
Abstract
In furtherance of our previous study on some dengue virus (DENV) NS-5 protease inhibitors, which provided us with a lead compound exhibiting high potency against DENV NS-5 protease. This study aimed to design more potent derivatives of the lead inhibitor with good pharmacokinetic properties and non-toxic utilizing a computationally based process that is precise and less expensive. By substituting the quinoline of the sulfonamide and the core phenyl ring of the lead compound identified in our previous study, eight more potent derivatives were discovered, which were docked with the DENV NS-5 receptor and then tested for drug-likeness, bioavailability, and toxicity. Quantum chemical calculations were performed on the designed compounds using density function theory (DFT) to obtain their global reactivity parameters. The docking scores of the newly designed DENV NS-5 protease inhibitors obtained using Vina-pyrx software ranged between − 8.40 and − 10.40 kcal/mol, outperforming both the lead and reported referenced inhibitors (Fenretinide and S-adenosyl-l-homocysteine), which had docking scores of − 8.3 and − 6.0 kcal/mol, respectively. Compounds 29J, 29N, and 29P had the best docking scores of − 10.00, − 10.40, and − 10.30 kcal/mol, respectively, with a good drug-likeness, oral bioavailability, and no probable toxicity. The band energy gap obtained for the design compound is in the order of 29i (2.85 eV) < Lead (3.03 eV) < 29O (3.38 eV) < 29P (3.6 eV) < 29G (3.7 eV) < 26i (3.94 eV) (eV) = 29N (3.94 eV) < 29K (3.95 eV) < 29H (3.98 eV) < 29b (4 eV) < (4.05 eV). According to this study, the proposed compounds could be useful in the development of more effective DENV treatments and as a suitable alternative to existing DENV protease inhibitors.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
DENV disease is one of the arthropod-borne diseases which could lead to dengue fever and the likelihood of progressing to a fatal stage of bleeding, having undesirable health impacts on children and previously infected persons [1].
DENV infection has been spreading rapidly in recent times, evident by its recurrent outbreak in various regions around the world, thereby putting a large population at greater risk with no treatment for such a disease. Recent outbreaks of DENV infection in tropical and subtropical regions around the world, as well as a reported annual infection rate of 390 million, are cause for great concern [2, 3].
An important promising target for its pharmacological treatment, the non-structural protein 5 (NS-5) is reported to be involved in the replication of the virus, hence constituting a druggable target [4,5,6,7]. Effective treatment strategies are urgently required to battle the growing rate of dengue fever cases. Among the ten known proteases of the DENV, NS5 is among the proteases identified as one of the ideal targets for drug discovery [2,3,4,5,6,7].
Advances in the computational field have paved the way for drug development approaches to become more efficient and cost-effective, through a variety of commonly used approaches such as structure and ligand-based. In the last 3 years, in-silico models, as well as the virtual screening of lead compounds utilizing computational-based tools with promising outcomes, have been reported in the literature [8,9,10,11,12,13,14,15]. Recent studies, including molecular docking and QSAR, have identified DENV NS5 and NS2B/NS3 inhibitors with high therapeutic potential for treating DENV-related infections [8, 12,13,14,15].
Molecular docking is the most extensively utilized computer tool in structure-based drug design. The basic goal of structure-based drug development is to design novel lead compounds that bind to their therapeutic targets as firmly as possible [16, 17]. Factual errors in scoring functions, which frequently ignore solvation effects and entropy, as well as problems estimating certain interactions, like water-mediated interactions, can lead to incorrect predictions. Although approaches to improving binding mode prediction have emerged, such as molecular dynamics (MD) simulations of the complex to confirm that the predicted binding mode is stable [18], the Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) method which calculates binding free energies for the ligand–protease complex by combining molecular mechanics calculations and continuum solvation models [19].
However, because of the inherent constraints of the typical generalized Born (GB) model used in MM/GBSA, reliable prediction of protease–ligand binding affinity remains difficult. Though, MD simulations with an explicit solvent model are frequently used to construct structural ensembles for binding free energy calculations [16, 20].
As a result, the search for new DENV inhibitors should be facilitated by employing a computational-based strategy that is precise, less expensive, and quick in obtaining new drugs with improved anti-DENV potential to overcome the aforementioned health concern given by DENV infection.
In this study, a structure-based design of potential DENV NS-5 protease inhibitors of DENV has been developed using a computational approach. This study targeted the design of more potent derivatives of lead compounds earlier identified in our previous work [14].
