
Abstract
Purpose of Review
The more our knowledge on cancer biology progresses, the more it becomes evident that studying the cancer cell isolated from the cancer environment is reductive. Therefore, a better understanding of cancer biology needs a better characterization of the interactions a cancer cell can establish with the surrounding environment. The purpose of this review is to focus on one of the most intriguing cancer/healthy tissue interactions, which occurs in bone during breast cancer-bone metastasis.
Recent Findings
Bone and bone marrow represent a very peculiar environment populated by a variety of cells that cross-communicate. Bone is also by far the most common metastatic site in breast cancer. Breast cancer cells not only colonize the bone, but also alter its metabolism inducing osteoclast-mediated osteolysis. Recent findings further support the relevance of the pathological cross-talk at the basis of the breast cancer-induced bone metastasis, called the vicious cycle. In fact, targeting molecules essential for this cross-talk is already an effective therapeutic strategy, proven to be more constructive than targeting the cancer cells alone.
Summary
We will dissect in this article the vicious cycle and describe the interactions that happen in bone metastasis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Clinical advancements surrounding the therapeutic targeting of breast cancer has strongly improved the survival of patients [1, 2••]. However, second only to lung cancer, breast cancer still remains one of the most commonly diagnosed cancers in USA and Europe, where it is the most diagnosed cancer in women [2••, 3, 4]. Breast cancer mortality in 90% of the cases is a consequence of cancer cell dissemination within the body and metastasizing to secondary sites [5•]. According to cumulative epidemiologic reports [6], bone is the most common metastatic site in breast cancer. Around 70% of breast cancer metastases occur at the bone, with the highest prevalence observed in the axial hematopoietic skeleton [7, 8]. This suggests that the bone specifically provides the necessary tools for disseminated cancer cells to survive and take advantage of homeostatic processes. Therefore, understanding how this volatile interaction between bone and cancer cells manifests itself is paramount.
The ability of cancer cells to ‘home to the bone’ was first recognized by Paget in 1889 [9]. More than 100 years later, we call this phenomenon ‘organ tropism’; however, the reasons behind breast cancer metastatic specificity are only materializing now [7]. The bone is a dynamic tissue central to conducting the homeostasis of the entire organism [10••]. The foundational elements of the bone consist of osteoblasts, osteoclasts, and osteocytes. These cells are the construction workers responsible for bone remodeling [10••, 11, 12]. The bone also hosts a range of red bone marrow cells differentiated from hematopoietic stem cells, and endothelial cells that constitute the capillaries, essential for the bone vasculature [13•]. Sharing the same home, it is reasonable to assume that these cell types are able to interact with and cross-regulate each other. However, the very same vasculature that supports the bone can also guide metastasizing cancer cells to this rich microenvironment [13•]. There is accumulating evidence that once these breast cancer cells reach the bone, they are able to participate in the crosstalk. This enables them to manipulate the homeostatic cross-talk occurring in the bone. In this review article, we will focus particularly on the interaction between breast cancer cells and the fundamental bone cells osteoblasts, osteoclasts, and osteocytes.
To describe the interactions between breast cancer cells and bone cells, it is instrumental to introduce the bone remodeling cycle or the virtuous cycle. During adulthood, there is a tightly regulated balance between the bone that is resorbed by the osteoclasts, formed by osteoblasts and afterwards populated by osteocytes. Moreover, there is a spatial and temporal organization of these events that strongly support the idea that these cell types cross-regulate each other in the area of the bone that undergoes remodeling, suggesting a paracrine/autocrine loop of signals. Since this regulation is beneficial for bone homeostasis, it is called virtuous cycle of bone remodeling. This is contraposed to the vicious cycle, that is characterized by the presence of breast cancer cells and a consequent imbalance to favor bone resorption. The aims of the present literature review are to decompose the vicious cycle, analyzing the one-to-one interactions between bone and cancer cells, and describe the treatment used in the clinical practice for breast cancer bone metastases in the light of their effect on the disruption of the vicious cycle.
However, it is our opinion that it is important for a full comprehension of the pathogenesis of breast cancer bone metastasis to consider in a larger portrait also the interactions of breast cancer cells with the other components of the bone/bone marrow tissue.
The Role of Osteoclasts in the Vicious Cycle
Osteoclasts are multinucleated giant cells arising from hematopoietic precursors [12]. They have the unique role to degrade both the inorganic (i.e., hydroxyapatite) and organic parts (i.e., collagens) of the bone matrix [12]. Macrophage colony-stimulating factor (M-CSF) and receptor activator of NFκB ligand (RANKL) drive monocyte/macrophage lineage cells to fuse and maturate in osteoclasts. Osteoprotegerin (OPG), a decoy receptor, counterbalances RANKL action. All three cytokines M-CSF, RANKL, and OPG are secreted by stromal cells, osteoblasts and osteocytes, and are able to regulate the recruitment, action, and survival of osteoclasts [12]. Osteoclasts in turn influence osteoblast differentiation through two main mechanisms: i) release of bio-active molecules from the mineralized bone matrix in the bone microenvironment surrounding the bone resorption area and ii) by direct secretion of factors that regulate the bone stromal cells in the surrounding area [11].
In the case of a breast cancer bone metastasis, the tumor cells can enter this cross-regulation and exacerbate bone resorption by directly secreting RANKL which leads to the release and secretion of bio-active molecules by the osteoclasts.
The first product of bone degradation is calcium. Calcium is released from bone by the acidification of the extracellular environment (resorption lacuna) by osteoclasts [12]. As tumor cells express the calcium-sensing receptor (CASR), they are able to respond to the increased calcium, which acts as a pro-survival stimulus [14]. Once calcium is released, the bone organic matrix is degraded by cathepsin K and matrix-metalloproteinases (MMPs) secreted by the osteoclasts [12]. The degradation of the bone organic matrix leads to the release in the microenvironment surrounding the bone resorption area of bioactive products. These bioactive degradation products are physiologically involved in the regulation of the virtuous cycle and stimulate tumor growth in case of a bone metastasis. Among them, TGFβ is of particular relevance and will be better discussed later on [15, 16].
