
Abstract
Trypanosoma cruzi causes chagas disease, a life threating disease in non-endemic and endemic regions globally, the life cycle of T. cruzi strictly depends on endogenous synthesis of sterol via 14-α-demethylase pathway. The available drugs for chagas disease treatment are currently resistance and parade unwanted side effects. Herein, molecular docking, QSAR, molecular mechanics/generalized born surface area (MM/GBSA) estimation, ADME screening, and molecular dynamics (MD) simulation were performed using Schrodinger suite to identify 14-α-demethylase protease antagonist from Ilex kudingcha. Density function theory of the hit ligands was carried out using Spartan 14 to investigate the molecular reactivity of the lead molecules. Nine (9) hit molecules were predicted as 14-α-demethylase protease inhibitors with binding energy range of − 7.632 to − 9.559 kcal/mol which was comparable to the standard drug (benznidazole = − 6.969), two lead molecules were further subjected to MD simulation over 50 ns predicting that kulactone and gallocatechin form stable interactions with vital residues at the catalytic site of the protein. DFT analysis revealed that, the hit ligands have the ability to donate and accept proton donating and accepting hence, effective as solubility and inhibitory agent and the ADME screening revealed, all the hit ligands obey Lipinski rule of five presenting them as drug candidate. The observations from this study predict kulactone and gallocatechin as putative antagonist of 14-α-demethylase protease and should be experimentally verify as a lead compound for chagas disease therapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Chagas disease (CD) common known as American trypanosomiasis is one of the neglected tropical diseases (NTDs) caused by Trypanosoma cruzi, this protozoan is spread by kissing bug [1], the disease constitute one of the serious public health issues in most endemic countries [2]. The World Health Organization (WHO) reported that almost 6–7 million individuals globally are victim of CD. It is common in the rural areas of Latin American nations, where it is contacted by humans and other mammals through contact with the waste or urine of kissing bug (triatomine bugs) [3]. The disease has spread beyond the Latin American nations due to migration from these endemic countries to Japan, the United States, Canada, and Australia [4].
Contact with T. cruzi, the host is permanently infected with chagas disease which has three (3) stages: acute, indeterminate, and chronic [5]. The acute stage of chagas disease occurs immediately after infection, and the symptoms are difficult to detect due to the relatively non-specific clinical symptoms observed in the majority of infected patients, as well as the ancient and subjective methods available [6]. This delay in diagnosis has an impact on the infected by impeding treatment and thus the cure [7].
nifurtimox and Benznidazole have been the initial-line treatments for CD for many years. CD regarded as a neglected disease, therefore there is lack of competition among drug manufacturers to meet production quality criteria. It is also worth noting that the current and only CD medications are prohibitively expensive, making them inaccessible to CD patients [8]. Production of sterols by 14-α-demethylase is a vital pathway in the life cycle of T. cruzi [1]; sterol 14-α-demethylase protease is in charge of mammalian cholesterol biosynthesis. T. cruzi wholly relay on biologically active sterols for proliferation and survival, they cannot use host cholesterol; therefore, the parasite's sterol biosynthetic pathway is particularly appealing for drug development [9].
The unique biochemical activities and health benefits of phytochemicals pre-dates to human existence and most of the available pharmaceuticals are isolated from medicinal plants which have proven to possess herbal remedies [10]. Ilex, commonly known as holly, belongs to the family aquifoliaceae. Kudingcha, a bitter spike tea from Ilex latifolia and Ilex kudingcha is taken as a medicinal herb in southern China. Kudingcha is referred to as a dietetic drink and is becoming well-known with trade names green-golden tea, beauty-slimming tea, clearing-heat tea and longevity tea in China. Ilex kudingcha has been observed to possess anti-inflammatory, lipid metabolism, antioxidant and antitumor properties [11, 12]. Soniran et al., [13] has reported the in vitro anti-trypanosomal activities of methanol and chloroform extracts of Ilex kudingcha with their effects on the hepatocytes. The anti-trypanosomal mechanisms of the phyto-compounds from Ilex kudingcha have not been studied. Therefore, we aim to explore computational approaches to predict inhibitory activities of phyto-compounds from Ilex kudingcha against T. cruzi 14-α-demethylase protease.
