
Abstract
Olefins are by far the most valuable petrochemical intermediates. Historically thermal steam cracking of naphtha, though primarily used for the production of ethylene, is also a source of other olefins as coproduct of ethylene production. Another source has been fluid catalytic cracking process widely used in petroleum refining, providing abundance of propylene and butenes. Ethylene and propylene are No. 1 and 2, respectively largest volume petrochemical intermediates. Higher molecular weight olefins, with carbon number in the range of C8–C14 are used for the production of plasticizers, synthetic lubricant and detergent products. As the growth of petrochemicals outpaced the refining, these conventional sources for the olefins could not meet the demand and a search for alternate routes began. Catalytic dehydrogenation plays an important role in production of light (C3–C4 carbon range), detergent range (C10–C13 carbon range) olefins and for ethylbenzene dehydrogenation to styrene. This paper describes development of two routes for the production of olefins, one is catalytic dehydrogenation of paraffins to olefins and second is conversion of methanol to olefins. Both routes were developed by scientists and engineers at UOP LLC, now a Honeywell company, where the author of this paper played a major role in the development and commercialization of these technologies. The author also includes description of competitive processes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Olefins and aromatics are basic raw materials for production of petrochemical products. Naphtha, typically C5–C10 carbon range hydrocarbon, has played major role in production of these raw materials. The catalytic naphtha reforming process widely used in petroleum refining for higher octane transportation fuel contains ample quantity of aromatics that can be extracted to meet demand of benzene, toluene and xylenes. This naphtha when thermally cracked produces large quantity of ethylene, propylene and butenes.
Another important olefinic raw material is in C8–C12 carbon range for the production of plasticizers and detergent intermediates. Initially oligomerization of ethylene, propylene, butenes met the demand of these higher molecular weight olefins. Oligomerization of propylene and butenes employing solid phosphoric acid catalyst produced C6–C12 range branched olefins. Due to poor biodegradability of detergents produced employing C10-C12 branched olefins, during 1960s industry changed to linear C10–C12 olefins. Ethylene oligomerization could meet this need, however, cost of ethylene was increasing, one had to find alternate sources. Therefore, catalytic dehydrogenation of linear C10-C12 paraffins to corresponding linear olefins was developed during 1960s.
Ethylene and propylene are the two largest volume petrochemical intermediates. Ethylene demand increased from 60 million metric tons per year (MTA) in 1990 to over 200 million MTA in 2020 (Eramo 2016). Propylene demand also has similar growth standing at 140 million MTA in 2020. Until 1990 propylene demand was met by the conventional naphtha cracking process, as a coproduct of ethylene production. From 1990 onwards growth of propylene demands outpaced that of ethylene. Initially, fluid catalytic cracking (FCC) process, widely used in petroleum refinery, provided additional propylene. Increased quantity of propylene can be produced by modifying use of zeolitic base catalysts, such as ZSM-5. However, there is limit to it, as one has to balance the demand of transportation fuel and propylene. Also, such higher severity FCC operation also increases lower value methane production. Therefore, a need for catalytic dehydrogenation of propane to propylene came about.
From early days tetraethyllead (TEL) was used to boost octane number of gasolines that was phased out during 1970s. Refineries needed alternate source to increase the octane number of the gasoline. Methyl-tertiary-butene (MTBE), having research octane number of 112 was considered ideal blending component. MTNE can be produced by etherification of isobutene with methanol. This created significant demand growth for isobutene during 1980s. Therefore, a need of catalytic dehydrogenation of isobutane to isobutene was developed.
Ethylene and propylene demand is primarily driven by polyolefin. Other derivatives, such as ethylene oxide, ethylene dichloride, propylene oxide, acrylonitrile and others consume about 30–40%. The majority of the light olefins used for petrochemical applications are produced by the steam cracking of ethane, naphtha or other gas liquids, as shown in Table 1 (Vora et al. 2015).
Cost of feedstock plays major part in economics, so locations for new capacity are strongly influenced by the availability of cost-advantaged feedstocks. This is evident in the large capacity build-up of ethane-based ethylene production in the Middle East. Large amounts of ethane produced in association with crude oil production and countries provide incentives for ethane utilization. Availability of ethane has also increased with the discovery and production of shale gas in North America. Therefore, use of naphtha for production of ethylene has declined as seen in Table 2, with gains in use of LPG, ethane and the new entry of coal to olefins (CTO) or methanol to olefins (MTO). As of 2020, all CTO/MTO-based ethylene and propylene production takes place in China (see Fig. 1).

Ethylene and propylene demand
Economic analyses done by consulting firms have shown that Middle East ethane crackers and the remote gas MTO have the lowest cash cost of ethylene production, followed by North American ethane crackers, based on ethane recovered from shale gas. Figure 2 shows the cash cost of ethylene production according to Chemical Market Resources, Inc (2013). In China, cash cost of ethylene production from naphtha and via CTO route re similar. It is also seen that the production of ethylene from sugarcane or corn-derived ethanol in Brazil and the USA is not economical. A similar analysis done by IHS-CMAI (HIS Chemical 2013) for the cash cost of production of existing capacity on a geographic basis is shown in Fig. 3. It is seen that Middle East a significant advantage, followed by North America in ethylene production.

