
Abstract
Citrus tristeza virus (CTV) infected plant samples from the North Eastern states of India namely Arunachal Pradesh (AR1), Assam (AS1), Nagaland (NL1), Meghalaya (M1), Sikkim (SK1) and Tripura (TR1) were examined through double antibody sandwich enzyme linked immunosorbent assay and reverse transcriptase polymerase chain reaction using coat protein (CP) gene specific primers, yielding amplicon of 672 base pair for all the isolates. The sequence analysis revealed nucleotide (nt) and amino acid (aa) sequence similarity with other exotic CTV isolates. AR1 and AS1 shared similarity with T3 isolate (Florida) at 98–99% and 95–99% for nt-aa respectively, whereas, M1 showed 99% similarity with stem pitting isolate B165 (Bangalore). SK1 and NL1 were 95–97% and 97–99% similar with Hawaiian isolate HA at nt-aa level. TR1 showed 97% homology with seedling yellowing Israeli isolate VT. The CP gene sequence based phylogenetic analysis grouped present isolates into four clades with reference severe isolates (i.e. VT, B165, HA and T3), indicating their probable evolution. TR1 and NL1 are the putative recombinant isolates and ASI and SK1 were the major and minor parents respectively. This might be due to the conserved nature of CP gene. Moreover, the CP gene sequences of present isolates showing nucleotide identity to the VT, T3 and B165 isolates also suggest the prevalence of severe CTV isolates in Khasi mandarin growing areas comes under North East (NE) region of India.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Citrus tristeza virus (CTV) is one of the most vicious pathogen of citrus. This phytopathogen has been reported to affect millions of citrus plants all over the world since the last 70 years (Bar-Joseph and Dawson 2008). It belongs to the genus Closterovirus and Rutaceae family. The CTV virion particle appears to be long and filamentous in structure (2000 × 10–12 nm) (Dawson and Mooney 2000). It contains single stranded positive sense RNA (~ 19.3 kb), which is the largest genome of any known plant virus. Approximately, 97% of the virion body is encapsidated by a major coat protein (CP), whereas a minor CP (CPm) coats the 5′ end of tail (Satyanarayana et al. 2004). The CTV genome consists of 12 open reading frames (ORFs) along with 5′ and 3′ terminal untranslated regions (UTRs) of about 107 and 273 nt respectively (Karasev et al. 1995). During the past few years seven CTV reference genotypes such as T3, T30, T36, VT, HA16-5, B165 and trifoliate resistance breaking (RB) have also been reported (Vives et al. 1999; Harper et al. 2010; Melzer et al. 2010; Roy et al. 2013).
This phloem-limited Closterovirus, has only been documented in only two host plant genera i.e. Citrus and Fortunella (Moreno et al. 2008). The virus has a complex nature probably because of the long life span of the host plant or due to continuous vertical and horizontal viral transmission through grafting and aphids respectively (Vives et al. 2005; Hilf et al. 2005). Different methods have been used to differentiate and characterize the CTV isolates (Niblett et al. 2000; Roy et al. 2003). The phenotypic observation on the CTV induced symptoms revealed a greater variation in the intensity of CTV infestation in different citrus hosts which differ according to the type, biological characteristics and aphid transmissibility of different CTV isolates (Hilf et al. 2005). But, the phenotypic observations based on certain qualitative and quantitative parameters alone cannot conclusively determine CTV infection and differentiation between different isolates or strains is difficult (Ruiz-Ruiz et al. 2007). Therefore, the genomic sequence analysis is a more appropriate and globally accepted procedure for CTV isolates differentiation and genetic variation studies (Rubio et al. 2001). The nt sequences of the CP gene of diverse strains of CTV has been determined and sequence variability has been found to be related to the types of symptoms caused by CTV strains (Barzegar et al. 2006; Jiang et al. 2008; Harper et al. 2009). Although, these studies highlighted new strategies for plant virology, only few of the researchers has done work in that direction and the molecular determinants of different symptoms still remain unknown.
