
Abstract
This review focuses on new aspects of extracellular roles of the calgranulins. S100A8, S100A9 and S100A12 are constitutively expressed in neutrophils and induced in several cell types. The S100A8 and S100A9 genes are regulated by pro- and anti-inflammatory mediators and their functions may depend on cell type, mediators within a particular inflammatory milieu, receptors involved in their recognition and their post-translational modification. The S100A8 gene induction in macrophages is dependent on IL-10 and potentiated by immunosuppressive agents. S100A8 and S100A9 are oxidized by peroxide, hypochlorite and nitric oxide (NO). HOCl generates intra-chain sulfinamide bonds; stronger oxidation promotes cross-linked forms that are seen in human atheroma. S100A8 is >200-fold more sensitive to oxidative cross-linking than low-density lipoprotein and may reduce oxidative damage. S100A8 and S100A9 can be S-nitrosylated. S100A8–SNO suppresses mast cell activation and inflammation in the microcirculation and may act as an NO transporter to regulate vessel tone in inflammatory lesions. S100A12 activates mast cells and is a monocyte and mast cell chemoattractant; a G-protein-coupled mechanism may be involved. Structure–function studies are discussed in relation to conservation and divergence of functions in S100A8. S100A12 induces cytokines in mast cells, but not monocytes/macrophages. It forms complexes with Zn2+ and, by chelating Zn2+, S100A12 significantly inhibits MMPs. Zn2+ in S100A12 complexes co-localize with MMP-9 in foam cells in atheroma. In summary, S100A12 has pro-inflammatory properties that are likely to be stable in an oxidative environment, because it lacks Cys and Met residues. Conversely, S100A8 and S100A9 oxidation and S-nitrosylation may have important protective mechanisms in inflammation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Inflammation is a central component of acute responses to infection and wounding, and to numerous chronic human diseases including rheumatoid arthritis (RA), inflammatory bowel disease (IBD), atherosclerosis and allergic inflammation. Neutrophils and mast cells are important mediators of the acute innate immune response, but also contribute to the pathogenesis of chronic inflammatory diseases such as RA, asthma and other chronic lung disorders. Leukocyte recruitment to sites of infection/inflammation is mediated by generation of chemokines and other chemoattractants, and by cytokine stimulation of the vascular endothelium to facilitate leukocyte adhesion and extravasation. Initial activation of the innate immune system is usually self-limiting, but if this fails, chronicity ensues. In chronic inflammation, monocytes accumulate where they mature to macrophages that play an inflammatory and tissue destructive role. Activation of these cells by structures on pathogens known as pathogen-associated molecular patterns (PAMPs), through receptors such as the Toll-like receptors (TLRs) [reviewed in (Mogensen 2009)], modulates inflammatory processes via induction of inflammatory cytokines, reactive oxygen species (ROS) and nitric oxide (NO). However, macrophages are also important in resolution and interleukin (IL)-10 (which is produced by activated macrophages) and can induce anti-inflammatory effects via suppression of cytokine signaling 3 (SOCS3) (Gordon 2003).
In recent years, a subgroup of the S100 family (S100A12, S100A8 and S100A9) has been associated with acute/chronic inflammatory disorders (Foell et al. 2004a, b) and with various cancers (Salama et al. 2008). Collectively, these proteins are termed calgranulins, reflecting their calcium-binding properties and high expression in granulocytes. Despite intense research, the exact functions of these proteins remain enigmatic and disparate functional roles are proposed. This review will focus on the extracellular roles of these proteins in leukocyte migration and chemotaxis, leukocyte activation, oxidant scavenging and their relevance in inflammatory processes.
Expression and gene regulation of calgranulins
The calgranulins are abundant neutrophil proteins; S100A8 and S100A9 comprise ~45% of the cytosol (Edgeworth et al. 1991). Constitutive expression is restricted to neutrophils, myeloid-derived dendritic cells, platelets, osteoclasts, hypertrophic chondrocytes and in trophoblasts in developing embryos. Myeloid cell expression is dependent on the stage of differentiation (Koike et al. 1992; Sellmayer et al. 1992). Monocytes constitutively express low levels of S100A8 and S100A9 (Hessian et al. 1993; Hsu et al. 2005), but this is reduced during their extravasation from blood; normal tissue macrophages do not express these proteins (Lagasse and Clerc 1988; Zwadlo et al. 1988). S100A8 and S100A9 are often strongly expressed in acute and chronic inflammatory lesions and one or both can be induced by appropriate stimulants in several cell types (see Table 1).
In addition to neutrophils, macrophages are often S100A8/A9 positive in inflamed tissues (Odink et al. 1987; Delabie et al. 1990; Goebeler et al. 1994; McCormick et al. 2005; Terasaki et al. 2007). Murine (m)S100A8 mRNA is induced in macrophages by TLR ligands, such as LPS (Hu et al. 1996) and dsRNA (Endoh et al. 2009), and by the cytokines, TNFα, transforming growth factor β (TGF-β) and interferon-γ (IFNγ) (Xu and Geczy 2000) and by IFNγ (Endoh et al. 2009). The stress response modulator norepinephrine also induces S100A8 and S100A9 in human monocytoid cells, suggesting their regulation by stress (Suryono et al. 2006).
S100A8 induction by LPS and polyIC (a double-stranded RNA mimetic) is maximal at ~24-h post-stimulation and is strongly dependent on the earlier induction of the anti-inflammatory cytokine, IL-10. IL-10 alone has little effect, but is essential for, and potentiates, TLR ligand-induced responses (Xu et al. 2001; Endoh et al. 2009). S100A8 is not induced in stimulated macrophages from IL-10-null mice. We also demonstrated a crucial and convergent role for protein kinase R (PKR) in S100A8 induction by LPS and dsRNA, probably by virtue of its role in IL-10 induction (Xu et al. 2001; Endoh et al. 2009). TLR4, but not TLR3-mediated induction of mS100A8 mRNA, is also dependent on cyclo-oxygenase 2 (COX-2) via production of prostaglandin E2 and cyclic AMP (cAMP) (Xu and Geczy 2000; Hsu et al. 2005). Stimulation of MAP kinases ERK1/2 and p38 pathways is required to generate responses to both agonists (Hsu et al. 2005; Endoh et al. 2009). Interestingly, although S100A8 and S100A9 are said to require co-expression for stability (Hobbs et al. 2003; Manitz et al. 2003), we find that murine S100A9 (mS100A9) is not induced in macrophages by any of these stimuli. In contrast, human S100A8 and S100A9 are generally co-induced in human monocytes and macrophages (Hsu et al. 2005; Endoh et al. 2009).
