
Abstract
Purpose
Fibrosis is the only histological feature reflecting the severity and prognosis of nonalcoholic steatohepatitis (NASH). We aim to explore novel genes associated with fibrosis progression in NASH.
Methods
Two human RNA-seq datasets were downloaded from the public database. Weighted gene co-expression network analysis (WGCNA) was used to identify their co-expressed modules and further bioinformatics analysis was performed to identify hub genes within the modules. Finally, based on two single-cell RNA-seq datasets from mice and one microarray dataset from human, we further observed the expression of hub genes in different cell clusters and liver tissues.
Results
7 hub genes (SPP1, PROM1, SOX9, EPCAM, THY1, CD34 and MCAM) associated with fibrosis progression were identified. Single-cell RNA-seq analysis revealed that those hub genes were expressed by different cell clusters such as cholangiocytes, natural killer (NK) cells, and hepatic stellate cells (HSCs). We also found that SPP1 and CD34 serve as markers of different HSCs clusters, which are associated with inflammatory response and fibrogenesis, respectively. Further study suggested that SPP1, SOX9, MCAM and THY1 might be related to NASH-associated hepatocellular carcinoma (HCC). Receiver operating characteristic (ROC) analysis showed that the high expression of these genes could well predict the occurrence of HCC. At the same time, there were significant differences in metabolism-related pathway changes between different HCC subtypes, and SOX9 may be involved in these changes.
Conclusions
The present study identified novel genes associated with NASH fibrosis and explored their effects on fibrosis from a single-cell perspective that might provide new ideas for the early diagnosis, monitoring, evaluation, and prediction of fibrosis progression in NASH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nonalcoholic fatty liver disease (NAFLD) currently affects about 25% of the global population and is likely to become the leading cause of end-stage liver disease in the coming decades, with nearly a quarter of NAFLD patients progressing to NASH [1]. As a progressive inflammatory phenotype of NAFLD, NASH is closely associated with hypertension, obesity, dyslipidemia, type 2 diabetes and metabolic syndrome, and its progression involves multiple pathogenic pathways [2,3,4].
Fibrosis is an important concern in the NASH field and a major determinant of patient clinical outcomes [5,6,7,8]. Progressive fibrosis reflects the ongoing regrowth of repeated useless liver frameworks, and these defective liver regrowth increases the risk of cirrhosis and primary liver cancers [9]. Apart from lifestyle interventions, more and more treatment options are expected to be used to limit fibrosis and NASH. As a peroxisome proliferator-activated receptor agonist, Lanifibranor improves NASH by regulating metabolism, inflammation, and fibrogenesis [10]. In addition, GLP-1 RAs is considered a promising treatment option for NASH and deserves further investigation [11, 12]. However, it is also necessary to find new biomarkers, therapeutic targets, and ideas for fibrosis progression in NASH and NASH-associated HCC. As a common bioinformatics analysis method, WGCNA can effectively explore the relationship between genes and clinical characteristics. The significant advantage of WGCNA is that it links sample characteristics with changes in gene expression by clustering genes into co-expression modules so as to identify modules related to phenotype, and finally identify genes in disease pathway for further verification [13].
In this study, we aimed to identify susceptibility modules and genes associated with fibrosis. We used two RNA-seq datasets from the Gene Expression Omnibus (GEO) database to select gene expression data associated with NASH. The GEO database is an international public repository, which provides valuable high-throughput microarray and next-generation sequence functional genomic datasets that can be downloaded free of charge in a variety of formats and for further integration analysis [14]. WGCNA was used to construct gene co-expression network, select important modules, and screen hub genes through protein–protein interaction (PPI) network. Single-cell RNA-seq analysis was integrated to observe the expression of hub genes in different cell clusters and we further analyzed whether these genes are involved in the malignant progression of NASH based on another microarray dataset.
Materials and methods
Data download and processing
The framework of this study was shown in Fig. 1. Two RNA-seq datasets (GSE135251 and GSE162694), one microarray dataset (GSE164760), and two single-cell RNA-seq datasets (GSE166504 and GSE182365) were systematically extracted from the GEO database (Supplementary file 1). The datasets we selected were described in Table 1. GSE135251, GSE162694 and GSE164760 were sequenced from human liver tissues, while GSE166504 and GSE182365 were from mice fed with high fat diet. The clinical characteristics of the human samples were described in the datasets uploader's articles [15,16,17]. GSE135251 included 51 NAFL, 155 NASH and 10 controls. GSE162694 comprised of 31 normal and 112 NASH liver tissues. We selected gene expression and clinical information related to NASH in GSE135251 and GSE162694 to prepare for further analysis. One outlier sample (548nash100) was removed by cluster analysis (Supplementary file 2). Finally, 155 NASH samples from GES135251 and 111 NASH samples from GSE162694 were identified. We used the ClusterProfiler R package to perform gene symbol conversion on the downloaded counts data [18]. According to the suggestions of WGCNA, we processed the data with variance-stabilizing transformation and logarithmic transformation, screening out the top 5000 genes of variance variation in NASH samples respectively for WGCNA analysis.

Flow diagram of this study
WGCNA analysis and identification of modules
We used WGCNA R package to construct a co-expression network for the top 5000 genes mentioned above [13]. Firstly, the R function pickSoftThreshold was used to calculate soft threshold power from 1 to 20. Secondly, hierarchical clustering and dynamic tree cut package were used to divide modules (abline = 0.25). Thirdly, correlation between module eigengenes and clinical traits was analyzed to identify modules of interest. Finally, excluding the grey modules that did not cluster successfully, we selected modules in two networks that are significantly positively correlated with fibrosis progression.