In India, citrus is the third most important fruit crop after banana and mango (Anonymous 2014). It is cultivated in all of the four geographical zones: North East, North West, Central and South, with an acreage of 91,518 hectare and production of 1,171,500 tonnes (FAOSTAT 2017). In these zones, more than one million citrus trees are reported to be CTV infected with an estimated disease incidence of 16–90% (Biswas 2008). NE regions of India including the countries bordering this area are supposed to be the probable origin of CTV and its vector Toxoptera citricidus (Ahlawat 2007). Most of the citrus species are believed to be native of Southeast Asian tropical and subtropical regions which also include the NE part of India and the region between China and India (Bar-Joseph and Dawson 2008; Moreno et al. 2008). Khasi mandarin is a high value crop cultivated around 1600 ha area of NE Himalayan region including the Darjeeling district of West Bengal and contributes significantly to the small farm economy (Ghosh and Singh 1993). But, it has got very less attention towards CTV infection and very limited information as well as experimental data is available for the incidence and spread of this virus. In spite of the probable origin place of CTV only three reports are available for CTV in NE states; out of which one is from Sikkim (Kishore et al. 2010) and two reports from Assam and Meghalaya (Biswas et al. 2014; Borah et al. 2014). Moreover, the molecular characterization and determination of genetic diversity of CTV in Khasi mandarin from Assam, Arunachal Pradesh, Meghalaya, Manipur, Mizoram, Nagaland, Tripura and Sikkim has not yet been reported.
The present study also describes molecular characterization of CTV infecting Khasi mandarin of Tripura, Nagaland and Arunachal Pradesh States which is the first extensive study of CTV in NE Khasi mandarin to the best of our knowledge. Findings presented in this study will be helpful in formulating molecular based management strategy against CTV in India.
Materials and methods
Samples collection
Leaf samples and twigs of Khasi mandarin suspected to be infected with CTV were collected from six NE states of India viz., Arunachal Pradesh, Meghalaya (from Khasi Hills Division), Assam, Nagaland, Sikkim and Tripura. One twig from suspected CTV infected plant of Khasi mandarin (Citrus reticulata Blanco) showing symptoms of leaf yellowing and vein clearing characteristics of CTV (Borah et al. 2012) were collected.
ELISA and RT–PCR based detection
The double antibody sandwich enzyme linked immunosorbent assay (DAS-ELISA) was performed (Clark and Adams 1977) using polyclonal antisera (Agdia, USA) of CTV following the manufacturer’s protocol. The absorbance of the reaction was measured at 405 nm. Leaves and tender bark tissues of all ELISA positive samples were grounded to fine powder in liquid nitrogen and total RNA was isolated using RNeasy® Plant Mini Kit (Qiagen Inc., Valencia, CA, USA) following the manufacturer’s protocol. Subsequently, RT-PCR was done to amplify the CP gene (ORF7) of CTV with the help of a specific primer pair (Biswas 2010) KLM543 5′-CTCTAGATCTTTTGAATTATGGACGAC-3′ (forward primer) and KLM544 5′- CGCGAATTCAACAGATCAACGTGTGT -3′ (reverse primer) synthesized from Xcelris genomics, Ahmedabad, India. For the first strand cDNA synthesis, 10 µM reverse primer was added to 1 µg RNA in a PCR tube and mixed well by pipetting. The mixture was incubated at 70 °C in water bath for 5 min and immediately chilled on ice for 5 min. After that, 5× 1st strand buffer in final concentration of 1×, 10 mM each of dATP, dCTP, dGTP and dTTP, 200U M-MLV RT enzyme each from (Xcelris genomics, Ahmedabad, India), 20U RNase inhibitor from (Promega, Madison), WI and 3 μl nuclease free water were added in the same PCR tube to make up the total reaction volume of 20 µl. The above reaction mixture was incubated at 42 °C for 1 h and then 72 °C for 10 min in a Thermal Cycler (Applied Biosystems, USA) to synthesize the cDNA. The reaction mixture was transferred to ice immediately. The PCR amplification was performed with reaction mixture containing 5 μl of cDNA, 5 Unit Taq DNA polymerase (Xcelris genomics, Ahmedabad, India), 12.5 mM MgCl2, 2.5 mM each of dNTPs and 10 μM each of the above mentioned gene specific forward and reverse primers and nuclease free water was added to make 50 μl final reaction volume. The reaction components were carefully added and mixed well. PCR was performed following the method used by Biswas 2010 using Veriti® 96-well Thermal Cycler (Applied Biosystems, USA). The obtained PCR product was analysed on 1% agarose gel.
