
Abstract
Purpose
Our study aimed to uncover the crucial genes and functional pathways involved in the development of non-alcoholic steatohepatitis (NASH).
Methods
Liver transcriptome datasets were integrated with Robust rank aggregation (RRA) method, and transcriptomic signatures for NASH progression and fibrosis severity in NAFLD were developed. The functions of transcriptomic signatures were explored by multiple bioinformatic analyses, and their diagnostic role was also evaluated.
Results
RRA analyses of 12 transcriptome datasets comparing NASH with non-alcoholic fatty liver (NAFL) identified 116 abnormally up-regulated genes in NASH patients. RRA analyses of five transcriptome datasets focusing fibrosis severity identified 78 abnormally up-regulated genes in NAFLD patients with advanced fibrosis. The functions of those transcriptomic signatures of NASH development or fibrosis progression were similar, and were both characterized by extracellular matrix (ECM)-related pathways (Adjusted P < 0.05). The transcriptomic signatures could effectively differentiate NASH from NAFL, and could help to identify NAFLD patients with advanced fibrosis. Gene set enrichment analysis and weighted gene co-expression network analysis further validated the key role of ECM-related pathways in NASH development. The top 10 up-regulated genes in NASH patients were SPP1, FBLN5, CHI3L1, CCL20, CD24, FABP4, GPNMB, VCAN, EFEMP1, and CXCL10, and their functions were mainly related to either ECM-related pathways or immunity-related pathways. Single cell RNA-sequencing analyses revealed that those crucial genes were expressed by distinct cells such as hepatocytes, macrophages, and hepatic stellate cells.
Conclusions
Transcriptomic signatures related to NASH development and fibrosis severity of NAFLD patients are both characterized by ECM-related pathways, and fibrosis is a main player during NASH progression. This study uncovers some novel key genes involved in NASH progression, which may be promising therapeutic targets.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a chronic liver disease characterized by hepatic fat accumulation and hepatocyte injury [1]. It is a common disease with a prevalence of >20%, and has caused a serious economic burden worldwide [2]. NAFLD can increase risk of cardiovascular diseases, and some patients can progress to liver cirrhosis or hepatocellular carcinoma (HCC) [1, 3]. Multiple factors such as environmental risk factors, insulin resistance, and genetic predisposition are involved in its pathogenesis [4]. NAFLD is histologically classified into non-alcoholic fatty liver (NAFL), borderline steatohepatitis, and non-alcoholic steatohepatitis (NASH) [5]. NASH is accompanied by lobular inflammation and hepatocyte ballooning with or without fibrosis [5]. NAFL is an early stage of NAFLD and if not timely and appropriately controlled, some NAFL patients can progress to NASH rapidly and are at high risk of liver cirrhosis and HCC [4]. Though some therapeutic inventions have been proposed to treat NASH, effective management for NASH patients is still difficult [1, 4].
A better understanding of the pathogenesis of NASH is critical to developing new therapy for NASH, but the molecular mechanism underlying disease progression from NAFL to NASH is still largely elusive [6]. The progression from NAFL to NASH is extremely complex, and some possible explanations have been proposed for this progression [6]. Insulin resistance and oxidative stress have been proposed to be closely related to NASH [7]. Cell stress, apoptosis, and lipotoxicity have also been found to promote NASH progression, and endoplasmic reticulum (ER) stress also has a role in this process [8,9,10]. In addition, abnormal immune response and inflammation have also been found to exert important roles in NASH progression [11, 12]. However, the crucial genes and functional pathways determining the progression from NAFL to NASH are still not clearly defined. Elucidation of the key functional pathways and genes involved in NASH progression is critical to the development of new therapy for NASH. This study aimed to uncover those crucial genes and functional pathways involved in disease progression from NAFL to NASH through analyzing liver transcriptome data. Those genes and functional pathways involved in fibrosis progression of NAFLD patients were also analyzed.
Methods
Liver transcriptome datasets of NAFLD patients
We searched Gene Expression Omnibus (GEO) and ArrayExpress databases to identify liver transcriptome datasets of NAFLD patients. Datasets meeting the following criteria were included: (1) Transcriptome data of liver tissues; (2) More than 30 differentially expressed genes (DEGs) were identified between the two groups; (3) Whole-genome expression profiles were available in the GEO or ArrayExpress; (4) Samples must contain both NAFL and NASH patients, or contain NAFLD patients with either advanced fibrosis or mild fibrosis. Advanced fibrosis was defined to have fibrosis stages 3 or 4, while mild fibrosis was defined to have fibrosis stage 0 or 1. Only transcriptome data comparing NASH liver tissues with NAFL liver tissues and transcriptome data assessing the transcriptomic difference between NAFLD liver tissues with obvious fibrosis and those without obvious fibrosis were used, while those transcriptome data from controls without NAFLD were not analyzed.
Robust rank aggregation (RRA) analysis
In order to effectively integrate transcriptome data from multiple datasets, we adopted RRA [13]. DEGs between NAFL and NASH were calculated, and were then integrated using the RRA method. Microarray data were analyzed using R package of “limma” [14]. DESeq2 was used to determine DEGs in RNA sequencing (RNA-seq) dataset [15]. In the RRA analysis, those genes with an adjusted P < 0.05 and an absolute value of log2 fold change (log2FC) >0.30 were considered to be statistically significant genes. Transcriptomic signatures for NASH and fibrosis severity in NAFLD were developed with the top up-regulated genes in RRA analyses.
Gene set enrichment analysis (GSEA)
GSEA is a powerful and widely used bioinformatic method in transcriptome research to detect the concordant change of gene sets between two phenotypes [16]. To identify key functional pathways involved in NASH development or fibrosis progression in NAFLD patients, GSEA was performed based on the DEGs lists from RRA analyses. Functional pathways with a normalized enrichment score (NES) >1.0 and a false discovery rate (FDR) q < 0.25 were deemed as significantly enriched pathways. To explore the possible pathways through which those top 10 up-regulated crucial genes promoted NASH progression, GSEA was performed in GSE49541 and top 15 significantly enriched pathways were analyzed. Both gene ontology (GO) biological process gene sets and hallmark gene sets were used in GSEA analyses.