Although osteoclasts are mostly known for being directly involved in tumor growth-associated bone resorption, they also secrete miRNAs that modulate tumor growth without direct bone resorption [15,16,17,18]. Osteoclasts secrete exosomes containing miRNAs, including miR-378 which promotes tumor growth, angiogenesis, and tumor cell survival through the repression of tumor suppressors SuFu (suppressor of fused homolog) and Fus-1 (fusion protein 1) [19]. Additionally, osteoclast-derived miR-21 enhances tumor cell proliferation, by inhibiting the apoptotic pathway [20,21,22,23].
Finally, breast cancer metastatic cells also release factors contributing directly to bone degradation without the involvement of osteoclasts. In fact, tumor cells can acidify the extracellular microenvironment and it is shown that cathepsin K is produced by tumor cells [24••]. Cathepsin K promotes tumor cell invasiveness and may also contribute to bone degradation.
The Role of Osteoblasts in the Vicious Cycle
The osteoblasts derive from the differentiation of the mesenchymal stem cells in the bone. Their main functions are to produce the bone organic matrix, to facilitate its mineralization [11, 12], and to regulate the function of the osteoclasts. Osteoblasts undergo a multistep differentiation process, and in a healthy bone/bone marrow, it is possible to identify cells at different stages of differentiation, all of them contributing in a different ways to the virtuous/vicious cycle. Starting from the mesenchymal stem cells, the first step in differentiation is represented by the pre-osteoblasts. These are immature cells, poorly or non-polarized [11], populating the bone marrow in tight contact with hematopoietic cells. They are characterized by the expression of the early differentiation markers OSTERIX and runt-related transcription factor 2 (RUNX2), and are under the control of the WNT/β-CATENIN pathway that regulates their differentiation. Pre-osteoblasts have been described [25, 26] as high RANKL producers, and therefore, strongly contribute to osteoclast-mediated bone resorption [12]. In bone metastases, they can therefore stimulate pathological osteolysis [27]. Moreover, according to some reports, RANKL directly stimulates breast cancer proliferation in bone, further fuelling the vicious cycle [28••, 29, 30•]. In addition, pre-osteoblasts produce and secrete other factors influencing the bone metastasis for example, the hepatocyte growth factor (HGF). HGF, in turn stimulates the MET pathway eventually promoting the migration and homing of breast cancer cells in the bone [31•]. Pre-osteoblasts can also secrete different pro-inflammatory cytokines known to stimulate breast cancer progression . These include, interleukin 6 (IL6) [32], lipocalin 2 (LCN2) [33•, 34, 35] and tumor necrosis factor alpha (TNFα) [36,37,38], which also act on the microenvironment stimulating the osteoclast differentiation and function [39]. Mature osteoblasts differ from pre-osteoblasts in terms of their localization, that is typically at the endosteal surface [11], and in terms of their morphology being cuboidal, with pronounced polarization and characterized by an intense cytoplasmic basophilia, a sign of intense protein synthesis [40]. The mature osteoblasts largely contribute to the virtuous/vicious cycle with direct and indirect effects. In fact, during bone formation, osteoblasts release in the bone matrix, a variety of bioactive molecules that exert their function only after they are released from the bone matrix during the osteoclast bone resorption, as mentioned before. Among them, there are growth factors, such as bone morphogenetic proteins 2 and 4 (BMPs), insulin-like growth factor-1 (IGF-1), transforming growth factor-beta1 (TGFβ1) and vascular endothelial growth factor (VEGF) [41] that stimulate tumor growth and neo-angiogenesis. On the other hand, bone-specific proteins such as osteopontin [42], known to stimulate cell adhesion, migration, the bone sialoprotein [43], of which the over-expression can enhance bone metastasis. Osteoblasts release in the bone matrix also poorly studied non-collagenous proteins that can interfere as inhibitors of the virtuous/vicious cycle. In the same vein we can mention the proline and leucine-rich-end repeat protein (PRELP) and (chondroadherin) CHAD. Both proteins have been discovered and described by the group of Prof. Heingard [44, 45], and later on characterized in different articles. During osteoclast bone resorption, the degradation fragments of the PRELP and CHAD can be released and partake in a negative feedback loop, impairing bone resorption [46, 47]. Thus, suggesting a strategy of preservation of the bone matrix that directly inhibits its degradation and can partially explain the tropism for specific bone areas for metastasis.
To complete the complex role of mature osteoblasts in regulating osteoclast activity, we should mention several secreted factors, starting from RANKL and OPG, but also of particular interest is the secretion of cytokines such as IL6 and TNFα. After bone deposition, mature osteoblasts can differentiate into osteocytes, bone lining cells or undergo apoptosis. While the role of osteocytes in the vicious cycle will be described more extensively in the following paragraph, here we will briefly describe the few known effects of bone lining cells and apoptotic osteoblasts in the vicious cycle. Bone lining cells cover the quiescent bone surface and are characterized by a flat morphology. Their function is poorly described, but the flat morphology suggests a low biosynthetic activity. According to Matic and colleagues [48•], the bone lining cells are the major source of osteoblasts during adulthood, and have elevated expression of the pro-osteoclast genes RANK-L and M-CSF but also produce high levels of matrix-metalloproteinase 13 (MMP13) levels. MMP13 is under the transcriptional control of OSTERIX [49]. Once secreted, MMP13 is able to stimulate osteoclast differentiation and serves as a pro-survival stimulus for breast cancer cells colonizing the bone [50]. Apoptotic osteoblasts are able to influence the healthy and pathological bone turnover [51]. Apoptosis of osteoblasts is not a random event, but temporally and spatially tuned. It is essential for ensuring the proper bone size and architecture. A plethora of molecules have been identified as inducers of osteoblast apoptosis, such as TNFα, BMP2, RANKL, and OPG, and according to several reports [51], metastasizing breast cancer cells induce osteoblast apoptosis. However, the effect of apoptotic osteoblasts on the surrounding healthy bone cells is still poorly described. We can speculate that the effect of apoptotic osteoblasts mirrors the effect of apoptotic cancer stroma. For example, in colorectal cancer, stromal cell apoptosis is associated with a still poorly understood mechanism to decreased survival in patients, suggesting a pro-cancer effect [52]. Similarly, according to the elegant theory of Rui-An Wang et al. [53], apoptosis of a surrounding cell can be a trigger to stimulate cancer progression by a mechanism probably involving extracellular vesicle signaling.