2 Materials and Methods
2.1 Ligands and Protein Structure Preparation
Ninety-eight (98) compounds from Ilex kudingcha gotten from literature search were retrieved from database of PubChem (https://pubchem.ncbi.nlm.nih.gov), the compounds were prepared by Ligprep tool from Schrodinger suite according to Omoboyowa [14].
The crystallographic structure of sterol-14α-demethylase protease was downloaded from databank (pdb: 4CKA) (https://www.rcsb.org/). This biomolecule was prepared with protein preparation wizard to rectify all errors including assigning of bond orders and missing hydrogen atoms. Receptor grid generation tool was used to generate the target glide grid file at the binding site of the co-crystalized ligand which automatically generate coordinates x = 0.52, y = 26.38, z = 459.97.
2.2 Quantum Chemical Calculation
The lead molecules were minimized by the DFT/Becke Three Lee Yang Parr/6-31G(d) level of theory, restricted hybrid Hartree Fock-DFT self-consistent field calculation with Pulay’s direct inversion of the iterative sub-space and geometric direct minimization was employed using Spartan 14 computational chemistry software. The HOMO and LUMO energies (EHOMO and ELUMO) were calculated with the reactivity descriptors extrapolated from the EHOMO and ELUMO values [15].
2.3 Virtual Screening by Molecular Docking
Ninety-eight (98) ligands from I. kudingcha and reference drug (Benznidazole) were screened through HTVS docking precision. Twenty (20.4%) of the lead ligands were further screened by SP and XP docking precision, the top-scored ligands with higher docking score than the reference drug were finally selected. Validation of the screening protocol was carried out by extracting, preparing and re-docking of the co-crystalized ligand into the original catalytic site of 4CKA to validate the reliability of the docking procedure [14].
2.4 Calculation of the MM/GBSA
The docked ligands-sterol-14α-demethylase protease complexes were optimized by local optimization feature in prime, binding energies (∆bind) for the ligands-sterol-14α-demethylase protease complexes were determined by the OPLS3 force field. The MM/GBSA estimation was performed on the docked complexes using the formula:
2.5 Screening for ADME Profile
The pharmacokinetic and drug-likeness profile of the lead ligands from I.kudingcha was predicted by the Qikprop tool of Schrodinger suite.
2.6 Development of Automated QSAR Model
The experimental data with pIC50 of 14α-demethylase protease antagonists were retrieved from the database of CHEMBL via www.ebi.ac.uk/chembl/ by blasting of the protein FASTA sequence. The inhibitors were converted to sdf format using DataWarrior v.2 [1]. The file saved as sdf was uploaded onto the workspace of the software and prepared by macromodel minimization. QSAR model for 14α-demethylase protease was developed based on the inhibitors’ pIC50. Mlr_2 model was preferred based on the ranking outcomes.
2.7 Molecular Dynamic Simulation
Molecular dynamics simulation tends to compute the movement of atoms based on time by incorporating the classical equation of Newton’s motion. The simulations were performed for a period of 50 ns with Desmond tool of Schrödinger suite. The ligand–protein docking complexes were the initial stages of the simulation; this was carried out to determine the binding status of the ligands in physiological state.
The pre-process of the protein–ligand complexes were performed using protein preparation wizard of Schrodinger suite which involve the minimization and optimization of complexes. All systems were prepared by the System Builder tool according to the protocol described by Omoboyowa et al. [15].
3 Results and Discussion
3.1 Post-Docking Analysis and Calculation of MM/GBSA
Molecular docking remains an important and established computational structural based virtual screening method employed in drug discovery and design. It predicts potential drug targets and molecular ligand-target interactions at the atomic level [16]. In this study, the molecular docking protocol was validated by preparation of extracted co-crystalized ligand, re-docked it into the active site of the target. Figure 1 represents the overlapping of the re-docked ligand, the ligand deviated from its original geometry with RMSD value of 0.74 Å (> 2.0 Å), suggesting the reproducibility and reliability of the protocol [17].