CP gene cloning, sequencing and analysis
Amplified DNA was eluted from the gel with the help of DNA Gel/PCR Purification Mini Kit (Xcelris genomics, Ahmedabad, India) as per manufacturer’s instructions, ligated into pGEM-T Easy Vector (Promega, Madison, WI) and transformed into Escherichia coli JM109 according to the protocol of Sambrook et al. (1989). The plasmid DNA from recombinant E. coli cell was isolated by the boiling method (Holmes and Quigley 1981) and digested with restriction enzyme EcoRI restriction enzyme (Supplementary Fig. 1) for further confirmation of the insert before sequencing. The sequencing of the positive clones was performed commercially at Xcelris Genomics, Ahmedabad, India using the SP6 and T7 Universal primers. Database searches for the homology of sequences of cDNA clones were done with the help of BLASTn tool version 2.8.0 (Zhang et al. 2000) Multiple sequence alignments were done using the program Multalin programme (http://multalin.toulouse.inra.fr/multalin/) (Corpet 1988). Pairwise alignments of nt and aa sequences were done using clustalW program (version 1.83) available at http://www.genome.jp/. Evolutionary Divergence between nt sequences and aa sequences of CTV isolates obtained in this study and reference isolates were calculated on the basis of distance matrix plot using the Poisson correction model (Zuckerkandl and Pauling 1965). Unrooted bootstrapped parsimonious phylogenetic tree (Nei and Kumar 2000) was constructed with 1000 bootstrap replicates using Mega6 software. The sequence sources and Gen Bank accession numbers of the CTV CP gene (Table 1) used for the construction of phylogenetic trees. Recombination analysis was done using the latest recombination detection program RDP4 Beta 4.88 and programs therein (Martin et al. 2015) using default parameter values.
Results and discussion
DAS-ELISA has confirmed the presence of CTV in all the suspected survey samples of Khasi mandarin grown in NE States of India. The ELISA reading was considered as positive when the OD value obtained for samples was three times more than the healthy samples value as reported by Lbida et al. (2005). RT-PCR using CP gene specific primers KLM543 and KLM544 gave an amplification of expected size approximately 672 bp (Fig. 1) from leaves and bark tissues of Khasi mandarin (Biswas 2010), which further confirmed that plant samples were infected with CTV. Amplified products were eluted from all the isolates, cloned (Supplementary Fig. 1) and sequenced. These sequences were submitted to the EMBL database under accession numbers LN997804, LT576374, LT576375, LN997805, LT626941 and LT671630 for the isolate AR1, AS1, NL1, M-1, SK1 and TR1 (Table 1). The obtained sequences of the CP gene from CTV isolates were compared with each other and also with other reference isolates of the virus reported from other countries. The details of the various isolates used for analysis are shown in Table 1.

Approximately 672 base pair Coat Protein gene (CP gene) amplification of Citrus tristeza virus). Lane M represents 100 bp DNA ladder (Xcelris genomics, Ahmedabad, India)
In amino acid sequence alignment (Fig. 2) it was interesting to observe that the valine (V) and phenylalanine (F) residues at aa position 122 and 124 were also present in all the six isolates of this study which was reported to be conserved among severe CTV isolates that cause either decline, stem-pitting, or seedling yellows in citrus (Pappu et al. 1993a, b; Suastika et al. 2001; Barzegar et al. 2006; Jiang et al. 2008). However, these aa residues were replaced by isoleucine (I) and tyrosine (Y) in mild strains (Rocha-Pena et al. 1995). It was further evident from the phylogenetic analysis that all of the presently studied isolates were clustered into groups with well-characterized severe strains B165, NZ-B18, T318A, SY568, NuagA, VT p346, T3 and HA16-5 that had aa V and aa F at the positions 122 and 124, respectively, in their CP and confirm that these two aa are conserved in severe strains.

Multiple alignment of the deduced aa sequences of the coat protein gene (ORF7) of CTV isolates from North Eastern Hill states of India and representative severe and mild strains. Dots indicate identical aa residues. Consensus sequence was shown in bottom row. Boxes highlight the positions where specific aa were conserved for either North Eastern or severe representative isolates
Pair-wise sequence identities of the CP gene among CTV isolates
The nt and translated protein sequences were compared, which showed 91–99% at nt level and 96–100% at aa level identities within the NE isolates of present study, whereas 94–100% nt level and 94–100% aa sequence identities were found among all CTV reference isolates On state-wise analysis, it was found that isolate AR1 from Arunachal Pradesh showed closest relationship as 99% and 100% at nt and aa level was with AS1 from Assam. Isolate AR1 showed maximum sequence homology with T3 isolate from Florida. At aa level, AR1 showed maximum aa sequence similarity with A18 isolate from Thailand. The isolate M1 from Meghalaya showed closest relationship as 95% at nt level with TR1 and 100% at aa level with AS1. The nt sequence of isolate M1 was 99% similar at nt and aa level with exotic isolates NZ-B18 and A18 respectively. The isolate SK1 from Sikkim showed closest relationship 95% at nt level with isolate NL1 and 96% at aa level with isolates TR1 and AS1. The nt sequence of SK1 isolate was 95% similar with exotic isolate HA16-5. At aa level it showed closest relationship 97% with HA16-5 and A18. The isolate TR1 from Tripura showed closest relationship 95% at nt level with isolate M1 and 97% at aa level with isolates M1 and AS1. The isolate TR1 showed its closest relationship 98% with NuagA, T3 and A18. The isolate AS1 from Assam showed maximum similarity 99% and 100% at nt and aa level was showed with isolate AR1. On comparison with other reference isolates maximum similarity 99% was shown with isolate NZRB-TH30 at nt level. Whereas, at aa level its closest relationship 99% was found with isolate A18. For isolate NL1 from Nagaland showed maximum similarity 95% with isolate SK1 at nt level. At aa level maximum similarity was showed with isolates ARP 1, M-1 and AS1. On comparison with other reference isolates it showed maximum similarity with isolates HA16-5 at nt level, while at aa level maximum homology 99% was found with the isolate HA16-5 (Supplementary Table 1).
