
Abstract
Quantitative Structure–Activity Relationship (QSAR) is known as one of the most suitable and applicable approaches in computer-aided drug discovery field. Establishing QSAR involves difficulties and challenges in its different sections. One open challenge QSAR dealing with is the existence of compounds with activity cliff property in datasets. Drug compounds with activity cliff property have very similar structures, but the values of their activities are different. In this study, we propose a method called PCAC to apply a genetic neural network algorithm as feature selection method in order to discover the most proper descriptors. In order to predict compounds with activity cliffs, an ensemble machine learning method employed random forest algorithms has been utilized. This paper aims to find drug compounds with activity cliff used Structure–Activity Landscape Index (SALI), and compounds are classified as cliff and non-cliff based on their SALI values. Experiments are conducted on three datasets, including Costanzo, Dai, and Kalla. PCAC is compared with baselines, results show that the proposed method has better performance in most cases in comparison with baselines in terms of the correlation coefficient, root-mean-square error and mean absolute error.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Quantitative Structure–Activity Relationship (QSAR) is a widely-used, proper and applicable strategy in computer-aided drug design (CADD) areas for extracting information from big datasets [1]. QSAR is an indirect Lig-based approach that models the structure properties of biological activity. QSAR model has been applied to propose new compounds with improved activity and candidate compounds for a special drug goal [2]. The Idea of QSAR is based on mold molecules’ properties such as solving in water, fats, oils and Lipophilicity solvent and balance electronic features and biologic activity of molecules. This relation can be simplified in Eq. (1):
where BR is a biologic response like IC50, ED50, LD50, and Ki. rn are molecules descriptors, including mathematical molecules features. Main problems with QSAR are when applications relying on biologic activity and the matrix of molecules descriptors are non-linear. Another problem arises when the number of computed variables is larger than the number of compounds in datasets [3]. To deal with the non-linear issue, non-linear modeling framework are used, and the problem of increase in dimension is solved by feature selection, selecting the descriptors with highest effect on activity of a set of compounds. One reason leading failure in QSAR is associated to the nature of SARThe essential assumption of QSAR and methods based on similarity is SAR continuity [4]. Thus that, gradual changes in a structure must bring to gradual changes in activity. However, the systematic and quantitative properties of different compound sets indicate that most SARs are heterogeneous [5] in a way that the plot of their activity has had slight descends and in some points witnesses sharp and low deep cliffs. Activity cliffs or totally property cliff, are pairs of compounds with high similarity in structure and against what expected have large differences in activity value [6]. We consider machine learning to detect compounds with this property. Our method has contributions in two parts: (1) Feature selection with genetic neural network, (2) Applying ensemble learning algorithm.
The rest of this paper is organized as follows: in Sect. 2 related works are reviewed. In Sect. 3 our method called PCAC is presented, in Sect. 4 experimental settings and empirical results are reported, and we conclude in Sect. 5.
2 Related work
Several studies focused on QSAR applying machine learning [7]. These models include transforming molecular structures to mathematical descriptors, descriptors selection, extracting a function as a relationship between descriptors and biologic characteristics and model validation [8].
2.1 Transforming molecular structures to mathematical descriptors
A molecular descriptor is statistical representations of a molecule [9], which is the result of an algorithm employed to molecular representation or an experimental procedure [10]. These descriptors are always derived from the x, y, z cartesian coordinates of the molecule atoms; thus, they are called 3D-molecular descriptors [11]. Molecular descriptors are divided into two classes: experimental measurements, such as log P, molar refractivity, dipole moment, polarizability, and Physico-chemical properties, and theoretical molecular descriptors, which are derived from a symbolic representation of the molecule and are classified according to the different types of molecular representation. Information on molecular geometry has experienced an explosion as a result of augmentation of topological representation of a molecule. Several geometrical descriptors are extracting from the three dimensional spatial coordinates of a molecule, including shadow indexes, charged partial surface area descriptors, WHIM descriptors, gravitational indexes, EVA descriptors, 3D-MoRSE descriptors, and GETAWAY descriptors [10].