To circumvent likely drawbacks at the advanced stage of drug development, bioavailability [21], and ADMET (absorption, distribution, metabolism, excretion, and toxicity) [22] predictions were further targeted on our designed compounds, utilizing these proficient computational tools to design less toxic and potent DENV inhibitors, thus avoiding drug development failure.
2 Materials and Methods
2.1 Data
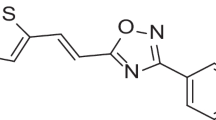
Based on findings from our previous work [14], some potent inhibitors of dengue were identified through virtual screening from sets of leads reported by Yokokawa and co-workers [6]. Compound 29 was identified as the lead owing to its binding score as well as good biological activity. During our screening compound, 29 was also found within our developed domain of applicability for the QSAR model [14]. Other derivatives of the leads not involved in our previous study reported by Lim and co-researchers [7] in one of their articles were also included in this study to provide a basis for the outcome of our findings. The lead compound was reported to have better pharmacological activity in cell culture assays at every concentration [6] (Fig. 1) .
2.2 Insilco Design of Hypothetical Compounds
The high expense of lead identification for subsequent development of other derivatives utilizing the classic drug discovery approach has been a major source of concern for scientists. Progress in drug development computation tools has assisted many stages of drug development at a reduced cost [8,9,10,11,12,13,14,15]. The approach of structure-based design was adopted for the design of the novel derivatives of lead based on the information obtained from our previous report on the interaction of the lead compound with the target of interest [14].
Hereafter, a systematic substitution of the quinoline at the terminal of the sulfonamide moiety and on the central phenyl ring resulted in the derivatives with modifications at the terminal of the sulfonamide and central phenyl ring. As described earlier regarding the selection of compound 29 from our previous work [14], towards this end, about eight derivatives of compound 29 [6], were designed. The 2-dimensional chemical structures (2D) of the compounds were drawn by the use of Chemdraw [23], which are presented in Table 1.
2.3 Target and Ligand Preparation for Docking
The 2D drawn chemical structures of the designed compounds were subjected to energy minimization to obtain their best conformation as well as convert them into recognisable files utilised by the docking algorithm. The energy minimization was accomplished using density functional theory (DFT) [24].
The DENV NS-5 protease crystal structure was taken from the protein data bank (PDB) (http://www.rcsb.org/pdb) [25], with the identification code (PDB ID: 6KR3). The protease was obtained in a complex with other ligands and water molecules, which was subsequently prepared into the required docking format using a standard protocol and the use of Discovery Studio Software [26].
2.4 Molecular Docking
To obtain the putative binding score of the interaction between the designed compounds and the protease, molecular docking calculations were accomplished using Vina-pyrx [27]. The top models with the most negative docking scores (binding energy in kcal/mol) and low root mean square deviation were selected to investigate the binding interactions using Discovery Studio Visualizer [26].
2.5 In Silico ADMET Predictions of the Designed DENV Inhibitors
The ADMET parameters of the designed compounds were assessed with the use of the Swiss-ADME [28] and pkCSM—pharmacokinetics [29] free online tools. Flops in various drug designs and development have long been ascribed to poor pharmacokinetics and bioavailability qualities as well as weak efficacy. The method involved using the SwissADME and pkCS pharmacokinetics (http://biosig.unimelb.edu.au/pkcsm/), which have been described [28, 29].
2.6 Quantum Chemical Reactivity Analysis of the Design Compounds
Some quantum chemical properties from the geometrically optimized designed compounds using the DFT with a 6-31G* basis set, such as the energy of the highest occupied (εHOMO) and the lowest unoccupied molecular orbital (εLUMO), were obtained. The εHOMO and εLUMO of each compound were used to compute other properties such as chemical hardness (η), chemical potential (μ), electronegativity (χ), softness (S), and electrophilicity index (ω) using Eqs. 1–5, respectively [30]. The 3D chemical structure of the geometrically optimized lead compound is presented in Fig. 2.
Using Eq. 1, researcher [31, 32] have described the principle of absolute maximum for a system with N electrons and total energy E.
In Eq. 1 and A have been estimated in terms of the εLUMO) and (εLUMO) according to Koopman’s approximation [32]. Also, the global softness can be expressed as using Eq. 3 as the inverse of chemical hardness.