Various regions such as 3′ and 5′ end, ORF, CP and CPm of the genome has been utilized to differentiate different isolates of CTV (Xiao-yun et al. 2012). However, diagnosis with the coat protein alone is a historical legacy, as it is conserved and least variable with 91% nt identity and 95% aa identity between isolates examined in this study. In summary, CP is the most highly conserved and any mutation in this region makes it significant (Nolasco et al. 2009). Moreover, Niblett et al. (2000), suggested that single nt change in the highly conserved viral capsid protein gene were sufficient to predict symptoms and to differentiate strains simultaneously.
Phylogenetic relationship between various CTV isolates
For the selection of appropriate CTV representative accessions for an expressive analysis with the NE representative CTV isolates, database CTV accessions were analysed phylogenetically on their own. The analysis identified seven distinct clades associated with the T30, T36, VT, B165, RB, HA 16-5 and T3 CTV genotypes (data not shown). The CP gene sequences of 19 Gen Bank CTV accessions representing the seven identified phylogenetic clades were selected for phylogenetic analysis.
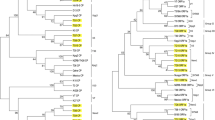
Phylogenetic relationships between the six isolates of NE region, reported for this study and 19 previously characterized CTV reference isolates (Table 1) were inferred from the nt sequence alignment by maximum parsimony tree using the Subtree-Pruning-Regrafting (SPR) algorithm with a search level of 1 in which the initial trees were obtained by the random addition of 10 sequences (Nei and Kumar 2000) at 1000 bootstrap replications (Fig. 3). Based on the resulting tree the isolates were tentatively classified into seven groups. Based on their placements related to reference sequences of CTV isolates (Harper 2013) these groups were designated as group 1 B165, group 2 VT, Group 3 T3, Group 4 T30, group 5 T36 group 6 RB and group 7 HA 16-5 (Fig. 3). The sequences of CTV isolates from NE region segregated into four groups, suggesting no correlation between the genetic relationship and geographic origin of CTV isolates as reported earlier (Rubio et al. 2001; Martin et al. 2009). According to the generated tree, the isolate M1 from Meghalaya was found very similar to the isolate B165 and clustered together to form group 1 with isolates NZ-B18, T318A, SY568 and NuagA in the phylogenetic tree, which are severe isolates of CTV and responsible to cause stem pitting and seedlings yellowing (Roy and Brlansky 2010). The isolate TR1 from Tripura was found similar to VT p346 isolates and clustered to form group 2 VT p346 responsible to cause seedlings yellowing (Mawassi et al. 1996). The isolates ARP1 and AS1 from Arunachal Pradesh and Sikkim were found very similar to T3 isolate and clustered to form group 3 T3 along with isolates A18 from Thailand which further confirm that T3-like isolates have been found in citrus samples from around the world but are particularly prevalent in the Asia–Pacific region and responsible to cause Quick decline (Hilf et al. 2005). The isolates SK1 and NL1 from Sikkim and Nagaland was found very similar to a non-standard Hawaii isolate HA16-5 and clustered to form group 4 HA16-5 (Fig. 3) and also closely similar with VT isolates (Melzer et al. 2010; Wang et al. 2013). The unusual placement of HA16-5 in the phylogenetic tree as a new genotype might be because of its partial recombinant nature (Melzer et al. 2010).

