosome 2p13.1, encompassing 5 exons. MOGS is expressed in the endoplasmic reticulum and is involved in the trimming of N-glycans by cleaving the distal alpha 1,2-linked glucose residue from the Glc3Man9GlcNAc2 oligosaccharide precursor [14]. This step is critical to the subsequent processing of N-glycans.
Analysis of serum transferrin isoelectric focusing (TIEF) was performed to help the diagnosis of CDGs; however, normal or inconclusive transeferrin isofocusing has been observed in various CDG subtypes [11, 15]. Mass spectrometry applied to enzymatically removed N-glycans or to entire apoC-III is also a valuable tool in the diagnosis and definition of CDG-II [16, 17]. In recent years, broader accessibility and higher rates of conclusive diagnosis have made next-generation sequencing a preferred approach for the solving undiagnosed genetic disorders [18]. Herein, we report a new Chinese patient with developmental delay, seizures, intellectual disability, and low levels of immunoglobulins. Two compound heterozygous mutations in MOGS gene were identified in this patient by using whole exome sequencing (WES). In order to investigate the effect of these two mutations, we examined the expression of MOGS gene and proteins in the peripheral blood of patient and Hela cells, respectively. Our results showed that these mutations were associated with lower expression of MOGS gene and protein, decreased the cell proliferation, probably via inhibiting Wnt/β-catenin signaling pathway.
Patient and methods
Patient information
The patient is the only child of healthy nonconsanguineous parents without personal or familial medical history. She was born at 40 weeks of gestation by cesarean delivery without complications. Her birth weight was 3200 g. She had no significant perinatal and postnatal problems. During the first month, she developed infantile spasms that required hospital admission. She was treated with levetiracetam and steroid showed slight improvement. She was noticed that she had low vision and was unable to follow objects. She experienced surgery left inguinal hernia repair at age of 4 months. She had recurrent upper respiratory infection and septicopyemia. She was first seen in the Precision Medical Clinic at age of 1 year. She had dysmorphic features, poor interaction, developmental delay, hypotonia, and low vision (Table 1). Dysmorphic features included full moon face, narrow forehead, small palpebral fissure, broad nose, and generalized edema (Fig. 1a). She was unable to raise her head steadily and roll over. She did not show interest in strangers or surroundings. She was extensively investigated by blood biochemistry examinations, chromosomal microarray, whole exome sequencing, echocardiography, electroencephalogram, and brain MRI. At the age of 4 years, she was unable to sit and turn over, and she seldom got viral or bacterial infection. This study has been approved by the institutional review board of Wuhan Children’s Hospital, Tongji Medical College, Huazhong University of Science & Technology.

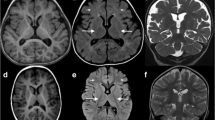
a Abnormal facial appearance; b hypoplasia of the corpus callosum and slightly wide sulci at bilateral frontal parietal lobes in brain MRI; c Sanger sequencing of MOGS mutations in the family in our study; d Conservation analysis of MOGS protein among different species. The position of the mutations at amino acids 540 and 565 is indicated by a blue bar and highly conserved throughout all indicated species; eMOGS RNA level was lower in peripheral blood of the patient compared with that of normal control. Both wide-type and mutated MOGS were expressed in Hela cells, and the mutant MOGS expression decreased, RNA level f, protein level g. P: patient, N, normal controls; WT, wild type. The results of three independent experiments are expressed as the mean + standard deviation (*P < 0.05; **P < 0.01)
Chromosomal microarray assay and whole exome sequencing
Genomic DNAs of the patient and his parents were extracted from leukocytes of whole blood samples using the QIAamp Blood DNA mini kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Genome-wide copy number analysis was performed using Illumina Human Cyto-SNP12 BeadChip (Illumina, San Diego, CA). The data from the images were analyzed using KaryoStudio v1.4. WES and subsequent data analysis were conducted with the help of the third party medical testing laboratory (Chigene Lab, Beijing, China) on the patient. Candidate variants were confirmed by Sanger sequencing using self-designed primers. We conducted a conservative analysis of mutation sites using MEGA software (https://www.megasoftware.net/).