S100A8 and S100A9 induction in other cell types may be somewhat stimulus specific. In murine microvascular endothelial cells (ECs), mS100A8 is induced by LPS and IL-1β, peaking 24 and 16-h post-stimulation, respectively, but not by TNFα or IFNγ. In contrast, mS100A9 is induced earlier by IL-1β (maximal 8-h post-stimulation) and is also induced by TNFα in these cells (Yen et al. 1997). Co-stimulation with TNFα enhanced LPS responses, but not IL-1β-mediated induction in vitro and in vivo. The S100s are not expressed in normal vessels, but mS100A8 is seen in microcapillaries in delayed-type hypersensitivity lesions in mice (Yen et al. 1997). Human S100A8 and S100A9 are expressed in neovessels, but not in large vessels or arteries in human atheroma (McCormick et al. 2005). ECs in lungs of mice bearing metastatic tumors also express these proteins, and vascular EC-derived growth factor and TNFα are implicated in their induction (Hiratsuka et al. 2006).
In murine fibroblasts, LPS, FGF-2 and IL-1β, but not TNFα or IFNγ, induce mS100A8; induction by FGF-2 is markedly enhanced by heparin, but suppressed by TGF-β (Rahimi et al. 2005). Murine S100A9 was not induced with any stimuli tested (Rahimi et al. 2005). As for microvascular EC (Yen et al. 1997), the degree of cell confluence was important for expression in fibroblasts in vitro (Rahimi et al. 2005), indicating that cell-cell contacts may provide additional signals for optimal induction in these cell types.
S100A8/A9 is not found in normal skin, but the pattern of S100A8 expression in fibroblasts in rat dermal wounds suggests its involvement in fibroblast-to-myofibroblast differentiation at sites of inflammation and in repair/remodeling. In keeping with gene regulation studies, TGF-β mediates the later stages of wound healing when S100A8 levels decline (Rahimi et al. 2005). S100A8/A9 expression has been associated with the hyperproliferative responses typical of psoriasis (Gabrielsen et al. 1986) and with wound healing (Thorey et al. 2001). We also found S100A8, but not S100A9 induction in UVA-irradiated skin of mice, and upregulation in a keratinocyte cell line by peroxide (Grimbaldeston et al. 2003). Both responses were suppressed by anti-oxidants, confirming ROS-mediated induction (Grimbaldeston et al. 2003). Cultured human keratinocytes also express S100A8 and S100A9 in response to TNFα, IFNγ (Mork et al. 2003) and IL-1α (Hayashi et al. 2007). TGF-β and retinoic acid reduced induction, suggesting a relationship with keratinocyte differentiation (Hayashi et al. 2007). The late induction of these genes in keratinocytes also suggests that they are secondary-response gene products.
Our finding that S100A8 induction in macrophages is IL-10-dependent suggested that this protein may have anti-inflammatory properties. In keeping with this, we found that glucocorticoids (GC) directly upregulated S100A8 and S100A9 in human monocytes, and S100A8-positive macrophage numbers increased in rheumatoid synovium in RA patients treated with high-dose GC (Hsu et al. 2005). The synthetic GC, dexamethasone (DEX), strongly enhances LPS-induced mS100A8 induction in murine macrophages (Hsu et al. 2005). Since the promotor region of the mS100A8 gene lacks a steroid-response element, and because anti-IL10 suppresses induction by DEX, this response is likely dependent on IL-10 induction by GC (Hsu et al. 2005). Increased levels of mS100A8 were also detected in the lungs of DEX-treated mice after intranasal administration of LPS (Bozinovski et al. 2005). GC also induced S100A8 in human dendritic cells (Kumar et al. 2003) and in a human keratinocyte cell line and potentiated its induction by IL-17 (Bai et al. 2007). However, DEX suppressed mS100A8 and mS100A9 expression associated with phorbol ester-induced skin inflammation in wild-type, but not in c-fos−/− mice, suggesting that these are negatively regulated c-fos/AP-1-target genes in skin (Gebhardt et al. 2002). We also found that DEX potentiated S100A8 gene induction by LPS in primary murine fibroblasts and microvascular EC (Hsu et al. 2005). Because IL-10 and GC are anti-inflammatory and immunosuppressive [reviewed in (Couper et al. 2008) and (Wilckens 1995)], this pattern of gene expression indicates that S100A8 may be involved in the resolution phase of inflammation and may be more consistent with the proposed oxidant-scavenging role (Lim et al. 2009) than the pro-inflammatory role recently proposed via ligation of TLR4 (Vogl et al. 2007).
S100A12 is constitutively expressed in neutrophils at low levels (~5% of cytosolic protein) (Guignard et al. 1995), and expression patterns in myeloid cell lines (Vogl et al. 1999a, b) suggest that it is expressed later in myeloid cell differentiation than S100A8 and S100A9 (Robinson and Hogg 2000). PolyIC, LPS and TNFα induce S100A12 in monocytes (Yang et al. 2001; Endoh et al. 2009). Induction by TNFα is more rapid than for S100A8 and S100A9, with optimal S100A12 mRNA induction at 6–12 h (Yang et al. 2001). IL-6 also upregulates S100A12 expression in differentiated THP-1 macrophages and is dependent on the JAK-STAT pathway and de novo protein synthesis (Hasegawa et al. 2003). Pioglitazone, a peroxisome proliferator-activated receptor-γ (PPAR-γ) agonist, inhibited S100A12 mRNA induction in these cells (Hasegawa et al. 2003). Ligand-bound PPAR-γ represses signal-dependent transcription of many inflammatory cytokines through interactions with promoter-bound co-regulatory proteins (Pascual and Glass 2006). The rapid upregulation by pro-inflammatory stimuli and repression by PPARγ agonists is consistent with a pro-inflammatory role for S100A12. In keeping with this, and in contrast to S100A8 and S100A9, we found that GC does not upregulate the S100A12 gene in activated human monocytes (K. Hsu, C. Geczy, unpublished data).
S100A12 is expressed by monocytes and/or macrophages in giant cell arteritis (Foell et al. 2004a, b), atherosclerotic lesions (Goyette et al. 2009) and macrophages in asthmatic lung (Yang et al. 2007), rheumatoid synovuim (Yang et al. 2001) and inflamed mucosa in IBD (Foell et al. 2003; Leach et al. 2007). S100A12 mRNA is detected in fetal bovine esophagus and skin (Hitomi et al. 1996), protein found in corneal fibroblasts (keratocytes) (Gottsch et al. 1995, 1999a, b), differentiating esophageal epithelial cells (Hitomi et al. 1998) and in human keratinocytes in psoriatic lesions, but not normal skin (Mirmohammadsadegh et al. 2000). IL1α and TNFα increase S100A12 expression in bovine keratocytes (Gottsch et al. 1999a, b). Although circulating eosinophils do not express S100A12, eosinophils in the airways of asthmatic lung express S100A12 (Yang et al. 2007), indicating that it is also inducible in these cells and the mechanisms are currently being characterized by us.
Calgranulins and clinical conditions
There has been significant interest in realizing the clinical diagnostic potential of monitoring calgranulin concentrations in body fluids. Unlike acute-phase reactants primarily produced by the liver (e.g., C-reactive protein), these proteins are mostly expressed in cells in inflammatory lesions. Thus, calgranulin levels in the circulation are elevated in inflammatory conditions characterized by infiltration of neutrophils or activation of monocyte/macrophages or other stromal cells. Since leukocyte recruitment and activation is common to many inflammatory conditions, circulating levels may not be appropriate for diagnosis of any particular condition, although calgranulins are suggested to be non-specific markers of phagocyte activation (Foell et al. 2004a, b, 2007). On the other hand, samples derived directly from an inflammatory source (e.g., feces, sputum) may be more reliable markers of inflammatory disorders such as IBD (Foell et al. 2009) or eosinophilic asthma (Yang et al. 2007).