2.2 Descriptors selection
Having a large number of molecules descriptors, the interpretation of the process can face with problems. A model with few numbers of descriptors can lead to a better rather than with a large number of ones. A solution to this issue is feature selection including filter, wrapper and floating approach [12]. Filter methods based on intent properties of data try to find the relationships between features. For each property, a score is computed and features with fewer scores are removed. This model is simple, quick and independent from the classification. In [13] correlation-based method and in [14] information- theory are employed as filter feature selection. Wrapper methods are utilized with mapping algorithms and a subset of features according to the efficiency of machine learning and its error for that subset is selected. Genetic algorithm [15], Particle Swarm Optimization [16] and Simulated annealing [17] are used as wrapper methods. Floating approach has more than one forward and backward in subsets of feature set. A dynamic backward search provides reliable results. In [18] sequential forward floating selection is used as a floating approach.
2.3 Extracting a function as a relationship between descriptors and biologic characteristics
Detecting compounds with activity cliffs leads to designing new compounds. The issue can be interpreted as a classification problem. Support Vector Machine, Gaussian Process, Artificial Neural Network, Naïve Bayesian, Decision Trees and Ensemble learning are applied for activity cliffs detection. SVM algorithms rooted in the concepts of structural risk minimization and statistical learning theory. SVM in QSAR is known as a robust and highly accurate classification technique. In [19], the classification of compounds to cliff and Non-cliff has been down with SVM by defining kernel function considering pairs of compounds instead of individual compounds. GP method for nonlinear regression is proper for a large number of descriptors. GP enjoys sound properties such as selection of the important descriptors, handling overtraining, and estimating likelihood in predictions. Authors in [20] extend the application of GP for classification and to indicate the predictive accuracy of GP compared it with decision trees, random forest, SVM, and probit partial least squares. NN are consisting of several parallels distributed processor units, resemble the brain. NNs, including one input layer, one output layer and one or more hidden layers. Each layer consists of nodes and nodes in each layer are connected to the nodes from their previous and next layer. In [21] the uses of NNs in QSAR models generated from large diverse dataset is elucidated. NB classifier is utilized for learning tasks where each instance x is described by a conjunction of attribute values and where the target function f (x) can take on any value from some finite set. In [22] developed large scale human ligand–protein predictive models based on bioactivity data using NB. In [23] a method employing RF is presented, assumed that compounds with activity cliff properties have a large value of SALI. Therefore, pairs of compounds are detected indirectly via predicting the SALI value.
2.4 Model validation
Validation is the last stage of every learning model including QSAR. In validation, the reliability and validity of a learned model is assessed. Validation methods are categorized to internal and external models [24]. Statistical methodologies applied to ensure that the generated models are sound and unbiased have been introduced as external techniques. The methods of least square fit (R2), cross-validation (Q2), adjusted R2 (R2adj), chi-squared test (χ2), root-mean-squared error (RMSE), bootstrapping and scrambling (Y-randomization) are internal methods. It has been suggested the only way to estimate the true predictive power of a QSAR model is to compare the predicted and observed activities of an (sufficiently large) external test set of compounds that were not used in the model development [25]. Statistical characteristics: correlation coefficient R between the predicted and observed activities; coefficients of determination (R2) (predicted vs. observed activities r02, and observed vs. predicted activities r0′); slopes k and k′ of the regression lines through the origin are external methods [26].
3 Proposed method for predicting compounds with activity cliff property (PCAC)
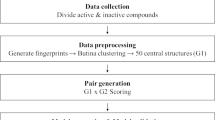
We proposed a novel method to predict compounds with activity cliff property called PCAC, which is based on the idea of predicting SALI value of components’ pairs. PCAC consists of feature reduction, descriptors calculations, computing SALI value for compounds’ pair feature selection and learning (Fig. 1).

Block diagram of PCAC model for predicting compounds with activity cliff in QSAR approach
3.1 Feature reduction
Similar to [23], two simple strategies are applied to reduce the number of features. Features having constant values or near-constant values and features with high correlation according to R2 correlation coefficient are deleted from datasets. R2 is a statistical metric that describes how close the data are and defined as the following in Eq. (2):
To determine similar features, a cutoff of 0.8 is considered.