MOGS plasmid construction and cell transfection
Hela cells were grown in DMEM supplemented with 10% fetal bovine serum (Gibco). The MOGS coding sequence was amplified from Hela cells with the oligonucleotides 5′-ATG GCT CGG GGC GAG CGG-3′ and 5′-TCA GTA GTC TTC AGC CAT GGC CAG-3′ using Pfu DNA polymerase (Transgen Biotech). The wild type MOGS (WT-MOGS) construct was obtained by inserted the amplified fragment into the pCMV.2b expression plasmid using HindIII and Xho I restrictions sites. To generate MOGS-mutated proteins (R540H-MOGS, R565Q-MOGS), site-directed mutagenesis was performed with the oligonucleotides 5′-GAA AGG CCT TGC CCC ACC TG C ATG CCT GG-3′ and 5′-GCT GGC GGG GAC AGG ACC C GCC TTA C-3′ using overlap PCR. All the positive clones were verified for the correct sequence by Sanger sequencing. Hela cells cultured in a 6-well plate were transfected with 2 μg plasmids using the Lip2000 (Invitrogen) according to the manufacturer’s instructions.
MOGS expression analysis using real-time PCR
Peripheral blood mononuclear cells (PBMCs) were purified using a gradient of Ficoll-Hypaque, and total RNA was extracted from PBMCs by using Trizol Reagent (Invitrogen, USA). The first complementary DNA was synthesized from RNA using reverse transcriptase (TAKARA, Dalian). Real-time PCR was performed using SYBR Green PCR Kit (TaKaRa, Dalian) and β-actin as an internal control.
Western blot
Cells were lysed in 1% NP-40 lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, and 0.5% sodium deoxycholate) for 30 min on ice, and then centrifugated at 12000 rpm for 10 min. Protein concentration was determined by BCA assay (Thermo Fisher Scientific), and 20 mg total protein was separated by 10% SDS-PAGE and subsequently transferred to PVDF membrane. After blocking in 3% BSA (bovine serum albumin), membranes were probed with the following Abs: anti-Flag, anti-cMyc, anti-Cyclin D1 (Santa Cruz Biotechnology, Inc.), anti-AKT, anti-N-cadherin (Cell Signaling Technology, Inc.), and Bound Abs were detected using secondary Abs (Southern Biotech) and enhanced chemiluminescence (ECL, Thermo Fisher Scientific).
Confocal microscopy
To examine the localization of MOGS, a co-localization analysis of MOGS with endoplasmic reticulum(ER) marker (PDI) was performed. Cells adhered to poly-L-lysine-coated slides were fixed for 15 min with 3% paraformaldehyde, and then permeabilized for 15 min in 0.1% Triton X-100/PBS (phosphate buffer saline). After blocking for 60 min in 3% BSA/PBS, cell spots were incubated with the anti-MOGS and anti-PDI antibody (Proteintech) for 60 min. Cells were washed in PBS for 3 times and then incubated with goat anti–rabbit secondary Ab conjugated to Alexa Fluor 488 and Alexa Fluor 555 (Invitrogen). Cells were then counterstained with DAPI and examined by confocal microscopy (LSM800, ZEISS).
Proliferation analysis by cell counting kit-8 assay
Cells (2000 cells/well) were seeded into the 96-well plates and then incubated at 37 °C in 5% CO2 for 0 h, 6 h, 12 h, 24 h, 36 h, and 48 h. Cells were added with 10 μl cell counting kit (CCK)-8 solutions (CCK-8, Beyotime, China) and incubated at 37 °C for 2 h according to the manufacturer’s protocol. Optical density was detected by a microplate reader (Bio-Rad, Hercules, CA, USA) at 450 nm.