Gene set variation analysis (GSVA) of transcriptomic signatures
GSVA can evaluate the changes of functional pathways or gene sets at whole genome transcriptome level [17]. To validate the changes of transcriptomic signatures in NASH patients or NAFLD patients with advanced fibrosis, GSVA analysis of NASH transcriptomic signature or fibrosis transcriptomic signature in NAFLD liver tissues was performed. Four validation cohorts were used in the assessment of NASH transcriptomic signature (GSE130970, GSE126848, EMTAB4856, and GSE83452), and two validation cohorts were used in the assessment of fibrosis transcriptomic signature (GSE130970 and GSE49541).
Validation of the differential expression of crucial genes
The abnormal expressions of top 10 up-regulated crucial genes between NASH patients and NAFL patients were validated using a dataset using RNA-seq (GSE130970), and the abnormal expression of top 10 up-regulated crucial genes in NAFLD patients with advanced fibrosis were also validated. Transcripts per million (TPM) values for all genes were calculated, which could provide a more accurate measurement of mRNA abundance [18]. The TPM values of those top 10 up-regulated crucial genes were then extracted and were compared.
Pathway enrichment analysis
In order to uncover the possible functions of those crucial genes, GO enrichment analysis from the Database for Annotation, Visualization, and Integrated Discovery (DAVID) was used [19, 20]. The significant enriched pathways were then shown.
Protein–protein interaction (PPI) analysis
Network-based analyses such as PPI can provide valuable information for constructing biological networks and predicting gene function, and have demonstrated good effectiveness in exploring complex diseases including NAFLD [21,22,23]. Those genes with intensive interactions with other molecules often contribute greatly to certain disease phenotype, and thus can be considered as hub genes. Finding those hub genes from numerous DEGs through PPI will help to identify the key molecules involved in the pathogeneses of complex diseases and uncover candidate therapeutic targets. To uncover the interaction networks of those crucial genes and reveal the hub genes involved in NASH progression or fibrosis progression, PPI analysis was performed with STRING (version 11.0; https://string-db.org/) [24]. The outcomes from STRING database were further analyzed by Cytoscape (version 3.4), and candidate hub genes were retrieved with the “cytoHubba” plug‑in of Cytoscape software [25, 26]. Top 20 nodes in the network ranked by Maximal Clique Centrality (MCC) were identified as candidate hub genes involved in NASH progression. In the transcriptomic analyses on fibrosis progression, top 15 nodes in the network ranked by MCC were identified as candidate hub genes involved in fibrosis progression.
Weighted gene co-expression network analysis (WGCNA)
To further define the possible functional pathways of those crucial genes identified in RRA analysis, WGCNA was performed [27]. To ensure the power of enrichment analysis, gene co-expression network of those top 3000 genes ranked by the expression variance in GSE49541 together with those crucial genes from RRA analysis was constructed using the R package of “WGCNA”. In addition, the functions of those crucial genes were inferred from the functions of relevant gene co-expression modules, which were analyzed through GO enrichment analysis.
Single cell RNA-sequencing (scRNA-seq) analyses
The expression patterns of those key genes were explored by scRNA-seq analyses of human liver tissues from GSE136103 [28]. scRNA-seq data of four samples including two healthy liver samples (GSM4041156 and GSM4041159) and two NAFLD liver samples (GSM4041162 and GSM4041163) were analyzed with Seurat approach [29]. The types of cells in liver tissues were annotated with the combination of findings from human liver cell atlas and the annotation analyses with SingleR [30, 31]. The expression patterns of those top 10 up-regulated genes in RRA analysis on NASH and RRA analysis on fibrosis were analyzed through the findings above together with evidence from Human Protein Atlas (https://www.proteinatlas.org/).
Statistical analysis
Difference in gene expression levels between groups was determined by Mann–Whitney U test. The enrichment scores from GSVA were compared between groups using unpaired t test. Receiver operating characteristic (ROC) analysis was also performed. P < 0.05 was considered statistically significant. R software (Version 3.6.1) was used in data analyses.
Results
Transcriptomic signature of NASH development
Based on the predefined inclusion criteria, 12 liver transcriptome datasets were included, and the main characteristics of these datasets were shown in Supplementary Table 1. These 12 datasets contained a total of 281 NASH patients and 252 NAFL controls.
RRA analysis identified 141 crucial genes involved in the progression from NAFL to NASH, 116 of which were significantly up-regulated in NASH liver tissues (Supplementary Table 2, 3). Those top 10 up-regulated genes included SPP1, FBLN5, CHI3L1, CCL20, CD24, FABP4, GPNMB, VCAN, EFEMP1, and CXCL10 (Fig. 1A). SPP1 was the most significant gene aberrantly expressed between NAFL and NASH (adjusted P = 2.29E-15), followed by FBLN5 (adjusted P = 7.24E-13) and CHI3L1 (adjusted P = 1.62E-12) (Supplementary Table 2, 3).

Identification of transcriptomic signature of NASH development. Figure 1-A, Heatmap in the RRA analyses of 12 liver transcriptome datasets comparing NASH with NAFL. The number was for the log2 value of fold change. The red color indicated up-regulation in the NASH liver tissues, while the green color indicated down-regulation in the NASH liver tissues. Figure 1-B, GO enrichment analyses of those crucial genes involved in the progression from NAFL to NASH. The significance of enrichment was expressed by the log10 value of Benjamini adjusted P value together with the color transition from white to red. The gene number was for the number of enriched genes in certain functional pathway, while the percentage was for the proportion of enriched genes among total genes in certain functional pathway. Figure 1-C, PPI networks of those crucial genes identified in RRA analysis of NASH. Figure 1-D, Comparison of the difference in the GSVA enrichment score of NASH transcriptomic signature between NASH patients and NAFL patients
GO analyses of those 141 crucial genes suggested that their functions were characterized by multiple extracellular matrix (ECM)-related pathways, such as Extracellular matrix (Benjamini P = 2.71E-14) and Extracellular space (Benjamini P = 5.42E-14) (Fig. 1B; Supplementary Table 4). Several non-ECM-related pathways were also enriched such as Chemokine receptor binding (Benjamini P = 0.005) and Immune response (Benjamini P = 0.04). The outcomes above suggested that ECM-related pathways were the main functional pathways involved in the progression of NASH, and immunity-related pathways were also involved. PPI analysis suggested there were intensive interaction networks among those crucial genes, and some candidate hub genes were identified such as SPP1, LUM, and COL3A1 (Fig. 1C).