The Role of Osteocytes in the Vicious Cycle
The final differentiation stage of osteoblasts is represented by the osteocytes [11]. They are post-mitotic stellate cells, completely immersed in the mineralized bone matrix with a small lenticular cell body containing a dense nucleus, surrounded by plasma membrane protrusions forming dendrites. Dendrites run into bone canaliculi and connect the osteocytes in a complicated network through GAP junctions. Although their morphology suggests a poor cell activity, the osteocyte functions are surprisingly complex and contribute to regulating not only the bone metabolism but also kidney metabolism and the phosphate plasma levels [10••]. Their physical localization and their distribution in the bone make these cells good candidates for regulating bone remodeling, and in bone metastases to fuel the vicious cycle. Indeed, according to recent reports [54•, 55], the osteocytes are important producers of RANKL. However, they can also control the metastatic growth by secreting factors such as DMP1 (dentin matrix protein-1). DMP1 in turn activates breast cancer cell proliferation by epithelium growth factor receptor (EGFR) binding [56•]. DMP1 is also found in primary breast tumor and appears to be implicated also in metastasization in patients [57].
The Role of Breast Cancer Cells in the Vicious Cycle
The switch between virtuous and vicious cycle is mediated by the presence of cancer cells. They use several strategies to enter, alter, and exacerbate the virtuous cycle, and use it for i) stimulating their growth and ii) removing the physical barrier represented by the mineralized bone matrix. The removal of mineralized matrix requires the osteoclast action. However, osteoclast formation and function are under the tight control of osteoblasts, pre-osteoblasts, osteocytes, and other bone marrow cells not discussed in this review. Therefore, a tumor cell needs to secrete factors able not only to regulate the osteoclasts, but also to interfere with the regulation of osteoclasts by other cell types.
In this scenario, TGFβ is one of the main actors [58]. TGFβ activates directly tumor growth and inhibits T cell proliferation and natural killer cell activation, suppressing the immune system [58,59,60]. TGFβ induces the release of parathyroid hormone-related protein (PTHrP) and interleukin 11 (IL-11) from tumor cells, which in turn stimulates osteoclast activation and bone resorption [15, 21]. PTHrP alters the RANKL/OPG ratio in favor of RANKL and is one of the most important mediators of osteoclast activation [21]. PTHrP is overexpressed in most of the patients suffering from breast cancer that has metastasized to the bone [58, 60]. Thus, suggesting PTHrP to be a bio-marker for predicting bone metastasis in patients with breast cancer [61]. Breast cancer cells are also able to boost bone resorption engaging directly with OC precursors. In fact, breast cancer cells express the vascular cell-adhesion molecule 1 (VCAM-1), which recruits and activates OC precursors [62]. Tumor cells are also reported to secrete several factors that inhibit osteoblast differentiation such as dickkopf-1 (DKK-1) and sclerostin (SOST) [59], suppressing bone formation and indirectly contributing to bone degradation.
One more piece in the complex puzzle of the virtous/vicious cycle is represented by the extracellular vesicles (EVs) [63•]. Several reports suggest the involvement of EVs in bone metabolism and bone metastasis. EVs are cell-derived ‘smart’ delivery particles containing a plethora of molecules, such as miRNAs, mRNAs, and proteins. Tumors can use EVs to educate the bone microenvironment to sustain cancer progression [63•]. Breast cancer-derived EVs are reported to promote a myofibroblastic phenotype of adipose tissue-derived mesenchymal stem cells, resulting in increased expression of the tumor-promoting factors TGF-β, VEGF, stromal cell-derived factor 1 (SDF-1), and C–C motif chemokine ligand 5 (CCL5) [64].
Tumor EVs have been implicated in organotropism of metastatic tumor and in the formation and evolution of the premetastatic niche through their integrin cargo [65••]. Other studies highlighted that breast cancer-associated fibroblasts secrete exosomes that promote dissemination of cancer through the Wnt-planar cell polarity (Wnt-PCP) signaling pathway [66]. EVs are also reported to promote osteoclast differentiation directly through RANKL/RANK axis, and PTH is able to increase the release of RANKL-EVs [67, 68••]. Since PTHrP (released from tumor) shares the same receptor with PTH, it is reasonable to think that the tumor may induce EV production from osteoblasts and increase osteoclastogenesis.
On the other hand, the tumor could weaken the bone and endorse its degradation.
Therapeutic Strategies
Treatment plans for breast cancer bone metastases include different strategies affecting the tumor or the interaction with microenvironment such as hormonal therapy, chemotherapy, radiotherapy, and antiresorptive coadjutants [69•, 70••]. While the complexity of the biological basis (hormonal sensitiveness, genomic profile, chemoresistance) for selecting the best treatment option lies outside the purposes of this review, we will describe the most commonly used treatments focusing on their effects on the cancer cells and on the vicious cycle.