4CKA co-crystalized ligand overlapped at its binding domain
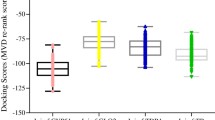
The binding affinity of the nine (9) lead molecules and the standard drug (structures shown on figure S1) are presented in Fig. 2, the result reveals that, the nine hit compounds have higher binding affinity compared with the standard drug (benznidazole) (− 6.979 kcal/mol). Kulactone showed the highest binding affinity of − 9.559 kcal/mol at the binding domain of the target, since the negativity of the binding energy reveal the intensity of the interaction [15]. Molecular docking has been reported to form a key method used in the drug discovery process. The pharmaceutical industries currently rely on this computational model to screen large library of compounds to identify novel molecules for design of new therapeutic agent [18]. From Table 1 and Fig. 3, (−)(−)epicatechin, Luteolin and Quercetin showed three H-bonds interaction with the residues at the binding domain of the protein while the standard drug (benznidazole) showed 2 H-bonds interaction with TYR 116. The interaction of small molecules with the catalytic site amino acid of protein is necessary for their inhibitory activity [1]. Five hit compounds were observed from Table 1 to form H-bond with CYS 422. Cysteine (CYS) has been observed to act as a switch to control the activity of proteins including metabolic enzymes, transcription factors and protein kinases [19, 20]. Thus, the functional importance and high nucleophilicity and of CYS makes it attractive for development of targeted covalent ligands to modulate the function of diverse proteins [21].

Representation of the binding affinity and MM/GBSA (ΔGbind) of lead compounds viz: 15560423: Kulactone; 65084: (+)(−)Gallocatechin; 72276: (−)(−)Epicatechin; 101761: Erythrodiol; 5281605: Baicalein; 72281: Hesperetin; 5280445: Luteolin; 4788: Phloretin; 5280343: Quercetin; 31593: Benznidazole

2D interaction of lead molecules at the binding domain of 14α-demethylase viz: 15560423: Kulactone; 65084: (+)(−)Gallocatechin; 72276: (−)(−)Epicatechin; 101761: Erythrodiol; 5281605: Baicalein; 72281: Hesperetin; 5280445: Luteolin; 4788: Phloretin; 5280343: Quercetin; 31593: Benznidazole
The MM/GBSA protocol is a popular model to calculate the binding free energy of small ligands binding to biological macromolecules. They are intermediate in accuracy and computational effort of strict alchemical perturbation methods and empirical scoring, which have been applied to a large number of systems with varying degree of success [22]. Herein, the free binding energy values (Δbind) of the hit compounds-protein complexes were presented in Fig. 2, the result reveals that, kulactone (− 50.047) has the highest negative value for the MM/GBSA. Erythrodiol (− 30.203), baicalein (− 29.783) and hesperetin (33.933) were observed to have lower negative MM/GBSA values compared with the standard drug (− 40.840), with other compounds presenting higher negative values than the standard drug.
3.2 Pharmacokinetics and Drug-Likeness Screening
Pharmacokinetic profiling relate to the metabolism and excretion of drug candidates. These features are predicted after drug discovery protocol; however, with computational tools such absorption, distribution, metabolism and excretion (ADME) profiles can be carried out at the early stage of drug design. Optimization of the ADME profiles of small molecules is often the most challenging part of drug discovery process which also has a major impact on the probability of success of drug candidates [23]. Herein, the nine (9) top-scored compounds from the post-docking analysis were screened for their ADME properties. From Table 2, Lipinki’s rule of five was use to predict the drug likeness of the compounds which includes H-bond donor (> 5.00), H-bond acceptor (> 10), and molecular weight (> 500 KD) [24]. Interesting, the entire lead compound obeyed Lipinski rule of five with at most one violation, this suggest that the lead compounds are drug candidates.
The pharmacokinetic profiles of the hit compounds were analyzed for QPPcaco, QPlogHERG, QPPMDCK, QPlogBB, QPlogkp and QPlogkhsa parameters using Qikprop of Schrodinger suite. From Table 3, the result showed that, the hit ligands are within the recommended range for brain/blood partition coefficient (− 1.3 to 1.2) and the IC50 QPlogHERG (> − 5). The results obtained reveal that the hit ligands have low QPlogHERG values suggesting that, the hit compounds could block hERG channels therefore more cardiotoxic [25]. Penetration of the blood–brain barrier is often reported as a significant barrier in the neurological drug development stages [26]. This has been reported to obstruct the entry of drug molecules into the central nervous system (CNS), hence poses as a serious challenge in drug discovery for the CNS disorders [1]. Only kulactone and erythrodiol showed great calcium carbonate (Caco) cell permeability of 1507.214 nm/s and 1905.837 nm/s respectively observed to be greater than the reference value of 25 nm/s. The Caco cell line is use as a model of human intestinal absorption of new therapeutic agents [27]. The Madin-Darby Canine kidney (MDCK) cells are used for the rate of drug active transport, permeability and efflux in drug design [28]. (+)(−)Gallocatechin (6.689 nm/s) and Quercetin (7.210 nm/s) were observed with low QPPMDCK values.