A NASH transcriptomic signature consisting of those 116 significantly up-regulated genes in NASH liver tissues was developed. In all 4 validation cohorts, the GSVA enrichment score of NASH transcriptomic signature was significantly higher in NASH patients (P < 0.05; Fig. 1D). ROC analyses suggested the NASH transcriptomic signature could effectively differentiate NASH from NAFL, and the Area under ROC curve (AUC) in 3 datasets were >0.75 (Supplementary Fig. 1).
Transcriptomic signature of fibrosis progression in NAFLD patients
There were five liver transcriptome datasets that compared the transcriptomic changes between NAFLD patients with advanced fibrosis (Fibrosis stage 3 or 4) and those with mild fibrosis (Fibrosis stage 0 or 1) (Supplementary Table 5). These five datasets contained a total of 63 NAFLD patients with advanced fibrosis and 169 NAFLD patients with mild fibrosis (Supplementary Table 5).
RRA analysis of those five datasets identified 106 crucial genes involved in fibrosis progression among NAFLD patients, 78 of which were significantly up-regulated (Supplementary Tables 6, 7). Those top 10 up-regulated crucial genes included AKR1B10, AEBP1, STMN2, CCL19, EPCAM, DCDC2, ITGBL1, EFEMP1, PDZK1IP1, and CCL21 (Fig. 2A). AKR1B10 was the most significant gene aberrantly expressed in NAFLD patients with advanced fibrosis (Adjusted P = 1.17E-12), followed by AEBP1 (Adjusted P = 1.06E-08) and STMN2 (Adjusted P = 6.86E-08) (Supplementary Table 6). Intriguingly, 50 of those 78 up-regulated genes in RRA analysis on fibrosis were among those 116 up-regulated genes in RRA analysis on NASH (Supplementary Fig. 2), which suggested that a large part of those crucial genes involved in fibrosis progression and NASH progression were overlapping and fibrosis was a hallmark change during the progression from NAFL to NASH.

Identification of transcriptomic signature of fibrosis progression in NAFLD patients. Figure 2-A, Heatmap in the RRA analyses of 5 liver transcriptome datasets comparing NAFLD patients with advanced fibrosis and those with mild fibrosis. The number was for the log2 value of fold change. Figure 2-B, GO enrichment analyses of those crucial genes involved in fibrosis progression in NAFLD patients. Figure 2-C, PPI networks of those crucial genes identified in RRA analysis of fibrosis. Figure 2-D, Comparison of the difference in the GSVA enrichment score of fibrosis transcriptomic signature between NASH patients with advanced fibrosis and those with mild fibrosis. Figure 2-E, Assessment of the diagnostic role of fibrosis transcriptomic signature through ROC method
GO analyses of those 106 crucial genes suggested that their functions were characterized by multiple ECM-related pathways such as Extracellular matrix (Benjamini P = 9.98E-09) and Extracellular space (Benjamini P = 3.91E-11) (Fig. 2B; Supplementary Table 8), which were similar to the functions of those crucial genes involved in NASH progression. PPI analysis suggested there were intensive interaction networks among those crucial genes involved in fibrosis progression (Fig. 2C), and most of those hub genes could also be found in the PPI analysis of the crucial genes involved in NASH progression.
A fibrosis transcriptomic signature consisting of those 78 significantly up-regulated genes in liver tissues of NAFLD patients with advanced fibrosis was developed. In both validation cohorts, the GSVA enrichment score of fibrosis transcriptomic signature was significantly higher in patients with advanced fibrosis (P < 0.01; Fig. 2D). ROC analyses suggested the fibrosis transcriptomic signature could effectively identify NAFLD patients with advanced fibrosis (Fig. 2E).
Crucial functional pathways involved in NASH progression
GSEA analysis of the up-regulated DEGs list from RRA analysis on NASH suggested multiple ECM-related pathways were significantly enriched in the liver tissues of NASH patients such as Collagen fibril organization (NES = 1.92, FDR q = 0.008) and Extracellular matrix assembly (NES = 1.69, FDR q = 0.10) (Fig. 3A and Supplementary Fig. 3). Several immunity-related pathways were also enriched in the liver tissues of NASH patients such as TNF-α signaling via NF-κB (NES = 1.68, FDR q < 0.001) and Monocyte chemotaxis (NES = 1.70, FDR q = 0.11). The outcomes above suggested that ECM-related pathways were the main pathways involved in NASH progression, and immunity-related pathways also had key roles in this transition.

Enriched pathways in the GSEA analyses of the up-regulated DEGs lists from RRA analysis on NASH and RRA analysis on fibrosis. Figure 3-A, Main enriched pathways in the GSEA analysis of the up-regulated DEGs lists from RRA analysis on NASH. The significance of enrichment was expressed by the log10 value of FDR q value together with the color transition from white to red, and the gene number was for the number of enriched genes in certain functional pathway. Figure 3-B, Main enriched pathways in the GSEA analyses of the up-regulated DEGs lists from RRA analysis on fibrosis
GSEA analysis of the up-regulated DEGs list from RRA analysis on fibrosis suggested multiple ECM-related pathways were significantly enriched in the liver tissues of NAFLD patients with advanced fibrosis such as Epithelial-mesenchymal transition (NES = 1.63, FDR q < 0.001) and Extracellular matrix assembly (NES = 1.49, FDR q = 0.20) (Fig. 3B; Supplementary Fig. 4). Several immunity-related pathways were also enriched such as TNF-α signaling via NF-κB (NES = 1.31, FDR q = 0.018). The outcomes above suggested that both ECM-related pathways and immunity-related pathways were involved in fibrosis progression among NAFLD patients.
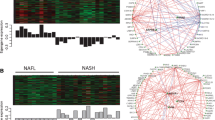
The co-expression pattern of genes in the liver transcriptome data of NAFLD patients was successfully analyzed by WGCNA (Fig. 4A-D; Supplementary Fig. 5). Brown co-expression module was the most significant module related to NASH severity (Correlation coefficient = 0.90, P = 1.0E-18). Brown module was also the most significant module related to advanced fibrosis (Correlation coefficient = 0.81, P = 1.0E-12). The function of Brown module was characterized by multiple ECM-related pathways such as Extracellular matrix and Extracellular matrix assembly (Fig. 4C).