S100 proteins lack a signal peptide for secretion via the Golgi-mediated pathway and there is some debate regarding whether the high levels derived from inflammatory lesions are due to active secretion or passive release as a consequence of neutrophil necrosis. S100A8/A9 release correlates with loss of neutrophil viability (Voganatsi et al. 2001) and because these are short-lived, necrosis probably represents a significant extracellular source, particularly in situations such as acute inflammatory lesions or chronic conditions such as cystic fibrosis or RA, where neutrophil infiltration is significant. However, activation of protein kinase C by pro-inflammatory stimuli and elevation of [Ca2+]i by contact with activated endothelium (Frosch et al. 2000) can also provoke S100A8/A9 release from phagocytes. LPS from several bacterial species (Kido et al. 2005) and chemoattractants such as C5a (Hetland et al. 1998) cause rapid release from neutrophils. Although mechanisms are unclear, secretion of S100A8 and S100A9 from monocytes and neutrophils is tubulin dependent (Rammes et al. 1997; Ryckman et al. 2004). Secretion following activation of neutrophils with monosodium urate crystals involves activation of CD11b, CD16, Src kinases, Syk and tubulin polymerization (Ryckman et al. 2004).
Secretion may also be cell-type specific. For example, S100A8 and S100A9 in activated microvascular ECs, fibroblasts and keratinocytes are mainly cell associated (Yen et al. 1997; Grimbaldeston et al. 2003; Ravasi et al. 2004; Rahimi et al. 2005), implying that these proteins have predominantly intracellular roles in these cells. In contrast, stimulated monocytes and macrophages readily secrete the three calgranulins (Rammes et al. 1997; Xu and Geczy 2000; Xu et al. 2001; Hasegawa et al. 2003; Hsu et al. 2005; Endoh et al. 2009).
Extracellular functions
Calgranulins are implicated in numerous intracellular functions in phagocytes (reviewed in (Nacken et al. 2003; Roth et al. 2003; Gebhardt et al. 2006; Foell et al. 2007; Lim et al. 2009)), however this review will focus on some newer aspects of their extracellular roles. Tables 2 and 3 summarize some key extracellular functions proposed for human and murine S100A8, S100A9, the heterodimer (S100A8/A9) and for S100A12. However, there are several conflicting reports regarding their activities, making assessment of some functions difficult. Because the S100A8 and S100A9 genes are regulated by pro- and anti-inflammatory mediators, together with their emerging anti-inflammatory properties, the functions of these proteins may depend on several factors including the cell type and mediator involved in their induction in a particular inflammatory milieu, the receptors involved in their recognition and their post-translational modification. For this reason, we propose that they are mislabeled as ‘pro-inflammatory’ and in some circumstances we believe that their high expression levels may actually mediate host protection. For example, S100A8 and S100A9 expression patterns in incision wounds in skin lead to the suggestion that elevated levels may have a therapeutic effect against inflammation in wound healing (Wu and Davidson 2004), and their positive expression in skin may be crucial for the protection against bacterial infections (Champaiboon et al. 2009) and UVA-mediated oxidative damage (Grimbaldeston et al. 2003). Interestingly, S100A8/A9 in stools from healthy newborns is elevated over 3–30 days and may be involved in the normal response to microbial infection (Baldassarre et al. 2007). Although S100A8/S100A9 has been associated with chondrocyte-mediated cartilage destruction in RA (van Lent et al. 2007), anti-inflammatory properties of the injected proteins are indicated in rodent models of arthritis (Brun et al. 1995), autoimmune myocadritis (Otsuka et al. 2009) and endotoxin-mediated inflammation (Ikemoto et al. 2007). In addition, S100A9 has antinociceptive properties that are effective in various acute inflammatory pain models, and the use of a C-terminal peptide may represent a potential therapeutic (Paccola et al. 2008). Taken together with the pro-inflammatory properties described for these proteins, their ability to modulate inflammation should also be considered. Functions may be governed by the receptors that are activated by the calgranulins. In addition, post-translational modifications may occur in oxidative environments and other factors such as proteolytic cleavage by proteases released as a consequence of phagocyte activation (Greenlee et al. 2006), and zinc binding may also have important functional ramifications.
Endogenous ligands of TLR4?
Extracellular S100A8/S100A9 is reported to bind numerous receptors on cells. One of the most important may be the ability of murine S100A8 to bind TLR4; interaction between immobilized mS100A8 and recombinant TLR4 was demonstrated by surface plasmon resonance (Kd ~ 11–25 nM) (Vogl et al. 2007). Stimulation of murine bone marrow cells (a population normally comprising ~30% neutrophils, immature myeloid cells and haematopoietic stem cells) with mS100A8 activated nuclear factor κB (NFκB) via the MyD88-dependent pathway and secretion of TNFα. In contrast, mS100A9 or the mS100A8/A9 complex did not stimulate TNFα release, although the complex enhanced TNFα production in response to LPS (Vogl et al. 2007). Bone marrow cells from mS100A9−/− mice are less responsive to LPS stimulation and the mice are resistant to LPS-induced septic shock (Vogl et al. 2007). The latter is not surprising, considering the fact that neutrophils from these mice do not migrate normally to various chemotactic stimuli (Manitz et al. 2003; Vogl et al. 2004; McNeill et al. 2007; Schnekenburger et al. 2008). The authors claim that mS100A9−/− mice are also deficient in mS100A8 (essentially mS100A8−/−) because this is absent in circulating neutrophils (Hobbs et al. 2003; Manitz et al. 2003; Nacken et al. 2005) and thus implicate mS100A8/A9 in LPS-induced septic shock. However, mS100A8 was found in bone marrow cells in mS100A9−/− mice (Manitz et al. 2003) and mS100A8 levels in serum, or from activated macrophages from these mice, which has not been reported.
In contrast to these findings, Ikemoto et al. (2007) showed that intraperitoneal injection of S100A8/A9 into rats 1 or 3.5 h after LPS injection significantly reduced serum IL-6 and nitrite levels, whereas administration of anti-S100A8/A9 IgG 1 h after LPS injection slightly increased IL-6 levels. This study suggests that the S100 complex bound pro-inflammatory cytokines and also reduced NO levels. The latter would concur with our studies showing that S100A8 and mS100A8 are readily S-nitrosylated on Cys41/42 (Lim et al. 2008) by NO donors and these may have scavenged the NO generated in vivo.
