
Abstract
Purpose of Review
Increase in neutrophil counts that are associated with cardiovascular risk factors including myocardial infarction (MI) and early development of heart attack is a major concern among all age groups in recent years. Neutrophil production in response to heart failure is mediated by various ways including recruitment of immune cells at the site of injury, release inflammatory molecules, and continuous supply of leucocytes.
Recent Findings
Recent evidences proved the importance of neutrophil-derived alarmin molecule S100A8/A9 in provoking inflammation after MI. Besides, clinical trials with increases in serum level of circulating calgranulins and major adverse cardiac events (MACE) in MI subjects were demonstrated which implicated that targeting neutrophils or their inflammatory alarmins could be effective in reducing heart attack. On the other hand, neutrophils also found to involved in the regression of inflammation and atherosclerotic plaques.
Summary
In this review, we discuss the overview of S100 protein family in various metabolic diseases with a main focus on cardiac inflammation. We also discuss the recent evidences on neutrophil-derived S100A/A9 which triggers inflammation and improves cardiac function after MI. We also insist that neutrophils could be a better biomarker and therapeutic targets for various inflammatory diseases including cardiovascular disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Increasing incidence of cardiovascular risk factors and acute coronary syndrome (ACS) is considered as one of leading cause of death in recent years. Rapid advancement in early diagnosis, predicting early and late development of heart failure and myocardial infarction (MI), has not only greatly reduced the death rate, but also for those subjects who are at high risk of developed heart injury and requires future heart transplants [1, 2]. Heart attack followed by MI is mainly determined by cardiomyocyte death that triggers a reparative response, scar formation, dilated remodeling of the left ventricle, and elicited inflammatory response. Cardiac repair with initial recruitment of neutrophils and monocytes/macrophages however prolonged increase in immune response which can also cause adverse ventricular remodeling and heart attack [3, 4]. Moreover, various research groups have insisted that the recurrence of ACS could be prevented by attenuating inflammation; however, indiscriminate targeting of particular inflammatory pathway remains unsuccessful. On the contrary, American College of Cardiology Foundation (ACCF)/American Heart Association (AHA) Task Force on practice guidelines reported that use of non-steroidal anti-inflammatory drug and the selective cyclooxygenase II enzyme (COX-2) inhibitors is also not recommended in acute phase of MI [5]. Additionally, suppressing the soluble signaling molecules like IL-1β, macrophage migration inhibitory factor (MIF), and NLRP3 dampens inflammatory leucocyte production thereby reducing the incidence of cardiovascular events after MI [6,7,8,9]. Quite recently, investigations by Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) and Colchicine Cardiovascular Outcomes Trail (COLCOT) showed reduction of heart failure due to diminishing inflammatory molecules [10, 11]. In close contrast, neutrophil count also attributed as biomarker for inflammation and associated clinical illness including ACS and escalated neutrophil count is interrelated with size of the infarct and reduction in left ventricular ejection fraction (LVEF). Different strategies aimed at reducing the inflammation by suppressing the neutrophil recruitment and activation have been reported in regression of heart injury; however, it is less effective in terms of clinical therapies. Lately, neutrophil-derived alarmins—S100A8/A9—have significant role in not only dictating the inflammatory response after MI, but also involved in the resolution of inflammation. It is necessary for ameliorative response and considered as main signaling molecules that can manage further complication after MI. Therefore, it is imperative to recognize the prime role of S100A8/A9 released from neutrophils, its kinetics, turnover, and signaling mechanisms that orchestrate divergent function, for future development of novel therapeutics. This review will provide a comprehensive and detailed overview into the multitude of S100A8/A9 and its role in inflammation, myelopoiesis, expression, and regulation of S100A8/A9 molecule in other metabolic diseases. Pharmacological strategies aimed at targeting S100A8/A9 would be potential candidate for therapeutic applications.
S100 Calcium-Binding Proteins (S100A8/A9)
S100A8 and S100A9 are commonly known as myeloid-related proteins MRP-8 and MRP14 that belong to S100 family, mainly expressed in cells of myeloid lineage, also called as calgranulin A and calgranulin B, or calprotectin due to their Ca2+ binding efficiency and their antimicrobial properties [12, 13]. Human S100A8/A9 found to have 93/113 amino acids forming a complex with molecular weight of 13.2 kDa/12.7 kDa and genes encoding these molecules are located as cluster of chromosomes 1q21. It may exist as homodimers, heterodimers, and tetramers, either intracellularly or extracellularly depending on the biological function of the protein (Table 1). Secreted S100 proteins can also function as damage-associated molecular patterns (DAMPs) to transmit signals through a variety of cell surface receptors. This binding will activate signaling pathways that influence cytokine/ROS generation, cell cycle, death, adhesion molecule expression, cytoskeletal remodeling, and migration. During the early reperfusion stage of myocardial ischemia-reperfusion (MI/R) injury, S100A8/A9 is significantly upregulated [14]. Also following a myocardial infarction, S100A8 and S100A9 are mostly released by activated myeloid cells (neutrophils and monocytes). Toll-like receptor-4 (TLR4) and receptor of advanced glycation end products (RAGEs) are activated by these factors to activate signaling cascades which in turn will lead to the recruitment of inflammatory cells at the site of injury. Elevated circulation of S100A8/A9 causes severe effects including myocarditis, acute myocardial infarction, thrombosis, peripheral arterial disease, and early development of heart failure [15, 16]. In addition, S100A8/A9 has been found to modulate the activity of vascular smooth muscle cells and endothelial cells in a paracrine fashion. Due to its pivotal role in cardiovascular biology, S100A8/A9 is a promising target for cardiovascular-related diseases such as atherosclerosis and hypertension (Table 2).