Unrooted maximum Parsimony tree using the Subtree-Pruning-Regrafting (SPR) algorithm with a search level of 1 in which the initial trees were obtained by the random addition of 10 sequences at 1000 bootstrap replications. The CTV isolate mentioned in both the phylogram indicating their GenBank accession numbers followed by the isolate name and geographic location. North Eastern isolates obtained in this study were indicated by their accession numbers followed by the country Name (India) and isolate name based on province of origin in parenthesis (AR1, Arunachal Pradesh; AS1, Assam; NL1, Nagaland; M-1, Meghalaya; SK1, Sikkim; TR1, Tripura) and indicated by black circle. Scale bar represent the expected nt substitutions per site for a unit branch length. Branch lengths are proportional to the genetic distances. Both the trees were constructed using the MEGA6 program
Moreover, the segregation of NE isolates of India into different phylogenetic clusters indicated that they are extensively diverse with each other suggesting no correlation between the genetic relationship and geographic origin of CTV isolates. This could be due to the perennial nature of citrus, chronic infection for many years with “continuous crop-host availability” coupled with multiple infestations by aphid vectors which facilitate the high gene flow between distant geographical regions, the possible selection pressure and multiple infections by successive aphid inoculations could have contributed to the recombination events between variant sequences (Weng et al. 2007; Roy and Brlansky 2009).
Another possible explanation for segregation of NE isolates in different clusters is according to the general hypothesis of virus transmission. CTV and its vector T. citricidus originated in the NE regions of India including the countries bordering this region (Ahlawat 2007) and citrus is also believed to be native of the same area (Bar-Joseph and Dawson 2008; Moreno et al. 2008). Therefore, the grouping of these reference isolates (B165, VT p346, T3 and HA16-5) with NE isolates of India and making different clusters in phylogenetic tree is an effort to trace the strains from which they originated. Previously, in most citrus growing countries, CTV and its vector both moved along with the planting material from this region (Ahlawat 2005). Though the CP gene is not a reflective of the complete genome, but it has been utilized to differentiate among the separate isolates of CTV (Xiao-yun et al. 2012) in modern phytopathology.
Evolutionary divergence between nt sequences and aa sequences of CTV isolates obtained in this study and other well characterized isolates form other part of world were calculated on the basis of distance matrix plot. The overall average distances along with corresponding standard error were calculated on the basis of number of base differences and aa substitutions per site between sequences using analytical formulas (Supplementary Table 2). Analyses for aa were conducted using the Poisson correction model (Zuckerkandl and Pauling 1965). Evolutionary analyses were conducted in MEGA6 (Tamura et al. 2013).
However, on careful observation of the data obtained from the evolutionary divergence (Supplementary Table 2), minimum distance was found among isolates from Assam and Arunachal with isolate T3 from Florida, isolates from Meghalaya and Bangalore with NZ-B18, isolates from Sikkim, Nagaland with HA 16-5 and between isolates from Tripura and Israel.. These informations were quite similar to phylogenetic analysis and can be easily correlated to the findings of sequence analysis. However, the data strengthen our findings regarding the presence of severe isolates of CTV similar to B165, VT, T3 and HA16-5 which can cause either decline, stem-pitting, or seedling yellows in citrus in NE region of India. The association between CP gene sequence variability and symptom severity has already been described by Yang et al. (1999).
All the sequences used for phylogenetic analysis were analysed for putative recombination through recombination detection program RDP4 Beta 4.88 and programs therein (Martin et al. 2015). No recombination event has been detected on comparison of our isolates with other well characterized isolates (Data not shown) further supporting an ancient recombination event that occurred before the recombinants spread worldwide, as previously suggested for the CP gene (Rubio et al. 2013). However, two recombination events have been detected for the isolate when we compare only our isolate with each other (Fig. 4a). In event 1 isolate NL1 showed presence of recombination with isolate AS1 and SK1 as major and minor parents with break point position 83- 636 of isolate NL1 CP gene (Fig. 4b) whereas in event 2 the TR1 isolate showed presence of recombination with isolate M-1 and SK1 as major and minor parents with break point position 45-212 of isolate TR1 CP gene (Fig. 4c). Both the putative recombinants were detected with two programme SISTER SCAN (Gibbs et al. 2000), and the 3Seq method (Boni et al. 2007). The presence of very rare recombination in CP region might be due to the highly conserved nature of CP gene and also corroborate the findings of Guan-Wei et al. (2015).