3.3 Analysis of Auto QSAR Model
Quantitative structure–activity relationship (QSAR) is vital tool in computational model that reveals the connection between chemical molecules and structural features [29]. AutoQSAR is a plugin in Schrodinger suite with various topological descriptors with independent variable for generation of models [1]. From Table 4, the autoQSAR splits the dataset into 23% test and 77% train set as computed from the predictive model (mrl_2) for the experimental data. The parameters generated from the model includes: standard deviation (S.D) (0.7123), R2 (0.5265), root mean square error (RMSE) (0.6855) and the predictive squared correlation coefficient (Q2) (0.5202) as shown in Table 5. Figure 4 showed the scatter plot of the observed activity and predicted activity of the dataset which revealed more train set (blue colour) than the test set (red colour). The result on the plot was consistent with the data on Table 4. The inhibitory capacity (pIC50) of the lead molecules and reference ligand were predicted base on the generated best predictive model is presented in Table 6. IC50 is the half maximal inhibitory concentration of an inhibitor required for 50% inhibition of proteins in-vitro [30]. All the hit compounds were observed to posess lower PIC50 values compared with the standard drug.

Analysis of the scatter plot for the predictive model
3.4 Density Functional Theory Analysis
Density functional theory (DFT) has been estimated through quantum mechanical methods and showed competence to be employed in pharmacological studies. Hence, DFT has formed a vital protocol in drug designing process [31]. The energies of highest occupied molecular orbital (HOMO) and Least unoccupied molecular orbital (LUMO) are vital orbital descriptors of small molecules, play vital roles in the electric properties, optical and quantum chemistry [32]. Other descriptors such as energy band gaps (Eg), chemical hardness (η), softness (δ), Electronegativity (χ) and chemical potential (Cp) were obtained from the HOMO and LUMO energies as shown in Table 7.
The EHOMO and ELUMO give reveals the reactivity of the compounds, while EHOMO reveals the electron donating ability of molecules; ELUMO predicts the electron acceptance ability. Hence, the higher the EHOMO the more readily the molecule donate electrons and the lower the ELUMO the more readily the molecule accept electrons, this predict the reactivity of molecules [32]. From Table 7 and Fig. S2, the value of EHOMO (− 5.48 eV and − 6.69 eV) showed that the molecules would interact readily through donation of electrom. The ELUMO values (− 0.78 eV and − 2.76 eV) indicated that the molecules would be an electron acceptor. The standard drug (benznidazole) showed higher EHOMO and ELUMO values (− 6.69 and − 2.76 respective) compared with the hit compounds, suggesting that the standard would readily donate and accept protons than the hit compounds. The reactivity of the compounds was further ascertained by the values of other descriptors derived from the EHOMO and ELUMO such as chemical hardness, softness, electronegativity and chemical potential (Table 7).