Physiological Regulation of Neutrophil Production
Neutrophils are among the first innate immune cells found to reach at the site of inflammation, where there is any inflammatory response elicited. The production of more neutrophils is termed as granulopoiesis, characterized by significant increase in neutrophil production by the de novo pathway, faster cellular turnover, and the discharge of immature and mature neutrophils into the bone marrow (BM) reserve, the post-mitotic pool [17]. During a localized bacterial infection, only a small percentage of the entire BM neutrophil reserve is discharged into circulation, which is known as “steady-state granulopoiesis.” In systemic bacterial infection, the neutrophils are consumed in large quantities. In order to counteract the neutrophil diminution after the hindrance of the first-line defense mechanisms, and to deliver the urgently needed neutrophils to battle systemic bacterial spread, a hematopoietic response gets initiated termed as “emergency granulopoiesis” which is robust de novo production of neutrophils from myeloid progenitors in BM. But, there will be depletion of BM lymphopoiesis with increase in BM granulopoiesis. Granulocyte colony-stimulating factor (G-CSF) is the major cytokine that mediates granulopoiesis. Both immature and fully developed neutrophils respond favorably to G-CSF mediated through the G-CSF receptor present in precursor cells of BM, which promotes their survival, proliferation, differentiation, and activation. G-CSF receptors are maintained at high concentrations on neutrophil cell surfaces from an early stage of development [18, 19].
In addition, mature neutrophils exhibit low amounts of CXC chemokine receptor 4 (CXCR4), a G-protein coupled receptor on their cell surface. IL-3, granulocyte-macrophage colony-stimulating factor (GM-CSF), and IL-6 are a few more hematological cytokines that drive granulopoiesis in vivo [20]. Neutrophil production from the BM can also be stimulated by a wide variety of substances, such as cytokines, chemokines, leukotrienes, bacterial products, and other inflammatory mediators [21, 22]. In the case of emergency granulopoiesis, there are two different strategies involved immediately to kick off the process in particular phase of an infection—direct activation: sensing by hematopoietic multipotential stem and progenitor cells (HSPCs) and indirect activation: sensing by mature hematopoietic and non-hematopoietic cells. In both circumstances, pathogen sensing via pattern recognition receptors (PRRs) like TLRs is required to switch from steady state to emergency granulopoiesis, which is then followed by molecular translation and the commencement of emergency granulopoiesis [17]. In close contrast, neutrophils reside in two compartments—the marginal compartment (those neutrophils attached to the endothelium of the blood vessel) and the circulating compartment (those circulating in the blood vessels along with other cells). These two pools are in dynamic balance, but harmless stimuli such as exercise, physiological stress, or a surge in glucocorticoids could disrupt this equilibrium [23]. Though it is well known that glucocorticoids enhance the concentration of neutrophils in the circulatory compartment, raising the WBC count, demargination is also one of the processes implicated in their anti-inflammatory characteristics. However, the exact mechanisms which induce demargination remain unclear, but found ectodomain shedding of L-selectin (CD62L) on the surface of neutrophils by Adam 17, a disintegrin and cysteine metalloproteinase [24,25,26]. Moreover, the post-mitotic transit time in humans is estimated to be around about 4–6 days; having completed their development in the BM, neutrophils are then retained until they are released into the bloodstream. Within inflamed tissue, neutrophils either die off through apoptosis/necrosis/NETosis are eliminated from the body after migrating across the epithelium [21]. A major characteristic of the immune system is the concurrent existence of pro-inflammatory and anti-inflammatory processes by recruiting the cells through positive and negative feedback cascades, thereby maintaining the homeostasis. Neutrophil homeostasis involves more than regulation of extravasation, also includes tight control of granulopoiesis, the release of mature neutrophils from the bone marrow into the circulation, and clearance of apoptotic or senescent neutrophils [23].
Myelopoiesis and Granulopoiesis After Myocardial Infarction: Role of S100A8/A9
In response to myocardial infarction, neutrophil count raises high in blood and also recruited at the site of infarct within few hours, but the exact mechanism which influences the increase in circulation neutrophils after MI remains scary. Earlier studies have reported that acute mobilization of neutrophils from BM sinusoidal endothelium is mediated by disrupting the CXCR4-SDF-1 axis and CXC chemokines. On the other hand, activation of the sympathetic nervous system (SNS) or an increase in norepinephrine (NE) levels following MI could also trigger neutrophilia production via CXCR4-SDF1 disruption, indicating that stress plays a major role in the myocardial infarction-induced granulopoiesis. These SNS signals together with the local signals from the infarction provoke the release of hematopoietic stem cell (HSCs) from the BM, which promotes extramedullary hematopoiesis either in the spleen or in the liver. Thus, in response to MI, both medullary and extramedullary granulopoiesis may contribute to the overall circulatory pool of neutrophils [6, 16, 27].
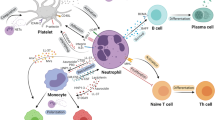
Quite recently, soluble factors (e.g., IL-1β, S100A8/A9) from necrotic cells appear in the myocardium within hours after an ischemic injury and can stimulate granulopoiesis by interacting with their specific receptors on myeloid cells in the BM. These soluble factors are mostly derived from leukocytes, of which neutrophils were found to be the most predominant one. Most pioneering studies have shown the increased expression of various chemokines like CxCl1, Cxcl2, Ccl3, Cxcl5, and CCL2, inflammatory cytokine line tumor necrosis factor alpha (TNF-α) and IL-6, along with alarming molecules like S100A8 and S100A9, after ischemic injury. Recently, findings from cell sorting methods with qRT-PCR and confocal imaging revealed that neutrophils are the primary source of S100A8/A9 in the infarct (Fig. 1) During myocardial infarction (MI), neutrophils are recruited at the site of ischemic heart where they undergo NETosis and release the alarming molecules S100A8/A9. These released calgranulins bind specifically to TLR4 receptor on naïve neutrophils and activated the inflammasome pathway and release IL-1β, where it binds to IL1R on myeloid cells in BM and thus granulopoiesis get stimulated. On the other hand, following exposure to SARS-CoV-2 infection induces aberrant neutrophil subsets, where S100A8/A9 get release from the neutrophils when interacts with TLR4 and there is an imbalance of immune response. Short-term treatment with paquinimod and resatorvid ameliorates MI and viral load by decreasing S100A8/A9-induced inflammation, which reduced aberrant neutrophil subsets [28].