a Recombination analysis by RDP4 identified positions of unique recombination events in relation to the CP gene of CTV from 6 North Eastern isolates sequence alignment. The corresponding isolate and the minor parents name are given to the top and side of the each colour box, respectively. Total of 2 unique recombination events were identified, that are demarcated by the boxes below the sequences of each CTV isolate. Event 1 shows the recombination in isolate NL1 (LT576375) where isolate AS1 (LT576374) and isolate SK1 (LT626941) contribute as a major and minor parent between nt positions 83 to 636 of Nagaland isolate CP gene. Event 2 shows the recombination in isolate TR1 (LT671630) where isolate M-1 (LN 997805) and isolate SK1 (LT626941) contribute as a major and minor parent between nt positions 45–212 of Tripura isolate CP gene. b UPGMA Tree (Jukes Cantor model 1969, transition: transversion ratios estimated from the data, 1000 bootstrap replicates) constructed using recombination portions of the NL1 coat protein gene (CP) sequence alignment for event 1. C. UPGMA Tree (Jukes Cantor model 1969, transition: transversion ratios estimated from the data, 1000 bootstrap replicates) constructed using recombination portions of the TR1 coat protein gene (CP) sequence alignment for event 2
Summary and conclusion
The sequence analysis reveals the presence of severe strains of CTV in NE region of India on the basis of sequence and phylogenetic analysis. The presence of severe strains of CTV in this region where sour orange are being used as a rootstocks by local people along with some exotic rootstocks, places the citrus industry of this entire region at high risk (both in terms of yield and percentage incidence) for an imminent, destructive outbreak of CTV, especially with the presence of the vector T. citricida in this area. CP gene sequencing, phylogenetic analysis and estimation of evolutionary distance in CTV isolates suggested relationships of NE isolates with B165, VT, T3 and HA16-5 reference isolates. The random distribution of CTV infected citrus planting materials as a means of virus dissemination is common in NE region of India. However, the segregation of NE isolates into groups B165, VT, T3 and HA revealed close sequence homology to these group isolates, which strengthen the general hypothesis about the origin of CTV in NE regions of India along with the countries bordering this region and citrus was also believed to be native of the same area (Ahlawat 2007; Bar-Joseph and Dawson 2008; Moreno et al. 2008). Thus, sanitation and replanting with virus free propagative materials will be an effective method to reduce the economic losses of citrus. Furthermore, the sequence analysis of large number of CTV isolates and phylogenetic studies may lead to develop a molecular management strategy targeting conserved sequence of the virus through gene silencing.
References
Ahlawat YS (2005) Virus and virus—like diseases affecting citrus in India—a step forward to management of die—back complex. Indian Phytopathol 3:257–268
Ahlawat YS (2007) Citrus decline: problems and prevention. Indian Phytopathol 60(1):1–12
Anonymous (2014) Horticultural database. National Horticultural Board, Gurgaon
Bar-Joseph M, Dawson WO (2008) Citrus tristeza virus. Encycl Virol 1:520–525
Barzegar A, Sohi HH, Rahimian H (2006) Characterization of Citrus tristeza virus isolates in northern Iran. J Gen Plant Pathol 72:46–51
Biswas KK (2008) Molecular Diagnosis of Citrus tristeza virus in Mandarin (Citrus reticulata) orchard of Dargiling hills of west Bengal. Indian J Virol 19(1):26–31
Biswas KK (2010) Molecular characterization of Citrus tristeza virus isolates from the North Eastern Himalayan region of India. Arch Virol 155:959–963
Biswas KK, Tarafdar A, Sharma SK, Singh JK, Dwivedi S, Biswas K, Jayakumar BK (2014) Current status of Citrus tristeza virus incidence and its spatial distribution in citrus growing geographical zones of India. Indian J Agric Sci 2:184–189
Boni MF, Posada D, Feldman MW (2007) An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 176:1035–1047
Borah M, Nath PD, Saikia AK (2012) Serological detection of Citrus tristeza virus affecting citrus tree species in Assam. Indian Phytopathol 3:289–293
Borah M, Nath PD, Saikia AK (2014) Biological and serological techniques for detection of Citrus tristeza virus affecting Citrus species of Assam, India. Afr J Agric Res 52:3804–3810
Clark MF, Adams AN (1977) Characteristics of the microplate method of enzyme-linked immunosorbent assay for the detection of plant viruses. J Gen Virol 34:475–483