Cost of ethylene production, $/MT

Cash cost of ethylene, $/MT
When it comes to propylene, naphtha crackers and refinery FCC units play an important role, supplying nearly 90% of the demand in 2012. Because of the increasing use of ethane in place of naphtha for the production of ethylene, coproduct propylene is less, thus, combined production of propylene from naphtha crackers and FCC units falls short of meeting the propylene demand. Therefore, since 1990, alternate sources for propylene, such as propane dehydrogenation and metathesis, have emerged to meet the propylene supply gap (Vora 2012).
Catalytic Dehydrogenation Historical Developments
Ipatieff and co-workers at UOP (Grosse and Ipatieff 1940; Universal dehydro Process U 1947) developed dehydrogenation of butanes over a chromia/alumina catalyst. UOP’s Catalytic Condensation Process for olefins dimerization or oligomerization over a solid phosphoric acid (SPA) catalyst was invented by R. E. Schaad and V. N. Ipatieff, and was first commercialized in 1933 (Egloff and Davis 1951). The production of C6-C12 range higher octene material by butene oligomerization made use of During World War II. Catalytic dehydrogenation of butanes over a chromia/alumina catalyst was practiced for the production of butenes which were Oligomerized to yield a high-octane aviation fuel. The first UOP-designed plant came on stream at ICI in Billingham in 1940 and was soon followed by two other units in Heysham in 1941 (Hornaday et al. 1961). These pioneering efforts were soon followed by other companies [e.g., Phillips Petroleum’s multi-tubular dehydrogenation reactor near Borger, Texas, in 1943 (Waddams 1980)]. Most significant development was made by Houdry employing operation at lower pressure, under vaccum for dehyfrogenation. This provided higher per-pass conversions. Houdry commercialized two stage n-butane dehydrogenation chromia/alumina system, known as Catadiene process, for the production of butadiene (Waddams 1980). The Houdry Catadiene process was used extensively for the production of butadiene, either by itself (n-butane to butadiene) or in conjunction with the Oxo-D catalytic process for the oxydehydrogenation of n-butene to butadiene that was commercialized by what was then known as Petro-Tex Chemical Corp. A similar oxydehydrogenation approach for the production of butadiene was followed by Phillips Petroleum in their O-X-D process (Waddams 1980). Growth of petroleum refining started during 1940s and accelerated during 1950s. As demand for intermediates, light olefins and BTX aromatics for petrochemicals started to rise during 1960s, extensive use of naphtha reforming and naphtha cracking began, providing abundant supply of transportation and aviation fuel, as well as butadiene, leading to shutdown of these most of Houdry Catadiene plants.
Development of Noble metal Platinum Catalysis in Dehydrogenation
Large quantity of ethylene, propylene and butenes are produced via thermal or catalytic processes that met the demand of petrochemical industry. These olefins over the solid phosphoric acid (SPA) catalyst produced dimers, trimers and tetramers via condensation process. Propylene tetramer, branched C12 olefins can be produced via propylene oligomerization. The branched dodecenes were then alkylated with benzene produces branched alkylbenzene, a raw material for synthetic detergent production, This material when sulfonated and neutralized produces alkylbenzenesulfonate (ABS), that showed good properties as a surfactant material, and was introduced in 1940 for the production of Synthetic detergents.
By 1960 people saw large quantity of detergent foam floating over lake and river waters and realized that the ABS based detergent was less biodegradable, causing pollution of lake and river waters. One can see detergent foam floating over the Rhine river (Water Pollution in North Rhine-Westphalia 1958) during late 1950s. Now it was necessary to find an alternate raw material. The cause of poor biodegradation was attributed to highly branched structure of propylene tetramer. A linear carbon to carbon molecule will make easier for bacterial digestion and thus search for linear alkene began.
Kerosene fraction of the crude oil consists of C10–C13 range hydrocarbons and of that 15–30%, depending on the source of crude oil, are linear paraffins. Selective separation of linear paraffins was already practiced in petroleum refining for improving octane number of motor fuel gasoline. Recovered linear paraffins having lower octane number were isomerized and reused in gasoline blending to increase the octane number of the final gasoline product. Thus, Selective separation of linear paraffins from other hydrocarbons, naphthene, aromatics, branched paraffins in higher, C10-C12 range required minimal new developments. The next step, dehydrogenation of paraffins to olefins required a major catalytic and engineering process design development.
Vladimir Haensel developed noble metal (Pt) catalysts for catalytic reforming of naphtha and was commercialized in 1949. He also demonstrated that Pt-based catalysts had high activity for the dehydrogenation of paraffins to the corresponding olefin (Haensel 1952). In the 1960s Dr. Herman Bloch (Bloch 1969) developed Pt-based catalysts for selective dehydrogenation of linear paraffins to the corresponding internal mono-olefins. A new process, dehydrogenation of C10–C12 paraffins to corresponding C10-C12 detergent range olefins, the UOP Pacol™ process was developed. UOP has licensed this Pacol process in combination of benzene alkylation for the production of linear alkylbenzene (LAB), a raw material for the production of biodegradable detergent (Vora et al. 2006). By year 2020 there are more than 50 commercial plants were in operation. Independently Roth and Shaefer at Monsanto developed a paraffin dehydrogenation catalyst and commercialized dehydrogenation alkylation combination for production of linear alkylbenzenes (Roth 2023). There was only one plant built at Louisiana and now owned by Huntsmann Chemicals.
Light Olefins
A mentioned earlier chromia-alumina catalyst was commercialized for butane dehydrogenation during 1940s and extended to two stage butane dehydrogenation for production of butadiene. During 1980s, application of chromia/alumina catalysts was extended to the dehydrogenation of propane to propylene and isobutane to isobutene, and over the two decades there were several new units were built for this purpose. These units operate on the same cyclic principles as in the former Catadiene process, and the new process application is named Catofin (Weiss 1970; Craig et al. 1986). The Catofin process technology is currently owned by Süd-Chemie and is offered for license by Lummus Technology Group.
Recent catalytic developments use platinum or modified platinum catalysts. In about 1959 an alternative chromia/alumina catalytic dehydrogenation process employing fluidized bed reactor system was introduced in the former Soviet Union. This reactor configuration is similar to the fluid catalytic cracking (FCC) process used in refineries (Sanfilippo et al. 1998). However, back mixing common to dense fluidized bed operations results in poor selectivity and increases the formation of heavies, sometimes called “green oils”. Circulating regenerated catalyst is used to provide the heat of reaction in the riser and spent catalyst is reheated by carbon burn in a regenerator. A larger scale isobutane dehydrogenation unit using this principle was commercialized by Snam Progetti in Saudi Arabia based on technology from Yarsintez in Russia (Sanfilippo et al. 1998).
During 1960s and 1970s, UOP engineers Lestor, Carson, and others also worked on development of fluid-bed catalytic dehydrogenation employing chromia alumina catalyst but the technology was not commercialized (Lester, et al. 1968; Vesely et al. 1972; Carson, et al. 1973; Gantt, et al. 1974). Chromia/alumina catalysts pose a significant health risk in case of spillage or by exposure to the plant operators during maintenance or catalyst changeover. Chromia/alumina catalysts always contain a significant proportion of Cr(VI), principally in the regenerated catalyst; Cr(VI) is a well-known carcinogen and its adverse health effects have been well documented (Occupational exposure to chromium (VI) 1975; Hearings start in Diet on chromium pollution 1975). Chromia/alumina catalysts sinter much more rapidly than alumina when exposed to high temperatures; the replacement of spent catalyst often requires special; safety precautions with strenuous and lengthy use of manual labor (Poole and MacIver 1967; Poole et al. 1962).
The dehydrogenation of ethylbenzene to styrene over an iron or an iron-chromium catalyst that usually also contains potassium in the form of potassium carbonate is commercially practiced. At elevated temperatures various complex mixed carbonates and oxides are formed; e.g., KFeO2. Typical operating temperatures are in the order of 630 °C, and steam is added to lower the partial pressure of the reactants. Because the reaction is strongly endothermic multiple stages with inter-stage injection of superheated steam are employed.
Development of Platinum Catalysts for Light Paraffin Dehydrogenation
Since the process for dehydrogenation of C10–C13 paraffins was already developed and successfully commercialized, one would think that its extension to propane and butane dehydrogenation would be simple and easy. However, this is not the case. To minimize losses of high value feed paraffins due to thermal cracking, dehydrogenation of C10–C13 paraffins typically below 500 °C, at low per-pass conversions under higher H2 partial pressure. Propane and butane are generally used as a fuel and thus are of much lower cost relative to products propylene or butenes. Propane/propylene equilibrium and need for higher conversion dictated by economics require operating temperature range above 600 °C. Such severe operating conditions compared to C10–C13 paraffin dehydrogenation required innovation of a new catalyst and design of reactor system as well product recovery.
Paraffin dehydrogenation is an endothermic reaction that is limited by chemical equilibrium and, according to Le Chatelier’s principle, higher conversion will require either higher temperatures or lower pressures. In a somewhat abbreviated form for the production of mono-olefins, this can be expressed as follows:
where, xe is the equilibrium conversion, P is the total pressure in atmospheres absolute and Kp is the equilibrium constant for the dehydrogenation reaction. The equilibrium constant can be easily calculated from Gibbs free energies as tabulated in the API 44 report or in similar sources of thermodynamic data. Figures 4 and 5 illustrate the equilibrium conversion levels that can be obtained for propane at 1 atm. abs. and at 0.23 atm. abs. (175 torr), respectively.