Using recombinant human or murine S100A8, S100A9 or the complex, we cannot induce significant levels of pro-inflammatory cytokines, in the presence/absence of LPS, in monocytes/macrophages from the relevant species (K. Hsu, C. Geczy, unpublished data). Taken together with the mechanisms regulating S100A8 gene expression that suggest an anti-inflammatory role, it is difficult to reconcile our results with a strictly pro-inflammatory role. The proteins used in the study showing that mS100A8 binds TLR4 were endotoxin tested, and heat treatment reduced activity, but polymyxin B (which binds to, and neutralizes LPS) did not, indicating that the activity was unlikely due to LPS contamination (Vogl et al. 2007). However, these recombinants were generated using a bacterial expression system, and contamination with other bacterial components, such as lipoproteins, is possible (Tsan and Gao 2004). Similar issues with contamination have been problematic for research characterizing endogenous ligands of TLR, such as heat shock proteins (Bausinger et al. 2002; Gao and Tsan 2003a, b; Reed et al. 2003) and HMGB1 (Tian et al. 2007). Clearly, more research is needed to clarify the role of TLR4 in mediating extracellular functions of S100A8.
EN-RAGE and the ‘S100/RAGE pro-inflammatory axis’
When S100A12 was found to bind the receptor for advanced glycation end products (RAGE), Hofmann et al. named it extracellular newly identified RAGE-binding protein (EN-RAGE) to signify the importance of this receptor to its function (Hofmann et al. 1999). These studies were supported by the observation that apoS100A12 binds RAGE in vitro with very low affinity (Kd ~ 140 μM), which increases > 1,000 fold when S100A12 is in the Ca2+- (Xie et al. 2007) or Zn2+-bound hexameric state (Moroz et al. 2009). The hypothesis that RAGE is the sole receptor mediating the pro-inflammatory functions of S100A12 has enjoyed wide support and is often referred to as the ‘RAGE/S100 pro-inflammatory axis’. This theory can be summarized as follows: RAGE ligation by S100A12 presents as a consequence of inflammatory conditions, triggers a signaling cascade in microvascular ECs, macrophages (inferred from results using BV-2, a microglial cell line) and lymphocytes (also cell lines), culminating in NFκB activation. This amplifies inflammation and, since the promoter region of the RAGE gene contains an NFκB binding site (Li and Schmidt 1997), RAGE itself is consequently upregulated. Thus, RAGE ligation initiates a feed-forward loop that could potentiate inflammation and some authors refer to RAGE as a ‘master switch’ in chronic inflammation (Schmidt et al. 2001; Bierhaus et al. 2005).
This theory set the scene for further studies by several groups investigating the role of RAGE in various functions provoked by numerous S100 proteins. Because of their structural similarities, S100s generally, and particularly the calgranulins, have been proposed to bind RAGE. This has often led to the erroneous use of the term ‘S100/calgranulins’ to refer to any or all S100s, perhaps implying functional redundancy. However, although the S100 proteins are structurally related, differences in expression profiles and subtle differences in amino acid sequences within functional domains may underlie non-redundant functions. For example, gene deletion of S100A8 in the mouse results in rapid and synchronous embryo resorption by day 9.5 of development, indicating a necessary function in trophoblast invasion and/or maternal recognition (Passey et al. 1999), whereas mice carrying a deletion of the S100A9 gene are apparently normal, but have somewhat compromised neutrophil function (Hobbs et al. 2003; Manitz et al. 2003; Vogl et al. 2004; McNeill et al. 2007). Moreover, particular S100 proteins have very different extracellular functions that can be cell type and/or concentration dependent such as those reported for S100B, which has RAGE-dependent and RAGE-independent activities (Sorci et al. 2003, 2004; Bianchi et al. 2007, 2008). Taken together, these observations do not support a theory for a common receptor for all S100 proteins.
We showed that S100A12 activates mast cells in vitro and in vivo, but failed to detect RAGE mRNA/protein in these cells, indicating an alternate receptor (Yang et al. 2007). In keeping with this, we showed that the chemotactic activity of S100A12 for monocytes and mast cells is RAGE independent and likely mediated by a G-protein-coupled receptor (Yan et al. 2008). Importantly, and in marked contrast to the finding that S100A12 induces IL-1β and TNFα in a murine microglial cell line stimulated with bovine S100A12 (Hofmann et al. 1999), we failed to reproduce these results with human S100A12 and primary human monocytes or macrophages (Goyette et al. 2009). This, together with the fact that neutralizing anti-S100A12 antibodies would be ineffective in murine models used to confirm its role in cell-mediated immunity and IBD because S100A12 is not expressed in the rodent genome (Ravasi et al. 2004), indicates that the central role for RAGE may need to be re-evaluated.
In addition to TLR4, other receptors such as heparin, heparan sulfate (Robinson et al. 2002), CD36 (Kerkhoff et al. 2001) and N-glycans (Srikrishna et al. 2001) are implicated in extracellular S100A8/A9 functions. Thus, some functions attributed to RAGE may depend on cell type. RAGE is implicated in proliferation of human estrogen receptor-negative breast cancer cells (Ghavami et al. 2008a, b) and in S100A8/A9-mediated cardiomyocyte dysfunction (Boyd et al. 2008), but not in activation of prostate cancer cells (Hermani et al. 2006) or monocytes (Sunahori et al. 2006). Results from different binding assays are somewhat contradictory. Because S100A8/A9 protected the VC1 domain of RAGE (recombinant) from protease digestion (Ghavami et al. 2008a, b), binding is implicated although S100A8/A9 does not bind RAGE-transfected CHO cells (Robinson et al. 2002) or interact with RAGE in surface plasmon resonance studies (Vogl et al. 2007). In contrast, RAGE was co-immunoprecipitated with anti-S100A8 and anti-S100A9 from LPS-stimulated cardiac myocytes (Boyd et al. 2008) and colocalized with S100A8/A9 in prostate cancer cells (Hermani et al. 2006). Recent evidence indicates that the glycosylation status of RAGE is important. RAGE purified from various sources is glycosylated and a small proportion of molecules (~1–5%) contain epitopes recognized by an N-glycan-specific mAb (mAbGB3.1) (Turovskaya et al. 2008). S100A8/A9 bound a similarly small fraction (~1%) of RAGE molecules; binding was abolished by deglycosylation of RAGE and blocked by mAbGB3.1, indicating that these novel N-glycans mediate the interaction (Turovskaya et al. 2008). These results may explain some of the variability in previous studies since N-glycans may be present on other membrane proteins, and the extent of RAGE glycosylation may differ between cell types.
As proposed for HMGB1, it is possible that some RAGE-dependent extracellular calgranulin functions may depend on a co-stimulus to activate co-operative signaling pathways or enhance receptor affinity. For example, HMGB1 augments CpG-stimulated IFNα production in DCs, and this is dependent on RAGE and TLR9 (Tian et al. 2007). Stimulation of transfected HEK293 cells with HMGB1–CpG complexes induced association of RAGE and TLR9 indicating that HMGB1 and RAGE may aid in CpG presentation to TLR9 (Tian et al. 2007). Alternatively, functions may be dependent on pre-activation of cells to induce other necessary gene products. For example, in EC pre-treated with AGE-modified albumin for 72 h, S100A8/A9 upregulate IL-6, ICAM-1, VCAM-1 and MCP-1, whereas only MCP-1 expression was enhanced in untreated cells (Ehlermann et al. 2006). AGE pre-treatment also upregulates RAGE expression in these cells and the authors assume that S100A8/A9 functions through this receptor (Ehlermann et al. 2006), though another putative RAGE ligand, S100B (Leclerc et al. 2007), did not have similar effects (Ehlermann et al. 2006). Another interpretation is that AGE pre-treatment provided a second signal necessary for S100A8/A9-mediated upregulation of pro-inflammatory genes in EC.