The electron affinity can also be used alongside the ionization energy to obtain electronic chemical potential, μ as shown in Eq. (2). The negative of electron affinity (− χ) was defined [31], as the characteristic of electronegativity of chemical compound:
The electrophilicity index (ω), introduced by Parr and co-researchers [32,33,34]; this can be obtained from the electronic chemical hardness, η, and chemical potential, μ, as presented in Eq. 5
3 Results
3.1 Molecular Docking Calculation and Visualisation
Following the docking of the designed compounds using the Vina-pyrx software, the binding affinity score of each docked compound was obtained (Tables 1 and 2). The binding score indicates how tightly bound the interaction of the compounds with the therapeutic target is. The binding affinity score is expressed in the unit of kcal/mol and the higher the value, the better the interaction (Tables 1 and 2).
The interaction of the various amino acid residues of the active and allosteric sites of the protease and the compounds (Fig. 3A–J), as well as the nature and the types of interactions (Table 2), were viewed using the Discovery Studio.
3.2 In silico ADMET predictions
The designed compounds, after successful docking screening, were further evaluated for drug-likeness and pharmacokinetics worthiness, as well as toxicity prediction. The results of the ADMET are presented in Tables 3 and 4.
4 Discussion
4.1 Molecular Docking of the Designed Potent Compounds with the Target
Hereto, our previous study on some derivatives of sulfonamide was conducted to identify potent inhibitors of DENV. This screening hinted at an outstanding lead compound 29 (Fig. 1), which turned out to be highly potent with a good binding score of − 8.3 kcal/mol with the target (PDB ID: 6KR3) [14].

The lead compound (29) 5-(5-(3-Hydroxyprop-1-yn-1-yl) thiophen-2-yl)-4-methoxy-2-methyl-N-(quinolin-8-ylsulfonyl) benzamide as a template for the design [6]
Hereafter, a systematic substitution of the quinoline at the terminal of the sulfonamide moiety and the central phenyl ring was accomplished, with modifications at the terminal of the sulfonamide and central phenyl ring. These modifications resulted in eight derivatives of the lead with enhanced anti-DENV inhibitory properties through their molecular docking binding scores (Table 2) and favourable interactions with the target (Fig. 3A–J).

Geometrically optimized structure of the lead compound (29) [6]


2-dimensional interactions of the designed compounds with the target protease (6KR3)
After this, DENV NS-5 protease was docked with the eight designed compounds (29B, 29G, 29H, 29J, 29K, 29N, 29O, and 29P), and also, 26i and 29i [14] (Table 1) were considered. The docking was done using Vina-pyrx software using the standard protocol earlier described in our previous work [14, 35].
The compounds with the best scores (compound 29N) had a binding energy score of − 10.40 kcal/mol, whereas the remaining designed derivatives gave binding affinity values in the range of − 8.70, − 10.10 kcal/mol (Table 2). Based on the binding scores obtained for the designed compounds (Table 2) and those for the standards (Fenretinide and S-adenosyl-l-homocysteine) [14], with reported binding score values of − 8.3 and − 6.0 kcal/mol, respectively, it could be concluded that the designed compounds have better activity than the selected standard inhibitor of DENV and hence are more potent.
The 2D interactions for the compounds are shown in Fig. 2A–J. None of the compounds, including the standards, showed unfavourable bonds except compound 29G, as such unfavourable bonds affect the stability of ligand-protease complexes due to repulsive forces within the complex.
Compound 26i interacted with Lys-95, Asn-96, and Asp-257 (2.382, 2.618, and 2.068 Å) protease's amino acid residues by forming favourable conventional hydrogen bonds (C–H-bonds) and carbon-hydrogen bonds (C–H bonds) with Pro-73, Asn-69, and Glu-67, whereas hydrophobic interactions were observed with Lys-356, Pro-299, and Lys-356 amino acid residues. This could explain the complexes' stability, as indicated by the lack of unfavorable interactions.
Compound 29i, which is regarded as derivative of the designed compounds reported by Lim et al. [7], was discovered to interact with Gly-148 and Asp-79 (2.84856 and 2.6592 Å) residues via C H-bond and hydrophobic interaction with Glu-111, Ile-147, Trp-87, His-110, and Lys-105 through Pi-Anion, Pi-Sigma, Pi-Sigma, Pi-Pi T-shaped and Pi-Alky respectively.
Among the designed compounds, compound 29B had the highest number of C–H-bond interactions of seven involving Thr-361, Thr-540, Arg-541, Arg-541, Arg-599, Ala-536 and Gly-537 (2.019, 2.723, 2.472, 2.454, 2.079, 3.020, and 2.731 Å), with two carbon–hydrogen bonds involving Lys-358 and Asp-539 amino acid residues of the protease. Additionally, hydrophobic interaction through Pi-Sigma and Pi-Sulfur, (Amide-Pi Stacked), Alkyl, Alkyl, Alkyl and Alkyl involving Lys-358, His-52, (Ala-536, Gly-537), Val-359, Ala-473, Ala-536, Lys-358, and Lys-95 residues were observed, respectively.