Hormonal therapy represents the first frontier to treat hormone-sensitive breast cancer bone metastases. Under the generic name ‘hormonal therapy’ there are different drugs that can either lower the amount of the estrogen available for the tumor (aromatase inhibitors, such as anastrozole, exemestane letrozole) [70••, 71•] or block their action (selective estrogen receptor modulators, such as tamoxifen, raloxifene, and toremifene) [71•, 72]. These drugs affect the bone metastatic breast cancer cells, but they also dramatically affect the bone microenvironment. In fact, both osteoblasts and osteoclasts are sensitive to estrogens [73] and the reduction of estrogen in women is associated with osteoporosis [74]. Therefore, even if the block of estrogen signaling is demonstrated to be effective in the treatment of breast cancer bone metastases, it has a pro-bone resorption effects, and paradoxically can facilitate the vicious cycle. However, according to clinical data, blocking the estrogen is effective in slowing or inhibiting the growth of breast cancer bone metastasis. There is no conclusive explanation for this contradictory evidence, and thus, we believe that it is an important field of investigation for future studies. A possible explanation that can be speculated is that bone metastases affect mostly women during post-menopause [75, 76]. In this condition, bone remodeling is already unbalanced in favor of bone resorption, and possibly the pharmacological estrogen inhibition does not further enhance this unbalanced condition. Therefore, it does not fuel the vicious cycle. To test this hypothesis, it would be of extreme interest to characterize the effects on breast cancer bone metastases of the hormonal therapy on pre-menopausal woman.
Chemotherapy is a common approach to treat metastatic breast cancer. Several drugs, used as single treatment or in combination, are clinically approved. Doxorubicin, taxanes (paclitaxel, docetaxel), cyclophosphamide, carboplatin, fluorouracil, gemcitabine just to cite some, are commonly used to fight bone metastatic breast cancers [72, 77, 78••, 79•]. All these drugs affect breast cancer cell proliferation, slowing their growth in the bone. At the same time, even if not well characterized, they should be able to affect the vicious cycle at different levels, by impairing pre-osteoblast proliferation and, according to some reports taxanes [80] and doxorubicin (personal observation) by affecting directly osteoclast function and survival.
Radioisotopes also play a role in the treatment of bone metastasis, especially for multifocal and painful metastatic lesions [70••, 81]. Radioisotopes are often conjugated to ligands that can direct the radioisotope to the bone. The main group of clinically approved radioisotopes consists of α-emitters, which have low penetration power in tissues [82]. Radium-223 is the most commonly used radioisotope and was demonstrated to increase the overall median survival by 3.6 months [82, 83]. Compared to other isotopes, Radium-223 appears to be less myelosuppressive with only mild thrombocytopenia [83, 84].
Beside the previous described remedies, mainly affecting the tumor cells, other drugs hit cells of the microenvironment, especially controlling the exacerbated bone resorption. Antiresorptive strategies for bone metastasis are currently based on inhibitors of osteoclast action, bisphosphonates (BPs) and denosumab. These drugs are used to reduce osteolysis and skeletal-related events (SREs), such as pathological fractures and spinal cord compression, and malignancy-induced hypercalcaemia and bone pain.
BPs are analogs of inorganic pyrophosphate PPi involved in mineralization. Due to their affinity for hydroxyapatite, BPs concentrate inside the mineralized bone matrix. Upon bone resorption, BPs are internalized into osteoclasts, where they interfere with ATP or melavonate pathway [85] inducing osteoclast apoptosis. From this point of view, BPs are meant to be a supportive treatment for bone metastasis in order to avoid SREs.
Intriguingly, some class of BPs showed in preclinical studies a direct antitumoral effect, alongside the anti-osteoclastic action. Some clinical data corroborates these conclusions, even if there is not yet a unanimous consensus on these results [86,87,88].
Denosumab is a fully humanized IgG2 monoclonal antibody which binds to and blocks RANKL [89]. A large randomized clinical trial showed the superiority of denosumab in delaying the time to first SRE and time to subsequent SREs in metastatic breast cancer patients. However, overall survival, disease progression, and rate of adverse events were similar to those observed in the bisphosphonate-treated group. Only a modest improvement in health-related quality of life was noted, favoring the use of denosumab [90]. We can speculate that the better effect of denosumab is due to its ability to block a molecule playing a central role in the vicious cycle.
The side effect profiles of bisphosphonates and denosumab are clearly established, and both are generally well tolerated [91, 92]. There is a small risk of osteonecrosis of the jaw with bisphosphonates and denosumab. This appears to increase with cumulative long-term therapy. Atypical fractures, usually subtrochanteric and diaphyseal fractures of the femur, are also reported with chronic treatment [93,94,95].
The biological complexity of virtuous/vicious cycle and the multiple pathogenetic players offers other potential target strategies [96]. For example, blockade of TGFβ and PTHrP production could be a potential approach for the treatment of bone metastases [6]. Indeed, in preclinical studies in animal models of bone metastasis, abrogation of TGFβ signaling by stable expression of dominant-negative TGFβ receptor 2 (DNTbRII), or by a TGFβ receptor 1 kinase inhibitor, suppresses bone metastasis [16, 97]. Cathepsin K is also excellent candidate for antiresorbing therapy. There is preclinical and clinical evidence showing that the treatment of bone metastases with a cathepsin K inhibitor (odanacatib, AFG-495) partially blocks bone degradation [98] and consequently osteolysis.
Conclusions
Advancing the understanding of cancer cell biology and the interactions with healthy cells is dramatically changing the approach researchers and clinicians are using to investigate cancer biology. Cancer cannot be considered anymore as a simple parasite that colonizes and destroys healthy tissues, but it should be rather considered as a smart group of cells that enter in the host tissue, interact with the resident cells, and use the signals important for manipulating the tissue homeostasis for their own advantage (Fig. 1). Using a military similitude, we can imagine the metastatic cancer not as an aggressive grenadier that conquers and destroys the host tissue, but better a fine and smart cryptographer, which interprets and uses at his own advantage signals from target tissue. Eventually, they destroy the host tissue, but this can be considered a consequence of their high ability in exploiting the interactions with the healthy tissue than the final aim of their colonization. This review article considers only part of the possible interactions that breast cancer cells can exploit to survive and proliferate in the bone; interactions with nerves, vasculature, and marrow cells (stromal and hematopoietic) should be taken into account for a comprehensive view of the breast cancer bone metastasis pathogenesis.