Proposed model showing how neutrophil-derived S100A8/A9 maintains inflammation, cardiac injury, SARS-CoV-2 infection, and regression of inflammation
Multifaceted Roles of S100A8/A9 in Cardiovascular Disease
Cardiac Inflammation and Vascular Inflammation
S100A8/A9 is a key regulator of cardiovascular homeostasis and inflammation. In response to ischemic injury, this S100A8/A9 get released from activated myeloid cells and binds to TLR4 and or RAGE receptors, thereby activating mitogen-activated kinase (MAPK) and NF pathways, resulting in recruitment of inflammatory cells, cytokine and chemokine production, and elevated extracellular matrix (ECM) proteins and proteases [29,30,31]. Overexpression of S100A8/A9 showed RAGE-dependent reduction in calcium flux and decrease in cardiomyocyte contractility [32]. Furthermore, S100A8/A9 involved in enhanced phagocyte NADPH oxidase activation by transferring the cofactor arachidonic acid to NADPH complex [33]. Being multifunctional protein, S100A8/A9 differentially modify the phenotypic states of different cell population either by intracellular or extracellular mechanisms (Table 1). Most growing evidence have proved that these calgranulin proteins are mandatory for neutrophil accumulation and recruitment of immune cells in inflamed tissue on multiple diseases including cardiac and vascular inflammation. Besides, robust increased expression of S100A8/A9 was noted in monocytes and activated macrophages in atherosclerotic lesions of humans and mice, and found to be associated with plaque instability and rupture (Table 2). It also regulates leukocyte adhesion on vascular inflammation, which reduced cytokine production via Mac-1 expression on S100a9−/− neutrophils. Intracellular expression of S100A8/A9 in non-myeloid cells, including endothelial cells and vascular smooth muscle cells (VSMCs), revealed impairment in membrane integrity and increased chemotactic factor expression. Alternatively, S100A8/A9 can function extracellularly by recruiting monocytes, neutrophil, and macrophages during inflammatory response and act as chemoattractant by activating and binding to receptors on leukocytes, trans-endothelial migration of phagocytes. Reorganization of microtubes is also controlled by modulating S100A8/A9 protein complex, where phosphorylation of S100A9 antagonist stabilizes microtubes, which subsequently activates small GTPases Rac1 and Cdc42 and thus transmigration of leukocytes is facilitated [34]. In close contrast, S100A8/A9 triggers the expression of vascular cell adhesion molecules like VCAM-1 and ICAM-1 on endothelial cells for leukocyte adhesion and rolling. Upon binding of S100A8/A9 on RAGE and TLR4 receptor, it aggravates multiple cytokine production that accelerates inflammation [35]. Increased serum level of these calgranulins can induce CD11b on circulating monocytes, activated macrophages, and dendritic cells (DCs) in diabetic patients. On the other side, during atherosclerotic progression, it can induce inflammatory leukocytes through differentiation of myeloid progenitor cells in the BM and increases the proliferation of inflammatory of Ly6Chi monocytes and neutrophils [36, 37].
Crosstalk Between S100A8/A9 and Atherosclerosis
S100A8/A9 released from circulating neutrophils found to hasten up atherogenesis via continuous recruitment of activated monocytes and neutrophils in the arterial wall and atherosclerotic lesions, and it has been extensively established in both humans and mice. Theranostic approach on targeting S100A8/A9 in vivo via synthetic gadolinium immuno nanoprobes revealed substantial delineation in plaque formation [38]. Proatherogenic effect and reduction in atherosclerotic plaque size and attenuated vascular inflammation were noted in TLR4, RAGE, and myeloid differentiation factor 88 (MyD88) KO mice, which implies that binding of S100A8/A9 over TLR4 and RAGE receptor meditated signaling. Seminal work proved in diabetic ApoE−/−-deficient mice when rendered with streptozotocin showed increased S100A8/A9 in blood plasma, elevated VCAM-1, MCP-1, and progression in atherosclerotic lesion [39, 40]. Several clinical studies also supported that S100A8/A9-mediated cardiovascular disease (CAD) associated with type 1 and type 2 diabetic patients; carotid intima-media thickness (IMT) with CAD; systemic lupus erythematosus (SLE); and other cardiovascular (CV) risk factors [41, 42]. In addition to this, smoking, hyperlipidemia, hyperglycemia, and obesity promote S100A8/A9 production by stimulating neutrophilia and monocytosis, either directly or indirectly. S100A8/A9 augments phagocyte production in bone marrow and expedites their recruitment into vascular wall through endothelial activation that elevates the expression and affinity of Mac-1. These effects are initially mediated by RAGE, and accelerated by hyperglycemia. S100A8/A9 binding to TLR4 triggers phagocyte activation and secretion of inflammatory cytokines, which induce phagocyte recruitment and atherogenesis in the vascular wall. In addition, S100A8/A9-mediated ROS production may contribute to vascular damage and plays a role in the pathogenesis of cardiovascular disease[28].
S100A8/A9 as a Biomarker
Recent days, biomarkers with their presence as primary endpoints in clinical trials have now become a common practice, indicating the medical state of a patient when observed from the outside. They can be defined as an objective and quantifiable characteristic of biological processes.
Biomarker for Various Inflammatory Diseases
A promising biomarker in early diagnosis, evaluation of prognosis and risk stratification were noted in increased S100A8/A9 level of septic shock group when compared with control group, systemic inflammatory response syndrome group. In addition to this, it is studied as a marker of cardiovascular risk and vascular inflammation in humans [12, 43, 44]. On the other side, clinical trials were also conducted to clarify whether serum S100A8/A9 can be a sensitive biomarker for inflammatory bowel disease (IBD). The blood samples were acquired with informed consent obtained from all patients following all ethical guidelines. Two monoclonal antibodies (mAbs: mAb2B9 and mAb3D2) specific for h-S100A8/A9 were used to establish an ELISA assay for the heterodimer. The results of the test showed that the serum S100A8/A9 acted as a more sensitive and specific biomarker for IBD than C-reactive protein (CRP) [45]. Neutrophil-derived S100A8/A9 proteins can be used as surrogate biomarkers for lung inflammation in active tuberculosis patients. Additionally, increased levels of S100A8/A9 proteins were associated with other inflammatory diseases such as rheumatoid arthritis (RA), cystic fibrosis, periodontitis, autoimmune synovitis, and autoimmune diseases [12].