Propane dehydrogenation at 1 Atm P

Propane dehydrogenation at 0.23 Atm P
The equilibrium constant for paraffin dehydrogenation increases significantly as the carbon number increases. The temperature required for the dehydrogenation of light paraffins is much higher than for heavy paraffins. For example, at 1 atm absolute pressure for 40% conversion, the dehydrogenation of propane requires a temperature of at least about 580 °C, while dodecane can be theoretically dehydrogenated to the same extent at only 450 °C. Higher conversion or the higher temperature accelerates thermal cracking and other side reaction, and thus lowers yield. Economic considerations dictate Per pass conversion. Of Course, knowledge of Pt catalysis for C10–C13 provided good starting point, due to severe operating conditions and required development of a new catalyst. Separation of coproduct hydrogen from reaction product was also much more complicated compared to higher molecular weight paraffin dehydrogenation. Higher conversion required multi-stage reactor system with inter-stage heat input to balance endothermic heat of reaction. All together propane dehydrogenation over Pt catalyst became a long research and development project.
Variety of feedstocks, ethane, LPG, naphtha to heavy vacuum gas oil are used in thermal steam cracking process for manufacture of ethylene, which also provides significant quantity of coproducts, such as, propylene, butenes and butadiene. Modern steam cracker have capacities for up to 1.5 million MTA ethylene. Steam cracking process employ multiple parallel cracking furnaces. A large furnace can produce up to 200,000 MTA ethylene, at very high severities in excess of 800 °C. Selectivity for ethylene from ethane cracking is close to 80 wt%, while that from naphtha feed drops to around 30 wt%. Use of ethane feed produces very little propylene or other byproducts. Propylene yield employing LPG and heavier feed range between 13 and 17 wt%. Naphtha feed also give 8–11 wt% butenes and about one half of that is butadiene (Chauvel and Lefebvre 2001).
Economic considerations dictate that per pass conversion of light paraffins in catalytic dehydrogenation in range of 30–50%, with selectivity to corresponding light olefins close to 90wt% or higher. Long-term catalyst stability and catalyst regenerability over multiple cycles also very important. Thus, developing such catalyst and the process is a challenging undertaking.
Continuous catalyst regeneration (CCR) technology was introduced by UOP during early 1970s for catalytic naphtha reforming. This process is widely used in petroleum refineries for the production of higher-octane gasoline, as well as, aromatics. In CCR technology a small amount of the catalyst is removed periodically from bottom of the reactor and sent to a separate regeneration vessel where carbon burning and platinum redistribution are completed. This regenerated catalyst is returned to the top of the reactor. By adjusting the rate of catalyst withdrawal and return, one can maintain a steady state catalyst activity and thus continuous constant conversion-selectivity can be maintained for a long time. Employing this CCR technology to catalytic dehydrogenation made it possible to operate catalyst at high severity without fear of coking. This approach provides steady state continuous operation with better design compared to cyclic multi-stage reactor system.
The combination of CCR technology and Pt-catalysis operating at high severity has made it possible to design, build, and operate very large catalytic dehydrogenation units (Gregor et al. 1999). The world propylene production capacity based on the use of catalytic dehydrogenation of propane has increased steadily over the past 30 years (Honeywell 2020) with 2020 production of propylene via propane dehydrogenation at 10 million metric tons per year. Dehydrogenation of ethane to ethylene employing Pt-alumina catalyst has also been investigated but, to date, the economics do not are not favorable because of the low equilibrium conversion and the need to operate under more sever condition. With significantly lower conversion relative to steam cracking of ethane, the cost of fractionating ethylene in an ethane-ethylene splitter is also too high. Dow Chemical has recently been awarded a patent (World patent 1996) for the dehydrogenation of ethane over a metal-mordenite catalyst complex at relatively low-conversions in which the product ethylene is selectively recovered from the dilute ethylene-ethane stream by alkylating it with benzene.
Another approach is to use steam dilution to lower paraffin partial pressure and thus can operate at a higher per pass conversion or get better selectivity at little lower temperature. Two other potential benefits are that superheated steam can be used as a heat carrier to supply heat for the dehydrogenation reaction and that steam interacts with coke deposits to maintain the catalyst free of coke and active.
This approach is used by Phillips Petroleum in developing their STAR technology for isobutene dehydrogenation employing Pt-type catalyst and multi-tubular reactor design. This reactor design resembles a typical steam reformer that is operated until the catalyst deactivates as a result of coke deposition. This reactor is taken out of service for catalyst regeneration while a spare muti-tubular reactor is brought on-stream. The STAR technology is currently owned and licensed by Krupp-Uhde.
Steam dilution is also employed in dehydrogenation of ethylbenzene to styrene. Bricker and associates developed steam stable catalysts for propane and isobutene dehydrogenation in a high steam environment (Bricker, et al. 1968, 1992).
Process Chemistry
Primary reaction in paraffin dehydrogenation is to form corresponding mono-olefin. However, consecutive reaction forming diolefins, triolefins also happens. For longer chain. C10–C13 paraffins, dehydrogenation-cyclization reaction can occur forming aromatics. In case of propane or butane dehydrogenation, this can also occur via dimerization-cyclization. In addition, side reactions of thermal cracking, isomerization, dimerization, polymerization could occur. Reactions that could take place on platinum (Pt) and acid (A) sites in the dehydrogenation of light and heavy paraffins are shown in Figs. 4 and 5, respectively. Challenging task for Catalysis scientist is to modify the Pt and acid sites with promoters and attenuators to minimize undesired side reactions.
The consecutive reactions, the dehydrogenation of mono-olefins to diolefins and tri-olefins, are catalyzed on the same active site as the dehydrogenation of paraffins to mono-olefins. Therefore, the consecutive reactions cannot be eliminated but can be suppressed not only by catalyst modification but also by controlling the reaction kinetics. The conversion of tri-olefins to aromatics is a very fast reaction and thermodynamically favorable. Thus, the formation of aromatics from tri-olefins must also be suppressed kinetically.
Dehydrogenation Catalysts and Modifiers
Role of a good dehydrogenation catalysts is to promote primary reaction while suppressing or minimizing the consecutive and side reactions. Unmodified Pt on alumina catalysts are active but are not selective forming numerous by-products, as shown in Figs. 6 and 7, can also form. In addition, formation of heavy hydrocarbons increases fouling and coke formation causing rapid deactivation of the catalyst. Therefore, the properties of platinum and the alumina support need to be modified to suppress these side reactions.

Reactions by platinum and acid sites with unmodified catalyst in light paraffin dehydrogenation

Reactions by platinum and acid sites with modified catalyst in heavy paraffin dehydrogenation
On Pt-alumina catalyst olefins more strongly adhered relative to paraffins. A role of attenuator and modifier is to reduce Pt activity balancing olefin diffusion. Meaning, as soon as the olefins are formed, they are detached from the catalyst to avoid consecutive reaction and increase Pt-paraffin interactions.
The consecutive dehydrogenation rate of mono-olefins and diolefins is decreased by this modification without lowering the rate of paraffin dehydrogenation significantly. The modifier also improves catalyst stability due to less coke forming precursors. Acid sites of alumina support promote skeletal isomerization, cracking, oligomerization, and polymerization of olefinic materials, and enhances "coke" formation. Therefore, acid sites are modified or neutralized to eliminate acidity.
Since first commercial operation of C10–C13 paraffin dehydrogenation in 1968, several catalytic advances have been made. Imai, Bricker and coworkers developed next generation modified Pt catalyst showing significantly lower coking and doubling the catalyst stability (Imai et al. 1988, 1989). Diffusion of products from the catalyst surface is or pores is very critical for good selectivity. Pore size of alumina support needs to adjusted according to the size of paraffin feed. Meaning a support with larger pores is needed for a long-chain paraffin feed compared to light olefins, smaller molecules like propane, isobutane. Jensen, Bricker and associates further advanced the catalysis by introducing finely dispersed thin Pt layer over an inert core and thus prepared next generation dehydrogenation catalyst (Jensen et al. 2001a, b). No catalyst development is complete unless a sound process design is wrapped around it that overcomes the techno-economic barrier for a commercial success. Vora and Scott developed a new radial flow reactor design (Vora et al. 1988, 1989) that minimizes pressure drop across the catalyst bed and also allows smooth removal and addition of catalyst.
TA good modified catalyst described gives desired activity and high selectivity to mono-olefins. The formation of diolefins can be controlled kinetically. The "coke" formation is also suppressed, and therefore, stability is greatly improved. In this case the reaction pathway becomes simpler as shown in Fig. 8.