Main findings in the WGCNA analysis of liver transcriptome data from GSE49541. Figure 4-A, Clustering dendrogram showed the co-expression pattern of genes in the liver transcriptome data of NAFLD patients. One cluster was for one co-expression gene module and was marked with one specific color. Figure 4-B, Heatmap showed the module-trait relationship in the WGCNA analysis. The correlation coefficient and P values were shown as numbers in each table together with the color transition from green to red. Figure 4-C, Enriched pathways of the Brown co-expression module in the WGCNA analysis. Figure 4-D, Heatmap showed the intensive correlation relationship in the expression levels of those hub genes from the Blown co-expression module. The correlation strength was positively shown as the color shape
Validation of the differential expression of top key genes
The differential expression of top 10 up-regulated genes was further validated. The significantly aberrant expressions of 9 genes were confirmed between NAFL and NASH patients such as SPP1 (P = 0.014), FBLN5 (P = 0.004), and CHI3L1 (P = 0.015) (Fig. 5A). The significantly aberrant expressions of top 10 up-regulated genes from RRA analysis on fibrosis were confirmed in NAFLD patients with advanced fibrosis (P < 0.05) (Fig. 5B).

Validation the differential expression of top key genes in RNA-seq transcriptome dataset of liver tissues. Figure 5-A, Validation of the aberrant expression of those top 10 up-regulated genes in RRA analysis on NASH in the liver tissues of NASH patients. The TPM values of those top 10 up-regulated genes were extracted from GSE130970 and were then compared between NASH and NAFL. Figure 5-B, Validation of the aberrant expression of those top 10 up-regulated genes in RRA analysis on fibrosis in the liver tissues of NAFLD patients with advanced fibrosis
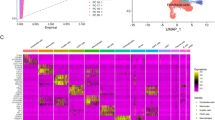
The expression patterns of those top up-regulated genes were analyzed by scRNA-seq analyses of liver tissues (Fig. 6; Supplementary Fig. 6). Those crucial genes were expressed by distinct cells such as hepatocytes, macrophages, and hepatic stellate cells (HSC) (Supplementary Table 9). The pathogenic roles of some genes such as SPP1, CHI3L1, and CCL20 had been confirmed by data from published literatures, but the roles of other genes such as FBLN5, CD24, EFEMP1, ITGBL1, and EPCAM had not been studied (Supplementary Table 9).

Assessment of the expression patterns of those key genes in different types of cells via scRNA-seq analyses of human liver tissues. Figure 6-A, Clustering liver cells by the unsupervised nonlinear dimension reduction method of t-Distributed Stochastic Neighbor embedding (t-SNE). Figure 6-B, Clustering liver cells by the unsupervised nonlinear dimension reduction method of Uniform Manifold Approximation and Projection (UMAP). Figure 6-C, Heatmap shows the expression patterns of those key genes in different types of liver cells. The expression levels of those key genes are expressed by the color transition from green to red
To uncover the possible functional pathways related to those crucial genes, GSEA was performed using transcriptome data of GSE49541. The outcomes suggested that those crucial genes may mediate the progression of NASH by regulating multiple functional pathways, and ECM-related pathways and immunity-related pathways were the main pathways (Supplementary Table 10–19). For example, FBLN5 and CHI3L1 may mediate NASH progression through multiple functional pathways, and ECM-related pathways such as extracellular structure organization were the main relevant pathways (Supplementary Table 11, 12). CXCL10 and GPNMB may exert their roles in NASH progression through immune response-related pathways (Supplementary Table 17, 18). Therefore, those top 10 up-regulated genes were likely to promote NASH progression mainly via ECM-related pathways and immunity-related pathways.
Discussion
The key genes and functional pathways involved in the progression from NAFL to NASH are still largely elusive. For the first time, our study used bioinformatic analyses to explore key molecular pathways and crucial genes involved in the progression from NAFL to NASH as well as those involved in the progression of fibrosis among NAFLD patients. A total of 116 abnormally up-regulated genes were identified as candidate crucial genes involved in NASH development, and 78 abnormally up-regulated genes were identified as crucial genes involved in fibrosis progression of NAFLD patients. The functions of those transcriptomic signatures of NASH development or fibrosis progression in NAFLD patients were similar, both of which were characterized by ECM-related pathways (Fig. 1 and 2). GSEA and WGCNA analyses further validated the key roles of ECM-related pathways in the progression from NAFL to NASH. Therefore, transcriptomic signatures related to NASH development and fibrosis severity of NAFLD patients are both characterized by ECM-related pathways, and fibrosis is the main player during NASH progression.
Among those up-regulated crucial genes from RRA analysis, SPP1, also known as osteopontin (OPN), was the most significant DEG in the NASH liver tissue, suggesting that it may exert a critical role in NASH progression. There is some evidence for the involvement of SPP1 in the development of NASH [32,33,34]. The expression of SPP1 in NASH patients was significantly higher than in healthy controls, and its expression was positively correlated with steatosis and insulin resistance in obese patients [32]. SPP1 knockout could inhibit hepatic steatosis, inflammation, and hepatic fibrosis in mice [33]. A recent study revealed that SPP1 was an essential player in the hepatocyte Notch activation of NASH [34]. The above findings suggest that SPP1 is a crucial factor in the development of NASH, and can promote the progression from NAFL to NASH through multiple mechanisms. Our study identified SPP1 as the most significant gene aberrantly expressed in the NASH liver tissue, which further proved SPP1 as a crucial therapeutic target for NASH. Moreover, GSEA analysis suggested that SPP1 could mediate the progression of NASH through multiple functional pathways such as extrinsic apoptotic signaling pathway and fibroblast proliferation-related pathway (Supplementary Table 10), which provided new explanations for the role of SPP1 in NASH. scRNA-seq analyses reveal that SPP1 is mainly expressed by hepatocytes, endothelial cells, and macrophages, suggesting the SPP1 can promote NASH progression via affecting the functions of multiple types of cells in liver tissues (Supplementary Table 9).