Alternatively, RAGE may act as a composite receptor. That is, RAGE may need to physically associate (perhaps directly bound or in lipid rafts) with another receptor/receptors to mediate certain functions. For example, RAGE and CD18/CD11b interact both in cis (on the same cell) and trans (between two cells) and interestingly HMGB1 requires both CD18/CD11b and RAGE to initiate NFκB transcriptional activity in neutrophils (Orlova et al. 2007). β2 integrins interact with other cell surface receptors in cis, including FcγRIIA (CD32), urokinase-type plasminogen activator receptor (CD87), CD14 and PDGFβ receptor (Petty et al. 2002), and are implicated as signaling partners for some of these (Petty et al. 1997; Todd and Petty 1997). Therefore, it is possible that CD18/CD11b and RAGE act as a composite receptor wherein RAGE binds the ligand and CD18/CD11b transduces the signal. Alternatively, RAGE and CD18/CD11b-initiated signals may both be required in close proximity, for example within lipid rafts. Such a composite receptor mechanism may explain why S100B requires the extracellular domain of RAGE, but not RAGE signaling, to stimulate NO production in BV-2 microglia (Adami et al. 2004) and similar mechanisms could mediate some of the responses of calgranulins.
Post-translational modifications of S100A8 and S100A9 by oxidants
S100A8 and S100A9 are exceptionally abundant in neutrophils and upregulated in activated macrophages, cell types that produce high levels of ROS. Myeloperoxidase (MPO) expressed in phagocyte granules is released following activation and generates the powerful oxidant, HOCl from H2O2 and Cl− (Klebanoff 2005). Large amounts (>100 μM) can be produced from 106 activated neutrophils within 2 h (Test and Weiss 1986). Activated macrophages also express high levels of MPO at chronic inflammatory sites such as in atheroma (Nicholls and Hazen 2005; Lau and Baldus 2006). Murine and human S100A8 are exceptionally sensitive to oxidation of their single, conserved Cys residue (Harrison et al. 1999). Mild oxidation with Cu2+ or low levels of H2O2 generate disulfide-linked dimers and activated neutrophils rapidly convert it to this form (Harrison et al. 1999). Intra- (between Lys34/35 and Cys41) and intermolecular (between Lys6, Lys76, Lys83 Lys87 and Cys41) sulfinamide bonds are generated with as little as 1:1 M equivalents of hypochlorite (HOCl) in vitro (Raftery and Geczy 2002; McCormick et al. 2005) and from PMA-stimulated neutrophils (Raftery et al. 2001). Covalent sulfinamide bonds are formed between thiol groups in Cys residues and the ε-amine group of Lys residues and, unlike disulfide bonds, cannot be broken by normal cellular reduction systems or the commonly used chemical reductant dithiothreitol (DTT) (Raftery et al. 2001). Some methionine residues in both proteins are also oxidized to methionine sulfoxides (Harrison et al. 1999). These processes are more fully covered in a recent review (Lim et al. 2009).
S100A8 and S100A9 are abundant at inflammatory sites and their sensitivity to modification by ROS/RNS may be important in protecting oxidant-sensitive proteins when oxidants are generated following phagocyte activation. In the context of atherosclerosis, oxidation by HOCl promotes aggregation of low-density lipoproteins (LDL), which are taken up more readily by macrophages than the unmodified form, facilitating formation of pro-atherogenic foam cells (Hazell and Stocker 1993). Lys and Met residues in apolipoprotein B are the main targets in LDL and these mediate aggregation, whereas lipid peroxidation is a quantitatively minor product (Hazell et al. 1994). An unsuspected oxidation pathway involving oxidative cross-linking of Cys and Lys residues was proposed to contribute to the pathogenesis of atherosclerosis (Fu et al. 2002). Appreciable amounts of aggregated LDL are generated with molar ratios HOCl to LDL, of 300:1, some 300-fold more than required for oxidative cross-linking of S100A8, which is induced by equimolar concentrations of HOCl (Raftery and Geczy 2002; McCormick et al. 2005). This exquisite sensitivity indicates that S100A8, and to a lesser extent S100A9, may protect against excessive oxidation in inflammatory conditions where activated neutrophils/macrophages express high levels of MPO, a property that may protect the host against severe tissue damage. DTT-stable cross-linked complexes (dimeric S100A82, S100A92, S100A8/A9 and S100A8/A92) were found in extracts from human atherosclerotic lesions (McCormick et al. 2005), indicating that this could be the case.
Leukocyte migration
Transmigration to inflammatory sites involves the process of rolling of leukocytes on activated endothelial cell surfaces mediated by binding of endothelial selectins to glycoprotein and glycolipid ligands on leukocytes (Sperandio 2006). Leukocyte integrins bind vascular cell adhesion molecules, thereby mediating tight adherence and immobilizing leukocytes, and allow subsequent transmigration through the endothelial layer toward chemotactic signals (Petri and Bixel 2006; Petri et al. 2008).
mS100A8 was the first S100 family member described as having potent chemotactic activity for murine neutrophils and monocytes and was initially named chemotactic protein 10 kDa (CP10) to reflect its function and molecular mass (Lackmann et al. 1992). Since then, several S100 proteins have been implicated in modulating migration of various cell types (Table 4). One feature is their potency; mS100A8 has maximal chemotactic activity for neutrophils and monocytes at 10−11 to 10−13 M and, in contrast to other chemoattractants (e.g. fMLP, IL-8), does not cause a Ca2+ flux or induce changes in integrin or L-selectin expression (Cornish et al. 1996). It causes profound shape changes in monocytes and may mediate initial events in diapedesis that are independent of changes in integrin expression (Cornish et al. 1996). These features are similar to those described for TGF-β, which is also considered to have anti-inflammatory functions (see (Cornish et al. 1996)).
Injection of mS100A8 into murine foot pads stimulates leukocyte recruitment (Lackmann et al. 1993) with kinetics typical of delayed-type hypersensitivity reactions elicited by injection of antigen into a sensitized animal, and with early recruitment of neutrophils followed by monocytes over 24 h (Devery et al. 1994; Lau et al. 1995). The mechanism through which mS100A8 exerts this function is unclear, but is possibly via a pertussis toxin-sensitive, G-protein-coupled receptor (Cornish et al. 1996).