Paraffin dehydrogenation on modified Pt catalyst
Dehydrogenation Catalyst Supports
Selection of a proper support material is critical to a good catalyst performance. Key features of a right support material are:
-
Promotes no adverse side reactions, or it id modified to suppress such reactions
-
Good thermal and physical strength under reaction and regeneration conditions
-
Appropriate surface area for Pt distribution
-
Appropriate pore size for the given feed and product molecules to minimize diffusion resistance
-
Appropriate shape and size for the chosen reactor system
Due to its high activity, very small quantity of Pt is required when uniformly dispersed over the support material. The high dispersion is also necessary to achieve high selectivity to dehydrogenation. For Pt catalyst alumina is a choice for the support, particularly, when properly modified for the reaction system to avoid side reactions.
The catalytic reaction rate is limited by the intraparticle mass transfer rate. When mass transfer rate is slow, activity and selectivity are lowered. To increase the mass transfer rate, the support must have a low pore diffusional resistance. Surface area of alumina can be adjusted by calcination at different temperatures. The strength of the support increase as the pore diameter decreases, and the pore diffusional resistance decreases as the pore diameter increases. Another important factor to be considered is stability of the surface area, pore size, as well as, physical strength when the process requires multiple frequent regenerations, that is carbon burn at high temperature.
Preliminary Catalyst Evaluation
In catalysis research, initial catalyst screen may be done employing several parallel micro reactors having 1 g to up to 5 g of catalyst. Often known as combinatorial chemistry set up. Such testing only good for identifying potential candidate. Next step is to test 50–200 g catalyst under desired temperature and pressure, which are determined from equilibrium data. Once a catalyst shows desired conversion and selectivity, one needs to determine its stability, that is length of operation. for maintaining the conversion and selectivity. At this point, it is good to have an experienced process engineer reviewing the data. Often, catalyst scientist may know have full understanding of overall process design, type of reactor system to be used and may continue looking for a more stable catalyst. As mentioned earlier, for development of a new catalyst for propane dehydrogenation, it was good start with the knowledge of commercially proven long-chain paraffin dehydrogenation. Long-chain paraffin dehydrogenation catalyst, under its operating conditions possesses stability of several months. So, if catalysis scientist is looking for a similar catalyst stability for propane dehydrogenation, due to sever operating conditions required, he may never find it. An experienced process engineer can select a type of reactor system that would allow operation in association with feasibility of frequent catalyst regeneration. Such early association of a process engineer with catalyst scientist significantly reduces time to commercialization.
Use of Pt catalyst requires co-feed of hydrogen for catalyst stability. Presence of hydrogen increases hydrogen partial pressure and minimizes catalyst coking. Reaction conditions, such as temperature, pressure, and hydrogen-to-paraffin feed ratio, are determined on the basis of chemical equilibrium analysis and testing of a reference catalyst. At preliminary evaluation stage the change of conversion as a function of time, as well as, changes in selectivity are observed and catalysts are compared to a reference catalyst. For instance, Fig. 9 shows the conversions of C10 to C13 n-paraffins with DeH-5™ and DeH-7™ Pacol™ dehydrogenation catalysts. DeH-7 catalyst is more stable than DeH-5 (Imai et al. 1989). The selectivity to the corresponding n-mono-olefins for DeH-7 catalyst is equivalent to DeH-5 catalyst, as shown in Fig. 10. The results indicate that DeH-7 has superior stability with similar initial activity to that of DeH-5 and maintains equivalent selectivity to DeH-5 catalyst. Therefore, using DeH-7 achieves a longer life under the same processing conditions or higher productivity with the same catalyst life under more severe conditions.

Tn-paraffin conversion comparison of catalyst stability conversion vs. hours on-stream

Linear olefin selectivity as a function of n-Paraffin Conv
Understanding the relationship between selectivity and the conversion is important. As conversion increases, concentration of product mono-olefin increases at some point reaches close to the equilibrium concentration. This means as the olefin concentration increases, the rate of reaction decreases due to closer approach to equilibrium. If the reaction was allowed to continue, it will mostly form undesired byproducts. The space velocity is chosen to avoid formation of such undesired byproducts, meaning olefin concentration in reactor effluent is in the range of 80 to 90% of equilibrium value at the reactor outlet conditions.
Figure 11 shows simulated selectivity to n-heptene and n-heptadiene for the dehydrogenation of n-heptane. In this simulation, the relative rate constants used are unity, which represents that the catalyst possesses perfect selectivity regarding consecutive dehydrogenation; the dehydrogenation rate of paraffin is equal to that of mono-olefin and diolefin. Experimental selectivity obtained over dehydrogenation catalyst show that the catalyst has virtually perfect selectivity for consecutive dehydrogenation steps, as seen in Fig. 11 (Imai et al. 1989).

Simulation of selectivity for dehydrogenation of n-Heptane
Catalyst stability and regeneration
Once an active and selective catalyst is identified, testing is done under operating conditions determined by process economics. Its activity, selectivity and stability are confirmed for the chosen reactor system. The catalyst scientist also confirms that the catalyst preparation method is reproducible by preparing and testing several batches prepared the same way. Based on catalyst stability, that is length of continuous steady state operation without loss of selectivity and activity, and economics determines whether periodic catalyst regeneration is necessary or not. For light paraffin dehydrogenation, catalyst deactivation is accelerated under high-temperature conditions, and therefore, frequent catalyst regeneration is necessary. This was not the case with long-chain paraffin dehydrogenation. Dehydrogenation processes based on use of chrome on alumina catalyst, both cyclic fix bed or fluidized bed reactor system is used to manage catalyst regeneration. The UOP Oleflex process with platinum catalyst employs moving bed rectors with Continuous Catalyst Regeneration (CCR) technology is applied to manage catalyst regeneration. Ultimate catalyst life of several years is achieved.
Other Research Activities in Alkane Dehydrogenation
In addition to catalytic dehydrogenation processes discussed above, other approaches have also been considered in the past. To date, none has reached to the level of commercialization. Some of the most notable are:
-
Oxydehydrogenation
-
Halogenated dehydrogenation
In oxydehydrogenation or oxidative dehydrogenation use of oxygen plays two different roles. Paraffins in general are inert, In one, oxygen activates paraffins and promotes dehydrogenation. In second it selectively oxidizes meaning combustion of hydrogen, from dehydrogenation, and thus to displace the dehydrogenation equilibrium to higher conversions. As mentioned earlier, this approach has also been used in ethylbenzene dehydrogenation to styrene as in the UOP Styro-Plus™ process or in the ABB Lummus/UOP SMART™ process. Vora has shown use of oxygen to partially combust hydrogen product in propane or butane dehydrogenation to balance endothermic hear of reaction. In this mode reactor can be operated at near isothermal condition, avoiding higher inlet temperatures (Vora 1983). For direct use of oxygen as a means of dehydrogenating, say, ethane to ethylene, some interesting work in this area was done by Prof. Lanny D. Schmidt (Schmidt and Huff 1996) and his colleagues at the University of Minnesota in the United States.
Pyrolysis of methane employing halogens has been worked on by Prof G. Olah (Olah et al. 1985; Olah and Bucsi 1992) at University of Southern California, Los Angeles and also by Prof. Eliseo Ranzi (Ranzi et al. 1992) and coworkers at the Polytechnic University of Milan, Ital. Prof. Sydney Benson (Bensen 1980) proposed it for the production of acetylene and ethylene. Waycullis and his associate at Marathon Oil Co. (Waycullis et al. 2023) did significant work on methane bromination for gas to chemicals (GTC) process, including a demonstration plant at one of their facilities in Texas. E. McFarland did a similar study at the University of California, Santa Barbara (McFarland 2012), and called the process gas reforming technology (GRT). Use of halogens for the dehydrogenation of paraffins has been proposed in different ways. For example, heavy paraffins were first chlorinated and then dehydrochlorinated to heavy olefins commercially in the past both by Shell and by Hüls, among others. Other chlorination/dehydrochlorination or bromination/dehydrobromination cycles have been proposed for the production of ethylene from ethane. None of these approaches have come to commercial reality. Halogens expensive alloys, or metallurgy like titanium or zirconium. Other difficulties are the cost of chlorine, and even more so of bromine, and the need to either dispose of or recycle vast quantities of hydrogen chloride or hydrogen bromide generated as a byproduct.
Conversion of Methanol to Olefins
Methanol-to-hydrocarbon conversion reactions were first discovered in the early 1970s by Chang and his associates at Mobil Oil Co. using ZSM-5 (MFI) catalysts (Chang and Silvestri 1977; Chang 1984). Mobil commercialized a methanol-to-gasoline process in New Zealand in the 1980s and also developed methanol to olefins employing a ZSM-5 catalyst. With support of DOE funding, a 100-barrels per/day demonstration unit was operated in Germany (). Edith Flanigen and associates (Wilson et al. 1982) during early 1980s at Union Carbide Corporation (UCC) discovered a new class of material, called silicoaluminumphosphate (SAPO). A particular structure with 3.8 Å pore opening known as SAPO-34, a silicon-aluminum-phosphorous-based molecular sieve, showed excellent properties for conversion of methanol to light olefins, primarily ethylene and propylene (Kaiser 1985, 1987). The small pore size of SAPO-34 restricts the diffusion of heavy and branched hydrocarbons, which leads to high selectivity to the desired olefins, ethylene and propylene. On the other hand, ZSM-5 molecular sieves produce very little ethylene, some C3–C4 olefins and large quantity of C5–C10 gasoline range products, primarily due to larger pore openings of about 5.5 Å in the MFI structure.
A comparison of ZSM-5 and SAPO-34 structures is shown in Fig. 12 (Vora et al. 2003). Another advantage of SAPO-34 is that the majority of the C4–C6 fraction is linear olefins. This C4–C6 fraction can be converted to light olefins via a catalytic cracking process and, thus, increase the production of C2 plus C3 olefins to near 90%. Reaction product distributions for methanol processed over the ZSM-5 and SAPO-34 catalysts are compared in Fig. 13 (Vora et al. 2006).