The quinoline of the sulfonamide was replaced by the 1, 3, 4-oxadiazole and 1, 2, 4-oxadiazole moiety to obtain compounds 29G and 29H.
Compound 29G formed four C H bonds (Lys-95, Asn-96, His-298, and Asp-257/2.357, 2.996, 2.039, and 2.256 Å), one C–H bond (Glu-67), and six hydrophobic contacts (Arg-63, Lys-356, Pro-299, Tyr-90, Pro-299, and Lys-356) via alkyl and pi-alkyl groups.
In compound 29H, only two conventional hydrogen bonds were observed with Val-66 and Pro-73 (2.432 and 2.468 Å) amino acid residues and two carbon–hydrogen bonds with Pro-73 and Glu-67, while the remaining interactions involving Arg-63, Lys-356, Pro-299, Tyr-90, Pro-299, and Lys-356 residues are hydrophobic. Despite its high binding score, compound 29H has an unfavorable bond interaction that could lead to complex instability due to repulsive force.
In contrast, only three C–H bonds involving Pro-299, Glu-67, and Phe-349 were observed in compound 29J, in which a halogen (Fluorine) interacting with two Pro-73 was observed, as well as Arg-63, Lys-356, Pro-299, Pro-299, Lys-356, and Leu-94 residues involved in the hydrophobic interaction. Despite the lack of a C H-bond, compound 29J had a higher binding score of 10.0 kcal/mol, which could be attributable to the presence of halogen [35].
Compound 29K, like compound 29J, did not create any C H-bonds with any amino acid residue, instead opting for hydrophobic interactions with two Tyr-607 residues by Pi-Pi Stacked, one involving Leu-512 via Alkyl, and His-711 and Cys-709 via Pi-Alkyl. Compound 29K's low binding score could be explained by the lack of a hydrogen bond acceptor on the phenyl ring of sulfonamide.
Compound 29N, with the best binding score (− 10.40 kcal/mol) among all the designed compounds, formed only one convention and carbon-hydrogen bonds with Gly-93 (2.208 Å) and Leu-258 respectively, whereas Glu-67, Glu-67, and Gln-352 residues were involved in the three halogen interactions and Lys-356, Arg-353, Tyr-90, Lys-356, and Leu-94 accounted for the five hydrophobic interactions through Alkyl and Pi-alkyl. The good binding energy score of compounds 29N is due to the presence of fluorine, hence it could be regarded as the most potent of the designed compounds.
Moreover, C H-bond interactions involving Ser-601, Thr-606, and Asp-539 (2.564, 2.117, and 2.459 Å) residues for compound 29O were seen, as well as a carbon-hydrogen bond with Gly-600 residue. Interestingly, compound 29O formed electrostatic interaction with the Asp-539 residue through P-Anion, whereas hydrophobic interaction was formed involving Trp-475, Ala-473, Trp-475, and Trp-475.
Compound 29P, despite having a better binding score, formed only one conventional hydrogen with Gly-93 (2.463 Å) and eight hydrophobic bond interactions (Arg-63, Lys-356, Arg-582, Pro-583, Arg-353, Phe-349, Phe-349, and Pro-299), through Alkyl, Alkyl, Alkyl, Alkyl, Alkyl, Pi-Alkyl, Pi-Alkyl, and Pi-Alkyl, respectively.
The stabilization of the complexes of the designed compound-protease was mostly through conventional hydrogen bond and hydrophobic bond interactions involving residues at the allosteric sites of the protease.
The designed compounds in this study all outperformed the selected conventional inhibitors (Fenretinide and S-adenosyl-l-homocysteine) [14] and some had better docking scores than the inhibitors reported by Lim and co-researchers [7] (26i and 29i) in terms of binding affinity with the targeted protease. The template from which the compounds were designed has been tested in vitro in DENV inhibitory experiments [6, 7]. This research established that the developed compounds are DENV antagonists.
4.2 Drug-Likeness, Pharmacokinetics, and ADME Prediction of the Designed Compounds
Chemical compounds with observed pharmacological or therapeutic activities are required to possess some desirable features such as good ADMET and drug-likeness properties to be identified as good drug candidates. The properties depend on some of their physicochemical properties, such as molecular weight of not less than 500, logP value of not greater than 5, hydrogen-bond donors of not greater than 5, hydrogen-bond acceptors of not greater than 10, and topological polar surface area (TPSA) of less than 140, as recommended by Lipinski [21]. The SwissADME and pkCSM online tool programs were used to assess the ADMET and drug-likeness features of the designed compounds.