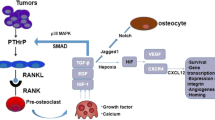
Breast cancer (BrCa) bone metastasis formation. In bone, BrCa cells secrete factors (purple arrows) that alter osteoblast differentiation and activity and increase RANKL release enhancing osteoclast formation. In addition, BrCa cells directly promote osteoclast differentiation and activity (purple arrows). Mature osteoclasts resorb bone, which releases bone-embedded growth factors in the surrounding micro-environment (red arrow). In addition, osteoclasts secret miRNAs/EVs promoting tumor growth. Osteoblasts and osteocytes finally release factors (blue arrows) feeding the tumor
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Kohler BA, et al. Annual report to the nation on the status of Cancer, 1975-2011, featuring incidence of breast Cancer subtypes by race/ethnicity, poverty, and state. J Natl Cancer Inst. 2015;107(6):djv048.
•• Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66(4):271–89. This paper presents a summary of epidemiology and statistics about cancer occurrence and success treatment rate.
Forman D, Bray F, Brewster DH, Gombe Mbalawa C, et al. Cancer incidence in five continents, vol. X. vol. 164. IARC Sci Publ; 2014.
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86.
• Raphael J, Verma S. Overall survival (OS) endpoint: an incomplete evaluation of metastatic breast cancer (MBC) treatment outcome. Breast Cancer Res Treat. 2015;150(3):473–8. This works provides an overview on pathogenetic and therapeutic aspects for breast cancer metastases.
Williams F, Jeanetta S, James AS. Geographical location and stage of breast Cancer diagnosis: a systematic review of the literature. J Health Care Poor Underserved. 2016;27(3):1357–83.
Brook N, Brook E, Dharmarajan A, Dass CR, Chan A. Breast cancer bone metastases: pathogenesis and therapeutic targets. Int J Biochem Cell Biol. 2018;96:63–78.
van der Pol CB, et al. Breast cancer and bone metastases: the association of axial skeleton MRI findings with skeletal-related events and survival. Breast Cancer Res Treat. 2014;146(3):583–9.
Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8(2):98–101.
•• Cappariello A, Ponzetti M, Rucci N. The ‘soft’ side of the bone: unveiling its endocrine functions. Horm Mol Biol Clin Investig. 2016;28(1):5–20. This paper highlights the endocrine role of skeleton and its function beyond mechanical properties.
Capulli M, Paone R, Rucci N. Osteoblast and osteocyte: games without frontiers. Arch Biochem Biophys. 2014;561:3–12.
Cappariello A, Maurizi A, Veeriah V, Teti A. Reprint of: the great beauty of the osteoclast. Arch Biochem Biophys. 2014;561:13–21.
• Coutu DL, Kokkaliaris KD, Kunz L, Schroeder T. Three-dimensional map of nonhematopoietic bone and bone-marrow cells and molecules. Nat Biotechnol. 2017;35(12):1202–10. This paper describes the spatial distribution of cellular and extracellular components of bone marrows other than hematopoietic lineage.
Sanders JL, Chattopadhyay N, Kifor O, Yamaguchi T, Butters RR, Brown EM. Extracellular calcium-sensing receptor expression and its potential role in regulating parathyroid hormone-related peptide secretion in human breast Cancer cell lines 1. Endocrinology. 2000;141(12):4357–64.
Buijs JT, Stayrook KR, Guise TA. The role of TGF-β in bone metastasis: novel therapeutic perspectives. Bonekey Rep. 2012;1:96.
Dunn LK, Mohammad KS, Fournier PGJ, McKenna CR, Davis HW, Niewolna M, et al. Hypoxia and TGF-β drive breast Cancer bone metastases through parallel signaling pathways in tumor cells and the bone microenvironment. PLoS One. 2009;4(9):e6896.
Sugatani T, Vacher J, Hruska KA. A microRNA expression signature of osteoclastogenesis. Blood. 2011;117(13):3648–57.
Ell B, Mercatali L, Ibrahim T, Campbell N, Schwarzenbach H, Pantel K, et al. Tumor-induced osteoclast miRNA changes as regulators and biomarkers of osteolytic bone metastasis. Cancer Cell. 2013;24(4):542–56.
Lee DY, Deng Z, Wang C-H, Yang BB. MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. Proc Natl Acad Sci. 2007;104(51):20350–5.
Si M-L, Zhu S, Wu H, Lu Z, Wu F, Mo Y-Y. miR-21-mediated tumor growth. Oncogene. 2007;26(19):2799–803.
Pollari S, Leivonen S-K, Perälä M, Fey V, Käkönen S-M, Kallioniemi O. Identification of MicroRNAs inhibiting TGF-β-induced IL-11 production in bone metastatic breast Cancer cells. PLoS One. 2012;7(5):e37361.
Zhu S, Si M-L, Wu H, Mo Y-Y. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1). J Biol Chem. 2007;282(19):14328–36.
Frankel LB, Christoffersen NR, Jacobsen A, Lindow M, Krogh A, Lund AH. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem. 2008;283(2):1026–33.
•• Clément-Demange L, Clézardin P. Emerging therapies in bone metastasis. Curr Opin Pharmacol. 2015;22:79–86. This paper gives an overview on new and potential therapeutical strategies for bone metastases.
Atkins GJ, Kostakis P, Pan B, Farrugia A, Gronthos S, Evdokiou A, et al. RANKL expression is related to the differentiation state of human osteoblasts. J Bone Miner Res. 2003;18(6):1088–98.
Galli C, Fu Q, Wang WF, Olsen BR, Manolagas SC, Jilka RL, et al. Commitment to the osteoblast lineage is not required for RANKL gene expression. J Biol Chem. 2009;284(19):12654–62.