PPI network construction and hub genes
The common genes within the modules were displayed by Venn diagram. To further understand the biological functions of common genes, ClusterProfiler R package was used for Gene Ontology (GO) enrichment, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis as well as ReactomePA R package was used for Reactome pathway analysis [19,20,21]. GO annotations include biological processes (BP), molecular functions (MF), and cellular components (CC). We presented the results of the top ten BP analysis, all KEGG and Reactome pathway analysis. Adjusted P < 0.05 indicates significant differences. We also used the STRING database to construct PPI network for overlapping genes and imported data with interaction score > 0.4 (medium confidence) into Cytoscape 3.8.2 software for visualization [22, 23]. Finally, CytoHubba and MCODE plugins were used to analyze the functional modules of PPI network and identify hub genes. CytoHubba plugin obtained top ten genes through MMC algorithm. MCODE plugin parameters were set as follows. Degree cut-off: 2, node score cut-0.2, cut-style: haircut, K-core: 2, Max. Depth: 100. Pearson’s correlation coefficient was used to evaluate correlations between hub genes.
Integrating single-cell RNA-seq analysis based on hub genes
Based on another dataset GSE166504, single-cell sequencing results of hepatocytes and liver non-parenchymal cells (NPC) in mice, we selected NPC during high-fat feeding and analyzed them using Seurat R package with default settings (Supplementary file 3) [24]. At the same time, we also performed cluster analysis on HSCs from high-fat fed mice in another dataset GSE182365 to observe gene expression in different cell clusters (Supplementary file 4). Combined with the CellMarker database and relevant literature, cell clusters were annotated [25,26,27,28].
Expression of hub genes in different liver tissues
GSE164760 is a human RNA-seq dataset including normal, cirrhotic, adjacent non-tumor NASH, and NASH-associated HCC liver tissues. Based on GSE164760, we further analyzed whether it was involved in NASH-HCC transformation. ROC analysis was performed using the R software package pROC to evaluate their accuracy in predicting HCC according to the expression of differential genes [29]. We also used the human protein atlas (HPA) database to compare differences at protein levels of corresponding genes (paired by antibody, sex, and age). HPA is a protein database which is committed to providing tissue and cell distribution information of a variety of human proteins and provides free public inquiries [30]. Finally, consensus clustering was performed for 53 NASH-associated HCC to compare the differences between different subtypes. Consensus Clustering, a common approach to the classification of cancer subtypes, allows for the discovery of new disease subtypes or comparative analysis of different subtypes by dividing samples into several subtypes based on different omics datasets [31]. The potential KEGG signaling pathway of HCC was explored by GSVA analysis [32]. The comparison between groups was performed by limma [33]. Benjamini & Hochberg was used for multiple comparison correction and adjusted P < 0.05 was considered statistically significant (Supplementary file 5).
Results
WGCNA and module identification
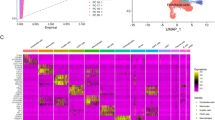
The top 5000 genes in NASH samples were selected to construct the co-expression network. According to the principle of scale-free network, 3 was selected as the soft threshold of network 1 and network 2 (scale free R^2 is 0.93 and 0.95 respectively) (Fig. 2a, b). Network 1 (from GSE135251) and Network 2 (from GSE162694) were divided into 11 modules respectively (Fig. 2c, d). We associated the models with fibrosis stage of NASH, looking for models that were most significantly associated with fibrosis progression (Fig. 2e, f). Excluding grey module (fail to cluster), we selected positive modules with P < 0.01 in NASH F4 and performed BP analysis on them. The blue module (GSE162694) is associated with the extracellular matrix, the red module (GSE162694) with immune inflammation, and the brown module (GSE135251) with both (Supplementary file 6).

WGCNA analysis (a, b) Scale-free fit index in network 1 and 2 (x-axis is soft threshold power, y-axis is signed R^2). (c, d) Clustering dendrogram of genes (visual comparison of modules based on dynamic tree cutting). (e, f) Heatmaps of correlation between module eigengenes and clinical traits
PPI network construction and functional annotation
There are 234 common genes, and a PPI network was constructed (Fig. 3a, b). According to BP, these genes are mainly involved in external encapsulating structure organization, extracellular structure organization and extracellular matrix organization (Fig. 3c). Based on KEGG pathway analysis, our results showed that these genes are mainly involved in wnt signaling pathway, extracellular matrix (ECM)-receptor interaction and viral protein interaction with cytokine and cytokine receptors (Fig. 3d). Reactome pathway analysis showed that these genes are mainly related to extracellular matrix organization (Fig. 3e). These results suggested that these genes mainly participate in fibrosis repair during NASH development.

PPI network of common genes and functional annotation (a) Venn diagrams of overlapping genes. (b) PPI network (Arranged by degree, the high degree (dark color) is in the center of the circle). (c) Bar chart of BP analysis results. (d, e) Bubble diagram of KEGG and Reactome analysis results
Hub genes in each fibrosis stage
The results of CytoHubba and MCODE were intersected to obtain seven hub genes (SPP1, PROM1, SOX9, EPCAM, THY1, CD34 and MCAM) (Fig. 4a). As we can see, there is a good correlation between hub genes, and the correlation is highly consistent in the two datasets (Fig. 4b). The line chart also showed an upward trend (Fig. 4c, d) (Supplementary file 5).