During the period of COVID-19, rather than the usage of commonly used inflammatory biomarkers such as CRP and calcitonin (CT) which require de novo protein biosynthesis, the S100A8/A9 proteins were more appealing as biomarkers. This was due its kinetic advantage as being the first host response to inflammatory conditions. Also, S100A8/A9 has a documented half-life of 5 h and is also stable at room temperature, suggesting that calgranulins are a potential biomarker for various inflammatory diseases, including COVID-19 [46]. Potential role of S100A8/A9 was also investigated in platelet activation and granule release post severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Observations suggested that S100A8/A9 played a minimal role in SARS-CoV-2-induced platelet activation. However, S100A8/A9 may play an indirect role in thromboembolism, and aberrant fibrinolysis by inducing inflammation remains unknown [47]. Lately, transcriptomic analysis from rhesus macaques and mice model infected with SARS-CoV-2 revealed that S100A8/A9 mediated TLR4 signaling pathway, which imbalanced immune response and robust increase in a group of aberrant immune neutrophils, suggesting as therapeutic targets for SARS-CoV-2 and other viral infections (Fig. 1) [48]. More interestingly, S100A8/A9-GPIbα axis was identified as a novel mechanism that triggered the formation of procoagulant platelets, independent of platelet aggregation. Platelet activation and high levels of S100A8/A9 were observed in plasma and lung autopsies infected with COVID-19. It is speculated that calprotectin deposition on vasculature in COVID-19 patients contributes to the formation of fibrin-rich thrombi[49].
S100A8/A9 in Metabolic Inflammation
The role of S100A8/A9 in metabolic inflammation is of clinical importance, since they have been detected in sera and sites of major metabolic inflammatory diseases, diabetes, obesity, gout, and a number of other chronic inflammatory diseases. Remarkably, many of these chronic inflammatory diseases such as Alzheimer’s disease, rheumatoid arthritis (RA), and cancer are also found to be associated with metabolic dysfunction [21].
Cancer
S100 family proteins are widely and specifically used in the progression, diagnosis, and treatment of cancer. Sixteen chromosomes of all the encoding family proteins are located in a tight cluster on human chromosome 1q21 in a region which is prone to deletions and rearrangements that cause dysregulation of S100 gene expression. For an example, in the formation of p53-dependent erythroid differentiation defect which is characterized by apoptosis at the transition from polychromatic to orthochromatic erythroblasts which causes age-dependent progressive anaemia, megakaryocyte dysplasia and loss of HSC quiescence. Besides, it actively involved in myeloid cytoskeletal rearrangements, arachidonic acid metabolism, cell migration, recruitment, and in the regulation of neutrophilic NADPH oxidase [50].
S100A9 and S100A8 mRNAs and proteins are easily detectable in enriched bone marrow HPC, but they gradually decline during culture with GM-CSF and IL-4 and are almost undetectable by day 7, when > 70% of cells in culture were DCs. The increase in S100A8/A9 in hematopoietic progenitor cells (HPCs) is the major reason for defective myeloid differentiation in cancer. The accumulation of myeloid-derived suppressor cells (MDSCs) and inhibition of DC differentiation are major characteristics of defective myeloid. STAT3 increases S100A8/A9 production under pathological conditions which results in the hyperproduction of MDSCs and dampens DC differentiation. But under physiological conditions, S100A9 is not necessarily essential for myeloid cell differentiation. There is also a speculation that the downregulation of S100A9 expression must be done by the myeloid cells so that the DC progression and macrophage differentiation occur and result in MDSC accumulation. Since S100A9 has a major role in regulating ROS, it may also play a role in DC differentiation [51].
Even though the upregulation of S100A8/A9 is found in acute myeloid leukemia (AML), S100A8 is linked to poor prognosis of AML, where S100A9 activates TLR4, leading to phosphorylation of p38 MAPK, ERK1/2, and JNK, which in turn activates CREB, c-JUN, and NF-κB [52].
Autoimmune Diseases
Increased serum level of S100A8/A9 proteins was noted in autoimmune diseases as well, mainly in the synovial fluid of patients who are suffering with psoriatic arthritis. In case of psoriatic epidermis, release of S100A8/A9 induces C3/CFB complement activation, leading to immune system dysfunction, angiogenesis, and keratinocyte hyperproliferation causing progression of psoriasis. Increased cell surface expression of S100A8/A9 on CD16+ proinflammatory monocytes, myeloid DCs (mDCs), plasmacytoid DCs (pDCs), and polymorphonuclear neutrophils (PMNs) was also remarkably observed with disease severity in systemic lupus erythematosus (SLE) patients [12, 53, 54].
Obesity
Obesity is a chronic condition characterized by inflammatory macrophage infiltration into obese adipose tissue with low-grade inflammation. The S100 proteins are found to be expressed in adipocytes like adipose tissue macrophages (ATMs) in white adipose tissue (WAT) and act as candidate biomarkers for obesity. S100A8/A9 serum concertation strongly corelated with altered body mass index (BMI), high sensitivity C-reactive protein (hs-CRP), TNF-α, and neutrophil count in obese subjects [55]. S100A4 is involved in regression of WAT inflammation, adipogenesis, and decrease in inflammatory factors in the setting of obesity. It has been reported that release of S100A8/A9 from adipose tissue macrophages in visceral adipose tissue (VAT) in turn heightened the production of neutrophils and monocytes via ATM-TLR4/MyD88 and NLRP3 signaling cascade of IL-1β production [56].