3.5 Molecular Dynamic Simulations of 14-α-Demethylase Protease-Ligand Complexes
The post-docking analysis, QSAR modeling and pharmacokinetic profiles of the lead compounds from Ilex kudingcha were carefully studied, kulactone and (+)(−)gallocatechin were observed as potent inhibitors of sterol-14-α-demethylase protease; therefore, to validate the ligands’ conformational stability at the binding pocket of the protein, the docked complexes of kulactone, (+)(−)gallocatechin and standard drug (benznidazole) with the target were used for 50 ns MD simulation. The docked complex stability is an important factor in studying the inhibitory mechanism of protein from molecular docking protocol [33]. Each compound’s root-mean-square fluctuation (RMSF), root-mean-square deviation (RMSD) and hydrogen bond mapping were analyzed. RMSD gives details about the protein concerning its structural backbone [34]. Figure 5 showed the RMSD plot of the target (Cα) and ligands fit protein over 50 ns simulation trajectory for kulactone, gallocatechin and standard drug. The Cα atoms in sterol-14-α-demethylase protease complexes with hit compounds exhibited mean deviation range was observed to be less than 4.0 Å (< 4.0 Å) which is acceptable for small globular proteins. The RMSD of sterol-14-α-demethylase protease and gallocatechin complex showed initial fluctuation between 1.0 and 1.50 Å within the 20 ns of simulation. After 20 ns of simulation, the trajectory was showed to be stable to the end of the simulation time. The RMSD of kulactone and sterol-14-α-demethylase protease complex showed stability for the first 10 ns of simulation with slight fluctuation of 1.5–2.1 Å differences. Between 10 and 20 ns, there was significant large difference in the RMSD value of Cα and Lig fit protein which indicates that the ligand has diffused away from its original binding domain but stability was restored after 30–50 ns of simulation. The standard drug (benznidazole) showed large fluctuation and RMSD difference between the Cα and Lig fit Prot which was restored before the end of the simulation period (40–50 ns). The result of the RMSD plot (Fig. 5) revealed that the hit compounds (kulactone and gallocatechin)-sterol-14-α-demethylase protease complexes are more stable with sight fluctuation compared with the benzidazole- sterol-14-α-demethylase protease complex.

RMSD calculation for Cα atoms (blue) sterol 14-demethylase protease and Ligands fit Protein (red) at 50 ns simulation period
3.6 Analysis of the Root Mean Square Fluctuation (RMSF)
The dynamic behavior of the amino acid of the ligand–protein complexes were evaluated by computing the atomic positional fluctuation of the amino acid backbone of the protein. Hence, the local changes along the sterol-14-α-demethylase residues were monitored by the RMSF value for 50 ns simulation time. The α-helices and β-strands of sterol-14-α-demethylase structure were shown to oscillate within 1.0–2.5 Å for the hit compounds and standard ligand (benzidazole) (Fig. 6). The secondary parameters like β-strands and α-helices are more stable than the unstructured portion of the protein, and thus show less fluctuation than the loop regions. The loop regions of the sterol-14-α-demethylase in all the complexes revealed high fluctuation above 4.5 Å.

Line representation of the evolution of root mean square fluctuation (RMSF) of sterol 14-demethylase protease Cα during 50 ns MD simulation
3.7 Interaction Mapping Between Protein–Ligand Complexes
The binding orientation of sterol-14-α-demethylase—ligand complexes established by docking model was further validated for stability of the intermolecular interaction, although simulation reveals the binding conformation by averaging all sterol-14-α-demethylase–ligand interactions obtained from individual frames of the trajectory and predicts the most favored contacts. From Fig. 7, all the docked compounds showed better interactions with amino acid in the selected site of sterol-14-α-demethylase structure during the simulation time. Interestingly, the ligands interacted with residues of the binding site of sterol-14-α-demethylase with hydrophobic bond, hydrogen bond and water bridges. Gallocatechin was shown to exhibits high H-bond interactions with vital amino acid residues in the binding site of sterol-14-α-demethylase at > 60% of the simulation interval compared to the standard drug. However, the hit compounds and benznidazole were observed to form H-bond interaction with ARG 361 with varying interaction fraction with kulactone forming above 1.2, gallocatechin forming 0.6 which are comparable to benznidazole (0.6). The standard drug was observed to exhibit more hydrophobic interaction with the amino acid of sterol-14-α-demethylase binding site.

Interaction of the protein–ligand mapping for sterol 14-demethylase protease
4 Conclusion
Sterol-14-α-demethylase is a key therapeutic target which has been identified in drug design and development against chagas disease. This study was performed to predict potent antagonists of sterol-14-α-demethylase from Ilex kudingcha compounds which can block the active site of the protein by employing computational models. Molecular docking, MM/GBSA calculation, quantum chemical calculation and virtual ADME screening of the molecules were carried out followed by MD simulation studies of hit compounds-sterol-14-α-demethylase complexes. The analysis predicted nine (9) lead compounds from the post-docking studies. Two top scored complexes were further validated by subjecting them to MD simulation study. Kulactone and gallocatechin satisfied all the parameters investigated including the MD simulation analysis. Therefore, these two compounds (kulactone and gallocatechin) were predicted as inhibitors of sterol-14-α-demethylase. However, further validation of our findings experimental analysis is suggested to establish the efficacy of the predicted compounds as drug candidate in the management of chagas disease.