Expression of hub gens in different fibrosis stages (a) Algorithm of CytoHubba and modules of MCODE. (b) Visualization of correlation between hub genes in different datasets. (c, d) Line charts of fibrosis stages (false discovery rate was used for multiple comparison correction in Deseq2, *adjusted P < 0.05; **adjusted P < 0.01; ***adjusted P < 0.001)
Hub genes in different liver NPC clusters from mice
Based on the expression of lineage-specific markers, we identified B cells, conventional dendritic cells (cDCs), cholangiocytes, cycling cells, endothelial cells, HSCs, hepatocytes, Kupffer cells, monocyte-derived macrophages (MDMs), NK cells and Plasmacytoid dendritic cells (pDCs) (Fig. 5a). As we can see, SPP1, PROM1, SOX9 and EPCAM are highly expressed in cholangiocytes, THY1 is in NK cells, and CD34 is in HSCs (especially in late period), while MCAM is not specifically expressed (Fig. 5b). The total expression of these hub genes increased gradually in NPC apart from MCAM (Fig. 5c).

Single-cell RNA-seq analysis of liver NPC from mice during high-fat feeding (a) t-SNE visualization of liver NPC clusters. (b) Violin plot of hub genes expression in liver NPC clusters. (c) Dot plot of total expression of hub genes at 15, 30 and 34 weeks during high-fat feeding
Hub genes in different HSCs clusters from mice
HSCs is the main source of ECM [34]. Through the clustering of HSCs from mice during high-fat feeding, we divided seven HSCs clusters (Fig. 6a, b). Cluster 7 is the mixed cholangiocytes based on annotation, which expresses most markers of cholangiocytes and has a small number of cells. We found that SPP1 and CD34 are markers of cell cluster 2 and 5, respectively (Fig. 6c). According to the BP analysis of their respective markers of cluster 2 and 5, they may have different biological functions. Cluster 2 expresses various cytokines, chemokines and receptors and is mainly involved in inflammation (Fig. 6d). Cluster 5 is mainly associated with extracellular matrix formation (Fig. 6e).

Single-cell RNA-seq analysis of HSCs from mice during high-fat feeding (a) Umap visualization of HSCs clusters. (b) Heatmap based on the top five markers of each cluster. (c) Violin plot of hub genes expression in HSCs clusters. (d, e) Circle diagrams of BP enrichment results of cluster 2 and 5
Correlation between hub genes and NASH-associated HCC
We further analyzed whether these hub genes were also involved in NASH-HCC transformation through another human dataset GSE164760. Four hub genes, SPP1, SOX9, MCAM and THY1, showed significant differences between NASH and NASH-associated HCC tissues, suggesting that they are likely to be associated with NASH-HCC, while no differences were observed in PROM1, EPCAM and CD34 (Fig. 7a). At the same time, ROC analysis indicated that the high expression of these differential genes (SPP1, SOX9, MCAM and THY1) can well predict the occurrence of HCC (Fig. 7b). Finally, we verified differences at protein levels using the HPA database. The results showed that the protein levels of these four genes in tumor tissues were higher than those in normal tissues (Fig. 7c). Taken together, these four genes play an important role in the transformation of NASH to HCC.

Expression of hub genes in different liver tissues (a) mRNA expression of hub genes in normal, cirrhotic, adjacent non-tumor NASH, and NASH-associated HCC liver tissues (**adjusted P < 0.01). (b) ROC analysis to evaluate expression of differential genes in predicting HCC. (c) Immunohistochemical staining of SPP1, SOX9, MCAM and THY1 in normal and HCC liver tissues
Comparison of different NASH-associated HCC subtypes
Through consensus clustering, it was found that 53 NASH-associated HCC samples from GSE164760 could be roughly divided into 2 subgroups (C1 and C2), which was well illustrated by PCA analysis (Fig. 8a, b). Compared with C1, 96 up-regulated pathways and 14 down-regulated pathways were found in C2 (Fig. 8c). Among them, these pathways are mostly related to metabolism (Fig. 8d). The expression of SOX9 was significantly downregulated in C2, while SPP1 and THY1 only showed a downward trend (Fig. 8e).

Difference between NASH-associated HCC subtypes (a) consensus clustering to divide subtypes. (b) PCA diagram of NASH-associated HCC samples. (c) Volcanic map of differential KEGG pathways. (d) Bar chart to show the top 50 most significant pathways. (e) Expression of SPP1, SOX9, MCAM and THY1 in C1 and C2
Discussion
NAFLD is represented by nonalcoholic fatty liver (NAFL) and NASH. Compared with NAFL, NASH progresses more rapidly in fibrosis, which can lead to cirrhosis and HCC [7, 34, 35]. Fibrosis is the only histological feature that can predict NASH clinical outcome and more and more noninvasive biomarkers are being attempted to reflect the severity of fibrosis [36,37,38,39]. In this study, we performed WGCNA on NASH samples from datasets GSE135251 and GSE162694, respectively, identified the modules of interest, and obtained 234 overlapping genes after intersection. BP analysis showed that, 234 overlapping genes are enriched in several biological terms, including external encapsulating structure organization, extracellular structure organization and extracellular matrix organization, confirming their association with NASH fibrosis progression. The most prominent KEGG and Reactome pathways are ECM-receptor interaction and extracellular matrix organization, respectively. NASH is associated with sustained activation of chronic HSCs (from stationary vitamin A-rich cells to fibrogenic, hyperplastic, and pro-inflammatory cells), resulting in accumulation of ECM and gradual replacement of liver parenchyma by fibrous tissue [5, 6, 34]. Taken together, these results suggested that these overlapping genes are well involved in the progression of NASH fibrosis.