Framework of SAPO-34 and ZSM-5 molecular sieves

Once-through hydrocarbon yields for ZSM-5 and SAPO-34
A number of technologies based on the use of ZSM-5 or SAPO-34 as a catalyst have been developed. These are the UOP/HYDRO MTO™ Process, which employs a catalyst based on SAPO-34 material (Vora et al. 1997; Chen et al. 2004), and the Lurgi MTP™ (“Methanol to Propylene”) Process, based on a ZSM-5 type catalyst (Gronemann 2005). Similar technologies also have been developed by Dalian and Sinopec in China (Ying et al. 2013). In China, one commercial unit of each of these technologies came into commercial operation during 2011, and by 2022 there are more than 15 units in operation and several more in design and construction. Both Chinese technologies use catalysts containing SAPO-34 in a fluidized bed reactor with continuous catalyst circulation and regeneration. In addition, there are two methanol to propylene units licensed by Lurgi in operation in China. The MTO Process mainly produces ethylene and propylene and some C4 olefins, while the MTP process mainly produces propylene with gasoline range C5-plus hydrocarbon byproduct. Honeywell’s UOP has licensed 8 or more units of eleven MTO units in China. The first unit at Wison, Nanjing, successfully came onstream during the 2013. By 2019, five UOP licensed units were in operation (Funk et al. 2014).
ZSM-5 catalysts allow larger gasoline range materials, to come out of the pores, thus, there is less formation of coke providing longer catalyst stability relative to SAPO-34, which only allows n-butene and lower molecular weight hydrocarbons. For the ZSM-5 based catalyst system, it is feasible to design a fixed bed reactors with cyclic regeneration, as used by the Lurgi in MTP design. On the other hand, SAPO-34-based catalyst systems employ a circulating fluidized bed reactor and regenerator similar to that used in the fluid catalytic cracking process (FCC) in petroleum refining.
Heat of Reaction
Above we discussed paraffin dehydrogenation, a highly endothermic, and methanol to olefins, a highly exothermic process. Heat of reaction plays important role in selection of reactor type. In paraffin dehydrogenation, for an adiabatic reactor, temperature in the catalyst bed continuously declines from inlet to outlet. The final reactor outlet conditions, pressure and temperature dictate paraffin-olefin equilibrium and thus reactor effluent composition. For a desired per pass conversion, one can calculate catalyst bed delt T. Adding this delta T value to outlet temperature determines required catalyst bed inlet temperature. In case of propane or isobutane dehydrogenation, for an economic conversion, one would see that the inlet temperature for a single stage is so high that significant quantity of the feed would thermally crack in the preheating zone before reaching to the catalyst. Such thermal cracking losses would not be acceptable, requiring lower inlet temperature. Thus, acceptable thermal cracking losses determine, the reactor inlet temperature, and to meet the total delta T of the heat of the reaction, multiple stages with inter-stage heating are used, when composition in the catalyst bed approaches the equilibrium composition, the reaction rate essentially comes to near zero. Any further reaction will only produce undesirable diolefins and other byproducts. This determines quantity of catalyst loading, in other words, optimum space velocity. Figures 14 and 15 illustrates conversion, equilibrium conversion, and temperature along the catalyst bed in a three-stage reactor system for the dehydrogenation of isobutane. IN UOP Oleflex propane dehydrogenation, a four-stage reactor system becomes more economical because higher average temperatures are needed (Imai et al. 1989).

Conversion, equilibrium and temperature relationship in a 3-stage adiabatic reactors

Temperature profile of a single stage and 3-stage reactor system
For methanol to olefins (MTO) processes, being exothermic reaction, reverse is the case, temperature increases as the reaction proceeds. Removing exothermic heat of reaction while reaction is progressing is very critical to avoid excessive temperature rise. Excessive temperature can cause runaway, uncontrolled reaction and would be of major safety concern. In case of methanol to propylene process offered by Lurgi, multistage adiabatic reactors with inter-stage cooling or quenching are used. For MTO process employing SAPO-34 catalyst, fluidized bed reactor system is chosen. In this case large quantity of catalyst is continuously circulated through a heat exchanger, commonly called as catalyst cooler, and steam is generated while catalyst is cooled and returned to the fluidized bed. SAPO-34 based commercial MTO plants currently in operation, either employing a process developed by Chinese companies or UOP, all use fluidized bed reactor system. Large amount of steam generated from the use of these heat exchangers can be used in the process for other energy requirement, such as, reboiler duty for distillation columns, or operate compressors employing steam turbine. Another advantage of fluidized bed reactor system is possibility of continuously removing catalyst for coke burning in a separate fluidized regenerator vessel and returning the regenerated catalyst back to the process rector. The coke burning is also an exothermic reaction and same principle as for the reactor is used to manage the heat of combustion of coke. This also generates large quantity of steam. The combined steam generation from process and regeneration provides energy required for downstream distillation, product recovery section.
Reactor Design Options
Catalyst stability plays important role in type of reactor selection. For a good long-term catalyst stability without any regeneration requirement, months to a year or longer, one would select a fixed bed reactor. As explained earlier, managing heat of reaction would dictate number of stages in series with inter-stage cooling or heating for exothermic or endothermic reaction, respectively. Fixed bed reactor provides plug flow through the catalyst bed. There are several design options for a plug flow design. When pressure drop across the catalyst bed is not an issue, which is usually the case with process operating at high pressure a downflow adiabatic reactor is appropriate. For processes operating at low pressure, one needs to minimize pressure drop across the catalyst bed, and one may choose a radial flow reactor. Another fixed bed type is a tubular heat exchanger type, where heat of reaction can be managed and thus operate in isothermal mode. Such tubular reactors also have high pressure drop and only selected for processes operation at high pressure, usually above 10 bar. For a very short catalyst stability catalyst, in matter of few minutes, one would select fluidized bed operation, as in this case large quantity of catalyst can be circulated between fluidized reactor and a regenerator. When catalyst stability is of few days to few weeks, acyclic reactor-regeneration, such as used in Catofin propane dehydrogenation process. An alternat approach of slow moving bed with a continuous catalyst regeneration, as used in UOP Oleflex propane dehydrogenation may be employed.The fluidized-bed reactor has several other advantages. Heat of reaction, endothermic or exothermic can be managed. For endothermic process regenerated hot catalyst can provide heat of reaction. If coke on spent catalyst is not sufficient to meet the required delta energy, one can burn an auxiliary fuel in the regenerator to meet the requirement. For exothermic process, one can remove heat of reaction by inserting cooling coils with steam generation in the fluidized bed, such as used in acrylonitrile, propylene ammoxidation process. Another approach is to use circulating catalyst through an heat exchanger, catalyst cooler, such as used in MTO or refinery FCC process. This reactor is approximately isothermal as a result of the high degree of mixing.
Its main disadvantage is that the reaction is not plug flow. That is, if back mixing of reaction products causes undesirable side product, the fluidized bed catalytic reactor is not the choice. In addition, there is continuous catalyst losses due to which requires periodic large catalyst additions, The following table summarizes the main characteristics of the five reactor systems discussed.
Downflow | Radial flow | Tubular | Fluidized bed | |
|---|---|---|---|---|
Low pressure drop | \(\bullet\) | \(\bullet\) | ||
Plug flow | \(\bullet\) | \(\bullet\) | \(\bullet\) | |
Catalyst addition or removal | \(\bullet\) | \(\bullet\) | ||
High heat transfer Near isothermal | \(\bullet\) | \(\bullet\) |
Commercial Catalytic Dehydrogenation Processes
Integrated C10-C13 range linear paraffins dehydrogenation to corresponding mono-olefins and benzene alkylation for the production of linear alkylbenzene is shown in Fig. 16, These processes are licensed by Honeywell/UOP. The paraffin dehydrogenation process consists of a radial-flow reactor and a product recovery section. The liquid phase alkylation process employs a downflow fixed bed reactor followed by product separation and recovery section. Worldwide more than 4 million MTA LAB is produced employing this process (Vora et al. 2009).