3.2 Descriptors calculations
Descriptors can be calculated for pairs of compounds rather than individuals [23]. Since the activity cliff concept and SALI are over the compounds’ pairs, first all compounds’ pairs in datasets are calculated and the new dataset is created. The initial dataset is altered to a pairwise dataset. For an initial dataset whit N number of compounds, the number of compounds’ pairs is N(N − 1)/2. Features for the compounds’ pairs are computed by following Eq. (3).
where f3i is set of i number of new descriptors, f2i and f1i are the descriptors of first and second compounds, OP is the operator, α and β are constants, if α = β = 1, OP is subtraction, if α = β = 1/2, OP is summation [27].
3.3 SALI value computation
For utilizing the compound data towards the machine learning strategies, Each molecule is numerically represented by a molecular fingerprint which is a binary descriptor. The standard approach to quantify similarity for AC assessment is the calculation of the Tanimoto coefficient on fingerprint descriptors [2]. This numerical similarity metric assesses whole-molecule similarity in ‘fingerprint space’ and requires the definition of threshold values. The 1051-bit BCI fingerprints form Digital chemistry or CDK19,20 1024-bit path fingerprint were applied to compute SALI values. Applying a simple index can detect compounds with cliff property. One typically uses either the raw activity value or the log of that value. The former is more appropriate for activity which is represented as percent inhibition; the latter, when the activity is a Ki or IC50.7. SALI index [28] is defined as follows Eq. (4):
where Ai, Aj are the activity values of the ith and jth molecule and sim is the correlation coefficient between these molecules. Compounds with cliff lead to large value of SALI. For two similar compounds, which the value of the correlation coefficient equals to one and the SALI value is infinite; the SALI value is set to the largest value of SALI.
3.4 Feature selection
A hybrid filter feature selection algorithm consisting of Genetic algorithm (GA) and Neural Network (NN) is applied. GA optimizes the prediction error of model on a set of features [29] to select features. A GA starts with generation population of N chromosomes then fitness function for each chromosome is calculated. Afterwards, new populations are generated by following iterative stages: selection of two parents according to their fitness, transition in order to create a child and mutation which modify a gen value of a chromosome with a probability and add new child to population. New population is applied over previous steps. This procedure is repeated until satisfied a convergence condition. NN [30] are mathematical models which are developed to have functions inspired by simple and idle biological neural. Each layer of NN has several neurons. A neuron receives signals from its input and computes activation level and sends it as output to the next layer. NN employed here to compute GA’s cost function.
3.5 Ensemble learning
Ensemble methods are efficient solutions to improve efficiency of learning models [31], which consist of generating some models and combining the results. First a number of classifiers are utilized sequentially or in parallel. Then results are combined with special methods to enjoy more accurate results. Ensembles can be learnt from different part of dataset, different learning algorithms, or different subset of features. Here, two random forest algorithms [32], which are ensemble models utilizing several random trees as basic learners, as classifier are applied. RF with different feature sets, as a result of its robustness against overfitting to the training data is selected. The reason behind applying two learners is to use the benefit of feature selection while making use of all features’ information simultaneously. The elements in RF are as follows; for kth tree, a random vector of θk, independent from previous random vectors θ1, …, θk − 1 with identical distribution is produced. A tree grows with training data and θk and result in a classifier h(x, θk), where after creating a considerable number of tree votes to find the class. One of RF model works with initial dataset; another is conducted on an altered dataset after feature selection. The results are integrated by mean and absolute different as aggregation operators. The predicted SALI values are used to detect compounds with activity cliff. The compounds’ pair with SALI value higher than a threshold, are assigned as cliff and others as non-cliff.