Hyperglycemia
Increasing evidences have proved that elevated S100A8/A9 protein is associated with hyperglycemic condition with inflammation. In a study, diabetic mice with robust increase in monocytes and neutrophils were shown when mice induce with STZ. Interaction of these proteins from neutrophils with receptor for advanced glycation end-products (RAGEs) on common myeloid progenitor cells in the BM resulted in myelopoiesis. Subsequently, when hematopoietic progenitor cells (CD 34+) were stimulated with S100A8/A9, it showed expansion and proliferation of CD14+ monocytes in a dose-dependent manner, suggesting these proteins involved in recruitment of monocyte and neutrophils at the site of sterile inflammation. More interestingly, when glucose level is controlled with dapagliflozin and sodium co-transported 2 inhibitor (SGLT2i), it reduces circulating S100A8/A9 level and regression of atherosclerosis in diabetic mice models [37, 57].
Gout
S100A8/A9 is also implicated in the gout, where these proteins were detected in the synovial fluid (SF) and in blood plasma. An in vitro study proved that when neutrophils are incubated with monosodium urate (MSU) crystals, it triggers the release of S100A8/A9. Seminal studies showed recruitment of neutrophils and macrophages, MSU crystal-mediated inflammation, and IL-1β production in rodents [58, 59].
Neurodegenerative Diseases
Alzheimer’s disease is a chronic progressive neurological disorder characterized by neuron damage/atrophy due to the aggregation of intracellular neurofibrillary tangles and extracellular amyloid species. The S100A8/9 and other members of the S100 family are assisted in Alzheimer’s disease (AD) and Parkinson’s disease (PD). Calgranulins are upregulated in the brain of AD patients due to microglial activation, astrocyte, and aggregation of protein inclusions. S100A8/A9 triggers microglia resulting in increased TNF-α and interferon gamma (INF-γ) level, and also formation of amyloid plaques. It acts as a neuroprotective by scavenging Zn2+ in the brain. Within the perivascular region, lymphoid aggregates are formed in some tissues due the activation of immune response pathways lead by the imbalance of S100A8/A9 expression during aging [60, 61].
Anti-inflammatory Function of S100A8/A9
The S100 members of the family not only involved in inflammation, but also, this complex can exhibit anti-inflammatory properties which prevent tissue damage that might have been caused by overwhelming inflammation, which implicated having specific and particular role under certain conditions. The assignment of the S100 family is mediated with intracellular binding leading to oxidations, S-nitrosylation, and phosphorylation in post-translational modification, and their interlinks between the other members of the S100 family determines its function; these events play a vital role in mediating functionality of the S100 members with protective mechanisms in inflammation [62]. For example, the S100A8 plays a role in the negative regulation of leukocyte adhesion and the transmigration through reducing the p38 MAPK phosphorylation. And additionally, the S100A8 reduces antigen-presenting cells like dendritic cells by inhibiting the B7 expression and thus preventing the hyperactivation of the immune responses [12]. Neutrophils devoid of S100A9 reduced the expression of cytokine even after interacting with TLR4 receptors and involved in controlling leukocyte recruitment, deactivated the macrophages, and inhibited the secretion of IL-1β, IL-6, and TNF-α in cardiac inflammation. However, the complete molecular mechanisms remain enigmas.
In a rodent study, the calprotectin would diminish the inflammatory, redox, metabolic, or behavioral pattern changes in RSDS (repeated social defeat stress) in S100A9-deficient mice, which would lack functional calprotectin, but in fact, the loss of S100A9 exacerbated the circulation and T-lymphocyte inflammation and worsening the problem by altering specific behavioral patterns of RSDS, suggesting the protective role in inflammation [63].
Orally active chemical compounds like tasquinimod and quinoline-3-carboxanides with potent anti-inflammatory properties were studied in different models with autoimmune diseases like SLE, collagen arthritis, autoimmune and encephalomyelitis [12], and the S100A9 is an intricated target of quinoline-3-carboxamindes. Quinoline-3-carboxamindes and ABR-215757 bind both human and miceS100A8 and S100A9 in a calcium and zinc-dependent manner, and the interaction with TLR4 and RAGE is blocked. Besides, endotoxin-stimulated cardiomyocytes showed reduced RAGE-dependent signals that mediated contractile dysfunction but, in the healing, infract, systolic function of the protective effects of the S100A9 might involve the direct protective effects on the cytokine-stimulated cardiomyocytes that might reduce contractile dysfunction [64].
Summary and Conclusion
In summary, neutrophil-derived alarming molecules could be a better target in clinical settings that are linked with ACS. Since these molecules integrated in activating both inflammatory and acute MI, diminishing S100A8/A9 at the precise phase would be a simple strategy to improve cardiac function. But, it is impossible to say the exact therapeutic window phase, since patients with ACS will have multiple inflammatory responses with varying degrees of injuries and other risk factors. On top of that, there are no sensitive diagnostic markers to identify the exact phase of both pro-inflammatory and anti-inflammatory reactions. We propose that continuous monitoring of the serum level of S100A8/9 and blockade of these calgranulins during acute phase of inflammation might not only be beneficial in the setting of heart failure, but also in other metabolic diseases.
Data Availability
Not applicable.
References
Lewis EF, Moye LA, Rouleau JL, Sacks FM, Arnold JMO, Warnica JW, et al. Predictors of late development of heart failure in stable survivors of myocardial infarction: the CARE study. J Am Coll Cardiol. 2003;42(8):1446–53.
Melendo-Viu M, Abu-Assi E, Manzano-Fernández S, Flores-Blanco PJ, Cambronero-Sánchez F, Pérez DD, et al. Incidence, prognosis and predictors of heart failure after acute myocardial infarction. REC: CardioClinics. 2020;55(1):8–14.
Frangogiannis NG. The immune system and cardiac repair. Pharmacol Res. 2008;58(2):88–111.
Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo J-L, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204(12):3037–47.
O’gara PT, Kushner FG, Ascheim DD, Casey DE Jr, Chung MK, De Lemos JA, et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2013;127(4):529–55.