UOP Pacol dehydrogenation process
Houdry Catadiene process and the Catofin process make use of parallel fixed shallow bed horizontal cylinder type reactors. Due to short chromia/alumina catalyst stability of about 30 min, these reactors operate in cyclic mode. One in process cycle, second in purge mode, third in carbon burning mode, fourth in purge mode and back to process. Thus, minimum of 4 parallel reactors required some large units may have as many as 8 parallel reactors. In these shallow bed reactors various size of alumina or silica balls are used below and above the catalyst bed to support the catalyst and also to fill up the reactor. The short reaction cycle is due to large build of coke on catalyst requiring frequent carbon burn.
The hot catalyst and the inert material in the reactor provide part of the endothermic heat of reaction. Thus, it reduces adiabatic delta T across the catalyst bed making it a semi-adiabatic operation. Figure 17 illustrates a schematic of such a process (Catofin Dehydrogenation Process 2014). Phillips developed catalytic dehydrogenation with steam dilution known as STAR technology (Dunn et al. 1992) and is currently offered by Krupp-Uhde. It uses tubular reactor in a furnace operating at super-atmospheric pressure. In many respects it is similar in design to a steam reforming furnace with the heat of reaction provided by firing outside tubes and thus operating at near isothermal condition. As of 2010 there are two small operating plants, 118,000 MTA and 13,000 MTA, for the production of isobutylene from isobutene.

Catofin process flow diagram
Most widely used process for propane and isobutane dehydrogenation, the UOP Oleflex™ process is shown in Fig. 18. This process employs three or four radial flow reactors in series with inter-stage heat input to provide endothermic heat of reaction. The rectors are integrated with continuous catalyst regeneration (CCR) as shown in Fig. 19. Today more than 5 million MTA of propylene and 2 million MTA isobutene are produced via this route. By 2018 more than 40 operating UOP Oleflex™ units for propane to propylene and six more for isobutane to isobutene, with several more in construction and design (Gregor et al. 2004).

UOP Oleflex process

Oleflex regeneration section
Ethylbenzene Dehydrogenation
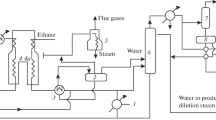
The ethylbenzene dehydrogenation reaction proceeds over an iron or an iron-chromium catalyst containing potassium in the form of potassium carbonate. The reaction takes place at 630 °C and sub-atmospheric pressure. Steam dilution is practiced to further lower the partial pressure of the reactants. Because the reaction is strongly endothermic, various reaction stages with inter-stage heating and interstage addition of superheated steam are normally employed. Figure 20 illustrates a typical process scheme for the dehydrogenation of ethylbenzene to styrene.

Typical ethylbenzene dehydrogenation unit for the production of styrene monomer (SM)
In a, that is hydrogen combustion giving exothermic heat to balance endothermic dehydrogenation reaction. In this mode the reactor in near isothermal. The hydrogen oxidation catalyst is very selective for hydrogen combustion only that there is practically no conversion or degradation of either ethylbenzene or styrene to CO2 (Woodle, et al. 2005). The alternative ethylbenzene dehydrogenation process, known commercially as SMART™(Styrene Monomer by Advanced Reheat Technology), was originally called the Styro-Plus process. The process was first demonstrated at Mitsubishi Chemicals, Kashima, Japan The SMART process is now licensed jointly by UOP/Honeywell and ABB Lummus Global Inc. In addition to supplying the heat of reaction, the SMART process benefits from the equilibrium displacement as a result of selective removal of one of the reaction products, hydrogen (Bricker 2012).
Commercial Methanol to Olefin Processes
Lurgi MTP process
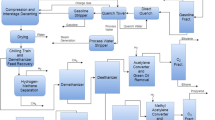
The Lurgi MTP process uses a fixed-bed ZSM-5 catalyst manufactured by Süd-Chemie AG. It provides high propylene selectivity in the range of 65–7-%. Due to large pore size of the ZSM-5 structure, significant amount, in the range of 20–30% gasoline range hydrocarbons are also produced. Ethylene yield is only in 5–10% range. Methanol, both fresh and recycled, recovered from aqueous streams in product recovery operation, is the feed to the MTP unit. Figure 21 (Wurzel 2006) shows a schematic process flow diagram. The methanol is vaporized, superheated, and fed to a single stage adiabatic reactor, where most of the methanol is converted to dimethyl ether (DME) on an alumina catalyst. The reaction is exothermic and closely approaches thermodynamic equilibrium. The DME reactor effluent is sent to three MTP reactors in parallel: Two of the reactors are in operation, while a third one is in regeneration or on stand-by. Each of these reactors employ multiple catalyst beds with feasibility to inject lower temperature steam in between to offset exothermic heat of reaction. Thus, it avoids excessive temperature rise. The operating temperature is about 450 °C and the operating pressure is 0.15 MPa (about 20 psia). As mentioned earlier, gasoline range hydrocarbons is a large byproduct of the side reactions. Also, water is a large co-product.

Lurgi MTP process flow scheme
The ZSM-5 catalyst used in MTP process is regenerated by a simple carbon bun. The MTP reactor effluent is cooled in a heat recovery system and, finally, through a quench section, in which the hydrocarbons are separated from the bulk of the water. The water is condensed and sent to the methanol and DME recovery column, from which they are recycled to the DME reactor. The water with traces of oxygenates is routed biodegradation ponds for safe disposal or other uses, such as agriculture. The hydrocarbon vapor from the quench section is compressed by a multistage centrifugal compressor with intercoolers and partial condensers. The liquid and vapor hydrocarbons are sent to the purification section employing multiple distillation columns. Product propylene and other light and heavy byproducts are recovered.
UOP/HYDRO MTO process
As mentioned earlier, Edith Flanigen and her associates at UCC discovered SAPO-34 material and showed it to be a good catalyst for methanol to olefins reaction during the early 1980s. This group at UCC, then known as Catalyst, Adsorbent and Process Systems, was merged with UOP LLC, Des Plaines, Illinois. As a result, further development for the MTO technology took place at UOP. In 1992 UOP formed a partnership with Norsk Hydro of Norway for further joint development of the technology. A fluidized bed reactor-regenerator demo for a one ton per day methanol feed was built and operated at Norsk Hydro facility in Norway for several years. At the end of 1995, a joint team of UOP and Norsk Hydro first presented their data at the World Natural Gas Symposium held in South Africa, and proposed a process employing a fluidized bed reactor and regeneration design (Vora et al. 1997).
Though UOP engineers were confident of their design having 40 plus years of experience of fluid bed catalytic cracking (FCC) process widely used in petroleum refinery, was not sufficient to convince potential licensees viability of the design. Two issues were of main concern; scale-up of reactor data from a small fixed and fluid bed reactors to such large, 2–5 m in diameter commercial reactor. T The ethylene produced is from an oxygenate feed methanol, which was different than conventional ethane or naphtha feed, raising concern that the product ethylene may contain unknown oxygenate impurities that can cause adverse effects to downstream polymerization catalyst.
To address these issues, a partnership with Total of France was formed and a large fully integrated MTO demonstration unit was built with high purity ethylene and propylene recovery, including polymerization reactor for polyolefins. With successful demonstration by 2010, UOP licensed its first unit in China. The Total partnership was expanded to include Total’s Olefins Cracking Process (OCP), a process for cracking higher C4–C6 olefins to propylene and ethylene. Integration of this process with MTO allowed UOP to increase ethylene-propylene yield to near 90% and the integrated process is called UOP Advanced MTO Process.
The UOP/HYDRO MTO Process can use “crude” methanol, “fuel-grade” methanol, Grade AA methanol, or even DME as feed. The choice of feedstock generally depends on project-specific situations. Figure 22 illustrates a simple flow diagram for the UOP/HYDRO MTO Process (Eng et al. 1998). The MTO process utilizes a circulating fluidized bed reactor that offers a number of advantages over both fixed bed reactors and other types of fluidized bed reactors. The circulating fluidized bed reactor provides better mass transfer than bubbling bed fluidized bed reactors as well as better temperature control than riser and fixed bed reactors, especially given the highly exothermic nature of the methanol-to-olefins reactions. This type of reactor has been widely used in the FCC process units in petroleum refineries.