Based on these overlapping genes, we constructed a PPI network and identified seven hub genes (SPP1, PROM1, SOX9, EPCAM, THY1, CD34 and MCAM) through Cytoscape plugins. According to single-cell RNA-seq analysis of hub genes, we found that they are expressed primarily in different cell clusters such as cholangiocytes, NK cells, and HSCs. NAFLD with cholestasis is characterized by ductal inflammation, bile duct loss and swelling and bile duct hyperplasia, and more prone to bridging fibrosis and cirrhosis [40]. Cholangiocytes injury is an important factor to reflect the severity of NAFLD [41, 42]. At the same time, persistent biliary fibrosis can create an environment of liver tissue that promotes regeneration of hepatocytes [43]. SPP1 is located on human chromosome 4 and encodes a protein called osteopontin (OPN), OPN is closely related to chronic liver disease and involved in liver steatosis, inflammation, and fibrosis [44, 45]. A clinical trial found that serum OPN level increased progressively with the progression of NAFLD fibrosis [46]. SOX9 is a transcription factor involved in ECM production during liver fibrosis, which regulates the wnt pathway and its downstream target protein OPN [47, 48]. PROM1 is considered as a marker of endothelial progenitor cells, hematopoietic stem cells and other stem cells and participates in the expansion of cholangiocytes known as the ductular reaction [49, 50]. PROM1 also has a strong correlation with the expression of biliary fibrosis related genes, such as KRT19 and COL1A1 [51]. It was found that HSCs expressing stem/ progenitor cell marker PROM1 exist in the liver, show the characteristics of progenitor cells, and participate in the process of liver injury and fibrosis [52]. EPCAM is a type I transmembrane glycoprotein, which is also a marker of cholangiocytes [26, 53]. It is involved in a variety of biological processes such as cell adhesion, signal transduction, migration and proliferation [53]. In the mouse model of alcoholic hepatitis, silencing EPCAM can inhibit liver fibrosis and HSCs proliferation [54]. CD34 is considered a marker of microvascular formation [55]. In this study, we found that the expression of CD34 was significantly increased in HSCs during high-fat feeding. HSCs is the main source of ECM. When the liver is damaged, HSCs is activated and transformed into fibroblasts to participate in liver fibrosis and reconstruction of intrahepatic structure [34]. Our results suggested that CD34 is likely to influence the formation of NASH fibrosis by participating in the activation of HSCs. A study also found that CD34 positive microvessels were more common in areas with higher fibrosis and significantly associated with fibrosis in NASH [56]. As an important regulator of cell–cell and cell–matrix interactions, THY1 plays an important role in nerve regeneration, inflammation, metastasis, and fibrosis [57]. Through the single-cell analysis of liver NPC in high-fat diet-induced mice, we found that THY1 is a marker of NK cells. NK cells usually have anti-fibrosis properties including killing activated HSCs by IFNγ and inducing HSCs apoptosis by expressing death receptor ligands [58]. NK cells also help clear senescent activated HSCs to reduce fibrosis [59]. However, there have been no studies linking THY1 to NASH fibrosis, but THY1 has been shown to be involved in fibrosis formation in a mouse model of cholestatic liver injury, and a bioinformatics study has found that THY1 may be a potential key regulator of NAFLD progression [60, 61]. MCAM is an adhesion molecule of immunoglobulin superfamily [62]. Cell adhesion molecules are excellent biosensors with specific contributions to the liver, including leukocyte recruitment, cell differentiation and survival, matrix remodeling or angiogenesis, and are unique anti-fibrosis therapeutic targets [63]. Interestingly, we found that the total expression of MCAM was decreased in liver NPC, which may be because we did not consider the influence of liver parenchymal cells and MCAM may only be activated in specific cell clusters. Together, we discovered that the expression of these hub genes may be involved in the progression of fibrosis and verified them from a single-cell perspective. However, the progression of NASH fibrosis is affected by a variety of clinical characteristics (age, gender, diabetes, etc.), and the effect of these factors on gene expression remains to be further explored [64,65,66].
HSCs are the central driver of fibrosis in liver injury [58]. Based on the cluster analysis of HSCs in another dataset, we found that SPP1 and CD34 are highly expressed on cluster 2 and 5, respectively. Further BP analysis of clusters 2 and 5 suggested that they might be closely related to the progression of fibrosis. Cluster 2 is closely associated with inflammation, showing leukocyte chemotaxis and the release of cytokines and receptors. HSCs need to strictly regulate autocrine and paracrine crosstalk to rapidly respond to changes in extracellular matrix content [67]. Cytokines are important for the initiation and duration of HSCs activation, causing ECM production and contractility, respectively [68]. In addition, cluster 5 showed the properties of myofibroblasts (activated HSCs), participating in the formation of hepatic fibrosis and reconstruction of intrahepatic structure through proliferation and secretion of extracellular matrix.
Finally, NASH has resulted in a dramatic increase in HCC prevalence [34]. We found that SPP1, SOX9, MCAM and THY1 were differentially expressed in NASH and NASH-associated HCC, suggesting that these four genes are involved not only in NASH fibrosis, but also in the malignant progression of NASH. In addition, further studies suggested that SOX9 may be associated with changes in metabolism-related aspects in NASH-associated HCC. However, this study also has some limitations. Firstly, due to the absence of clinical characteristics of the samples, we could not exclude the influence of clinical confounding factors on the expression of these genes that might be involved in the progression of fibrosis. Similarly, we did not correct these factors in ROC analysis for predicting the occurrence of HCC. Secondly, we did not explore whether these genes are associated with other forms of fibrosis, so it is not clear whether they share a common pathway. Finally, we also need to further verify the role of these hub genes in the progression of NASH through molecular biology experiments in the future.