Apart from SPP1, the other genes such as CCL20, GPNMB, CXCL10, FABP4, and CHI3L1 have also been reported to be possibly involved in NASH development. Several studies found that CCL20 was up-regulated in patients with NAFLD and may promote fibrogenesis in NAFLD as an important inflammatory mediator, thus participating in the progression of NAFLD [35, 36]. GPNMB was found to be up-regulated in the serum of NASH patients, and overexpression of GPNMB could ameliorate liver fat accumulation and fibrosis in mice [37]. CXCL10 was significantly up-regulated in NASH patients, and circulating CXCL10 level was associated with lobular inflammation, suggesting that it could play a role in the pathogenesis of NASH [38, 39]. Serum CHI3L1(YKL-40) has been reported to be a marker of liver fibrosis in NAFLD patients, but the molecular mechanism underlying the role of CHI3L1 in NASH has not been clearly defined [40, 41]. Therefore, half of those top 10 up-regulated crucial genes from RRA analysis have been reported to be up-regulated in NASH patients in previous studies, which prove the reliability of the findings in our study. Moreover, the molecular mechanisms underlying the roles of those genes in NASH have not been clearly defined. Our study reveals that CCL20, GPNMB, CXCL10, and CHI3L1 may exert their roles in the progression of NASH via ECM-related pathways and immunity-related pathways, which need to be validated by future studies.
Among those top 10 up-regulated genes, some genes have not been studied in NASH such as FBLN5, EFEMP1, VCAN, FABP4, and CD24. FBLN5 is a glycoprotein secreted by smooth muscle cells, fibroblasts, and vascular endothelial cells [42]. Studies using gene knockout mice have shown that FBLN5 is a key molecule of elastic fibers and plays a key role in the development of elastogenesis [43, 44]. Moreover, FBLN5 knockout mice showed aortic curvature at birth, adult pelvic organ prolapse, and destruction of elastic fiber morphology [45]. In our study, FBLN5 is the second most significant DEG in NASH, and GSEA analysis suggests that FBLN5 may play a key role in NASH progression through multiple functional pathways such as extracellular structure organization and fibroblast proliferation regulation pathway, which needs to be explored in further studies (Supplementary Table 11). scRNA-seq analyses reveal that FBLN5 is mainly expressed by hepatocytes, fibroblasts, and endothelial cells (Supplementary Table 9), which indicates that FBLN5 may promote NASH progression via its regulatory role in multiple types of cells. CD24 is an important co-stimulus molecule and is involved in the processes of inflammation, immune response, cancer biology, and autoimmune diseases [46,47,48]. Our study showed that CD24 was one of those top 10 up-regulated genes in NASH, and CD24 may be involved in NASH through fibrosis-related pathways such as regulation of fibroblast migration and mesenchymal cell differentiation (Supplementary Table 14). The roles of EFEMP1, VCAN, and FABP4 in the progression from NAFL to NASH have also not been well defined, and further studies are warranted.
Our study suggests that ECM-related pathways and immunity-related pathways are the main functional pathways involved in the progression from NAFL to NASH. ECM is a multi-molecule complex structure composed of collagen, elastin fibers, and structural glycoproteins, and is a critical player in fibrosis [49, 50]. ECM has proven to be closely related to progressive fibrosis and inflammation in NASH, and it has been proposed to be not only a consequence but also a driving factor in fibrosis [51,52,53]. Apart from ECM, immune responses have also been shown to be important in the progression of NASH [54,55,56]. For instance, cell death in NASH can trigger an inflammatory response through innate immune cells, which further promotes fibrosis progression via activating HSC [57, 58]. In the present study, GO enrichment analysis of those 141 significant crucial genes and WGCNA analysis both confirmed the critical roles of ECM-related pathways and immunity-related pathways in the progression from NAFL to NASH. Nevertheless, the exact molecular mechanisms for those two functional pathways in the development of NASH are still not well defined. It is no doubt that blocking ECM-related pathways or immunity-related pathways by suppressing drugs may help to prevent or reverse NASH progression, which is worth exploring in more future studies.
There were several limitations in this study. First, there was obvious heterogeneity in the clinical characteristics of NAFLD patients such as demographic data, disease severity, and disease duration, which undoubtedly could increase the risk of biased outcomes. Nevertheless, RRA is a rank aggregation method and is a rigorous way in detecting the statistically significant DEGs, which has been adopted in many studies to integrate transcriptome datasets from different technological platforms [13, 59, 60]. Second, our study identified that immunity-related pathways were important functional pathways involved in the progression from NAFL to NASH. However, the crucial types of immune cells involved in NASH progression are currently unclear and need to be explored in future studies. Finally, the expression pattern of those DEGs in a specific types of cells in both the progression of NASH and the progression of fibrosis is still difficult to be explored by transcriptome data from bulk tissues. More single-cell transcriptome data focusing on the progression from NAFL to NASH or fibrosis progression among NAFLD patients are recommended in future studies.
In summary, transcriptomic signatures related to NASH development and fibrosis severity of NAFLD patients are both characterized by ECM-related pathways, and fibrosis is the main player in NASH progression. ECM-related pathways and immunity-related signaling pathways are essential functional pathways determining the progression from NAFL to NASH. The molecular mechanisms underlying the roles of some genes such as FBLN5, EFEMP1, FABP4, and CD24 in the progression of NASH are still unclear and need to be elucidated in future studies, which may provide promising therapeutic targets for NASH.