Kozlow W, Guise TA. Breast Cancer metastasis to bone: mechanisms of Osteolysis and implications for therapy. J Mammary Gland Biol Neoplasia. 2005;10(2):169–80.
•• Rao S, Cronin SJF, Sigl V, Penninger JM. RANKL and RANK: from mammalian physiology to Cancer treatment. Trends Cell Biol. 2018;28(3):213–23. This work describes the importance and the role of RANK/RANKL axis in the organogenesis and the involvement in cancer progression.
Schuster C, Mo H, Shen CL, Gollahon L (2017) RANK/RANKL/OPG: The Axis of Breast Cancer Bone Metastasis Evil? Ann Breast Cancer Res 2(1): 1008
Nolan E, et al. RANK ligand as a potential target for breast cancer prevention in BRCA1-mutation carriers. Nat Med. 2016;22(8):933–9. This paper highlights the importance of RANKL in breast cancer onset and the potential therapeutics advantages of a target strategy.
• Vallet S, Bashari MH, Fan FJ, Malvestiti S, Schneeweiss A, Wuchter P, et al. Pre-osteoblasts stimulate migration of breast Cancer cells via the HGF/MET pathway. PLoS One. 2016;11(3):e0150507. This paper describes how important are the stromal-osteoblast cell in the distribution and migration of breast cancer cells during dissemination and metastasization processes.
Ishimi Y, et al. IL-6 is produced by osteoblasts and induces bone resorption. J Immunol. 1990;145(10):3297–303.
• Costa D, Principi E, Lazzarini E, Descalzi F, Cancedda R, Castagnola P, et al. LCN2 overexpression in bone enhances the hematopoietic compartment via modulation of the bone marrow microenvironment. J Cell Physiol. 2017;232(11):3077–87. This paper describes the effect of the proinflammatory cytokine lipocalin 2 in the context of vicious cycle of bone.
Yang J, Bielenberg DR, Rodig SJ, Doiron R, Clifton MC, Kung AL, et al. Lipocalin 2 promotes breast cancer progression. Proc Natl Acad Sci. 2009;106(10):3913–8.
Ören B, Urosevic J, Mertens C, Mora J, Guiu M, Gomis RR, et al. Tumour stroma-derived lipocalin-2 promotes breast cancer metastasis. J Pathol. 2016;239(3):274–85.
Chaudhary LR, Spelsberg TC, Riggs BL. Production of various cytokines by normal human osteoblast-like cells in response to interleukin-1 beta and tumor necrosis factor-alpha: lack of regulation by 17 beta-estradiol. Endocrinology. 1992;130(5):2528–34.
Abuna RPF, De Oliveira FS, Santos TDS, Guerra TR, Rosa AL, Beloti MM. Participation of TNF-α in inhibitory effects of adipocytes on osteoblast differentiation. J Cell Physiol. 2016;231(1):204–14.
Passeri G, Girasole G, Manolagas SC, Jilka RL. Endogenous production of tumor necrosis factor by primary cultures of murine calvarial cells: influence on IL-6 production and osteoclast development. Bone Miner. 1994;24(2):109–26.
Azuma Y, Kaji K, Katogi R, Takeshita S, Kudo A. Tumor necrosis factor-alpha induces differentiation of and bone resorption by osteoclasts. J Biol Chem. 2000;275(7):4858–64.
Liu F, Malaval L, Aubin JE. The mature osteoblast phenotype is characterized by extensive plasticity. Exp Cell Res. 1997;232(1):97–105.
Wildemann B, Kadow-Romacker A, Haas NP, Schmidmaier G. Quantification of various growth factors in different demineralized bone matrix preparations. J Biomed Mater Res Part A. 2007;81A(2):437–42.
Rodrigues LR, Teixeira JA, Schmitt FL, Paulsson M, Lindmark-Mänsson H. The role of osteopontin in tumor progression and metastasis in breast cancer. Cancer Epidemiol Biomark Prev. 2007;16(6):1087–97.
Zhang J-H, Tang J, Wang J, Ma W, Zheng W, Yoneda T, et al. Over-expression of bone sialoprotein enhances bone metastasis of human breast cancer cells in a mouse model. Int J Oncol. 2003;23(4):1043–8.
Bengtsson E, Mörgelin M, Sasaki T, Timpl R, Heinegård D, Aspberg A. The leucine-rich repeat protein PRELP binds perlecan and collagens and may function as a basement membrane anchor. J Biol Chem. 2002;277(17):15061–8.
Haglund L, Tillgren V, Önnerfjord P, Heinegård D. The C-terminal peptide of Chondroadherin modulates cellular activity by selectively binding to Heparan sulfate chains. J Biol Chem. 2013;288(2):995–1008.
Rucci N, Capulli M, Ventura L, Angelucci A, Peruzzi B, Tillgren V, et al. Proline/arginine-rich end leucine-rich repeat protein N-terminus is a novel osteoclast antagonist that counteracts bone loss. J Bone Miner Res. 2013;28(9):1912–24.
Rucci N, Capulli M, Olstad OK, Önnerfjord P, Tillgren V, Gautvik KM, et al. The α2β1 binding domain of chondroadherin inhibits breast cancer-induced bone metastases and impairs primary tumour growth: a preclinical study. Cancer Lett. 2015;358(1):67–75.
• Matic I, Matthews BG, Wang X, Dyment NA, Worthley DL, Rowe DW, et al. Quiescent bone lining cells are a major source of osteoblasts during adulthood. Stem Cells. 2016;34(12):2930–42. This paper describes the differentiation process of osteoblasts in adulthood from other source than mesenchymal stem cells.
Zhang C, Tang W, Li Y. Matrix metalloproteinase 13 (MMP13) is a direct target of osteoblast-specific transcription factor Osterix (Osx) in osteoblasts. PLoS One. 2012;7(11):e50525.