Conclusion
To investigate the association of gene expression with NASH fibrosis, WGCNA and single-cell RNA-seq analysis were used. Finally, we found seven related hub genes (SPP1, PROM1, SOX9, EPCAM, THY1, CD34 and MCAM), and single-cell RNA-seq analysis showed that cholangiocytes seemed to play an important role in NASH fibrosis. Further studies suggested that SPP1 and CD34 were highly expressed in different clusters of HSCs which perform different functions. Further studies suggested that SPP1, SOX9, MCAM and THY1 were associated with NASH-associated HCC, and that SOX9 may be related to changes in metabolism-related pathways between different subtypes of HCC.
Data availability
Data were collected from the GEO database.
References
Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J, Bugianesi E (2018) Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 15(1):11–20. https://doi.org/10.1038/nrgastro.2017.109
Sheka AC, Adeyi O, Thompson J, Hameed B, Crawford PA, Ikramuddin S (2020) Nonalcoholic steatohepatitis: a review. JAMA 323(12):1175–1183. https://doi.org/10.1001/jama.2020.2298
Ciardullo S, Grassi G, Mancia G, Perseghin G (2021) Nonalcoholic fatty liver disease and risk of incident hypertension: a systematic review and meta-analysis. Eur J Gastroenterol Hepatol. https://doi.org/10.1097/meg.0000000000002299
Morieri ML, Vitturi N, Avogaro A, Targher G, Fadini GP (2021) Prevalence of hepatic steatosis in patients with type 2 diabetes and response to glucose-lowering treatments. A multicenter retrospective study in Italian specialist care. J Endocrinol Invest 44(9):1879–1889. https://doi.org/10.1007/s40618-021-01501-y
Schwabe RF, Tabas I, Pajvani UB (2020) Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology 158(7):1913–1928. https://doi.org/10.1053/j.gastro.2019.11.311
Zhu C, Tabas I, Schwabe RF, Pajvani UB (2021) Maladaptive regeneration—the reawakening of developmental pathways in NASH and fibrosis. Nat Rev Gastroenterol Hepatol 18(2):131–142. https://doi.org/10.1038/s41575-020-00365-6
Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, Mills PR, Keach JC, Lafferty HD, Stahler A, Haflidadottir S, Bendtsen F (2015) Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 149(2):389–97.e10. https://doi.org/10.1053/j.gastro.2015.04.043
Vilar-Gomez E, Calzadilla-Bertot L, Wai-Sun Wong V, Castellanos M, Aller-de la Fuente R, Metwally M, Eslam M, Gonzalez-Fabian L, Alvarez-Quiñones Sanz M, Conde-Martin AF, De Boer B, McLeod D, Hung Chan AW, Chalasani N, George J, Adams LA, Romero-Gomez M (2018) Fibrosis severity as a determinant of cause-specific mortality in patients with advanced nonalcoholic fatty liver disease: a multi-national cohort study. Gastroenterology 155(2):443-457.e17. https://doi.org/10.1053/j.gastro.2018.04.034
Diehl AM, Day C (2017) Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N Engl J Med 377(21):2063–2072. https://doi.org/10.1056/NEJMra1503519
Francque SM, Bedossa P, Ratziu V, Anstee QM, Bugianesi E, Sanyal AJ, Loomba R, Harrison SA, Balabanska R, Mateva L, Lanthier N, Alkhouri N, Moreno C, Schattenberg JM, Stefanova-Petrova D, Vonghia L, Rouzier R, Guillaume M, Hodge A, Romero-Gómez M, Huot-Marchand P, Baudin M, Richard MP, Abitbol JL, Broqua P, Junien JL, Abdelmalek MF (2021) A randomized, controlled trial of the pan-PPAR agonist lanifibranor in NASH. N Engl J Med 385(17):1547–1558. https://doi.org/10.1056/NEJMoa2036205
Mantovani A, Petracca G, Beatrice G, Csermely A, Lonardo A, Targher G (2021) Glucagon-like peptide-1 receptor agonists for treatment of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: an updated meta-analysis of randomized controlled trials. Metabolites. https://doi.org/10.3390/metabo11020073
Morieri ML, Targher G, Lapolla A, D’Ambrosio M, Tadiotto F, Rigato M, Frison V, Paccagnella A, Simioni N, Avogaro A, Fadini GP (2021) Changes in markers of hepatic steatosis and fibrosis in patients with type 2 diabetes during treatment with glucagon-like peptide-1 receptor agonists. A multicenter retrospective longitudinal study. Nutr Metab Cardiovasc Dis 31(12):3474–3483. https://doi.org/10.1016/j.numecd.2021.08.049
Langfelder P, Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. https://doi.org/10.1186/1471-2105-9-559
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A (2013) NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res 41:D991–D995. https://doi.org/10.1093/nar/gks1193
Govaere O, Cockell S, Tiniakos D, Queen R, Younes R, Vacca M, Alexander L, Ravaioli F, Palmer J, Petta S, Boursier J, Rosso C, Johnson K, Wonders K, Day CP, Ekstedt M, Orešič M, Darlay R, Cordell HJ, Marra F, Vidal-Puig A, Bedossa P, Schattenberg JM, Clément K, Allison M, Bugianesi E, Ratziu V, Daly AK, Anstee QM (2020) Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aba4448