Cell cycle analysis
Cell cycle analysis was conducted using Cell Cycle and Apoptosis Analysis Kit (Beyotime, China) following manufacturer’s instructions. In brief, Hela cells were cultured in serum-free medium for 24 h, and then cultured in serum-containing medium for 48 h. Cells were harvested and fixed with 70% cold ethanol at 4 °C overnight. DNA was stained with propidium iodide for 30 min at a 37 °C incubator. Cell cycle distribution was then detected using flow cytometry (Beckman Coulter Inc., FL, USA). The percentages of cells in G0/G1, S, and G2/M phases were calculated using ModFitLT V5.0 Software.
Results
Clinical investigations
Blood biochemistry examinations showed abnormal liver function with 55 U/L of glutamic-oxaloacetic transaminase (reference, 10~44 U/L), abnormal myocardial function with 255 U/L of CK (reference, 20~250 U/L), and 68 U/L of CK-MB (0~25 U/L), low immunoglobulin (13.7 g/L; reference, 20~40 g/L), impaired lipid metabolism with decreased HDL (0.75 mmol/L; reference, 0.9~1.74 mmol/L) and ApoA1(0.92 g/L; reference, 1.1~1.95 g/L), increased urine acid (562.7umol/L; reference, 90~420 μml/L). Blood sugar and thyroid function were normal. Echocardiography showed patent foramen ovale or atrial septal defect, electroencephalogram indicated atypic hypsarrhythmia. Brain MRI displayed thin corpus callosum and slightly wide sulci at bilateral frontal parietal lobes (Fig. 1b).
Chromosomal microarray assay and WES analysis
Chromosomal microarray assay did not detect any microdeletion/microduplication in chromosomes. Further, WES and subsequent data analysis revealed two heterozygous mutations (c.1694G>A: R565Q and c.1619G>A:R540H) in exon 4 of MOGS (NM_006302.2) in the patient, and Sanger sequencing confirmed these two mutations in the patient and found that the parents were heterozygous for one of the each mutated alleles (Fig. 1C). These two variants is absent from the variation database (i.e., gnomAD, ClinVar, and 1000 genomes). Bioinformatic analysis showed that sites are conserved among different species (Fig. 1D) and the amino acid substitutions probably damaging, with a high score of PolyPhen-2 server (= 1) and a SIFT tolerance index of 0.0 and 0.003.
MOGS protein expression and subcellular localization
To investigate whether these two MOGS mutations affect the gene expression of MOGS in the patient, quantitative real-time PCR was performed by using RNA extracted from PBMCs of the patient and three age-matched normal controls. As shown in Fig. 1E, the gene expression of MOGS was lower in patient than that of controls. We further observed the effect of these two mutations on the expression and localization of MOGS protein by in vitro experiment. Four kinds of plasmids, control (pCMV.2b), WT-MOGS, R540H-MOGS, and R565Q-MOGS, were transfected into Hela cells, 24 h later, transfected cells were harvested and lysed or fixed for Western blot or immunofluorescence staining. As shown in Fig. 1f and g, these two mutants dramatically decreased the expression of MOGS gene and protein.
After examining the localization of mutated MOGS, we found that MOGS was co-localized with ER marker (PDI) (Fig. 2), indicating that these two mutations do not alter the cellular localization of MOGS protein.

MOGS protein expression and subcellular localization
MOGS mutations suppressed cell cycle and proliferation
Our patient displayed developmental delay, and brain MRI showed hypoplasia of brain, especially the corpus callosum, and the regulation of cell cycle and growth arrest play important roles in proliferation, differentiation and maturation of neurons; thus, we further investigate the effects of mutated MOGS on cell proliferation and cell cycle. Hela cells were transfected with mutated MOGS and wide-type plasmid, respectively, then analyzed for proliferation after incubating with CCK-8 solutions for 2 h. As shown in Fig. 3a, the proliferations of mutant MOGS (R540H and R565Q) were inhibited compared with wide-type controls.