References
N. Stefan, H.U. Haring, K. Cusi, Non-alcoholic fatty liver disease: causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet. Diabetes Endocrinol. 7(4), 313–324 (2019). https://doi.org/10.1016/S2213-8587(18)30154-2
Z. Younossi, Q.M. Anstee, M. Marietti, T. Hardy, L. Henry, M. Eslam, J. George, E. Bugianesi, Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 15(1), 11–20 (2018). https://doi.org/10.1038/nrgastro.2017.109
F. Baratta, D. Pastori, F. Angelico, A. Balla, A.M. Paganini, N. Cocomello, D. Ferro, F. Violi, A.J. Sanyal, M. Del Ben, Nonalcoholic fatty liver disease and fibrosis associated with increased risk of cardiovascular events in a prospective study. Clin. Gastroenterol. Hepatol. 18(10), 2324–2331 e2324 (2020). https://doi.org/10.1016/j.cgh.2019.12.026
A.M. Diehl, C. Day, Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N. Engl. J. Med. 377(21), 2063–2072 (2017). https://doi.org/10.1056/NEJMra1503519
N. Chalasani, Z. Younossi, J.E. Lavine, M. Charlton, K. Cusi, M. Rinella, S.A. Harrison, E.M. Brunt, A.J. Sanyal, The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67(1), 328–357 (2018). https://doi.org/10.1002/hep.29367
I. Pierantonelli, G. Svegliati-Baroni, Nonalcoholic fatty liver disease: basic pathogenetic mechanisms in the progression from NAFLD to NASH. Transplantation 103(1), e1–e13 (2019). https://doi.org/10.1097/TP.0000000000002480
A.J. Sanyal, C. Campbell-Sargent, F. Mirshahi, W.B. Rizzo, M.J. Contos, R.K. Sterling, V.A. Luketic, M.L. Shiffman, J.N. Clore, Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 120(5), 1183–1192 (2001). https://doi.org/10.1053/gast.2001.23256
A.E. Feldstein, A. Canbay, P. Angulo, M. Taniai, L.J. Burgart, K.D. Lindor, G.J. Gores, Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 125(2), 437–443 (2003). https://doi.org/10.1016/s0016-5085(03)00907-7
P. Puri, F. Mirshahi, O. Cheung, R. Natarajan, J.W. Maher, J.M. Kellum, A.J. Sanyal, Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 134(2), 568–576 (2008). https://doi.org/10.1053/j.gastro.2007.10.039
P. Hirsova, S.H. Ibrahim, G.J. Gores, H. Malhi, Lipotoxic lethal and sublethal stress signaling in hepatocytes: relevance to NASH pathogenesis. J. Lipid. Res. 57(10), 1758–1770 (2016). https://doi.org/10.1194/jlr.R066357
A.R. Mridha, F. Haczeyni, M.M. Yeh, W.G. Haigh, G.N. Ioannou, V. Barn, H. Ajamieh, L. Adams, J.M. Hamdorf, N.C. Teoh, G.C. Farrell, TLR9 is up-regulated in human and murine NASH: pivotal role in inflammatory recruitment and cell survival. Clin. Sci. (Lond) 131(16), 2145–2159 (2017). https://doi.org/10.1042/CS20160838
B. Cai, P. Dongiovanni, K.E. Corey, X. Wang, I.O. Shmarakov, Z. Zheng, C. Kasikara, V. Davra, M. Meroni, R.T. Chung, C.V. Rothlin, R.F. Schwabe, W.S. Blaner, R.B. Birge, L. Valenti, I. Tabas, Macrophage MerTK promotes liver fibrosis in nonalcoholic steatohepatitis. Cell. Metab. 31(2), 406–421 e407 (2020). https://doi.org/10.1016/j.cmet.2019.11.013
R. Kolde, S. Laur, P. Adler, J. Vilo, Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics 28(4), 573–580 (2012). https://doi.org/10.1093/bioinformatics/btr709
M.E. Ritchie, B. Phipson, D. Wu, Y. Hu, C.W. Law, W. Shi, G.K. Smyth, Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic. Acids. Res. 43(7), e47 (2015). https://doi.org/10.1093/nar/gkv007
M.I. Love, W. Huber, S. Anders, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome. Biol. 15(12), 550 (2014). https://doi.org/10.1186/s13059-014-0550-8
A. Subramanian, P. Tamayo, V.K. Mootha, S. Mukherjee, B.L. Ebert, M.A. Gillette, A. Paulovich, S.L. Pomeroy, T.R. Golub, E.S. Lander, J.P. Mesirov, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A 102(43), 15545–15550 (2005). https://doi.org/10.1073/pnas.0506580102
S. Hanzelmann, R. Castelo, J. Guinney, GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14, 7 (2013). https://doi.org/10.1186/1471-2105-14-7
G.P. Wagner, K. Kin, V.J. Lynch, Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci. 131(4), 281–285 (2012). https://doi.org/10.1007/s12064-012-0162-3
M. Ashburner, C.A. Ball, J.A. Blake, D. Botstein, H. Butler, J.M. Cherry, A.P. Davis, K. Dolinski, S.S. Dwight, J.T. Eppig, M.A. Harris, D.P. Hill, L. Issel-Tarver, A. Kasarskis, S. Lewis, J.C. Matese, J.E. Richardson, M. Ringwald, G.M. Rubin, G. Sherlock, Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25(1), 25–29 (2000). https://doi.org/10.1038/75556
G. Dennis Jr, B.T. Sherman, D.A. Hosack, J. Yang, W. Gao, H.C. Lane, R.A. Lempicki, DAVID: database for annotation, visualization, and integrated discovery. Genome. Biol. 4(5), P3 (2003)
M.P. Dobay, S. Stertz, M. Delorenzi, Context-based retrieval of functional modules in protein-protein interaction networks. Brief. Bioinform. 19(5), 995–1007 (2018). https://doi.org/10.1093/bib/bbx029
A.I. Amanatidou, G.V. Dedoussis, Dedoussis, Construction and analysis of protein-protein interaction network of non-alcoholic fatty liver disease. Comput. Biol. Med. 131, 104243 (2021). https://doi.org/10.1016/j.compbiomed.2021.104243
O. Philipp, A. Hamann, H.D. Osiewacz, I. Koch, The autophagy interaction network of the aging model Podospora anserina. BMC Bioinformatics 18(1), 196 (2017). https://doi.org/10.1186/s12859-017-1603-2