Data availability
The research data associated with this paper is available, it can be accessible on request from the authors.
References
Omoboyowa DA (2022) Exploring molecular docking with E-pharmacophore and QSAR models to predict potent inhibitors of 14-α-demethylase protease from Moringa spp. Pharmacol Res-Modern Chin Med 4:100147. https://doi.org/10.1016/j.prmcm.2022.100147
Lidani KCF, Andrade FA, Bavia L, Damasceno FS, Beltrame MH, Messias-Reason IJ, Sandri TL (2019) Chagas disease: from discovery to a worldwide health problem. Front Public Health 7:166. https://doi.org/10.3389/fpubh.2019.00166
Conlan J, Lal A (2015) Socioeconomic burden of foodborne parasites. In: Gajadhar AA (ed) Woodhead Publishing Series in food science, technology and nutrition, foodborne parasites in the food supply web. Woodhead Publishing, pp 75–98. https://doi.org/10.1016/B978-1-78242-332-4.00005-9
Ribeiro A, Nunes M, Teixeira M (2012) Diagnosis and management of Chagas disease and cardiomyopathy. Nat Rev Cardiol 9:576–589. https://doi.org/10.1038/nrcardio.2012109
Buckner FS (2008) Sterol 14-demethylase inhibitors for trypanosoma cruzi infections. Drug targets in kinetoplastid parasites. Springer, pp 61–80
Antas PR, Medrano-Mercado N, Torrico F, Ugarte-Fernandez R, Gómez F, Correa Oliveira R, Chaves AC, Romanha AJ, Araújo-Jorge TC (1999) Early, intermediate, and late acute stages in Chagas’ disease: a study combining anti-galactose IgG, specific serodiagnosis, and polymerase chain reaction analysis. Am J Trop Med Hyg 61(2):308–314. https://doi.org/10.4269/ajtmh.1999.61.308
Andrade DV, Gollob KJ, Dutra WO (2014) Acute chagas disease: new global challenges for an old neglected disease. PLoS Negl Trop Dis 8(7):e3010. https://doi.org/10.1371/journal.pntd.0003010
Pinheiro E, Brum-Soates L, Reis R, Cubides J (2017) Chagas disease: review of needs, neglect, and obstacles to treatment access in Latin America. Rev Soc Bras Med Trop 50(3):296–300. https://doi.org/10.1590/0037-8682-0433-2016
Lepesheva GI, Villalta F, Waterman MR (2011) Targeting Trypanosoma cruzi sterol 14α-demethylase (CYP51). Adv Parasitol 5:65–87. https://doi.org/10.1016/B978-0-12-385863-4.00004-6
Fabricant DS, Farnsworth NR (2001) The value of plants used in traditional medicine for drug discovery. Environ Health Perspect 109(Suppl 1):69–75
Li L, Xu LJ, Ma GZ, Dong YM, Peng Y, Xiao PG (2013) The large-leaved Kudingcha (Ilex latifolia Thunb and Ilex kudingcha C.J. Tseng): a traditional Chinese tea with plentiful secondary metabolites and potential biological activities. J Nat Med 67(3):425–437. https://doi.org/10.1007/s11418-013-0758-z
Sun Y, Xu W, Zhang W, Hu Q, Zeng X (2011) Optimizing the extraction of phenolic antioxidants from kudingcha made from Ilex kudingcha C.J. Tseng by using response surface methodology. Sep Sci Technol 78:311–320
Soniran O, Ngele K, Onyemeziri CA, Omoboyowa DA, Nnabude A (2018) Histopathological studies on the effects of chloroform and methanolic extracts of Ilex kudingcha in Trypanosoma brucei infected Albino Wistar Rats. Recent Adv Biol Med. 4:50–62. https://doi.org/10.18639/RABM.2018.04.735155
Omoboyowa DA (2022) Virtual screening of phyto-compounds from Blighia sapida as protein tyrosine phosphatase 1B inhibitor: a computational approach against diabetes. Chem Afr 5:1–11. https://doi.org/10.1007/s42250-022-00373-w
Omoboyowa DA, Iqbal MN, Balogun TA, Bodun DS, Fatoki JO, Oyeneyin OE (2022) Inhibitory potential of phytochemicals from Chromolaema odorata L against apoptosis signal-regulatory kinase 1: a computational model against colorectal cancer. Comput Toxicol 23:100235