Pivetta E, Scapolan M, Pecolo M, Wassermann B, Abu-Rumeileh I, Balestreri L, et al. MMP-13 stimulates osteoclast differentiation and activation in tumour breast bone metastases. Breast Cancer Res. 2011;13(5):R105.
Hock JM, Krishnan V, Onyia JE, Bidwell JP, Milas J, Stanislaus D. Osteoblast apoptosis and bone turnover. J Bone Miner Res. 2001;16(6):975–84.
Koelink PJ, Sier CFM, Hommes DW, Lamers CBHW, Verspaget HW. Clinical significance of stromal apoptosis in colorectal cancer. Br J Cancer. 2009;101(5):765–73.
Wang R-A, Li QL, Li ZS, Zheng PJ, Zhang HZ, Huang XF, et al. Apoptosis drives cancer cells proliferate and metastasize. J Cell Mol Med. 2013;17(1):205–11.
• Xiong J, Piemontese M, Onal M, Campbell J, Goellner JJ, Dusevich V, et al. Osteocytes, not osteoblasts or lining cells, are the main source of the RANKL required for osteoclast formation in remodeling bone. PLoS One. 2015;10(9):e0138189. This paper highlights the importance of osteocytes in controlling osteoclast differentiation and formation through production of RANKL.
Nakashima T, Hayashi M, Fukunaga T, Kurata K, Oh-hora M, Feng JQ, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17(10):1231–4.
• Lee K, Nam K, Oh S, Lim J, Kim YP, Lee JW, et al. Extracellular matrix protein 1 regulates cell proliferation and trastuzumab resistance through activation of epidermal growth factor signaling. Breast Cancer Res. 2014;16(6):479. This paper showed one important aspect of chemioresistance in cancer cells.
Bucciarelli E, Sidoni A, Bellezza G, Cavaliere A, Brachelente G, Costa G, et al. Low dentin matrix protein 1 expression correlates with skeletal metastases development in breast cancer patients and enhances cell migratory capacity in vitro. Breast Cancer Res Treat. 2007;105(1):95–104.
Suva LJ, Washam C, Nicholas RW, Griffin RJ. Bone metastasis: mechanisms and therapeutic opportunities. Nat Rev Endocrinol. 2011;7(4):208–18.
Weilbaecher KN, Guise TA, McCauley LK. Cancer to bone: a fatal attraction. Nat Rev Cancer. 2011;11(6):411–25.
Martin TJ. Manipulating the environment of cancer cells in bone: a novel therapeutic approach. J Clin Invest. 2002;110(10):1399–401.
Takagaki K, et al. Parathyroid hormone-related protein expression, in combination with nodal status, predicts bone metastasis and prognosis of breast cancer patients. Exp Ther Med. 2012;3(6):963–8.
Lu X, Mu E, Wei Y, Riethdorf S, Yang Q, Yuan M, et al. VCAM-1 promotes osteolytic expansion of indolent bone micrometastasis of breast cancer by engaging α4β1-positive osteoclast progenitors. Cancer Cell. 2011;20(6):701–14.
• Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular vesicles in Cancer: cell-to-cell mediators of metastasis. Cancer Cell. 2016;30(6):836–48. This paper describes the crosstalk of cancer cells based on extracellular vesicles as source of biological messagges in tumor microenvironment.
Lee K, Park H, Lim EH, Lee KW. Exosomes from breast cancer cells can convert adipose tissue-derived mesenchymal stem cells into myofibroblast-like cells. Int J Oncol. 2011;40(1):130–8.
•• Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527(7578):329–35. This work shows the involvment of extracellular vesicles and their pivotal role in determining the organotropism in the dissemination of a primary tumour.
Luga V, Zhang L, Viloria-Petit AM, Ogunjimi AA, Inanlou MR, Chiu E, et al. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast Cancer cell migration. Cell. 2012;151(7):1542–56.
Deng L, Wang Y, Peng Y, Wu Y, Ding Y, Jiang Y, et al. Osteoblast-derived microvesicles: a novel mechanism for communication between osteoblasts and osteoclasts. Bone. 2015;79:37–42.
•• Cappariello A, Loftus A, Muraca M, Maurizi A, Rucci N, Teti A. Osteoblast-derived extracellular vesicles are biological tools for the delivery of active molecules to bone. J Bone Miner Res. 2018;33(3):517–33. This paper shows the involvement of extracellular vesicle in the context of bone physiology between osteoblasts and osteoclasts.
• Harbeck N, Gnant M. Breast cancer. Lancet. 2017;389(10074):1134–50. This paper provides an overview on biological basis of breast cancer and potential therapeutical approaches.
•• McDonald ES, Clark AS, Tchou J, Zhang P, Freedman GM. Clinical diagnosis and Management of Breast Cancer. J Nucl Med. 2016;57(Suppl 1, no. Supplement 1):9S–16S. This work makes a summary on the main diagnostic and therapeutical strategies to treat breast cancer.
• Francis PA, Regan MM, Fleming GF, Láng I, Ciruelos E, Bellet M, et al. Adjuvant ovarian suppression in premenopausal breast Cancer. N Engl J Med. 2015;372(5):436–46. This paper reports the impact of estrogen-bloking chemioadjuvant on premenopausal breast cancer patients.
International Breast Cancer Study Group, et al. Tamoxifen after adjuvant chemotherapy for premenopausal women with lymph node-positive breast cancer: international breast Cancer study group trial 13-93. J Clin Oncol. 2006;24(9):1332–41.
Zallone A. Direct and indirect estrogen actions on osteoblasts and osteoclasts. Ann N Y Acad Sci. 2006;1068(1):173–9.
Klibanski, A., Adams-Campbell, L., Bassford, T., Blair, S. N., Boden, et al. Osteoporosis prevention, diagnosis, and therapy. JAMA, 2001;285(6), 785-795
Zhang Y, Kiel DP, Kreger BE, Cupples LA, Ellison RC, Dorgan JF, et al. Bone mass and the risk of breast Cancer among postmenopausal women. N Engl J Med. 1997;336(9):611–7.