4 Experiment
4.1 Experimental setting
Herein three datasets including Costanzo, Kalla and Dai dataset similar to [23] are used. The subset of molecules in each assay that had non-censored experimental values are applied. Costanzo contained 60, alpha-Ketoheterocycles as inhibitors of Thrombin. Kalla has 38, 8-(C-4-pyrazolyl) xanthines. Dai has 44, 3-aminoindazole. Table 1 summarized in datasets’ properties. Ndesc indicates the sizes of the descriptor pool after reduction.
In running experiments and evaluation tenfold cross-validation is applied. To evaluate methods metrics such as Correlation Coefficient, Mean Absolute Error (MAE) and Root Mean Squared Error (RMSE) are used.
Correlation Coefficient shows the amount and condition of linear relationship between two variables and is calculated by following Eq. (5):
where Cov (X, Y) is the covariance value of X and Y variables, and σx is the standard variance. The value result is in the range of (− 1, 1) where 1 indicates the direct relationship and − 1 expresses the indirect relationships of two variables. Zero means the lack of relationship between two variables.
Mean Absolute Error (MAE) reports the error of prediction for the proposed method and is defined by Eq. (6):
where fi is the predicted value, and yi is the real value.
Root Mean Squared Error (RMSE) is a popular metric in investigating the differences in real values and estimated values. This measure is defined as the following Eq. (7):
4.2 Experimental results
We compare PCAC with baselines including: random forest (RF) [23], the result of genetic neural network feature selection on RF (RF with GNN), neural networks (NN), neural network with genetic neural network feature selection (NN with GNN) and ensemble method on NN. The results of classifiers, are combined with the same aggregation operators, namely mean (shown as fmean) and absolute different (demonstrated by fdiff) applied in [30].
4.2.1 Experiment 1. Correlation coefficient
Table 2 shows the results of Correlation Coefficient over datasets. The highest value in Costanza is gained by PCAC applying diff as aggregation function. The second best correlation obtained by RF. Feature selection did not bring an increase in RF. NN methods led to less value. The feature selection leads to a slight improvement in on NN. Among NN models ensemble has the highest value. Models act differently from the aforementioned results when mean has been applied to integrate the data. In this case RF enjoyed the best value, feature selection cause a reduction. Ensemble witnesses an increase in comparison to RF with GNN. For NN models similar behavior have been observed. In Kalla dataset, all RF models and PCAC showed almost similar behavior with diff aggregation function. RF models obtain more correlation almost twice in comparison to NN methods. In the case of mean function, feature selection decreases model’s correlation and the ensemble model have risen in comparison to the GNN. The results on the Dai dataset show PCAC shows a slight improvement. It is 0.0091 larger than the value of RF when applying diff function and gained the highest rank between all models. For the same aggregation function, PCAC outperforms other methods.
4.2.2 Experiment 2. Mean average error (MAE)
Table 3 shows the results in terms of MAE for datasets. In Costanza, the least value is performed by PCAC and RF. RF with GNN experienced a growth in comparison to RF while NN with GNN had a reduction in comparison to NN. Ensemble NN proves the lowest error between NN algorithms. For both aggregations, the above results have been noticed, except for NN with GNN, which error did not fall in comparison with NN. When diff is aggregating function, all three RF models perform similarly in Kalla dataset. PCAC witnesses a small growth 0.0427 rather the values of RF, while still performing better in comparison with other models in case of applying mean. NN models act with more error value, but the error has reduced after feature selection and ensemble the results when diff aggregation is applied. The results on Dai dataset indicates PCAC similar to RF has the best results. RF with GNN experienced a growth in comparison to RF. NN after feature selection experience a growth. Finally, ensemble reduces the error over all models.