Inhibited cell proliferation a, cell cycle G1 arrest b, and decreased expression of cell cycle–related proteins, β-catenin, c-myc, and cyclinD1 c in Hela cells transfected with mutant MOGS, compared with wild type. GAPDH serves as a loading control; d scheme of the distribution of MOGS mutations. The two mutations in red were reported in present study. The results of three independent experiments are expressed as the mean + standard deviation (*P < 0.05; **P < 0.01 and NS, no significance)
In order to figure out whether the inhibition of cell proliferation was induced by impaired cell cycle, we conducted flow cytometric analysis for cell cycle, as shown in Fig. 3b, compared with control cell, the population of G1 phase cell was increased, accompanied with a concomitant decrease of cell population in S-phase in mutant MOGS cell lines (R540H and R565Q). Cell cycle-related proteins, such as β-catenin, Cyclin D1, and C-myc significantly decreased in mutated MOGS compared with controls (Fig. 3c). These results collectively demonstrated that mutant MOGS affected cell proliferation and G1 cell cycle arrest, which was accompanied by decreased cell cycle–related proteins.
Discussion
Glycosylation is an essential biological process for various protein or lipid modifications, and approximately 2% of human genes encode glycosylation-related proteins [19]; therefore, it is not surprising that CDGs are expanding rapidly, and comprised of more than 125 genetic disorders [9, 10]. CDGs affect many organs and lack uniformity in clinical manifestations, and it is very difficult to make a definitive diagnosis. However, next-generation sequencing offers an unprecedented opportunity to unravel of many undiagnosed cases and expand the genetic spectrum of CDGs [18]. In this study, we identified two novel variants in MOGS gene in a patient with infantile spasm as initial symptom by using WES. Bioinformatic analysis and further in vitro experiment suggested that these variants are pathogenic.
MOGS-CDG is very rare and only 7 cases from 4 families and a total of 12 variants (Table 1) were found [10,11,12,13], including our study. The clinical presentation in these patients seems relatively uniform regardless of the mutations in MOGS. Our patient had marked generalized hypotonia, hypomotility, and facial dysmorphism, including prominent occiput, small palpebral fissure, broad nose, retrognathia, and generalized edema, in accordance with previous report. Likewise, hypoimmunoglobulemia, reported in all patients with MOGS-CDG, was observed in our case, which could be due to a reduced circulating immunoglobulin half-life [14]. Despite hypogammaglobulinemia in patients with MOGS-CGD, they do not have clinical evidence of recurrent infections because the impaired N-glycosylation of the patients compromises their ability to support efficient replication and cellular entry of viruses [4]. However, the reason for the resistance to bacterial infections remains unclear. In addition, hepatomegaly or abnormal liver function, seizure, thin corpus callosum, and ophthalmological problem were presented in our patient. The age of the surviving patients ranged from a few months to 11 years at their last evaluation. One patient died at 74 days of age [11], and two patients, a 6-year-old sister and an 11-year-old brother from one family, had survived into late childhood [12]. The longer survival of these two individuals could attribute to mitotic intragenic recombination in compound heterozygous alleles of MOGS gene within somatic cells, which causes reversion of a wild-type allele and the survival of these cells with wild-type allele could ameliorate the clinical manifestations [12].
MOGS is located in endoplasmic reticulum, and it has a vital role in the processing of N-glycans by cleaving the outermost glucose from the Glc3Man9GlcNAc2. N-glycosylation is the most common covalent modification of proteins in human cells and it affects many biological processes, such as protein folding, stability, trafficking, cell-cell/cell-matrix/receptor-ligand interaction, interacellular signaling, cell growth and differentiation. Therefore, we hypothesized that MOGS mutations may result in decreased gene and protein expression or cellular localization of MOGS, and then affect these biological processes. To test this hypothesis, we first investigated whether the patient’s MOGS gene expression is lower than that of healthy controls. Because the fibroblast cells were not available, we obtained lymphocytes from peripheral blood, and we found that downregulated MOGS gene expression in our patient. Next, we further examined the expressions of gene and protein and localization of MOGS by transiently transfecting Hela cells using wild-type and mutant MOGS plasmids, and we found that these mutations resulted in lower expression levels of RNA and protein in Hela cells. However, we did not see obvious change in the cellular localization of mutant proteins. Developmental delay and hypoplasia of brain, especially the corpus callosum, were observed in our patient, and the dysregulation of cell cycle and growth arrest play important roles in proliferation, differentiation, and maturation of neurons [20]. Our study showed that MOSG mutations impaired cell cycle and led to growth arrest in G1, and several cell cycle-related proteins, including β-catenin, cyclin D1, and C-Myc, were downregulated in mutant cells. All these findings suggest that MOGS mutations exert negative effects on MOGS expression, cell proliferation, and cell cycle, which probably is related to β-catenin signaling pathway. As C-myc, cyclin D1 and β-catenin are cytosolic and O-glycosylated whereas MOGS is ER-localized and involved in the processing of N-glycans [21, 22]; thus, it seem unlikely that mutant MOGS protein directly affect these cell cycle-related proteins. However, Gallo et al. reported that abrogation of glucosidase I-mediated glycoprotein results in a distorted cell wall and in the absence of underlying ER membranes in yeasts [23]; therefore, we cannot exclude the possibility that mutant MOGS impaired ER membrane, and then affect the synthesis of proteins, including these cell cycle–related proteins. It needs to be further studied how MOGS mutations affect these proteins, and then interferer cell cycle via β-catenin signaling.