UOP/HYDRO MTO process flow scheme
Constant catalyst activity and product composition can be maintained via continuous regeneration of a portion of used catalyst by coke burning with air. UOP’s MTO catalyst has demonstrated the required selectivity, long term stability, and attrition resistance necessary for attractive economics with low operating costs.
The overall selectivity of the UOP/HYDRO MTO process is about 75–80% to ethylene and propylene on a carbon basis, and about 15% C4 plus hydrocarbons. The balance is C1-C3 paraffins plus coke on catalyst. The C4 plus material is mostly linear butenes and some pentenes. These olefins make an ideal feed to the OCP unit to further increase the yields of ethylene and propylene. Propylene to ethylene ratios in the product can be adjusted within the range of 0.80–1.33 (Fig. 23) to reflect the relative market demand and values for ethylene and propylene. The reactor temperature is the key variable for controlling propylene to ethylene ratios, with higher temperatures leading to a higher ethylene yield. The temperature requirements have to be balanced with higher coke formation at higher temperatures.

Olefin selectivity vs. operating severity of the UOP MTO process with and without olefin cracking process
The reactor pressure is normally dictated by mechanical considerations. Lower methanol partial pressure leads to higher selectivity to light olefins, especially ethylene. Therefore, a slight yield advantage occurs when using a crude methanol feed compared to high purity methanol. The reactor effluent is cooled and quenched to separate water from the product gas stream. The reactor provides very high conversion, so there is no need for a large recycle stream.
A small amount of unconverted oxygenates are recovered in the oxygenate recovery section, after which, the effluent is further processed in the fractionation and purification section. Conventional treating methods have been shown to be effective for removing by-products to the specification levels required for producing polymer-grade ethylene and propylene products. As shown in Fig. 16 (Senetar 2019), the total ethylene plus propylene yield can be further enhanced by incorporating a cracking process to convert C4 plus material to propylene and ethylene. Overall carbon selectivity for the integrated flow scheme approaches 90% ethylene plus propylene.
Integration of CTL/GTL with CTO/GTO
GTL and CTL processes offer large product market opportunities for natural gas and coal utilization but are challenged by high capital costs and the relatively low transportation-fuel product values. Since synthesis gas production is a common step in the manufacture of GTL and methanol, there are possibilities for integrated complexes. Figure 24 (Vora et al. 2010) illustrates such a complex, using coal or natural gas as feedstocks, and producing both olefins and liquid fuels. Both options—coal or natural gas liquid fuels (CTL, GTL) and coal or natural gas to polymers (CTP, GTP) facilities—incorporate sizeable front-end synthesis gas units for the processing of natural gas. Over 60% of the capital cost is related to the production of synthesis gas. These units are the major contributors to the relatively high investments required for these complexes. It follows that the integration of these facilities to co-produce fuels and chemicals could offer substantial synergistic savings.