The predicted ADME properties are presented in Table 3, from which it can be seen that our designed compounds passed Lipinski’s rule of five, which also suggested an orally active drug be a non-violator of not more than one of the criteria.
The effects of the designed compounds on some important enzymes involved in drug metabolism, such as CYP1A2, CYP2C9, and CYP2D6, were considered (Table 3). Except for compounds 29G and 26i, which appeared to inhibit CYP1A2 and CYP2D6, all of the designed compounds were found to be non-inhibitors of CYP1A2 and CYP2D6. Conversely, all the compounds were predicted to inhibit CYP2C9 except for compounds 29B, 29G, and 29H. Additionally, the compounds were revealed to be non-Pgp substrates [36].
The pan-assay interference compounds (PAINS) analysis was also performed, which finds compounds with false positive biological activity in assays. A compound that has a positive PAINS alert could be considered to have misleading biological activity. Table 3 shows the findings of the PAINS alert. No compound was found to have a positive PAINS alert, which means it would have positive biological activities if examined in a biological activity assay [37].
The bioavailability score of all the designed compounds reported in Table 3 was found to be 0.55 which is an indication of how active the compounds are. It has been suggested that when the score is greater than 0, the compound is recommended to be active, while a score between − 5.0 and 0.00 is regarded moderately active, and less than − 5.0 is considered inactive [38, 39]. All the designed compounds demonstrated excellent bioactive scores and good synthetic accessibility [40] (Table 3).
The gastrointestinal absorption of the designed compounds presented in Table 4 lies within the range of 57.15 to 89.90% (Table 4); however, the designed compounds have better and higher gastrointestinal adsorption, since compounds with percentage absorption of lower than 30% have been regarded as as poorly absorbed [29].
The toxicity evaluation of the designed compounds was accomplished with the aid of the pkCSM online tool to gain insight into the toxicity of the designed compounds, which could lead to the potential failure of the compounds as drug candidates. Because of their good drug-like properties, the toxicity study of all the designed compounds was carried out to ensure their safety.AMES toxicity, hERG (human ether-a-go-go gene) I inhibitor, Hepatotoxicity, Skin Sensitization, Oral Rat Acute Toxicity, Renal OCTS (Organic Cation Transporter 2) substrate are among the toxicity factors evaluated (Table 4).
The result of the Ames toxicity test, presented in Table 4, which assesses a compound's mutagenic potential, reveals a negative test for all the designed compounds. A positive test indicates that the substance is mutagenic and could cause cancer [29].
The inhibition of potassium channels encoded by hERG (human ether-a-go-go gene) is the primary cause of the established long QT syndrome, which leads to severe ventricular arrhythmia. The result of the test on the designed compounds to determine whether they are hERG I inhibitors is presented in Table 4. All of the designed compounds were found to be non-hERG I inhibitors [29].
If a chemical substance causes a pathological liver event associated with a disruption in normal liver function, it is considered hepatotoxic. All of the designed compounds (Table 4), except compound 26, demonstrated positive hepatotoxicity. Such compounds with positive hepatotoxicity can lead to a disruption in the liver's normal function [29].
None of the designed compounds had skin sensitization as seen in Table 4. Such a test reveals the tendency of chemical compounds to produce allergic reactions when they come into contact with the skin [29].
The results of the oral rat acute toxicity in lethal dosage (LD50) (Table 4) revealed the LD50 of the designed compounds in the range of 2.22 to 2.75 mol/kg. Such a test shows the amount of a substance given all at once that kills half of a set of test animals.
Renal OCTS is a drug-clearing renal uptake transporter. With co-administered OCTS inhibitors, OCTS substrate has the potential to cause an undesirable reaction. As observed in Table 4, no compound was seen to be Renal OCTS substrate.
Due to their shown negative values for the toxicity assessment criteria studied, it is possible to conclude that the proposed compounds are non-toxic based on their toxicity profiles (Table 4). In addition to their remarkable drug-likeness, all of the proposed compounds may be regarded as relatively safe based on their toxicity profiles [38].
4.3 Frontier Molecular Orbital Analysis
According to the frontier molecular orbital theory of chemical reactivity, the transition of an electron is due to interaction between the εHOMO and εLUMO of reacting species. The εHOMO is a measurement of a molecule's tendency to donate electrons. As a result, larger εHOMO values suggest a greater proclivity for electron donation [30].