Pinyol R, Torrecilla S, Wang H, Montironi C, Piqué-Gili M, Torres-Martin M, Wei-Qiang L, Willoughby CE, Ramadori P, Andreu-Oller C, Taik P, Lee YA, Moeini A, Peix J, Faure-Dupuy S, Riedl T, Schuehle S, Oliveira CP, Alves VA, Boffetta P, Lachenmayer A, Roessler S, Minguez B, Schirmacher P, Dufour JF, Thung SN, Reeves HL, Carrilho FJ, Chang C, Uzilov AV, Heikenwalder M, Sanyal A, Friedman SL, Sia D, Llovet JM (2021) Molecular characterisation of hepatocellular carcinoma in patients with non-alcoholic steatohepatitis. J Hepatol 75(4):865–878. https://doi.org/10.1016/j.jhep.2021.04.049
Pantano L, Agyapong G, Shen Y, Zhuo Z, Fernandez-Albert F, Rust W, Knebel D, Hill J, Boustany-Kari CM, Doerner JF, Rippmann JF, Chung RT, Ho Sui SJ, Simon E, Corey KE (2021) Molecular characterization and cell type composition deconvolution of fibrosis in NAFLD. Sci Rep 11(1):18045. https://doi.org/10.1038/s41598-021-96966-5
Yu G, Wang LG, Han Y, He QY (2012) clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16(5):284–287. https://doi.org/10.1089/omi.2011.0118
Thomas PD, Mi H, Lewis S (2007) Ontology annotation: mapping genomic regions to biological function. Curr Opin Chem Biol 11(1):4–11. https://doi.org/10.1016/j.cbpa.2006.11.039
Kanehisa M (2019) Toward understanding the origin and evolution of cellular organisms. Protein Sci 28(11):1947–1951. https://doi.org/10.1002/pro.3715
Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, Sidiropoulos K, Cook J, Gillespie M, Haw R, Loney F, May B, Milacic M, Rothfels K, Sevilla C, Shamovsky V, Shorser S, Varusai T, Weiser J, Wu G, Stein L, Hermjakob H, D’Eustachio P (2020) The reactome pathway knowledgebase. Nucleic Acids Res 48(D1):D498-d503. https://doi.org/10.1093/nar/gkz1031
Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, Jensen LJ, von Mering C (2017) The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 45(D1):D362-d368. https://doi.org/10.1093/nar/gkw937
Doncheva NT, Morris JH, Gorodkin J, Jensen LJ (2019) Cytoscape StringApp: network analysis and visualization of proteomics data. J Proteome Res 18(2):623–632. https://doi.org/10.1021/acs.jproteome.8b00702
Zhao X, Wu S, Fang N, Sun X, Fan J (2020) Evaluation of single-cell classifiers for single-cell RNA sequencing data sets. Brief Bioinform 21(5):1581–1595. https://doi.org/10.1093/bib/bbz096
Zhang X, Lan Y, Xu J, Quan F, Zhao E, Deng C, Luo T, Xu L, Liao G, Yan M, Ping Y, Li F, Shi A, Bai J, Zhao T, Li X, Xiao Y (2019) Cell Marker: a manually curated resource of cell markers in human and mouse. Nucleic Acids Res 47(D1):D721-d728. https://doi.org/10.1093/nar/gky900
MacParland SA, Liu JC, Ma XZ, Innes BT, Bartczak AM, Gage BK, Manuel J, Khuu N, Echeverri J, Linares I, Gupta R, Cheng ML, Liu LY, Camat D, Chung SW, Seliga RK, Shao Z, Lee E, Ogawa S, Ogawa M, Wilson MD, Fish JE, Selzner M, Ghanekar A, Grant D, Greig P, Sapisochin G, Selzner N, Winegarden N, Adeyi O, Keller G, Bader GD, McGilvray ID (2018) Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat Commun 9(1):4383. https://doi.org/10.1038/s41467-018-06318-7
Xiong X, Kuang H, Ansari S, Liu T, Gong J, Wang S, Zhao XY, Ji Y, Li C, Guo L, Zhou L, Chen Z, Leon-Mimila P, Chung MT, Kurabayashi K, Opp J, Campos-Pérez F, Villamil-Ramírez H, Canizales-Quinteros S, Lyons R, Lumeng CN, Zhou B, Qi L, Huertas-Vazquez A, Lusis AJ, Xu XZS, Li S, Yu Y, Li JZ, Lin JD (2019) Landscape of intercellular crosstalk in healthy and NASH liver revealed by single-cell secretome gene analysis. Mol Cell 75(3):644-660.e5. https://doi.org/10.1016/j.molcel.2019.07.028
Deczkowska A, David E, Ramadori P, Pfister D, Safran M, At The B, Giladi A, Jaitin DA, Barboy O, Cohen M, Yofe I, Gur C, Shlomi-Loubaton S, Henri S, Suhail Y, Qiu M, Kam S, Hermon H, Lahat E, Ben Yakov G, Cohen-Ezra O, Davidov Y, Likhter M, Goitein D, Roth S, Weber A, Malissen B, Weiner A, Ben-Ari Z, Heikenwälder M, Elinav E, Amit I (2021) XCR1(+) type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat Med 27(6):1043–1054. https://doi.org/10.1038/s41591-021-01344-3
Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez JC, Müller M (2011) pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. https://doi.org/10.1186/1471-2105-12-77
Thul PJ, Lindskog C (2018) The human protein atlas: a spatial map of the human proteome. Protein Sci 27(1):233–244. https://doi.org/10.1002/pro.3307
Li J, Xie L, Xie Y, Wang F (2020) Bregmannian consensus clustering for cancer subtypes analysis. Comput Methods Programs Biomed. https://doi.org/10.1016/j.cmpb.2020.105337
Gong J, Li R, Chen Y, Zhuo Z, Chen S, Cao J, Zhang Q, Chong Y, Hu B (2021) HCC subtypes based on the activity changes of immunologic and hallmark gene sets in tumor and nontumor tissues. Brief Bioinform. https://doi.org/10.1093/bib/bbaa427
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43(7):e47. https://doi.org/10.1093/nar/gkv007