D. Szklarczyk, A.L. Gable, D. Lyon, A. Junge, S. Wyder, J. Huerta-Cepas, M. Simonovic, N.T. Doncheva, J.H. Morris, P. Bork, L.J. Jensen, C.V. Mering, STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic. Acids Res. 47(D1), D607–D613 (2019). https://doi.org/10.1093/nar/gky1131
G. Su, J.H. Morris, B. Demchak, G.D. Bader, Biological network exploration with Cytoscape 3. Curr. Protoc. Bioinformatics. 47(8), 11–24 (2014). https://doi.org/10.1002/0471250953.bi0813s47
C.H. Chin, S.H. Chen, H.H. Wu, C.W. Ho, M.T. Ko, C.Y. Lin, cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 8 Suppl 4(Suppl 4), S11 (2014). https://doi.org/10.1186/1752-0509-8-S4-S11
A.P. Presson, E.M. Sobel, J.C. Papp, C.J. Suarez, T. Whistler, M.S. Rajeevan, S.D. Vernon, S. Horvath, Integrated weighted gene co-expression network analysis with an application to chronic fatigue syndrome. BMC Syst. Biol. 2, 95 (2008). https://doi.org/10.1186/1752-0509-2-95
P. Ramachandran, R. Dobie, J.R. Wilson-Kanamori, E.F. Dora, B.E.P. Henderson, N.T. Luu, J.R. Portman, K.P. Matchett, M. Brice, J.A. Marwick, R.S. Taylor, M. Efremova, R. Vento-Tormo, N.O. Carragher, T.J. Kendall, J.A. Fallowfield, E.M. Harrison, D.J. Mole, S.J. Wigmore, P.N. Newsome, C.J. Weston, J.P. Iredale, F. Tacke, J.W. Pollard, C.P. Ponting, J.C. Marioni, S.A. Teichmann, N.C. Henderson, Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 575(7783), 512–518 (2019). https://doi.org/10.1038/s41586-019-1631-3
A. Butler, P. Hoffman, P. Smibert, E. Papalexi, R. Satija, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36(5), 411–420 (2018). https://doi.org/10.1038/nbt.4096
N. Aizarani, A. Saviano, Sagar, L. Mailly, S. Durand, J.S. Herman, P. Pessaux, T.F. Baumert, D. Grun, A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 572(7768), 199–204 (2019). https://doi.org/10.1038/s41586-019-1373-2
D. Aran, A.P. Looney, L. Liu, E. Wu, V. Fong, A. Hsu, S. Chak, R.P. Naikawadi, P.J. Wolters, A.R. Abate, A.J. Butte, M. Bhattacharya, Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nat. Immunol. 20(2), 163–172 (2019). https://doi.org/10.1038/s41590-018-0276-y
A. Bertola, V. Deveaux, S. Bonnafous, D. Rousseau, R. Anty, A. Wakkach, M. Dahman, J. Tordjman, K. Clement, S.E. McQuaid, K.N. Frayn, P.M. Huet, J. Gugenheim, S. Lotersztajn, Y. Le Marchand-Brustel, A. Tran, P. Gual, Elevated expression of osteopontin may be related to adipose tissue macrophage accumulation and liver steatosis in morbid obesity. Diabetes 58(1), 125–133 (2009). https://doi.org/10.2337/db08-0400
F.W. Kiefer, S. Neschen, B. Pfau, B. Legerer, A. Neuhofer, M. Kahle, M. Hrabe de Angelis, M. Schlederer, M. Mair, L. Kenner, J. Plutzky, M. Zeyda, T.M. Stulnig, Osteopontin deficiency protects against obesity-induced hepatic steatosis and attenuates glucose production in mice. Diabetologia 54(8), 2132–2142 (2011). https://doi.org/10.1007/s00125-011-2170-0
C. Zhu, K. Kim, X. Wang, A. Bartolome, M. Salomao, P. Dongiovanni, M. Meroni, M.J. Graham, K.P. Yates, A.M. Diehl, R.F. Schwabe, I. Tabas, L. Valenti, J.E. Lavine, U.B. Pajvani, Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci Transl Med 10(468), (2018). https://doi.org/10.1126/scitranslmed.aat0344
X. Chu, Q. Jin, H. Chen, G.C. Wood, A. Petrick, W. Strodel, J. Gabrielsen, P. Benotti, T. Mirshahi, D.J. Carey, C.D. Still, J.K. DiStefano, G.S. Gerhard, CCL20 is up-regulated in non-alcoholic fatty liver disease fibrosis and is produced by hepatic stellate cells in response to fatty acid loading. J. Transl. Med. 16(1), 108 (2018). https://doi.org/10.1186/s12967-018-1490-y
A. Hanson, I.S. Piras, D. Wilhelmsen, C.D. Still, X. Chu, A. Petrick, G.S. Gerhard, J.K. DiStefano, Chemokine ligand 20 (CCL20) expression increases with NAFLD stage and hepatic stellate cell activation and is regulated by miR-590-5p. Cytokine 123, 154789 (2019). https://doi.org/10.1016/j.cyto.2019.154789
A. Katayama, A. Nakatsuka, J. Eguchi, K. Murakami, S. Teshigawara, M. Kanzaki, T. Nunoue, K. Hida, N. Wada, T. Yasunaka, F. Ikeda, A. Takaki, K. Yamamoto, H. Kiyonari, H. Makino, J. Wada, Beneficial impact of Gpnmb and its significance as a biomarker in nonalcoholic steatohepatitis. Sci. Rep. 5, 16920 (2015). https://doi.org/10.1038/srep16920
X. Zhang, J. Shen, K. Man, E.S. Chu, T.O. Yau, J.C. Sung, M.Y. Go, J. Deng, L. Lu, V.W. Wong, J.J. Sung, G. Farrell, J. Yu, CXCL10 plays a key role as an inflammatory mediator and a non-invasive biomarker of non-alcoholic steatohepatitis. J. Hepatol. 61(6), 1365–1375 (2014). https://doi.org/10.1016/j.jhep.2014.07.006
K. Tomita, B.L. Freeman, S.F. Bronk, N.K. LeBrasseur, T.A. White, P. Hirsova, S.H. Ibrahim, CXCL10-mediates macrophage, but not other innate immune cells-associated inflammation in murine nonalcoholic steatohepatitis. Sci. Rep. 6, 28786 (2016). https://doi.org/10.1038/srep28786
J.S. Johansen, P. Christoffersen, S. Moller, P.A. Price, J.H. Henriksen, C. Garbarsch, F. Bendtsen, Serum YKL-40 is increased in patients with hepatic fibrosis. J. Hepatol. 32(6), 911–920 (2000). https://doi.org/10.1016/s0168-8278(00)80095-1
E. Kumagai, Y. Mano, S. Yoshio, H. Shoji, M. Sugiyama, M. Korenaga, T. Ishida, T. Arai, N. Itokawa, M. Atsukawa, H. Hyogo, K. Chayama, T. Ohashi, K. Ito, M. Yoneda, T. Kawaguchi, T. Torimura, Y. Nozaki, S. Watanabe, M. Mizokami, T. Kanto, Serum YKL-40 as a marker of liver fibrosis in patients with non-alcoholic fatty liver disease. Sci. Rep. 6, 35282 (2016). https://doi.org/10.1038/srep35282