In this work, εHOMO and εLUMO and several quantum chemical descriptors such as the global reactivity factors were calculated (Tables 5). As expected, the εHOMO and εLUMO energies, along with the band gap energies of the designed compounds (Table 5), played an essential role in binding the molecular compounds to the protease [32, 34]. High values of εHOMO energy are an indication of the enhanced ability of the ligand to donate electrons to the neighboring compounds [41], whereas, the lower values of εLUMO energy imply that the studied molecular compounds have the ability to receive electrons from the neighboring compounds which has the ability to donate electrons [41].
The obtained energy band gab for the designed compounds presented in Table 5 is in the order of 29i (2.85 eV) < Lead (3.03 eV) < 29O (3.38 eV) < 29P (3.6 eV) < 29G (3.7 eV) < 26i (3.94 eV) (eV) = 29N (3.94 eV) < 29K (3.95 eV) < 29H (3.98 eV) < 29b (4 eV) < (4.05 eV) (Table 5), while their respective HOMO–LUMO energy diagrams are presented in Supplementary Fig. (SF) a–k. Because a lower energy band gap necessitates a higher probability for the compound to contribute electrons to the next compound, compound 29i was found to have the lowest energy gap and hence be more reactive than the others. The importance of the energy band gap in protease–ligand interactions is highlighted by this. Other compounds, such as those with a slightly wider gab than compound 29i, were shown to have a higher docking score, which could be due to their electron accepting capacity.
The energy gap (Δε) describes charge transfer interaction within a molecule, such that a region of the molecule with higher εHOMO contributes electrons to that region with higher εLUMO. This is a reflection of the chemical activity of the molecule [42].
Moreso, the localization of εHOMO and εLUMO at the same site has been reported to be associated with reduced reactivity of chemical compounds [43]. However, the lower binding scores observed for some of the compounds can be attributed to the localization of their εHOMO and εLUMO at the same sites (Fig. SF a–k).
The computed values of χ, S, η, ω, and μ of the global reactivity factors for the designed compounds [41], are presented in Table 5. The high value of μ entails that a compound is nucleophilic, whereas the low value of χ suggests that a compound is electrophilic and vice versa. The values of S and η of molecules can be described by their energy gaps. The molecules with large energy gaps are considered hard and hence less polarizable, whereas those with small energy gaps are regarded as soft, hence more polarizable.
The high value of ω defines the tendency to attract more electrons from a donor molecule. From Table 5, it can be seen that compound 29O has the highest value of ω, which means it attracts more electrons from the donor than the rest. It also measured the tendency of a species to accept electrons. Moreover, a highly reactive nucleophile is defined by a lower value of μ and ω, whereas a good electrophile is defined by a high value of μ and ω [32].
5 Conclusion
Based on the 5-(5-(3-Hydroxyprop-1-yn-1-yl) thiophen-2-yl)-4-methoxy-2-methyl-N-(quinolin-8-ylsulfonyl) benzamide hit identified in our previous study, we accomplished structure-based design of some series of DENV NS-5 inhibitor. Through structural modification of this lead, eight derivatives were designed to obtain a more potent derivative of the lead against DENV NS-5 as a therapeutic target. Through molecular docking studies of the designed derivative with the biological target for the inhibition of DENV NS-5 protease, the potency of the eight designed derivatives in terms of their binding scores and interactions with the amino acid residues of the protease through hydrogen bonding, hydrophobic and electrostatic interactions was better than the template, making them more active than the template as well as considered standard target inhibitors. The ADMET and drug-likeness studies using the Swiss-ADME webtool recommended the designed compounds as orally bioavailable, with good gastrointestinal absorption and better pharmacokinetic properties with low toxicity. Among the designed compounds, compounds 29J, 29N, and 29P were found to be promising in terms of docking score and drug-likeness, among others. This study proved that the designed derivative of lead could serve as a potent DENV inhibitor. It has been demonstrated that all the designed compounds show better pharmacological activity than the template and are not toxic. According to the reactivity analysis, the energy band gap plays a critical role in the ligand-protease interaction. As a result, it's reasonable to draw the conclusion that the developed compounds could be interesting therapeutic candidates for the treatment of DENV infection. This study highlighted the value of structure-based drug design in obtaining more effective pharmaceutical candidates at a lower cost and less time. In vivo studies is needed to understand the mechanism of action of the proposed compounds as well as their synthesis.
Availability of Data and Materials
Not applicable.