Similar to mS100A8, S100A12 may regulate leukocyte trafficking by acting as a chemoattractant and influencing expression of adhesion molecules. It upregulates CD11b integrin expression and L-selectin shedding on neutrophils, which enhances their adhesion to ECs (Rouleau et al. 2003). S100A12 may also induce ICAM-1 and VCAM-1 expression on ECs, thereby ensuring the tight adhesion required for extravasation (Hofmann et al. 1999). S100A12 is chemotactic in vitro for monocytes (Miranda et al. 2001) and mast cells (Yan et al. 2008) and has weak activity for neutrophils, but not lymphocytes (Yang et al. 2001), with optimal activities of about 10−9 to 10−12 M. Because of the functional similarities in leukocyte trafficking, we suggest that S100A12 may represent the human homolog of mS100A8 (Ravasi et al. 2004).
The chemotactic properties of human S100A8 and S100A9 are less clear. Ryckman et al. demonstrated their chemotactic activity for human neutrophils (Ryckman et al. 2003a, b), although other studies (Newton and Hogg 1998) found no activity. Ryckman et al. (2003a, b) suggest that because the human proteins were active in the picomolar range, and inactivated by HOCl treatment, differences could be due to oxidation. At the concentration of HOCl used (100 μM), S100A8 forms intermolecular sulfinamide-linked complexes (Harrison et al. 1999). We found no activity of disulfide-bonded mS100A8 dimers (Harrison et al. 1999), or of cross-linked complexes, although very mild oxidation with HOCl stabilized its function (Raftery et al. 2001). The hinge region of mS100A8 is responsible for its chemotactic acitivity (see below). The structure of human S100A8 is shown in Fig. 1 and the Cys and Lys residues highlighted are conserved in mS100A8. Figure 1 shows that Cys41 lies adjacent to the hinge region (colored red) and on opposite sides of the dimer. Disulfide bond formation may sterically hinder access of the hinge to its receptor, whereas the Lys34,35 are in closer proximity to Cys41 and intrachain sulfinamide bond formation may not disrupt access to the hinge and would stabilize the molecule.

Ribbon diagram structure of the S100A8 homodimer. The S100A8 homodimer is shown as a ribbon diagram with each subunit colored green or blue and the hinge regions colored red. Side chains forming inter- and intramolecular sulfinamide bonds are shown on both subunits, but labeled on only one. Images were produced with PyMOL and are based on the crystal structure of human S100A8 (PDB accession # 1MR8)
Others report a fugetactic (cell repulsion) activity of S100A8 and S100A9 for human neutrophils (Sroussi et al. 2006, 2007), which in S100A9 was inhibited by oxidation of Met63 and Met83 (Sroussi et al. 2007). Thus, it is clear that oxidation can alter S100A8 and S100A9 functions and may regulate their role in leukocyte recruitment. Moreover, these studies stress the requirements for appropriate storage and testing of these proteins for potential oxidative modifications before functional assays are performed. In contrast to S100A8 and S100A9, the lack of Cys and Met residues in S100A12 would make it more resistant to modification by oxidation and allow it to function in oxidative conditions typical of inflammatory sites.
S100A8 and S100A9 may also contribute to leukocyte trafficking by influencing integrin expression and affinity. One study showed that S100A9 induces the high-affinity epitope of the β2 integrin CD11b on neutrophils, without causing shedding of L-selectin or enhanced CD11b expression; this activity was negatively regulated by formation of the heterodimer with S1008 (Newton and Hogg 1998). However, another report (Ryckman et al. 2003a, b) showed that S100A8, S100A9 and S100A8/A9 cause l-selectin (CD62L) shedding and enhance CD11b expression on monocytes and neutrophils in addition to induction of its high-affinity epitope. Because of its ability to enhance CD11b affinity, S100A9 facilitates adhesion of neutrophils to fibrinogen (Ryckman et al. 2003a, b) and fibronectin (Anceriz et al. 2007), and of monocytes to EC (Eue et al. 2000a, b) in vitro. Taken together, these data suggest that S100A9 modulates leukocyte adhesion to activated endothelium at sites of inflammation. However, this may be regulated by formation of the S100A8/S100A9 complex and, because the complex has been seen on endothelium in areas of contact between adherent leukocytes in vivo (Robinson et al. 2002), the significance of these observations is unclear.
S100A9−/− neutrophils have reduced capacity to migrate through an EC monolayer in response to IL-8 or leukotriene B4, and in contrast to wild-type cells, surface expression of CD11b was not increased by IL-8 in vitro (Manitz et al. 2003). These results, together with the reduced Ca2+ transients observed after stimulation of S100A9−/− neutrophils with several chemokines (McNeill et al. 2007), indicate a role for S100A9 in response to chemotactic signals. It is proposed that S100A8/A9 regulates polymerization of the microtubule network in response to elevated [Ca2+]i, an event antagonized by p38 MAP kinase-dependent phosphorylation of S100A9 (Vogl et al. 2004). S100A8 is the active subunit and phosphorylation of S100A9 may modulate this, and deficiencies in S100A9−/− leukocyte migration may be due to dysregulation of cytoskeletal dynamics.
Some in vivo models also support a role for S100A8/A9 in leukocyte migration. In murine air pouch models, blockade of S100A8 and S100A9 by specific antibodies significantly suppressed neutrophil migration in response to LPS or monosodium urate crystals (Ryckman et al. 2003a, b; Vandal et al. 2003). Human S100A8/A9 injected into the murine air pouch recruits neutrophils, with maximal numbers 12-h post-injection, an effect independent of TLR-4 (Ryckman et al. 2003a, b). In S100A9-null mice, no differences in the accumulation or time course of leukocytes recruited in thioglycollate-induced peritonitis to the air pouch following LPS administration (Hobbs et al. 2003) were seen, whereas neutrophil infiltration into skin wounds (Vogl et al. 2004) and into the pancreas in a model of caerulein-induced pancreatitis (Schnekenburger et al. 2008) was reduced. Several responses of S100A9−/− neutrophils appear to be stimulus dependent; for example, the oxidative burst generated by S100A9−/− neutrophils stimulated with phorbol esters is similar to wild-type cells (Hobbs et al. 2003) whereas responses to ATP or platelet-activating factor are reduced (Nacken et al. 2005). It is proposed that S100A9 may regulate a specific signaling pathway, possibly diacylglycerol signaling (McNeill et al. 2007), and may explain why leukocyte influx is disrupted in some in vivo models while appearing normal in others.
The C-terminal region of S100A9 is identical to neutrophil immobilizing factor (NIF) (Freemont et al. 1989) that inhibits neutrophil migration and chemotaxis in vitro (Goetzl and Austen 1972; Goetzl et al. 1973). The question of whether NIF is proteolytically cleaved from S100A9 was posed over 15 years ago (Freemont et al. 1989). Unpublished results from our laboratory indicate that NIF is efficiently produced when S100A9 in digested with α-chymotrypsin, and to a lesser extent with cathepsin G (L. Miranda, M. Raftery & C.Geczy, unpublished data). This suggests that granular contents comprising numerous proteases from activated neutrophils (Raptis and Pham 2005) could generate NIF and this may represent one means of limiting neutrophil influx into inflammatory lesions. A C-terminal domain peptide within this region of S100A9 (H92-H102) is implicated in regulation of neuropathic pain (Paccola et al. 2008) and inhibits spreading and mannose-dependent phagocytic activity of adherent peritoneal macrophages (Pagano et al. 2005), indicating a regulatory role for this peptide in macrophage function.