GTO/GTL complex for production of liquid fuels and olefins from coal or natural gas
A rough rule of thumb is that the quantity of synthesis gas required to produce 25,000 barrels per day (BPD) liquid hydrocarbons could also be used to produce in excess of one million MTA light olefins. A typical liquid fuels plant is likely to have a capacity of 100,000 BPD or more. Thus, the addition of one million MTA light olefins represents roughly 25% additional synthesis gas production capacity. The incremental production will require substantially less capital cost than a stand-alone smaller unit. The integration of methanol and liquid fuels facilities combined seen in today’s oil refining sector, where there is increased focus on opportunities for petrochemicals production as some regional fuel demands change with the conversion of methanol to olefins, can provide cost saving synergies together with the production of high value-added olefins and polymer products. This would follow the current trend.
Direct Conversion of Syngas into Light Olefins
During the past decade China has built several MTO commercial plants, some based on indigenous methanol derived from coal and others on imported methanol from North America or Middle East. Because of success of SAPO-34 in selective conversion of methanol to light olefins, scientists at several Chinese academic and state institutions are exploring direct conversion of synthesis gas to olefins with dual function SPO-34 with various metal oxides. Sen Wang and his associates (Wang et al. 2020) at Institute of coal chemistry and Chinese academy of sciences has done significant work on direct conversion of syngas into light olefins over bifunctional catalysts. It was found that a zinc–cerium–zirconium solid solution (ZnxCe2–yZryO4) and a SAPO-34 mixture showed CO conversion, light olefin selectivity in hydrocarbons, and O/P ratios of about 7%, 83%, and 23, respectively, at 300 °C and 1 atm. Nevertheless, higher conversion at 380 °C led to producing large amounts of CO2 Another Chinese group, Peng Zhang and his associate (Zhang 2019) reported a bifunctional catalyst consisting of a Mn–Ga oxide and SAPO-34 was designed for the direct conversion of syngas to light olefins. A selectivity for olefins of 88.3% and 13.7% CO conversion were obtained. In both above studies conversion of synthesis gas is less than 20% compared to near 100% for methanol over SAPO-34, and therefore at present it is economically not viable option. Furthermore, plants for direct conversion of synthesis gas to olefins.
Must be located at the same site. Methanol being liquid and easily transportable gives flexibility of producing at the coal or natural gas reserves site and produce olefins at the location where olefins are consumed.
Conclusions
Catalytic dehydrogenation of paraffins and of ethylbenzene is a commercial reality in widespread use for numerous applications These include the production of light olefins, heavy olefins, and alkenyl aromatics. Oxydehydrogenation, on the other hand, is still in the developmental stage but if successful, holds great promise due to the potential energy savings of this process. For the production of heavy olefins, selective paraffin dehydrogenation over noble metal catalysts has proven to be the preferred and dominant route.
When only one or two light olefins are desired, in particular propylene or isobutylene or perhaps a mixture of propylene and isobutylene, catalytic dehydrogenation over noble metal catalysts has acquired a significant and growing market share. Though there are no direct routes for the conversion of coal or methane to olefins, these raw materials can be converted to ethylene and propylene via methanol as shown by MTP and MTO processes already in commercial use. These are the primary raw materials for a vast number of petrochemicals and polymer industry products, like PVC, polyethylene, polypropylene, acetic acid, acrylonitrile, formaldehyde, ammonia and many more. The vast resources of coal in areas of high demand, namely the USA, China, and India, could provide long-term raw material security.
Finally, the choice of reactor plays an important role in securing the success of a catalytic process. Pressure drop, heat transfer, and the ability to move or to regenerate the catalyst all must be taken into account in the process development and design stages.
Data availability
This is a review paper, no new research data were used.
References
Bensen SW (1980) U. S. patent 4,199,533, April 20, 1980
Bloch HS (1969) UOP discloses new way to make linear alkylbenzene. In: Oil and Gas Journal, 79–81, Jan. 16, 1967; U. S. patent 3,448,165, June 3rd, 1969
Bricker JC (2012) Advanced catalytic dehydrogenation technologies for production of olefins. Top Catal 55(19–20):1309–1314
Bricker JC et al (1968) U. S. patent 4,914,075, 5 December 1968
Bricker JC et al (1992) U. S. patent 5,233,118, 22 January 1992
Carson DB et al (1973) US Patent 3,799,868; 8 May 1973
Catofin Dehydrogenation Process (2014) CBI-Lummus; http://www.chemwinfo.com/private_folder/Uploadfiles2015_December/CBI_CatofinTechSheet_2014. Accessed 17 Apr 2023
Chang CD (1984) Chemicals from methanol. Catal Rev-Sci Eng 26(3&4)
Chang CD, Silvestri AJJ (1977) The conversion of methanol and other O-compounds to hydrocarbons over zeolite catalysts. J Catal 47:249–259
Chauvel A, Lefebvre G (2001) Sources of olefinic and aromatic hydrocarbons, Chapter 2, Table 2–2. In: Petrochemical Processes. Editions Technip, France, p 140
Chemical Marketing Reporter (CMR) inc. (2013) vol 7, no. 15
Chen JQ, Vora BV, Pujado PR, Gronvold A, Fuglerud T, Kvisle S (2004) Most recent development in ethylene and propylene production from natural gas using the UOP/Hydro MTO ProcessTM. Stud Surf Sci Catal 147:1–6
Craig RG, Spence DC (1986) Catalytic dehydrogenation of liquefied petroleum gas by the Houdry Catofin and Catadiene processes. In: Meyers R (ed) Handbook of petroleum refining processes. McGraw-Hill, Berlin (section <Emphasis Type="Bold">4.1</Emphasis>)
DOE (1986) Conversion of Methanol to Gasoline-Extended Project: Methanol to Olefins Demonstration Plant mile stone Report; US Department of Energy, DOE/ET/14914-H; DE86 015960, April 1986
Dunn RO, Brinkmeyer FM, Schuette GF (1992) The Phillips STAR Process for dehydrogenation of C3 C4 and C5 paraffins. In: NPRA Annual Meeting, New Orleans, LA 22–24 March 1992
Egloff G, Davis RF (1951) Polymerization of mono-olefins with solid phosphoric acid. In: XII International Congress of Pure and Applied Chemistry, New York, NY, September 10–13, 1951
Eng CN, Arnold EC, Vora BV, Fuglerud T, Kvisle S, Nilsen H (1998) Integration of the UOP/HYDRO MTO process into ethylene plants. In: AIChE Spring National Meeting, March 8–12, 1998, New Orleans, Louisiana
Eramo M (2016) Global ethylene market. In: IHS Chemical Asia Chemical Conference November 2016, Singapore
Funk G, Senetar J, Bozzano A, Gregor J (2014) Methanol to olefins a new and profitable route for the production of light olefins. In: AIChE Spring Meeting 2014
Gantt JE et al (1974) US Patent 3,799,868; 26 March 1974
Gregor J, Wei D (2004) UOP Oleflex process for light olefin production, Chapter 5.1. In: Meyers R (ed) Handbook of petroleum refining processes, 3rd edn. McGraw-Hill Handbooks
Gregor JH, Antonelli R, Foley TD, Arnold EC (1999) Increased opportunities for propane dehydrogenation. In: DeWitt World Petrochemical Review, Houston, Texas, March 23–25, 1999
Gronemann V (2005) 3 in 1—Lurgi Syngas to Propylene. In: Presented at 2005 World Methanol Conference, Miami, FL. USA, December 12–14
Grosse AV, Ipatieff VN (1940) Catalytic dehydrogenation of gaseous paraffins. Ind Eng Chem 32(2):268–272
Haensel V (1952) U. S. patent 2,602,772, July 8, 1952
Hearings start in Diet on chromium pollution (1975) The Daily Yomiuri (Japan), Sept. 10, 1975, and other similar Japanese newspaper articles concerning chromium pollution.
HIS Chemical (2013) World Light Olefins Analysis
Honeywell UOP (2020) Oleflex technology continues growth in China, Hydrocarbon Processing, 10 September 2020; https://www.hydrocarbonprocessing.com/news/2020/09/honeywell-uop-oleflex-technology-continues-growth-in-china. Accessed 2019
Hornaday GF, Ferrell FM, Mills GA (1961) Manufacture of mono- and diolefins from paraffins by catalytic dehydrogenation. In: McKetta Jr JJ (ed)Advances in petroleum chemistry and refining, Vol. 4. Interscience Publishers
Imai T et al (1988) U. S. patent 4,786,625, 22 November 1988
Imai T, Vora BV, Bricker JC (1989) Development of dehydrogenation catalysts and processes. Petroleum and Petrochemical College, Chulalongkorn University, Bangkok
Imai T et al (1989) U. S. patent 4,827,072, 2 May 1989
Jensen RH et al (2001) U. S. patent 6,177,381, 23 January 2001
Jensen RH et al (2001) U. S. patent 6,280,608, 28 August 2001
Kaiser SW (1985) US Patent 4 499 327
Kaiser SW (1987) US Patent 4,617,242
Lester GR et al (1968) US Patent 3,361,839; 2 January 1968
McFarland W (2012) Unconventional chemistry for unconventional natural gas. Science 308:340–342
Occupational exposure to chromium (VI) (1975) U.S. Dept. of Health, Education, and Welfare, Public Health Service, Center for Disease Control, National Institute for Occupational Safety and Health, 1975.
Olah G, Bucsi L (1992) Selective monochlorination of methane over solid acid catalyst and zeolite catalysts. Catl Lett 16:27–38
Olah G et al (1985) Methane activation by halogenation. J Am Chem Soc 107:7097–7105
Poole CP, Kehl WL, MacIver DS (1962) Physical properties of coprecipitated chromia-alumina catalysts. J Catal 1:407–415
Poole CP, MacIver DS (1967) The physical-chemical properties of chromia-alumina catalysts. Adv Catal 17:223–314
Ranzi E, Dente M, Rovaglio M, Faravelli T, Karra S (1992) Pyrolysis and Chlorination of small hydrocarbons. Chem Eng Commun 117(1):17–39
Roth (2023) US Patent 3,356,757
Sanfilippo D, Buonomo F, Fusco G, Miracca I (1998) Paraffins activation through fluidized bed dehydrogenation: the answer to light olefins demand increase. Stud Surf Sci Catal 119:919–924
Schmidt LD, Huff M (1996) World patent 96/13475; 9 May 1996
Senetar J (2019) Development of UOP’s advanced MTO process—the result of collaborative efforts catalyzed by Bipin Vora. In: Spring 2019 AIChE Meeting, F&PD Session 105 in honor of Bipin Vora, April 1–4, New Orleans, LA
Universal dehydro Process U (1947) Oil & Gas J. 22 March 1947, p 179
Vesely KD et al (1972) US patent 3,654,186; 4 April 1972
Vora BV (1983) US Patent 4,376,225; 8 March 1983
Vora B (2010) Chemicals from coal—a smart choice, chapter 15. In: Sikdar S, Princiotta F (eds) carbon management technologies. Taylor Francis Group, pp 321–341
Vora BV (2012) Development of dehydrogenation catalysts and processes. Top Catal 55:1297–1308
Vora BV et al (1988) U. S. patent 4,720,336, 19 January 1988
Vora BV et al (1989) U. S. patent 4,869,808, 26 September 1989
Vora BV, Marker TL, Barger PT, Nilsen HR, Kvisle S, Fuglerud T (1997) Economic route for natural gas conversion to ethylene and propylene. In: de Pontes M, Espinoza RL, Nicolaides CP, Scholz JH, Scurrel MS (eds) Stud. Surf. Sci. Catal., vol 107. Elsevier, Amsterdam, pp 87–98
Vora BV, Pujado P, Anderson J, Greer D (2003) Natural Gas Utilization-DME, GTL and polyolefins integration. Oil Asia J
Vora BV, Bozzano A, Foley T, Anderson J (2006) Utilize cost advantaged raw materials for light olefin production. In: Lee S (ed) 2006 ERTC Petrochemical Conference, February, Dusseldorf, Germany.
Vora BV, Peterson GA, Sohn SW, Riley MG (2009) Detergent alkylate and detergent olefins production. In: Hand book of detergents part F, surfactant science series Vol 142, p 39–48
Vora BV, Funk G, Bozzano A (2015) Chemicals from natural gas and coal. In: Treese SA, Pujado PR, Jones DS (eds) Handbook of petroleum processes, vol 1. Springer Reference, pp 883–904
Vora BV et al (2006) Detergent alkylate. In: Encyclopedia of chemical processing and design. Marcel Dekker, Inc., pp 15663–672
Waddams AL (1980) Chemicals from petroleum, 4th edn. Gulf Publishing Company, p 1978
Wang S et al (2020) Direct conversion of syngas into light olefins with low CO2 emission. ACS Catal 10(3):2046–2059
Water Pollution in North Rhine-Westphalia (1958) www.google.com/search?q=rhine+river+detergent+pollution
Waycullis J, Gadewar S, Thomas R (2023) GTC Technology, Marathon oil US 8802908B2
Weiss AH (1970) The manufacture of propylene. In: 158th Meeting of the American Chemical Society, Sept. 10–12, 1969. Refining Petroleum for Chemicals, Advances in Chemistry Series, Vol. 97, American Chemical Society, 1970
Wilson ST, Lok BM, Flenigen EM (1982) US Patent 4,310,440
Woodle G et al (2005) US Patent 6,858,769; 22 February 2005
World patent (1996) WO 96/34843, November 7, 1996
Wurzel T (2006) Lurgi megamethanol technology. In: Synthesis Gas Chemistry, DGMK Conference, October 4–6
Ying L, Ye M, Cheng Y, Li X (2013) A kinetic study of methanol to olefins process in fluidized bed reactor. In: The 14th International Conference on Fluidization—From Fundamentals to Products, January
Zhang P (2019) Excellent selectivity for direct conversion of synthesis gas to light olefins. Sci Technol 9:5571–5581
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Vora, B.V. Development of Catalytic Processes for the Production of Olefins. Trans Indian Natl. Acad. Eng. 8, 201–219 (2023). https://doi.org/10.1007/s41403-023-00401-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41403-023-00401-2