References
Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL (2013) The global distribution and burden of dengue. Nature 496(7446):504–507
Timiri AK, Sinha BN, Jayaprakash V (2016) Progress and prospects on DENV protease inhibitors. Eur J Med Chem 117:125–143
Barrows NJ, Campos RK, Liao K, Prasanth KR, Soto-Acosta R, Yeh S, Schott-Lerner G, Pompon J, Sessions OM, Bradrick SS, Garcia-Blanco MA (2018) Biochemistry and molecular biology of flaviviruses. Chem Rev 118(8):4448–4482
Low JG, Ooi EE, Vasudevan SG (2017) Current status of dengue therapeutics research and development. J infect Dis 215(2):S96-102
Nitsche C, Holloway S, Schirmeister T, Klein CD (2014) Biochemistry and medicinal chemistry of the dengue virus protease. Chem Rev 114(22):11348–11381
Yokokawa F, Nilar S, Noble CG, Lim SP, Rao R, Tania S, Wang G, Lee G, Hunziker J, Karuna R, Manjunatha U (2016) Discovery of potent non-nucleoside inhibitors of dengue viral RNA-dependent RNA polymerase from a fragment hit using structure-based drug design. J Med Chem 59(8):3935–3952
Lim SP, Noble CG, Nilar S, Shi PY, Yokokawa F (2018) Discovery of potent non-nucleoside inhibitors of dengue viral RNA-dependent RNA polymerase from fragment screening and structure-guided design. Adv Exp Med Biol 1062:187–198
Chen J, Jiang H, Li F, Hu B, Wang Y, Wang M, Wang J, Cheng M (2018) Computational insight into dengue virus NS2B-NS3 protease inhibition: a combined ligand-and structure-based approach. Comp Bio Chem 77:261–271
Adawara SN, Mamza P, Gideon SA, Ibrahim A (2020) Anti-dengue potential, molecular docking study of some chemical constituents in the leaves of Isatis tinctoria. Chem Rev Lett 3(3):104–109
Macalino SJ, Billones JB, Organo VG, Carrillo MC (2020) In silico strategies in tuberculosis drug discovery. Molecules 2(3):665
Bugeac CA, Ancuceanu R, Dinu M (2021) QSAR models for active substances against Pseudomonas aeruginosa using disk-diffusion test data. Molecules 26(6):1734
Kühl N, Graf D, Bock J, Behnam MA, Leuthold MM, Klein CD (2020) A new class of dengue and West Nile virus protease inhibitors with submicromolar activity in reporter gene DENV-2 protease and viral replication assays. J Med Chem 63(15):8179–8197
Shin HJ, Kim MH, Lee JY, Hwang I, Yoon GY, Kim HS, Kwon YC, Ahn DG, Kim KD, Kim BT, Kim SJ (2021) Structure-based virtual screening: identification of a novel NS2B-NS3 protease inhibitor with potent antiviral activity against Zika and Dengue viruses. Microorganisms 9(3):545
Adawara SN, Shallangwa GA, Mamza PA, Ibrahim A (2021) In-silico approaches towards the profiling of some anti-dengue virus as potent inhibitors against dengue NS-5 receptor. Sci Afr 13:e00907
Adawara SN, Shallangwa GA, Mamza PA, Ibrahim A (2021) In silico studies of oxadiazole derivatives as potent dengue virus inhibitors. Chem Afr 4:1–8
Wang E, Liu H, Wang J, Weng G, Sun H, Wang Z, Kang Y, Hou T (2020) Development and evaluation of MM/GBSA based on a variable dielectric GB model for predicting protein–ligand binding affinities. J Chem Inform Mod 60(11):5353–5365
Rifai EA, van Dijk M, Vermeulen NP, Yanuar A, Geerke DP (2019) A comparative linear interaction energy and MM/PBSA study on SIRT1–ligand binding free energy calculation. J Chem Inform Mod 59(9):4018–4033
Majewski M, Barril X (2020) Structural stability predicts the binding mode of protein-ligand complexes. J Chem Inform Mod 60(3):1644–1651
Hou T, Wang J, Li Y, Wang W (2011) Assessing the performance of the MM/PBSA and MM/GBSA methods. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inform Mod 51(1):69–82
Mishra SK, Koča J (2018) Assessing the performance of MM/PBSA, MM/GBSA, and QM–MM/GBSA approaches on protein/carbohydrate complexes: effect of implicit solvent models, QM methods, and entropic contributions. J Phy Chem B 122(34):8113–8121
Lipinski CA (2016) Rule of five in 2015 and beyond: target and ligand structural limitations, ligand chemistry structure and drug discovery project decisions. Adv Drug Deliv Rev 101:34–41