Mast cell regulation
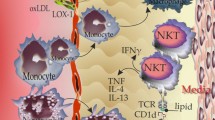
S100A12 was recently identified as a novel mast cell activator (Yang et al. 2007). In addition to a potential role in allergic inflammation, this function may have important ramifications in infection and in chronic inflammation such as in RA, IBD and atherosclerosis, where S100A12 is expressed and mast cell activation is implicated in pathogenesis, although endogenous activators in these circumstances are unclear [reviewed in (Galli et al. 2005; Bischoff 2007)]. S100A12 provoked degranulation of murine and human mucosal and tissue mast cells and significantly potentiated IgE-mediated degranulation (Yang et al. 2007). This is an important observation considering the functional heterogeneity of mast cells from different species (murine and human) and tissues (connective tissue and mucosal types) (Gurish and Austen 2001; Bischoff 2007). In marked contrast to its inability to induce pro-inflammatory cytokines in human monocytes/macrophages, S100A12 induced IL-6, IL-8, MCP-1 and MIP-1β from human cord blood-derived mast cells (Yang et al. 2007). Injection of S100A12 caused edema and leukocyte recruitment in mice; responses were not apparent in mast cell-deficient mice or wild-type mice injected with the mast cell stabilizer sodium cromoglycate. Leukocyte infiltration in mast cell-deficient mice was restored when these mice were reconstituted with bone marrow-derived mast cells from wild-type mice (Yang et al. 2007). Our recent studies show that S100A12 is also a mast cell chemoattractant in vitro and can sequester these cells when injected intraperitoneally (Yan et al. 2008). We propose that mast cells are a key target of S100A12 and their localized sequestration and activation would generate chemokines, histamine and cytokines (e.g., TNFα) that promote activation of the vasculature, which is necessary for more sustained leukocyte recruitment, particularly of monocytes.
Importantly, potentiation of IgE-mediated mast cell activation indicates a primary role in allergic inflammation. S100A12 is expressed by macrophages in the vicinity of tryptase-positive mast cells and in eosinophils in the airways of lung tissue of patients with asthma, but not in the normal lung. It is elevated in the sputum of patients with eosinophil asthma (Yang et al. 2007) indicating its release.
S100A8 is also implicated in the regulation of mast cell activation by acting as an NO shuttle (Lim et al. 2008). By covalent coupling to cysteine residues in particular proteins, NO mediates numerous processes including vascular homeostasis, smooth muscle function, neuronal development, anti-microbial defense and regulation of transcription and apoptosis via S-nitrosylation (the covalent coupling of NO to Cys residues) (Jaffrey et al. 2001). Relatively few proteins are targets for S-nitrosylation and specificity of this modification may be conferred by a local hydrophobic environment and acidic and basic residues in proximity to the Cys, which are important for reducing the thiol pKa (Perez-Mato et al. 1999; Hess et al. 2005). We showed that S100A8 is readily S-nitrosylated on Cys41 (Lim et al. 2008), which is conserved in human and murine S100A8, flanked by Glu40 and Arg46 residues, and lies within the hydrophobic pocket formed by the hinge and C-terminal regions. S100A8 is preferentially nitrosylated in the S100A8/A9 complex; S100A8–SNO was detected in normal human neutrophils and the levels increased in neutrophils treated with NO donors (Lim et al. 2008). The S100A8–SNO bond is relatively stable and able to trans-nitrosylate hemoglobin indicating that it may act as an NO shuttle. Importantly, S100A8–SNO inhibited mast cell degranulation stimulated by compound 48/80 in vitro, and leukocyte adhesion and transmigration triggered by mast cell activation in the mesenteric circulation of the rat (Lim et al. 2008). Since S-nitrosylated S100A8 can act as an NO donor, it may regulate other NO-dependent functions by trans-nitrosylating other NO targets. These properties indicate important functions of blood reflow in the microcirculation in resolving inflammatory lesions or in neovessels where S100A8 and S100A9 are expressed (McCormick et al. 2005).
Structure–function studies
S100s contain a low-affinity S100-specific N-terminal and a high affinity canonical C-terminal EF-hand Ca2+-binding domain (Gribenko and Makhatadze 1998; Heizmann and Cox 1998; Zimmer et al. 2003). The two EF hands are encoded by separate exons with an intron invariably splitting the coding sequence at the ‘hinge’ region between the two Ca2+-binding motifs. Thus, an S100 can bind two Ca2+ ions, each with different affinities. This may be functionally important in second messenger roles, since the lower Ca2+ affinity of the N-terminal EF hand indicates that it may only be occupied when intracellular Ca2+ levels are elevated. Moreover, the Ca2+ concentration in the extracellular space is sufficiently high to ensure that S100s remain in the Ca2+-loaded state (Heizmann and Cox 1998), suggesting this as the functionally relevant form in those S100 proteins with extracellular roles.
Under physiological conditions, S100s exist as non-covalent antiparallel dimers held together by hydrophobic interactions, with hydrophobic residues within α-helices I and IV predominantly responsible for the dimerization interface. S100s typically form homodimers, although heterodimers, trimers and tetramers of S100A8/A9 (Hunter and Chazin 1998; Propper et al. 1999; Vogl et al. 2006) and hexamers of S100A12 (Xie et al. 2007; Moroz et al. 2009) are reported. The calcium-binding domains within the EF hands contribute to intermolecular stability of the S100A8/A9 complexes (Vogl et al. 1999a, b). Heterodimerization is independent of the extended C-terminal domain of S100A9 (Champaiboon et al. 2009). Recent studies mutating glutamic acid residues within the Ca2+-binding domains of S100A9 ablated resistance to bacterial invasion of a keratinocyte cell line over-expressing the S100A8/S100A9 complex. This directly confirmed the functional importance of EF-hand domains in resistance, by controlling the number of bound bacteria that could be internalized by the cells. A loss of the Ca2+-induced negative molecular surface to a positive face in these mutants is proposed to alter its binding properties to cytoskeletal components, such as negatively charged tubulin that is involved in bacterial invasion (Champaiboon et al. 2009).
Some functions of S100A8 and S100A9 are negatively regulated by, and others dependent on, heterocomplex formation (see Table 2), indicating that homo-/heterodimer formation may be an important regulatory mechanism. Since dimerization generates two binding sites on opposite sides of each monomer, S100 proteins could bridge target proteins. In the case of heterodimers, two different target proteins may be brought together in close proximity, a property that may facilitate the function of S100 proteins (Donato 2003). For example, this could conceivably be involved in the Ca2+-dependent assembly of enzyme complexes, such as the role proposed for S100A8/A9 in the assembly of a functional nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex in phagocytes (Kerkhoff et al. 2005).