Loomba R, Friedman SL, Shulman GI (2021) Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 184(10):2537–2564. https://doi.org/10.1016/j.cell.2021.04.015
Teli MR, James OF, Burt AD, Bennett MK, Day CP (1995) The natural history of nonalcoholic fatty liver: a follow-up study. Hepatology 22(6):1714–1719
Ciardullo S, Ronchetti C, Muraca E, Oltolini A, Perra S, Bianconi E, Zerbini F, Cannistraci R, Manzoni G, Gastaldelli A, Lattuada G, Perseghin G (2020) Impact of using different biomarkers of liver fibrosis on hepatologic referral of individuals with severe obesity and NAFLD. J Endocrinol Invest 43(7):1019–1026. https://doi.org/10.1007/s40618-020-01188-7
Barchetta I, Ceccarelli V, Cimini FA, Barone E, Sentinelli F, Coluzzi M, Chiappetta C, Bertoccini L, Tramutola A, Labbadia G, Di Cristofano C, Silecchia G, Leonetti F, Cavallo MG (2021) Circulating dipeptidyl peptidase-4 is independently associated with the presence and severity of NAFLD/NASH in individuals with and without obesity and metabolic disease. J Endocrinol Invest 44(5):979–988. https://doi.org/10.1007/s40618-020-01392-5
Filardi T, Panimolle F, Tiberti C, Crescioli C, Lenzi A, Pallotta N, Morano S (2021) Circulating levels of fetuin-A are associated with moderate-severe hepatic steatosis in young adults. J Endocrinol Invest 44(1):105–110. https://doi.org/10.1007/s40618-020-01274-w
Alipoor B, Nikouei S, Rezaeinejad F, Malakooti-Dehkordi SN, Sabati Z, Ghasemi H (2021) Long non-coding RNAs in metabolic disorders: pathogenetic relevance and potential biomarkers and therapeutic targets. J Endocrinol Invest 44(10):2015–2041. https://doi.org/10.1007/s40618-021-01559-8
Sorrentino P, Tarantino G, Perrella A, Micheli P, Perrella O, Conca P (2005) A clinical-morphological study on cholestatic presentation of nonalcoholic fatty liver disease. Dig Dis Sci 50(6):1130–1135. https://doi.org/10.1007/s10620-005-2719-1
Chiba M, Sasaki M, Kitamura S, Ikeda H, Sato Y, Nakanuma Y (2011) Participation of bile ductular cells in the pathological progression of non-alcoholic fatty liver disease. J Clin Pathol 64(7):564–570. https://doi.org/10.1136/jcp.2011.090175
Natarajan SK, Ingham SA, Mohr AM, Wehrkamp CJ, Ray A, Roy S, Cazanave SC, Phillippi MA, Mott JL (2014) Saturated free fatty acids induce cholangiocyte lipoapoptosis. Hepatology 60(6):1942–1956. https://doi.org/10.1002/hep.27175
Yovchev MI, Locker J, Oertel M (2016) Biliary fibrosis drives liver repopulation and phenotype transition of transplanted hepatocytes. J Hepatol 64(6):1348–1357. https://doi.org/10.1016/j.jhep.2016.01.036
Song Z, Chen W, Athavale D, Ge X, Desert R, Das S, Han H, Nieto N (2021) Osteopontin takes center stage in chronic liver disease. Hepatology 73(4):1594–1608. https://doi.org/10.1002/hep.31582
Kazanecki CC, Uzwiak DJ, Denhardt DT (2007) Control of osteopontin signaling and function by post-translational phosphorylation and protein folding. J Cell Biochem 102(4):912–924. https://doi.org/10.1002/jcb.21558
Glass O, Henao R, Patel K, Guy CD, Gruss HJ, Syn WK, Moylan CA, Streilein R, Hall R, Mae Diehl A, Abdelmalek MF (2018) Serum interleukin-8, osteopontin, and monocyte chemoattractant protein 1 are associated with hepatic fibrosis in patients with nonalcoholic fatty liver disease. Hepatol Commun 2(11):1344–1355. https://doi.org/10.1002/hep4.1237
Kawai T, Yasuchika K, Ishii T, Miyauchi Y, Kojima H, Yamaoka R, Katayama H, Yoshitoshi EY, Ogiso S, Kita S, Yasuda K, Fukumitsu K, Komori J, Hatano E, Kawaguchi Y, Uemoto S (2016) SOX9 is a novel cancer stem cell marker surrogated by osteopontin in human hepatocellular carcinoma. Sci Rep. https://doi.org/10.1038/srep30489
Wang C, Deng J, Deng H, Kang Z, Huang Z, Ding Z, Dong L, Chen J, Zhang J, Zang Y (2020) A novel Sox9/lncRNA H19 axis contributes to hepatocyte death and liver fibrosis. Toxicol Sci 177(1):214–225. https://doi.org/10.1093/toxsci/kfaa097
Kordes C, Sawitza I, Müller-Marbach A, Ale-Agha N, Keitel V, Klonowski-Stumpe H, Häussinger D (2007) CD133+ hepatic stellate cells are progenitor cells. Biochem Biophys Res Commun 352(2):410–417. https://doi.org/10.1016/j.bbrc.2006.11.029
Fenlon M, Short C, Xu J, Malkoff N, Mahdi E, Hough M, Glazier A, Lee C, Asahina K, Wang KS (2020) Prominin-1-expressing hepatic progenitor cells induce fibrogenesis in murine cholestatic liver injury. Physiol Rep 8(14):e14508. https://doi.org/10.14814/phy2.14508
Zagory JA, Fenlon M, Dietz W, Zhao M, Nguyen MV, Trinh P, Adoumie M, Park A, Xu J, Mahdi E, Glazier A, Malkoff N, Mavila N, Wang KS (2019) Prominin-1 promotes biliary fibrosis associated with biliary atresia. Hepatology 69(6):2586–2597. https://doi.org/10.1002/hep.30550
Fujii T, Fuchs BC, Yamada S, Lauwers GY, Kulu Y, Goodwin JM, Lanuti M, Tanabe KK (2010) Mouse model of carbon tetrachloride induced liver fibrosis: Histopathological changes and expression of CD133 and epidermal growth factor. BMC Gastroenterol. https://doi.org/10.1186/1471-230x-10-79