Corpet F (1988) Multiple sequence alignment with hierarchical clustering. Nucl Acids Res 16(22):10881–10890
Dawson TE, Mooney PA (2000) Evidence for trifoliate resistance breaking isolates of Citrus tristeza virus in New Zealand. In: da Graca JV, Lee RF, Yokomi RK (eds) Proceeding of 14th conference international organization citrus virology. International Organization of Citrus Virologists, Riverside, pp 69–76
Food and Agriculture Organization (FAO) Statistical year book (2017) Published by Food and Agriculture Organization of the United Nations. www.fao.org/faostat/en/#data/QC
Ghosh SP, Singh RB (1993) Citrus in south Asia. FAO/RAPA publication no. 1993/24. Bangkok, Thailand, p 70
Gibbs MJ, Armstrong JS, Gibbs AJ (2000) Sister-Scanning: a Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 16:573–582
Guan-Wei W, Min T, Guo-Ping W, Feng-Yin J, Zuo-Kun Y, Li-Jing C, Ni H (2015) Genetic diversity and evolution of two capsid protein genes of citrus tristeza virus isolates from China. Arch Virol 160:787–794
Harper SJ (2013) Citrus tristeza virus: evolution of complex and varied genotypic groups. Front Microbiol 4:93. https://doi.org/10.3389/fmicb.2013.00093
Harper SJ, Dawson TE, Pearson MN (2009) Complete genome sequences of two distinct and diverse Citrus tristeza virus isolates from New Zealand. Arch Virol 154(9):1505–1510
Harper SJ, Dawson TE, Pearson MN (2010) Isolates of Citrus tristeza virus that overcome Poncirus trifoliata resistance comprise a novel strain. Arch Virol 155:471–480
Hilf ME, Mavrodieva VA, Garnsey SM (2005) Genetic marker analysis of a global collection of isolates of Citrus tristeza virus: characterization and distribution of CTV genotypes and association with symptoms. Phytopathology 95:909–917
Holmes DS, Quigley M (1981) A rapid boiling method for the preparation of bacterial plasmids. Anal Biochem 114:193–197
Jiang B, Hong N, Wang GP, Hu J, Zhang JK, Wang CX, Liu Y, Fan XD (2008) Characterization of Citrus tristeza virus strains from southern China based on analysis of restriction patterns and sequences of their coat protein genes. Virus Genes 37:185–192
Karasev AV, Boyko VP, Gowda S, Nikolaeva OV, Hilf ME, Koonin EV, Nibblet CL, Cline K, Gumpf DJ, Lee RF, Garnsey SM, Dawson WO (1995) Complete sequence of the Citrus tristeza virus RNA genome. Virology 208:511–520
Kishore K, Rahman H, Kalita H, Pandey B, Minika N (2010) Prevalence of Citrus tristeza virus in mandarin of Sikkim Himalayan Region. Ind J Virol 21:140–143
Lbida B, Bennani A, Serrhini MN, Zemzami M (2005) Biological, serological and molecular characterization of three isolates of Citrus tristeza Closterovirus introduced into Morocco. EPPO Bull 35:511–517
Martin S, Sambade A, Rubio L, Vives MC, Moya P, Guerri J, Elena SF, Moreno P (2009) Contribution of recombination and selection to molecular evolution of Citrus tristeza virus. J Gen Virol 90(1):527–538
Martin DP, Murrell B, Golden M, Khoosal A, Muhire B (2015) RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 1:1–5
Mawassi M, Mietkiewska E, Gofman R, Yang G, Bar-Joseph M (1996) Unusual sequence relationships between two isolates of citrus tristeza virus. J Gen Virol 77:2359–2364
Melzer MJ, Borth WB, Sether DM, Ferreira S, Gonsalves D, Hu JS (2010) Genetic diversity and evidence for recent modular recombination in Hawaiian Citrus tristeza virus. Virus Genes 40:111–118
Moreno P, Ambros S, Albiach-Marti MR, Guerri J, Pena L (2008) Citrus tristeza virus: a pathogen that changed the course of the citrus industry. Mol Plant Pathol 9:251–268
Nei M, Kumar S (2000) Molecular evolution and phylogenetics. Oxford University Press, New York, p 126
Niblett CL, Genc H, Cevik B, Halbert S, Brown L, Nolasco G, Bonacalza B, Manjunath KL, Febres VJ, Pappu HR, Lee RF (2000) Progress on strain differentiation of citrus tristeza virus and its application to the epidemiology of citrus tristeza disease. Virus Res 71:97–106
Nolasco G, Santos C, Silva G, Fonseca F (2009) Development of an asymmetric PCR-ELISA typing method for Citrus tristeza virus based on the coat protein gene. J Virol Methods 155:97–108
Pappu HR, Pappu SS, Manjunath KL, Lee RF, Niblett CL (1993a) Molecular characterization of a structural epitope that is largely conserved among severe isolates of a plant virus. Proc Natl Acad Sci USA 90:3641–3644
Pappu HR, Pappu SS, Niblett CL, Lee R, Civerolo E (1993b) Comparative sequence analysis of the coat protein of biologically distinct citrus tristeza Closterovirus isolates. Virus Genes 7:255–264