Ferreira L, Dos Santos R, Oliva G, Andricopulo A (2015) Molecular docking and structure-based drug design strategies. Molecules 20(7):13384–13421. https://doi.org/10.3390/molecules200713384
Omoboyowa DA, Singh G, Fatoki JO, Oyeneyin OE (2022) Computational investiga-tion of phytochemicals from Abrus precatorius seeds as modulators of peroxisome proliferator-activated receptor gamma (PPAR γ). J Biomol Struct Dyn 40:1–16. https://doi.org/10.1080/07391102.2022.2091657
Kitchen D, Decornez H, Furr J, Bajorath J (2004) Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 3(11): 935–949. https://www.nature.com/articles/nrd1549
Pace NJ, Weerapana E (2013) Diverse functional roles of reactive cysteines. ACS Chem Biol 8:283–296
Barford D (2004) The role of cysteine residues as redox-sensitive regulatory switches. Curr Opin Struct Biol 14:679–686
Maurias AJ, Weerapana E (2019) Reactive-cysteine profiling for drug discovery. Curr Opin Chem Biol 50:29–36
Genheden S, Ryde U (2015) The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 10(5):449–461
Ritchie TJ, Macdonald SJF, Peace S, Pickett SD, Luscombe CN (2013) Increasing small molecule drug developability in suboptimal chemical space. Med Chem Commun 4:673–680
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 46:3–26
Margulis E, Dagan-Wiener A, Ives RS, Jaffari S, Siems K, Niv MY (2021) Intense bitterness of molecules: machine learning for expediting drug discovery. Comput Struct Biotechnol J 19:568–576
Ghose AK, Herbertz T, Hudkins RL, Dorsey BD, Mallamo JP (2012) Knowledge-based, central nervous system (CNS) lead selection and lead optimization for CNS drug discovery. ACS Chem Neurosci 3(1):50–68
Van-Breemen RB, Li Y (2005) Caco-2 cell permeability assays to measure drug absorption. Expert Opin Drug Metab Toxicol 1(2):175–185
Jin X, Luong TL, Reese N, Gaona HV, Collazo-Velez C, Vuong B, Potter JC, Sousa R, Olmeda Q, Li L, Xie J, Zhang P, Zhang G, Reichard V, Melendez SR, Marcsisin BS (2014) Pybus, comparison of MDCK-MDR1 and Caco-2 cell based permeability assays for anti-malarial drug screening and drug investigations. J Pharmacol Toxicol Methods 70(2):188–194. https://doi.org/10.1016/j.vascn.2014.08.002
Kwon S, Bae H, Jo J (2019) Comprehensive ensemble in QSAR prediction for drug discovery. BMC Bioinform 20:521. https://doi.org/10.1186/s12859-019-3135-4
Ponmary PLD, Jeya SSD (2010) QSAR study for the prediction of IC50 and logP for 5-N-acetyl-beta-D-neuraminic acid structurally similar compounds using stepwise (multivariate linear regression. Int J Chem Res 2(1):32–38
Tandon H, Chakraborty T, Suhang V (2019) A brief review on importance of DFT in drug design. Res Med Eng Sci 7(4):791–795
Uzzaman MT, Mahmud T (2020) Structural modification of aspirin to design new potential cyclooxygenase (COX-2) inhibitors. In Silico Pharmacol 8:1
Balogun TA, Iqbal MN, Saibu OA, Akintubosun MO, Lateef OM, Nneka UC, Abdullateef OT, Omoboyowa DA (2021) Discovery of potential HER2 inhibitors from Mangifera indica for the treatment of HER2-Positive breast cancer: an integrated computational approach. J Biomol Struct Dyn 39:1–12
Baell JB, Congreve M, Leeson P, Abad-Zapatero C (2013) Ask the experts: past, present and future of the rule of five. Fut Med Chem 5:745–752
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Omoboyowa, D.A., Kareem, J.A., Saibu, O.A. et al. Identification of Phyto-Compounds from Ilex kudingcha as Inhibitors of Sterol-14α-Demethylase Protease: A Computational Approach Against Chagas Disease. Chemistry Africa 6, 1335–1347 (2023). https://doi.org/10.1007/s42250-022-00565-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42250-022-00565-4