Huang L, Yang Y, Yang F, Liu S, Zhu Z, Lei Z, Guo J (2018) Functions of EpCAM in physiological processes and diseases (review). Int J Mol Med 42(4):1771–1785. https://doi.org/10.3892/ijmm.2018.3764
Zhang Z, Wen H, Weng J, Feng L, Liu H, Hu X, Zeng F (2019) Silencing of EPCAM suppresses hepatic fibrosis and hepatic stellate cell proliferation in mice with alcoholic hepatitis via the PI3K/Akt/mTOR signaling pathway. Cell Cycle 18(18):2239–2254. https://doi.org/10.1080/15384101.2019.1642067
Rookmaaker MB, Verhaar MC, Loomans CJ, Verloop R, Peters E, Westerweel PE, Murohara T, Staal FJ, van Zonneveld AJ, Koolwijk P, Rabelink TJ, van Hinsbergh VW (2005) CD34+ cells home, proliferate, and participate in capillary formation, and in combination with CD34- cells enhance tube formation in a 3-dimensional matrix. Arterioscler Thromb Vasc Biol 25(9):1843–1850. https://doi.org/10.1161/01.Atv.0000177808.92494.14
Gill RM, Belt P, Wilson L, Bass NM, Ferrell LD (2011) Centrizonal arteries and microvessels in nonalcoholic steatohepatitis. Am J Surg Pathol 35(9):1400–1404. https://doi.org/10.1097/PAS.0b013e3182254283
Rege TA, Hagood JS (2006) Thy-1 as a regulator of cell-cell and cell-matrix interactions in axon regeneration, apoptosis, adhesion, migration, cancer, and fibrosis. Faseb j 20(8):1045–1054. https://doi.org/10.1096/fj.05-5460rev
Tsuchida T, Friedman SL (2017) Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 14(7):397–411. https://doi.org/10.1038/nrgastro.2017.38
Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW (2008) Senescence of activated stellate cells limits liver fibrosis. Cell 134(4):657–667. https://doi.org/10.1016/j.cell.2008.06.049
Katsumata LW, Miyajima A, Itoh T (2017) Portal fibroblasts marked by the surface antigen Thy1 contribute to fibrosis in mouse models of cholestatic liver injury. Hepatol Commun 1(3):198–214. https://doi.org/10.1002/hep4.1023
Zheng J, Wu H, Zhang Z, Yao S (2021) Dynamic co-expression modular network analysis in nonalcoholic fatty liver disease. Hereditas 158(1):31. https://doi.org/10.1186/s41065-021-00196-8
Joshkon A, Heim X, Dubrou C, Bachelier R, Traboulsi W, Stalin J, Fayyad-Kazan H, Badran B, Foucault-Bertaud A, Leroyer AS, Bardin N, Blot-Chabaud M (2020) Role of CD146 (MCAM) in physiological and pathological angiogenesis-contribution of new antibodies for therapy. Biomedicines. https://doi.org/10.3390/biomedicines8120633
Hintermann E, Christen U (2019) The many roles of cell adhesion molecules in hepatic fibrosis. Cells. https://doi.org/10.3390/cells8121503
Younossi ZM, Golabi P, de Avila L, Paik JM, Srishord M, Fukui N, Qiu Y, Burns L, Afendy A, Nader F (2019) The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: a systematic review and meta-analysis. J Hepatol 71(4):793–801. https://doi.org/10.1016/j.jhep.2019.06.021
Hagström H, Nasr P, Ekstedt M, Hammar U, Stål P, Hultcrantz R, Kechagias S (2017) Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J Hepatol 67(6):1265–1273. https://doi.org/10.1016/j.jhep.2017.07.027
Bellinato F, Gisondi P, Mantovani A, Girolomoni G, Targher G (2022) Risk of non-alcoholic fatty liver disease in patients with chronic plaque psoriasis: an updated systematic review and meta-analysis of observational studies. J Endocrinol Invest. https://doi.org/10.1007/s40618-022-01755-0
Friedman SL (2008) Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 88(1):125–172. https://doi.org/10.1152/physrev.00013.2007
Altamirano-Barrera A, Barranco-Fragoso B, Méndez-Sánchez N (2017) Management strategies for liver fibrosis. Ann Hepatol 16(1):48–56. https://doi.org/10.5604/16652681.1226814
Acknowledgements
We want to thank and acknowledge all participants of this study and the technical support provided by the Affiliated Hospital of Jiangsu University.
Funding
This work was supported by the National Natural Science Foundation of China (81870548, 81570721), the Social Development Project of Jiangsu Province (BE2018692), the Natural Science Foundation of Jiangsu Province, China (BK20191222, BK20151331), the High Caliber Medical Personnel Foundation of Jiangsu Province (LGY2016053), the Six Talent Peaks Project in Jiangsu Province (2015-WSN-006), and the Fifth “169 project” Scientific Research Project of Zhenjiang City, Jiangsu Province.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare that they have no competing interests.
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
No informed consent.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Wang, Z., Zhao, Z., Xia, Y. et al. Potential biomarkers in the fibrosis progression of nonalcoholic steatohepatitis (NASH). J Endocrinol Invest 45, 1379–1392 (2022). https://doi.org/10.1007/s40618-022-01773-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-022-01773-y