Powles TJ, Diem SJ, Fabian CJ, Neven P, Wickerham DL, Cox DA, et al. Breast cancer incidence in postmenopausal women with osteoporosis or low bone mass using arzoxifene. Breast Cancer Res Treat. 2012;134(1):299–306.
von Minckwitz G, Raab G, Caputo A, Schütte M, Hilfrich J, Blohmer JU, et al. Doxorubicin with cyclophosphamide followed by docetaxel every 21 days compared with doxorubicin and docetaxel every 14 days as preoperative treatment in operable breast cancer: the GEPARDUO study of the German breast group. J Clin Oncol. 2005;23(12):2676–85.
•• Cortazar P, Zhang L, Untch M, Mehta K, Costantino JP, Wolmark N, et al. Pathological complete response and long-term clinical benefit in breast cancer: the CTNeoBC pooled analysis. Lancet. 2014;384(9938):164–72. This paper reports the success rate and the beneficial effect on quality of life for breast cancer patients.
• Anampa J, Makower D, Sparano JA. Progress in adjuvant chemotherapy for breast cancer: an overview. BMC Med. 2015;13(1):195. This paper comment on the current precedures and pharmacological treatment in managing of brest cancer.
Takahashi M, Mizoguchi, T., Uehara, S. et al. Docetaxel inhibits bone resorption through suppression of osteoclast formation and function in different manners. J Bone Miner Metab (2009) 27(1):24–35
Wong M, N. P.-B. C. T. and Therapy, and undefined. Optimal management of bone metastases in breast cancer patients. 2011. ncbi.nlm.nih.gov.
Jadvar H, Quinn DI. Targeted α-particle therapy of bone metastases in prostate Cancer. Clin Nucl Med. 2013;38(12):1.
Takalkar A, Adams S, Subbiah V. Radium-223 dichloride bone-targeted alpha particle therapy for hormone-refractory breast cancer metastatic to bone. Exp Hematol Oncol. 2014;3(1):23.
Nilsson S, Franzén L, Parker C, Tyrrell C, Blom R, Tennvall J, et al. Bone-targeted radium-223 in symptomatic, hormone-refractory prostate cancer: a randomised, multicentre, placebo-controlled phase II study. Lancet Oncol. 2007;8(7):587–94.
Russell RGG. Bisphosphonates: the first 40years. Bone. 2011;49(1):2–19.
Mundy GR, Yoneda T. Bisphosphonates as anticancer drugs. N Engl J Med. 1998;339(6):398–400.
Santini D, Fratto ME, Galluzzo S, Vincenzi B, Tonini G. Are bisphosphonates the suitable anticancer drugs for the elderly? Crit Rev Oncol. 2009;69:83–94.
Coleman R, de Boer R, Eidtmann H, Llombart A, Davidson N, Neven P, et al. Zoledronic acid (zoledronate) for postmenopausal women with early breast cancer receiving adjuvant letrozole (ZO-FAST study): final 60-month results. Ann Oncol. 2013;24(2):398–405.
Bekker PJ, Holloway DL, Rasmussen AS, Murphy R, Martin SW, Leese PT, et al. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J Bone Miner Res. 2004;19(7):1059–66.
Martin M, Bell R, Bourgeois H, Brufsky A, Diel I, Eniu A, et al. Bone-related complications and quality of life in advanced breast cancer: results from a randomized phase III trial of Denosumab versus Zoledronic acid. Clin Cancer Res. 2012;18(17):4841–9.
Rosen LS, et al. Long-term efficacy and safety of zoledronic acid compared with pamidronate disodium in the treatment of skeletal complications in patients with advanced multiple myeloma or breast carcinoma: a randomized, double-blind, multicenter, comparative trial. Cancer. 2003;98(8):1735–44.
Bonomi M, Nortilli R, Molino A, Sava T, Santo A, Caldara A, et al. Renal toxicity and osteonecrosis of the jaw in cancer patients treated with bisphosphonates: a long-term retrospective analysis. Med Oncol. 2010;27(2):224–9.
Migliorati CA, et al. A systematic review of bisphosphonate osteonecrosis (BON) in cancer. Support Care Cancer. 2010;18(8):1099–106.
Bamias A, Kastritis E, Bamia C, Moulopoulos LA, Melakopoulos I, Bozas G, et al. Osteonecrosis of the jaw in Cancer after treatment with bisphosphonates: incidence and risk factors. J Clin Oncol. 2005;23(34):8580–7.
Saad F, et al. Incidence, risk factors, and outcomes of osteonecrosis of the jaw: integrated analysis from three blinded active-controlled phase III trials in cancer patients with bone metastases. Ann Oncol Off J Eur Soc Med Oncol. 2012;23(5):1341–7.
Roodman GD. Pathophysiology of bone metastases. Dordrecht: Springer; 2009. p. 31–50.
Yin JJ, Selander K, Chirgwin JM, Dallas M, Grubbs BG, Wieser R, et al. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J Clin Invest. 1999;103(2):197–206.
Lewiecki EM. Odanacatib, a cathepsin K inhibitor for the treatment of osteoporosis and other skeletal disorders associated with excessive bone remodeling. IDrugs. 2009;12(12):799–809.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Alfredo Cappariello reports having patent 16745158.1-1112 pending; Mattia Capulli declares no conflicts of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Molecular Biology of Bone Metastasis
Rights and permissions
About this article

Cite this article
Cappariello, A., Capulli, M. The Vicious Cycle of Breast Cancer-Induced Bone Metastases, a Complex Biological and Therapeutic Target. Curr Mol Bio Rep 4, 123–131 (2018). https://doi.org/10.1007/s40610-018-0099-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40610-018-0099-5