Rocha-Pena MA, Lee RF, Lastra R, Niblett CL, Ochoa- Corona FM, Garnsey SM, Yokomi RK (1995) Citrus tristeza virus and its aphid vector Toxoptera citricida Kirkaldy: serious threats to citrus production in the Caribbean Countries, Central America and North America. Plant Dis 79(5):437–445
Roy A, Brlansky RH (2009) Population dynamics of a Florida Citrus tristeza virus isolate and aphid-transmitted sub-isolates: identification of three genotypic groups and recombinants after aphid transmission. Phytopathology 99:1297–1306
Roy A, Brlansky RH (2010) Genome analysis of an orange stem pitting Citrus tristeza virus isolate reveals a novel recombinant genotype. Virus Res 151:118–130
Roy A, Ramachandran P, Brlansky RH (2003) Grouping and comparison of Indian citrus tristeza virus isolates based on coat protein gene sequences and restriction analysis patterns. Arch Virol 148(4):707–722
Roy A, Choudhary N, Hartung JS, Brlansky RH (2013) The prevalence of the Citrus tristeza virus trifoliate resistance breaking genotype among Puerto Rican isolates. Plant Dis 97:1227–1234
Rubio L, Ayllon MA, Kong P, Fernandez A, Polek M, Guerri J, Moreno P, Falk BW (2001) Genetic variation of isolates from California and Spain: evidence for mixed infections and recombination. J Virol 75:8054–8062
Rubio L, Guerri J, Moreno P (2013) Genetic variability and evolutionary dynamics of viruses of the family Closteroviridae. Front Microbiol 4:151
Ruiz-Ruiz S, Moreno P, Guerri J, Ambrós S (2007) A real-time RT-PCR assay for detection and absolute quantitation of Citrus tristeza virus in different plant tissues. J Virol Methods 145:96–105
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning—a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Satyanarayana T, Gowda S, Ayllón MA, Dawson WO (2004) Closterovirus bipolar virion: evidence for initiation of assembly by minor coat protein and its restriction to the genomic RNA 5′ region. Proc Natl Acad Sci USA 101:799–804
Suastika G, Natsuaki T, Terui H, Kano T, Ieki H, Okuda S (2001) Nucleotide sequence of Citrus tristeza virus seeding yellows isolate. J Gen Plant Pathol 67:73–77
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Vives MC, Rubio L, Lopez C, Navas-Castillo J, Albiach-Marti MR, Dawson WO, Guerri J, Flores R, Moreno P (1999) The complete genome sequence of the major component of a mild citrus tristeza virus isolate. J Gen Virol 80(3):811–816
Vives MC, Rubio L, Sambade A, Mirkov TE, Moreno P (2005) Evidence of multiple recombination events between two RNA sequence variants within a Citrus tristeza virus isolate. Virology 331:232–237
Wang J, Bozan O, Kwon S-J, Dang T, Rucker T, Yokomi RK, Lee RF, Folimonova SY, Krueger RR, Bash J, Greer G, Diaz J, Serna R, Vidalakis G (2013) Past and future of a century old Citrus tristeza virus collection: a California citrus Germplasm tale. Front. Microbiol 4:366. https://doi.org/10.3389/fmicb.2013.00366
Weng Z, Barthelson R, Gowda S, Hilf ME, Dawson WO, Galbraith DW, Xiong Z (2007) Persistent infection and promiscuous recombination of multiple genotypes of an RNA virus within a single host generate extensive diversity. PLoS ONE 2(9):E917
Xiao-yun W, Xiao-fei C, Lu L, Xiao-xia W (2012) Genetic diversity and global distribution of Citrus tristeza virus (CTV) strains. J Northeast Agric Univ 19:9–18
Yang ZN, Mathews DM, Dodds JA, Mirkov TE (1999) Molecular characterization of an isolate of Citrus tristeza virus that causes severe symptoms in sweet orange. Virus Genes 19:131–142
Zhang Z, Schwartz S, Wagner L, Miller W (2000) A greedy algorithm for aligning DNA sequences. J Comput Biol 7(1–2):203–214
Zuckerkandl E, Pauling L (1965) Evolutionary divergence and convergence in proteins. In: Bryson V, Vogel HJ (eds) Evolving genes and proteins. Academic, New York, pp 97–166
Acknowledgements
Authors acknowledge the laboratory as well as library assistance from BCKV, West Bengal. We further are thankful to the DBT for providing grant (BT/04/NE/2009) under the project Advanced level Institutional Biotech Hub, CHF, CAU, Pasighat.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
42360_2018_56_MOESM1_ESM.jpg
Eco RI digested CP gene insert cloned in pGEM-T Easy Vector. Lane M represents 100 bp DNA ladder (Xcelris genomics, Ahmedabad, India) (JPEG 94 kb)
Rights and permissions
About this article

{kind=link}
Cite this article
Singh, A.K., Meetei, N.T., Sutradhar, M. et al. Prevalence of severe Citrus tristeza virus infection in Khasi mandarin throughout the north-eastern states of India: an evidence study based on coat protein gene analysis. Indian Phytopathology 71, 435–443 (2018). https://doi.org/10.1007/s42360-018-0056-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42360-018-0056-5