In conclusion, we identified and characterized two compound heterozygous variants in MOGS gene in a patient with infantile spasm, developmental delay, and multi-organ involvement. Our work broadens the mutation spectrum of MOGS gene and also helps to understand the effects of these two novel mutations.
References
Van Scherpenzeel M, Willems E, Lefeber DJ (2016) Clinical diagnostics and therapy monitoring in the congenital disorders of glycosylation. Glycoconj J 33(3):345–358. https://doi.org/10.1007/s10719-015-9639-x
Freeze HH (2006) Genetic defects in the human glycome. Nat Rev Genet 7(7):537–551. https://doi.org/10.1038/nrg1894
Freeze HH, Eklund EA, Ng BG, Patterson MC (2012) Neurology of inherited glycosylation disorders. Lancet Neurol 11(5):453–466. https://doi.org/10.1016/S1474-4422(12)70040-6
Sadat MA, Moir S, Chun TW, Lusso P, Kaplan G, Wolfe L, Memoli MJ, He M, Vega H, Kim LJY, Huang YHN (2014) Glycosylation, hypogammaglobulinemia, and resistance to viral infections. Curr Opin Cell Biol 370(17):82–91. https://doi.org/10.1038/jid.2014.371
Correspondence P (2009) Conotruncal heart defects in three patients with congenital disorder of glycosylation type Ia (CDG Ia). J Med Genet 46(4):287–289. https://doi.org/10.1136/jmg.2008.057620
Marques-da-Silva D, Dos Reis Ferreira V, Monticelli M et al (2017) Liver involvement in congenital disorders of glycosylation (CDG). A systematic review of the literature. J Inherit Metab Dis 40(2):195–207. https://doi.org/10.1007/s10545-016-0012-4
Leroy JG (2006) Congenital disorders of N-glycosylation including diseases associated with O- as well as N-glycosylation defects. Pediatr Res 60(6):643–656. https://doi.org/10.1203/01.pdr.0000246802.57692.ea
Jaeken J, Hennet T, Matthijs G, Freeze HH (2009) CDG nomenclature: time for a change! Biochim Biophys Acta 1792:825–826. https://doi.org/10.1016/j.bbadis.2009.08.005
Freeze HH, Chong JX, Bamshad MJ, Ng BG (2014) Solving glycosylation disorders: fundamental approaches reveal complicated pathways. Am J Hum Genet 94(2):161–175. https://doi.org/10.1016/j.ajhg.2013.10.024
Kim YM, Seo GH, Jung E, Jang JH, Kim SZ, Lee BH (2018) Characteristic dysmorphic features in congenital disorders of glycosylation type IIb. J Hum Genet 63(3):383–386. https://doi.org/10.1038/s10038-017-0386-7
De Praeter CM, Gerwig GJ, Bause E et al (2000) A novel disorder caused by defective biosynthesis of N-linked oligosaccharides due to glucosidase I deficiency. Am J Hum Genet 66(MIM 266265):1744–1756. https://doi.org/10.1086/302948
Kane MS, Davids M, Adams C, Wolfe LA, Cheung HW, Gropman A, Huang Y, Ng BG, Freeze HH, Adams DR, Gahl WA, Boerkoel CF (2016) Mitotic intragenic recombination: a mechanism of survival for several congenital disorders of glycosylation. Am J Hum Genet 98(2):339–346. https://doi.org/10.1016/j.ajhg.2015.12.007
Li M, Xu Y, Wang Y, Yang X-A, Jin D (2019) Compound heterozygous variants in MOGS inducing congenital disorders of glycosylation (CDG) IIb. J Hum Genet 64(3):265–268. https://doi.org/10.1038/s10038-018-0552-6