4.2.3 Experiment 3. Root mean squared error (RMSE)
In Table 4 the results based on RMSE are summarized. PCAC has the least amount of RMSE in case of applying diff function for Costanza dataset. It can be seen by GNN values stayed steady on RF and NN, while the ensemble improves the results. When mean is applied as aggregation function RF with GNN showed considerable growth in comparison to RF. For NN models, similar phenomena are reported. The ensemble reduces the error for both RF and NN. PCAC did well and reduced 0.067 with applying difference function in the Kalla dataset. NN models did not work well on this dataset. Just in case of the ensemble, a few improvements are witnessed. The model for the Kalla dataset shows poor results. This performance can be rooted in the size of dataset which is small. It has been proved that conducting small dataset for learning models always cannot lead to high performance. Turns to the Dia dataset reduction of 0.0661 can be seen in PCAC, which is the lowest error between all models. Applying feature selection on RF deteriorates the performance of RF. Again NN models worked poorly on this dataset. The worst ranks are gained by NN with GNN. The merit behavior of PCAC rooted in the power of RF and aggregation of it with model’s result after feature selection.
In all datasets when applying diff function better results are provided in terms of correlation and RMSE. Just in case of NN the mean function has better performance according to MAE and RMSE. Altogether, our ensemble method applying RF outperforms other models almost in all cases. The results show that RF method outperformed NN. It can be seen that PCAC provided better performance in most cases using ensemble learning than RF with initial features. Using feature selection, it was expected the results indicate improvement, while merely feature selection did not satisfy this expectation. The ensemble model and feature selection leads to improvements both on RF and NN.
5 Conclusion and future works
This study focused on detecting compounds with activity cliff which is an open issue in QSAR approach. By predicting SALI value of pairs of compound the compounds with activity cliff are recognized. In this research Genetic Neural Network is employed in order to select proper descriptors. An ensemble machine learning algorithm is used to predict SALI value. These results are reported about the improvement in error and fair progress of the correlation coefficient in comparison to baselines. Considering the significant role of detecting compounds with activity cliff in order to enjoy more accurate QSAR models, future research will concern about (1) assessment of chemical structure of compounds and finding their shared cliff to apply them in automatic detection of these compounds (2) proposing a method for finding descriptors associated with compound pairs in calculating their SALI (3) extending current study to compound pairs to coordinated activity cliffs (4) considering an upper bound and lower bound of SALI in order to classifying compounds to cliff and non-cliff preciously according to predicted SALI index.
References
Hansch C, Fujita T (1964) A method for correlation of biological activity and chemical structure. J Am Chem Soc 86(8):1616–1626
Gasteiger J (2006) Chemo informatics: a new field with a long tradition. Anal Bioanal Chem 384(1):57–64
Caballero J, Fernandez M (2006) Linear, non-linear modeling of antifungal activity of some heterocyclic ring derivatives using multiple linear regression and Bayesian-regularized neural networks. J Mol Model 12:168–181
Maggiora GM (2006) On outliers and activity cliffs why QSAR often disappoints. J Chem Inf Model 46(4):1535–1535
Peltason L, Bajorath J (2007) SAR index: quantifying the nature of structure-activity relationships. J Med Chem 50(23):5571–5578
Medina-Franco JL, Yongye AB, López-Vallejo F (2012) Consensus models of activity landscapes. Statistical modelling of molecular descriptors in QSAR/QSPR, vol 2. Wiley, pp 307–326
Keyvanpour MR, Shirzad MB (2021) An analysis of QSAR research based on machine learning concepts. Curr Drug Discov Technol 18(1):17–30
Winkler DA, Burden FR (2002) Application of neural networks to large dataset QSAR, virtual screening, and library design, in Combinatorial Library. Springer, pp 325–367