W.S. Argraves, L.M. Greene, M.A. Cooley, W.M. Gallagher, Fibulins: physiological and disease perspectives. EMBO Rep. 4(12), 1127–1131 (2003). https://doi.org/10.1038/sj.embor.7400033
T. Nakamura, P.R. Lozano, Y. Ikeda, Y. Iwanaga, A. Hinek, S. Minamisawa, C.F. Cheng, K. Kobuke, N. Dalton, Y. Takada, K. Tashiro, J. Ross Jr, T. Honjo, K.R. Chien, Fibulin-5/DANCE is essential for elastogenesis in vivo. Nature 415(6868), 171–175 (2002). https://doi.org/10.1038/415171a
H. Yanagisawa, E.C. Davis, B.C. Starcher, T. Ouchi, M. Yanagisawa, J.A. Richardson, E.N. Olson, Fibulin-5 is an elastin-binding protein essential for elastic fibre development in vivo. Nature 415(6868), 168–171 (2002). https://doi.org/10.1038/415168a
C.L. Papke, H. Yanagisawa, Fibulin-4 and fibulin-5 in elastogenesis and beyond: Insights from mouse and human studies. Matrix. Biol. 37, 142–149 (2014). https://doi.org/10.1016/j.matbio.2014.02.004
C.Y. Chen, H. Kimura, M.A. Landek-Salgado, J. Hagedorn, M. Kimura, K. Suzuki, W. Westra, N.R. Rose, P. Caturegli, Regenerative potentials of the murine thyroid in experimental autoimmune thyroiditis: role of CD24. Endocrinology 150(1), 492–499 (2009). https://doi.org/10.1210/en.2008-0639
J.Q. Liu, J.W. Carl Jr, P.S. Joshi, A. RayChaudhury, X.A. Pu, F.D. Shi, X.F. Bai, CD24 on the resident cells of the central nervous system enhances experimental autoimmune encephalomyelitis. J. Immunol. 178(10), 6227–6235 (2007). https://doi.org/10.4049/jimmunol.178.10.6227
A.A. Barkal, R.E. Brewer, M. Markovic, M. Kowarsky, S.A. Barkal, B.W. Zaro, V. Krishnan, J. Hatakeyama, O. Dorigo, L.J. Barkal, I.L. Weissman, CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 572(7769), 392–396 (2019). https://doi.org/10.1038/s41586-019-1456-0
C. Bonnans, J. Chou, Z. Werb, Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell. Biol. 15(12), 786–801 (2014). https://doi.org/10.1038/nrm3904
J. Herrera, C.A. Henke, P.B. Bitterman, Extracellular matrix as a driver of progressive fibrosis. J. Clin. Invest. 128(1), 45–53 (2018). https://doi.org/10.1172/JCI93557
M.L. Decaris, K.W. Li, C.L. Emson, M. Gatmaitan, S. Liu, Y. Wang, E. Nyangau, M. Colangelo, T.E. Angel, C. Beysen, J. Cui, C. Hernandez, L. Lazaro, D.A. Brenner, S.M. Turner, M.K. Hellerstein, R. Loomba, Identifying nonalcoholic fatty liver disease patients with active fibrosis by measuring extracellular matrix remodeling rates in tissue and blood. Hepatology 65(1), 78–88 (2017). https://doi.org/10.1002/hep.28860
I.D. Munsterman, T.J. Kendall, N. Khelil, M. Popa, R. Lomme, J.P.H. Drenth, E. Tjwa, Extracellular matrix components indicate remodelling activity in different fibrosis stages of human non-alcoholic fatty liver disease. Histopathology 73(4), 612–621 (2018). https://doi.org/10.1111/his.13665
N. Sanchez-Romero, P. Sainz-Arnal, I. Pla-Palacin, P.R. Dachary, H. Almeida, C. Pastor, D.R. Soto, M.C. Rodriguez, E.O. Arbizu, L.B. Martinez, T. Serrano-Aullo, P.M. Baptista, The role of extracellular matrix on liver stem cell fate: a dynamic relationship in health and disease. Differentiation 106, 49–56 (2019). https://doi.org/10.1016/j.diff.2019.03.001
S. Schuster, D. Cabrera, M. Arrese, A.E. Feldstein, Triggering and resolution of inflammation in NASH. Nat. Rev. Gastroenterol. Hepatol. 15(6), 349–364 (2018). https://doi.org/10.1038/s41575-018-0009-6
K. Kazankov, S.M.D. Jorgensen, K.L. Thomsen, H.J. Moller, H. Vilstrup, J. George, D. Schuppan, H. Gronbaek, The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 16(3), 145–159 (2019). https://doi.org/10.1038/s41575-018-0082-x
M.A. Van Herck, J. Weyler, W.J. Kwanten, E.L. Dirinck, B.Y. De Winter, S.M. Francque, L. Vonghia, The differential roles of T cells in non-alcoholic fatty liver disease and obesity. Front. Immunol. 10, 82 (2019). https://doi.org/10.3389/fimmu.2019.00082
P. Kubes, W.Z. Mehal, Sterile inflammation in the liver. Gastroenterology 143(5), 1158–1172 (2012). https://doi.org/10.1053/j.gastro.2012.09.008
P. Hirsova, G.J. Gores,, Death receptor-mediated cell death and proinflammatory signaling in nonalcoholic steatohepatitis. Cell. Mol. Gastroenterol. Hepatol. 1(1), 17–27 (2015). https://doi.org/10.1016/j.jcmgh.2014.11.005
C. Andersson, H. Lin, C. Liu, D. Levy, G.F. Mitchell, M.G. Larson, R.S. Vasan, Integrated multiomics approach to identify genetic underpinnings of heart failure and its echocardiographic precursors: framingham heart study. Circ. Genom. Precis. Med. 12(12), e002489 (2019). https://doi.org/10.1161/CIRCGEN.118.002489
W. He, B. Wang, Q. Li, Q. Yao, X. Jia, R. Song, S. Li, J.A. Zhang, Aberrant expressions of co-stimulatory and co-inhibitory molecules in autoimmune diseases. Front. Immunol. 10, 261 (2019). https://doi.org/10.3389/fimmu.2019.00261
Acknowledgements
We would like to thank all the researchers who have shared their data in GEO database.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81870606).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
He, W., Huang, C., Zhang, X. et al. Identification of transcriptomic signatures and crucial pathways involved in non-alcoholic steatohepatitis. Endocrine 73, 52–64 (2021). https://doi.org/10.1007/s12020-021-02716-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-021-02716-y