Sager HB, Heidt T, Hulsmans M, Dutta P, Courties G, Sebas M, et al. Targeting interleukin-1β reduces leukocyte production after acute myocardial infarction. Circulation. 2015;132(20):1880–90.
Hettwer J, Hinterdobler J, Miritsch B, Deutsch M-A, Li X, Mauersberger C, et al. Interleukin-1β suppression dampens inflammatory leucocyte production and uptake in atherosclerosis. Cardiovasc Res. 2022;118(13):2778–91.
White DA, Fang L, Chan W, Morand EF, Kiriazis H, Duffy SJ, et al. Pro-inflammatory action of MIF in acute myocardial infarction via activation of peripheral blood mononuclear cells. PloS One. 2013;8(10):e76206.
Van Hout GP, Bosch L, Ellenbroek GH, De Haan JJ, Van Solinge WW, Cooper MA, et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur Heart J. 2017;38(11):828–36.
Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. New Engl J Med. 2017;377(12):1119–31.
Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ, et al. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet. 2018;391(10118):319–28.
Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100A8/A9 in inflammation. Front Immun. 2018;9:1298.
Katashima T, Naruko T, Terasaki F, Fujita M, Otsuka K, Murakami S, et al. Enhanced expression of the S100A8/A9 complex in acute myocardial infarction patients. Circ J. 2010;74(4):741–8.
Sreejit G, Nooti SK, Jaggers RM, Athmanathan B, Ho Park K, Al-Sharea A, et al. Retention of the NLRP3 inflammasome–primed neutrophils in the bone marrow is essential for myocardial infarction–induced granulopoiesis. Circulation. 2022;145(1):31–44.
Bai B, Xu Y, Chen H. Pathogenic roles of neutrophil-derived alarmins (S100A8/A9) in heart failure: from molecular mechanisms to therapeutic insights. Br J Pharmacol. 2023;180(5):573–88.
Sreejit G, Abdel-Latif A, Athmanathan B, Annabathula R, Dhyani A, Noothi SK, et al. Neutrophil-derived S100A8/A9 amplify granulopoiesis after myocardial infarction. Circulation. 2020;141(13):1080–94.
Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immun. 2014;14(5):302–14.
Richards MK, Liu F, Iwasaki H, Akashi K, Link DC. Pivotal role of granulocyte colony-stimulating factor in the development of progenitors in the common myeloid pathway. Blood. 2003;102(10):3562–8.
Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, et al. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. 1994;84(6):1737–46.
Kim HK, De La Luz SM, Williams CK, Gulino AV, Tosato G. G-CSF down-regulation of CXCR4 expression identified as a mechanism for mobilization of myeloid cells. Blood. 2006;108(3):812–20.
Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends Immun. 2010;31(8):318–24.
Christopher MJ, Link DC. Regulation of neutrophil homeostasis. Curr Opin Hematol. 2007;14(1):3–8.
Hajishengallis G, Chavakis T, Hajishengallis E, Lambris JD. Neutrophil homeostasis and inflammation: novel paradigms from studying periodontitis. J Leukocyte Biol. 2015;98(4):539–48.
Weber PS, Toelboell T, Chang LC, Tirrell JD, Saama PM, Smith GW, et al. Mechanisms of glucocorticoid-induced down-regulation of neutrophil L-selectin in cattle: evidence for effects at the gene-expression level and primarily on blood neutrophils. J Leukocyte Biol. 2004;75(5):815–27.
Ball CJ, Reiffel AJ, Chintalapani S, Kim M, Spector JA, King MR. Hydrogen sulfide reduces neutrophil recruitment in hind-limb ischemia-reperfusion injury in an L-selectin and ADAM-17 dependent manner. Plast Reconst Surg. 2013;131(3):487.
Romee R, Foley B, Lenvik T, Wang Y, Zhang B, Ankarlo D, et al. NK cell CD16 surface expression and function is regulated by a disintegrin and metalloprotease-17 (ADAM17). Blood, J Am Soc Hematol. 2013;121(18):3599–608.
Dutta P, Courties G, Wei Y, Leuschner F, Gorbatov R, Robbins CS, et al. Myocardial infarction accelerates atherosclerosis. Nature. 2012;487(7407):325–9.
Schiopu A, Cotoi OS. S100A8 and S100A9: DAMPs at the crossroads between innate immunity, traditional risk factors, and cardiovascular disease. Mediators Inflamm. 2013;2013:828354. https://doi.org/10.1155/2013/828354.
Gebhardt C, Riehl A, Durchdewald M, Németh J, Fürstenberger G, Müller-Decker K, et al. RAGE signaling sustains inflammation and promotes tumor development. J Exp Med. 2008;205(2):275–85.
Riva M, Källberg E, Björk P, Hancz D, Vogl T, Roth J, et al. Induction of nuclear factor-κ B responses by the S 100 A 9 protein is Toll-like receptor-4-dependent. Immunology. 2012;137(2):172–82.
Zhong A, Xu W, Zhao J, Xie P, Jia S, Sun J, et al. S100A8 and S100A9 are induced by decreased hydration in the epidermis and promote fibroblast activation and fibrosis in the dermis. Am J Pathol. 2016;186(1):109–22.
Boyd JH, Kan B, Roberts H, Wang Y, Walley KR. S100A8 and S100A9 mediate endotoxin-induced cardiomyocyte dysfunction via the receptor for advanced glycation end products. Circ Res. 2008;102(10):1239–46.
Kerkhoff C, Nacken W, Benedyk M, Dagher MC, Sopalla C, Doussiere J. The arachidonic acid-binding protein S100A8/A9 promotes NADPH oxidase activation by interaction with p67phox and Rac-2. FASEB J. 2005;19(3):1–28.
Vogl T, Ludwig S, Goebeler M, Strey A, Thorey IS, Reichelt R, et al. MRP8 and MRP14 control microtubule reorganization during transendothelial migration of phagocytes. Blood. 2004;104(13):4260–8.
Nishikawa Y, Kajiura Y, Lew JH, Ji K, Nagata T, Naruishi K. Calprotectin induces IL-6 and MCP-1 production via toll-like receptor 4 signaling in human gingival fibroblasts. J Cell Physiol. 2017;232(7):1862–71.