Monticelli M, Ferro T, Jaeken J, dos Reis Ferreira V, Videira PA (2016) Immunological aspects of congenital disorders of glycosylation (CDG): a review. J Inherit Metab Dis 39(6):765–780. https://doi.org/10.1007/s10545-016-9954-9
Scott K, Gadomski T, Kozicz T, Morava E (2014) Congenital disorders of glycosylation: new defects and still counting. J Inherit Metab Dis 37(4):609–617. https://doi.org/10.1007/s10545-014-9720-9
Barbosa EA, Fontes NC, Santos SCL et al (2019) Relative quantification of plasma N-glycans in type II congenital disorder of glycosylation patients by mass spectrometry. Clin Chim Acta 492(February):102–113. https://doi.org/10.1016/j.cca.2019.02.013
Bruneel A, Cholet S, Drouin-garraud V et al (2018) Complementarity of electrophoretic, mass spectrometric and gene sequencing techniques for the diagnosis and characterization of congenital disorders of glycosylation. Electrophoresis 39(24):3123–3132. https://doi.org/10.1002/elps.201800021
Jones MA, Rhodenizer D, da Silva C, Huff IJ, Keong L, Bean LJH, Coffee B, Collins C, Tanner AK, He M, Hegde MR (2013) Molecular diagnostic testing for congenital disorders of glycosylation (CDG): detection rate for single gene testing and next generation sequencing panel testing. Mol Genet Metab 110(1–2):78–85. https://doi.org/10.1016/j.ymgme.2013.05.012
Apweiler R (1999) On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta 1473(1):4–8
Kawauchi T, Shikanai M, Kosodo Y (2013) Extra-cell cycle regulatory functions of cyclin-dependent kinases (CDK) and CDK inhibitor proteins contribute to brain development and neurological disorders. Genes Cells:176–194. https://doi.org/10.1111/gtc.12029
Masclef L, Dehennaut V, Mortuaire M, Schulz C, Boyce M (2019) Cyclin D1 stability is partly controlled by O –GlcNAcylation. Front Endocrinol (Lausanne) 10(February):1–12. https://doi.org/10.3389/fendo.2019.00106
Stichelen SO, Dehennaut V, Buzy A et al (2014) O-GlcNAcylation stabilizes b-catenin through direct competition with phosphorylation at threonine. FASEB J 41:3325–3338. https://doi.org/10.1096/fj.13-243535
Gallo GL, Valko A, Aramburu SI, Etchegaray E, Völker C, Parodi AJ, D’Alessio C (2018) Abrogation of glucosidase I-mediated glycoprotein deglucosylation results in a sick phenotype in fission yeasts: model for the human MOGS-CDG disorder. J Biol Chem 293:19957–19973. https://doi.org/10.1074/jbc.RA118.004844
Funding
This research was supported in part by the National Key Research and Development Program of China (No. 2016YFC1306202), Wuhan Yellow Crane (Medicine and Healthcare) (2016-01), and Hubei Health and Family Planning Commission (WJ2015MB247).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interests
The authors declare that they have no competing interests.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Peiwei Zhao, Peng, X., Luo, S. et al. Identification and characterization of novel mutations in MOGS in a Chinese patient with infantile spams. Neurogenetics 21, 97–104 (2020). https://doi.org/10.1007/s10048-019-00590-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10048-019-00590-5