Roy S, Samant LR, Chowdhary A (2015) In silico pharmacokinetic analysis and ADMET of phytochemicals of Datura metel Linn. and Cynodon dactylon Linn. J Chem Pharm Res 7:385–388
Li Z, Wan H, Shi Y, Ouyang P (2004) Personal experience with four kinds of chemical structure drawing software: review on ChemDraw, ChemWindow, ISIS/Draw, and ChemSketch. J Chem Infor Comp Sci 44(5):1886–1890
Hehre WJ, Huang WW (1995) Chemistry with computation: an introduction to SPARTAN. Wavefunction. Inc, Irvine Google Scholar
Wu J, Ye HQ, Zhang QY, Lu G, Zhang B, Gong P (2020) A conformation-based intra-molecular initiation factor identified in the flavivirus RNA-dependent RNA polymerase. PLoS Pathol 16(5):e1008484
BIOVIA DS, DSME R (2016) Dassault Systèmes. San Diego, CA, USA
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comp Chem 31(2):455–461
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7(1):1–3
Pires DE, Blundell TL, Ascher DB (2015) pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem 58(9):4066–4072
Udhayakala P, Samuel AM, Rajendiran TV, Gunasekaran S (2013) DFT study on the adsorption mechanism of some phenyltetrazole substituted compounds as effective corrosion inhibitors for mild steel. Der Pharma Chemica 5(6):111–124
Zhou Z, Navangul HV (1990) Absolute hardness and aromaticity: MNDO study of benzenoid hydrocarbons. J Phys Org Chem 3(12):784–788
Oyewole RO, Oyebamiji AK, Semire B (2020) Theoretical calculations of molecular descriptors for anticancer activities of 1, 2, 3-triazole-pyrimidine derivatives against gastric cancer cell line (MGC-803): DFT, QSAR and docking approaches. Heliyon 6(5):e03926
Parr RG, Szentpály LV, Liu S (1999) Electrophilicity index. J Am Chem Soc 121(9):1922–1924
Oluwaseye A, Uzairu A, Shallangwa GA, Abechi SE (2020) Quantum chemical descriptors in the QSAR studies of compounds active in maxima electroshock seizure test. J King Saud Univ Sci 32(1):75–83
Adawara SN, Shallangwa GA, Mamza PA, Ibrahim A (2020) Molecular docking and QSAR theoretical model for prediction of phthalazinone derivatives as new class of potent dengue virus inhibitors. Beni-Suef Univ J Bas Appl Sci 9(1):1–7
Hollenberg PF (2002) Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Met Rev 34(1–2):17–35
Baell JB, Holloway GA (2010) New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 53(7):2719–2740
Olasupo SB, Uzairu A, Shallangwa GA, Uba S (2021) Unveiling novel inhibitors of dopamine transporter via in silico drug design, molecular docking, and bioavailability predictions as potential antischizophrenic agents. Future J Pharm Sci 7(1):1
Sharma CS, Shashank SM, Neeraj K, Hemendra PS, Shashi R (2018) In silico pharmacokinetic, bioactivity and toxicity study of some selected anti-asthmatic agents. Int J Pharm Sci Drug Res 10(4):278–282
Ertl P, Schuffenhauer A (2009) Estimation of synthetic accessibility score of drug-like molecules based on molecular complexity and fragment contributions. J Cheminform 1(1):1
Verma P, Srivastava A, Tandon P, Shimpi MR (2022) Experimental and quantum chemical studies of nicotinamide-oxalic acid salt: hydrogen bonding, AIM and NBO analysis. Front Chem 10:855132. https://doi.org/10.3389/fchem.2022.855132
Parthasarathi R, Subramanian V, Roy DR, Chattaraj PK (2004) Electrophilicity index as a possible descriptor of biological activity. Bioorg Med Chem 12(21):5533–5543
Galeazzi R, Marucchini C, Orena M, Zadra C (2002) Molecular structure and stereoelectronic properties of herbicide sulphonylureas. Bioorg Med Chem 10(4):1019–1024
Acknowledgements
The authors gratefully acknowledged the technical effort of Mr. Moses of the Department of Chemistry Ahmadu Bello University, Zaria.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
SNA designed and wrote the manuscript, GAS, PAM and AI supervised and carried out the statistical analysis. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Adawara, S.N., Shallangwa, G.A., Mamza, P.A. et al. Computer-Aided Drug Design and ADMET of Novel Potent Dengue Virus NS-5 Inhibitors. Chemistry Africa 5, 855–869 (2022). https://doi.org/10.1007/s42250-022-00361-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42250-022-00361-0