Bouma G, Lam-Tse WK, Wierenga-Wolf AF, Drexhage HA, Versnel MA. Increased serum levels of MRP-8/14 in type 1 diabetes induce an increased expression of CD11b and an enhanced adhesion of circulating monocytes to fibronectin. Diabetes. 2004;53(8):1979–86.
Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metabol. 2013;17(5):695–708.
Maiseyeu A, Badgeley MA, Kampfrath T, Mihai G, Deiuliis JA, Liu C, et al. In vivo targeting of inflammation-associated myeloid-related protein 8/14 via gadolinium immunonanoparticles. Arterioscl , Thromb Vasc Biol. 2012;32(4):962–70.
Cipollone F, Iezzi A, Fazia M, Zucchelli M, Pini B, Cuccurullo C, et al. The receptor RAGE as a progression factor amplifying arachidonate-dependent inflammatory and proteolytic response in human atherosclerotic plaques: role of glycemic control. Circulation. 2003;108(9):1070–7.
Soro-Paavonen A, Watson AM, Li J, Paavonen K, Koitka A, Calkin AC, et al. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes. 2008;57(9):2461–9.
Ionita MG, Catanzariti LM, Bots ML, de Vries J-PP, Moll FL, Sze SK, et al. High myeloid-related protein: 8/14 levels are related to an increased risk of cardiovascular events after carotid endarterectomy. Stroke. 2010;41(9):2010–5.
Tydén H, Lood C, Gullstrand B, Jönsen A, Nived O, Sturfelt G, et al. Increased serum levels of S100A8/A9 and S100A12 are associated with cardiovascular disease in patients with inactive systemic lupus erythematosus. Rheumatology. 2013;52(11):2048–55.
Sreejit G, Latif AA, Murphy AJ, Nagareddy PR. Emerging roles of neutrophil-borne S100A8/A9 in cardiovascular inflammation. Pharmacol Res. 2020;161:105212.
Gao S, Yang Y, Fu Y, Guo W, Liu G. Diagnostic and prognostic value of myeloid-related protein complex 8/14 for sepsis. Am J Emerg Med. 2015;33(9):1278–82.
Okada K, Okabe M, Kimura Y, Itoh H, Ikemoto M. Serum S100A8/A9 as a potentially sensitive biomarker for inflammatory bowel disease. Lab Med. 2019;50(4):370–80.
Bauer W, Diehl-Wiesenecker E, Ulke J, Galtung N, Havelka A, Hegel JK, et al. Outcome prediction by serum calprotectin in patients with COVID-19 in the emergency department. J Infect. 2021;82(4):84–123.
Gupta A, Qaisar R, Halwani R, Kannan M, Ahmad F. TFPI and FXIII negatively and S100A8/A9 and cystatin C positively correlate with D-dimer in COVID-19. Exp Biol Med. 2022;247(17):1570–6.
Guo Q, Zhao Y, Li J, Liu J, Yang X, Guo X, et al. Induction of alarmin S100A8/A9 mediates activation of aberrant neutrophils in the pathogenesis of COVID-19. Cell Host Micr. 2021;29(2):222–35. e4.
Colicchia M, Schrottmaier WC, Perrella G, Reyat JS, Begum J, Slater A, et al. S100A8/A9 drives the formation of procoagulant platelets through GPIbα. Blood J Am Soc Hematol. 2022;140(24):2626–43.
Schneider RK, Schenone M, Ferreira MV, Kramann R, Joyce CE, Hartigan C, et al. Rps14 haploinsufficiency causes a block in erythroid differentiation mediated by S100A8 and S100A9. Nat Med. 2016;22(3):288–97.
Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med. 2008;205(10):2235–49.
Laouedj M, Tardif MR, Gil L, Raquil M-A, Lachhab A, Pelletier M, et al. S100A9 induces differentiation of acute myeloid leukemia cells through TLR4. Blood J Am Soc Hematol. 2017;129(14):1980–90.
Soyfoo MS, Roth J, Vogl T, Pochet R, Decaux G. Phagocyte-specific S100A8/A9 protein levels during disease exacerbations and infections in systemic lupus erythematosus. J Rheumatol. 2009;36(10):2190–4.
Kang KY, Woo J-W, Park S-H. S100A8/A9 as a biomarker for synovial inflammation and joint damage in patients with rheumatoid arthritis. Kor J Intern Med. 2014;29(1):12.
Catalán V, Gómez-Ambrosi J, Rodríguez A, Ramírez B, Rotellar F, Valentí V, et al. Increased levels of calprotectin in obesity are related to macrophage content: impact on inflammation and effect of weight loss. Mol Med. 2011;17:1157–67.
Nagareddy PR, Kraakman M, Masters SL, Stirzaker RA, Gorman DJ, Grant RW, et al. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metabol. 2014;19(5):821–35.
Al-Sharea A, Murphy AJ, Huggins L, Hu Y, Goldberg IJ, Nagareddy PR. SGLT2 inhibition reduces atherosclerosis by enhancing lipoprotein clearance in Ldlr−/− type 1 diabetic mice. Atherosclerosis. 2018;271:166–76.
Ryckman C, McColl SR, Vandal K, de Médicis R, Lussier A, Poubelle PE, et al. Role of S100A8 and S100A9 in neutrophil recruitment in response to monosodium urate monohydrate crystals in the air-pouch model of acute gouty arthritis. Arthr Rheum Official J Am Coll Rheumatol. 2003;48(8):2310–20.
Ryckman C, Gilbert C, de Médicis R, Lussier A, Vandal K, Tessier PA. Monosodium urate monohydrate crystals induce the release of the proinflammatory protein S100A8/A9 from neutrophils. J Leukocyte Biol. 2004;76(2):433–40.
Kummer MP, Vogl T, Axt D, Griep A, Vieira-Saecker A, Jessen F, et al. Mrp14 deficiency ameliorates amyloid β burden by increasing microglial phagocytosis and modulation of amyloid precursor protein processing. J Neurosci. 2012;32(49):17824–9.
Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer's disease. Nat Rev Dis Prim. 2015;1(1):1–18.
Goyette J, Geczy CL. Inflammation-associated S100 proteins: new mechanisms that regulate function. Amino Acids. 2011;41:821–42.
Moshfegh CM, Elkhatib SK, Watson GF, Drake J, Taylor ZN, Reed EC, et al. S100a9 protects against the effects of repeated social defeat stress. Biol Psychiat Glob Open Sci. 2022; https://doi.org/10.1016/j.bpsgos.2022.12.002.
Frangogiannis NG. S100A8/A9 as a therapeutic target in myocardial infarction: cellular mechanisms, molecular interactions, and translational challenges. Eur Heart J. 2019;40(32):2724–6.
Hobbs JA, May R, Tanousis K, McNeill E, Mathies M, Gebhardt C, et al. Myeloid cell function in MRP-14 (S100A9) null mice. Mol Cell Biol. 2003;23(7):2564–76.
Cesaro A, Anceriz N, Plante A, Page N, Tardif MR, Tessier PA. An inflammation loop orchestrated by S100A9 and calprotectin is critical for development of arthritis; 2012. https://doi.org/10.1371/journal.pone.0045478.
Kerkhoff C, Sorg C, Tandon NN, Nacken W. Interaction of S100A8/S100A9− arachidonic acid complexes with the scavenger receptor CD36 may facilitate fatty acid uptake by endothelial cells. Biochemistry. 2001;40(1):241–8.
Robinson MJ, Tessier P, Poulsom R, Hogg N. The S100 family heterodimer, MRP-8/14, binds with high affinity to heparin and heparan sulfate glycosaminoglycans on endothelial cells. J Biol Chem. 2002;277(5):3658–65.
Kerkhoff C, Klempt M, Kaever V, Sorg C. The two calcium-binding proteins, S100A8 and S100A9, are involved in the metabolism of arachidonic acid in human neutrophils. J Biol Chem. 1999;274(46):32672–9.
Marinković G, Koenis DS, de Camp L, Jablonowski R, Graber N, de Waard V, et al. S100A9 links inflammation and repair in myocardial infarction. Circ Res. 2020;127(5):664–76.
Li Y, Chen B, Yang X, Zhang C, Jiao Y, Li P, et al. S100a8/a9 signaling causes mitochondrial dysfunction and cardiomyocyte death in response to ischemic/reperfusion injury. Circulation. 2019;140(9):751–64.
Otsuka K, Terasaki F, Ikemoto M, Fujita S, Tsukada B, Katashima T, et al. Suppression of inflammation in rat autoimmune myocarditis by S100A8/A9 through modulation of the proinflammatory cytokine network. Eur J Heart Fail. 2009;11(3):229–37.
Averill MM, Barnhart S, Becker L, Li X, Heinecke JW, LeBoeuf RC, et al. S100A9 differentially modifies phenotypic states of neutrophils, macrophages, and dendritic cells: implications for atherosclerosis and adipose tissue inflammation. Circulation. 2011;123(11):1216–26.
McCormick MM, Rahimi F, Bobryshev YV, Gaus K, Zreiqat H, Cai H, et al. S100A8 and S100A9 in human arterial wall: implications for atherogenesis. J Biol Chem. 2005;280(50):41521–9.
Ionita MG, Vink A, Dijke IE, Laman JD, Peeters W, van der Kraak PH, et al. High levels of myeloid-related protein 14 in human atherosclerotic plaques correlate with the characteristics of rupture-prone lesions. Arterioscl Thromb Vasc Biol. 2009;29(8):1220–7.
Peng WH, Jian WX, Li HL, Hou L, Wei YD, Li WM, et al. Increased serum myeloid-related protein 8/14 level is associated with atherosclerosis in type 2 diabetic patients. Cardiovasc Diabetol. 2011;10:1–7.
Cotoi OS, Dunér P, Ko N, Hedblad B, Nilsson J, Björkbacka H, et al. Plasma S100A8/A9 correlates with blood neutrophil counts, traditional risk factors, and cardiovascular disease in middle-aged healthy individuals. Arterioscl Thromb Vasc Biol. 2014;34(1):202–10.
Volz HC, Laohachewin D, Seidel C, Lasitschka F, Keilbach K, Wienbrandt AR, et al. S100A8/A9 aggravates post-ischemic heart failure through activation of RAGE-dependent NF-κB signaling. Basic Res Cardiol. 2012;107:1–16.
Altwegg LA, Neidhart M, Hersberger M, Müller S, Eberli FR, Corti R, et al. Myeloid-related protein 8/14 complex is released by monocytes and granulocytes at the site of coronary occlusion: a novel, early, and sensitive marker of acute coronary syndromes. Eur Heart J. 2007;28(8):941–8.
Morrow DA, Wang Y, Croce K, Sakuma M, Sabatine MS, Gao H, et al. Myeloid-related protein 8/14 and the risk of cardiovascular death or myocardial infarction after an acute coronary syndrome in the Pravastatin or Atorvastatin Evaluation and Infection Theraphy: Thrombolysis in Myocardial Infarction (PROVE IT-TIMI 22) trial. Am Heart J. 2008;155(1):49–55.
Ma L-P, Haugen E, Ikemoto M, Fujita M, Terasaki F, Fu M. S100A8/A9 complex as a new biomarker in prediction of mortality in elderly patients with severe heart failure. IntJ Cardiol. 2012;155(1):26–32.
Acknowledgements
The authors are grateful to the School of Life Sciences for providing necessary facilities for writing the article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
All the authors have given the consent to publish.
Conflict of Interest
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human and animals subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rao, R.S., Panbhukarasu, S., Waleed, M. et al. Neutrophil-Derived S100A8/A9 in Cardiovascular Disease and Beyond. Curr. Pharmacol. Rep. 9, 353–363 (2023). https://doi.org/10.1007/s40495-023-00328-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40495-023-00328-w