Grisoni F, Ballabio D, Todeschini R, Consonni V (2018) Molecular descriptors for structure-activity applications: a hands-on approach. J Methods Mol Biol 1800:3–53
Consonni V, Todeschini R (2010) Molecular descriptors. In: Puzyn T, Leszczynski J, Cronin M (eds) Recent advances in QSAR studies. Challenges and advances in computational chemistry and physics. Springer, Dordrecht
Todeschini R, Consonni V (2003) Descriptors from molecular geometry. In: Gasteiger J (ed) Handbook of chemoinformatics. Wiley-VCH, Weinheim
Moradi F, Gharaghani S, Keyvanpour M (2016) Molecular descriptors, their selection approaches and their role in upcoming QSAR applications. In: The 6th conference on bioinformatics, Tehran, Iran
Ahmadi M, Vogt M, Iyeer P, Bajorath J, Fröhlich H (2013) Predicting potent compounds via model-based global optimization. J Chem Inf Model 53(3):553559
Venkatraman V, Dalby AR, Yang ZR (2004) Evaluation of mutual information and genetic programming for feature selection in QSAR. J Chem Inf Comput Sci 44(5):1686–1692
Zhang H, Chen QY, Xiang ML, Ma CY, Huang Q, Yang SY (2009) In silico prediction of mitochondrial toxicity by using GA-CG-SVM approach. Toxicol In Vitro 23(1):134–140
Khajeh A, Modarress H, Zeinoddini-Meymand H (2012) Modified particle swarm optimization method for variable selection in QSAR/QSPR studies. J Struct Chem 24:1–9
Sutter JM, Dixon SL, Jurs PC (1995) Automated descriptor selection for quantitative structure-activity relationships using generalized simulated annealing. J Chem Inf Comput Sci 35(1):77–84
Chen Q, Wu L, Liu W, Xing L, Fan X (2013) Enhanced QSAR model performance by integrating structural and gene expression information. Molecules 18(9):10789–10801
Heikamp K, Hu X, Yan A, Bajorath J (2012) Prediction of activity cliffs using support vector machines. J Chem Inf Model 52(9):2354–2365
Obrezanova O, Segall MD (2010) Gaussian processes for classification: QSAR modeling of ADMET and target activity. J Chem Inf Model 50(6):1053–1061
Ma J, Sheridan RP, Liaw A, Dahl GE, Svetnik V (2015) Deep neural nets as a method for quantitative structure–activity relationships. J Chem Inf Model 55(2):263–274
Koutsoukas A, Lowe R, Kalantarmotamedi Y, Mussa HY, Klaffke W, Mitchell JB, Glen RC, Bender A (2013) In silico target predictions: defining a benchmarking data set and comparison of performance of the multiclass Naïve Bayes and Parzen-Rosenblatt window. J Chem Inf Model 53(8):1957–1966
Guha R (2012) Exploring uncharted territories: predicting activity cliffs in structure–activity landscapes. J Chem Inf Model 52(8):2181–2191
Veerasamy R, Rajak H, Jain A, Sivadasan S, Varghese CP, Agrawal RK (2011) Validation of QSAR models–strategies and importance. J Drug Des Discov 2(3):511–519
Kubinyi H, Hamprecht FA, Mietzner TJ (1998) Three-dimensional quantitative similarity-activity relationships (3D QSiAR) from SEAL similarity matrices. J Med Chem 41(14):2553–2564
Sachs L (1984) Applied statistics: a handbook of techniques. Springer-Verlag, BerlirdNew York
Novellino E, Fattorusso C, Greco G (1995) Use of comparative molecular field analysis and cluster analysis in series design. Pharm Acta Helv 70(2):149–154
Guha R, Van Drie JH (2008) Structure-activity landscape index: identifying and quantifying activity cliffs. J Chem Inf Model 48(3):646–658
Ozdemir M, Embrechts MJ, Arciniegas F, Breneman CM, Lockwood L, Bennett KP. Feature selection for in-silico drug design using genetic algorithms and neural networks. In IEEE mountain workshop on soft computing in industrial applications, Virginia Tech, Blacksburg, VA, 27 June 2001
Negnevitsky M (2001) Artificial intelligence: a guide to intelligent systems, 1st edn. Addison-Wesley, Boston
Zall R, Keyvanpour M (2015) MRE2C: a method for constructing multi relational ensemble classifier based on two step combining classifiers. Modares J Electr Eng 15:4
Breiman L, Friedman J, Olshen R, Stone C (1984) Classification and regression trees. Chapman & Hall/CRC, Boca Raton
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Keyvanpour, M.R., Barani Shirzad, M. & Moradi, F. PCAC: a new method for predicting compounds with activity cliff property in QSAR approach. Int. j. inf. tecnol. 13, 2431–2437 (2021). https://doi.org/10.1007/s41870-021-00737-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41870-021-00737-4