Amino acid sequences within the EF-hand regions of S100 proteins are generally highly conserved, whereas those within the hinge and C-terminal regions are the most divergent. Crystal and nuclear magnetic resonance (NMR) structures indicate that these regions form a hydrophobic pocket in the calcium-bound state, mediated by contraction of the C-terminal, high-affinity EF hand around the Ca2+ ion, which moves helix III into a position more perpendicular to helix IV and opens the hinge region. In contrast, the N-terminal EF hand undergoes very little Ca2+-dependent conformational change (Smith and Shaw 1998; Heizmann et al. 2002; Zimmer et al. 2003). Structures of S100B, S100A10 and S100A11 bound to target peptides indicate that hydrophobic residues exposed in the hinge and C-terminus as a result of Ca2+-ligation may mediate binding to target proteins (McClintock and Shaw 2003; Zimmer et al. 2003). The sequence conservation within the EF-hand regions indicates that this mechanism may be common to all S100s, whereas the sequence differences within the hinge and C-terminal regions could allow a diversity of target protein interactions that may determine the functions attributed to different family members (Kligman and Hilt 1988; Kube et al. 1992; Lackmann et al. 1993; Schafer and Heizmann 1996; Seemann et al. 1996; Heizmann et al. 2002; Ravasi et al. 2004; Yan et al. 2008).
Our earlier studies demonstrated that the chemotactic activity of mS100A8 for monocytes and neutrophils in vitro and in vivo was not found with human S100A8, which has only 57% amino acid identity, posing the question of whether the two proteins were orthologs (Lackmann et al. 1993) and whether there was equivalent chemotactic peptide in humans. We subsequently isolated a functional S100A8 homolog from human neutrophils, S100A12, which had potent monocyte chemotactic activity (Miranda et al. 2001; Yang et al. 2001) but failed to identify S100A12 in rodent genomes (Ravasi et al. 2004). The functional and sequence divergence suggested complex evolution of the S100 family in mammals. A synthetic peptide with sequence identical to the hinge region of mS100A8 (amino acids 42–55, mS100A842–55) mimicked the chemotactic activity of the full-length protein and was the first demonstration of an active hinge domain in the S100 protein family (Lackmann et al. 1993). However, synthetic mS100A842–55 elicited only transient neutrophil recruitment in vivo, indicating other essential structural domains.
In contrast to the positive activity of the murine hinge domain, an equivalent peptide segment corresponding to the hinge sequence of hS100A8 had no chemotactic activity (Lackmann et al. 1993). Comparisons of the amino acid sequences of m/h S100A8 and S100A12 are given in Fig. 1. Using structural models of the three proteins, we found that the S100A12 and hS100A8 templates produced successful models for mS100A8, although the total amino acid identity between hS100A8 and mS100A8 was higher than between S100A12 and mS100A8 (32%). Because of higher overall identity and the high surface hydrophobicity evident in the S100A12 model, the hS100A8 template was considered to be more accurate. However, the hinge regions of human and murine S100A8 have low homology, particularly in the C-terminal half. In the human S100A8 hinge, Gly49 would allow a level of flexibility not afforded by Asp49 in the mouse protein and consequently the murine hinge is unlikely to form the same kinked conformation described for hS100A8 (Ishikawa et al. 2000). Electrostatic potential maps indicated that the hinge regions of mS100A8 and S100A12 were structurally more similar. In contrast to the hinge regions of S100A12 and mS100A8, which are electro-negative, this region in hS100A8 is positively charged.
We next prepared a synthetic peptide based on the S100A12 hinge sequence, S100A1238–53, and showed that, like the full-length protein, this was chemotactic for human THP-1 monocytoid cells and for human monocytes (Ravasi et al. 2004). Because S100A12 consistently provokes THP-1 cell migration in a biphasic manner with two prominent peaks at 10−9 and 10−12 M, two receptors of different affinities are implicated. S100A1238–53 was also chemotactic at these concentrations, although somewhat less potent. Using alanine substitutions, we showed that Lys38, Leu40, Asn46, Ile47, Asp49, Lys50 and Ile53 were essential for monocyte chemotactic activity at 10−9 M, whereas Asn42 and Ile44 were important for activity at 10−12 M (Yan et al. 2008). In hydrophobic environments, the S100A1238–53 peptide adopts an α-helical conformation with the active hydrophobic residues on one side of the helix, suggesting that near the cell membrane (a hydrophobic environment), these residues may align to interact with a receptor (Yan et al. 2008). The hinge region of mS100A8 also has an α-helical structure in hydrophobic environments (Lazoura et al. 2000). As seen in Fig. 2, the sequence of S100A12’s hinge region is more similar to the hinge region of mS100A8 than the human protein, and Asn46 and Ile47 are conserved at similar positions in mS100A8 but not human S100A8, which may explain why the mS100A8 hinge region is chemotactically active, whereas the human S100A8 hinge is not (Yan et al. 2008).

Amino acid alignment of murine and human S100A8 and human S100A12. Positions of the helices, loops, hinge and C-terminal domains are indicated above the alignment. Residues involved in Zn2+-binding are highlighted in blue. Hydrophilic (orange) and hydrophobic (red) residues implicated in the chemotactic activity of S100A12 (and where they are conserved in human and murine S100A8) are also highlighted. Asterisk indicates a conserved residue, colon conservative substitution, dot weakly conservative substitution. Sequences used were: human S100A8 (P05109), murine S100A8 (P27005) and S100A12 (P80511)
The hinge region of S100A12 also mimicks the stimulation of mast cell degranulation and chemotactic activities of full-length S100A12. Hydrophobic residues (Ile44, Ile47 and Ile53) within this region are essential for monocyte chemotactic activity, mast cell activation and induction of edema in vivo. Mutation of Lys38 or Lys48 to Ala also reduced, but did not abolish edema caused by the S100A12 hinge region, indicating that these residues may contribute to the activity (Yan et al. 2008).
The redox-sensitive Cys residue in mS100A8 resides immediately before the hinge domain, but mutation to Ala did not alter its chemotactic activity in vitro or in vivo (Harrison et al. 1999). In keeping with its non-essential role in chemotaxis, S100A12 has no Cys residues (Fig. 2). In this respect, the chemotactic activity of S100A12, in contrast to mS100A8 (in which disulfide bond formation causes loss of activity), would be resistant to covalent modification by oxidants. S100A12 also has no Met residues and, because a number of classical chemoattractants are susceptible to oxidative inactivation (Harrison et al. 1999), S100A12 may be important in propagating monocyte migration into chronic inflammatory lesions such as in RA (Yang et al. 2001) and atherosclerosis (Goyette et al. 2009) where ROS are continuously generated.
The S100 protein family is a good example that amplification occurs by gene duplication followed by divergence from the original duplicated gene. We propose that a duplication and divergence of S100A8 in humans, compared to rodents, has permitted the separation of two functions. In mice, S100A8 is abundant and acts as an anti-oxidant, whereas it is chemotactic within the picomolar range, but its chemotactic activity can be compromised by sensitivity to ROS (Harrison et al. 1999). Interestingly, rabbit S100A12 has a single Cys residue at position 30, whereas porcine and bovine S100A12 have no Cys residues. In human, and in addition to the numerous functions ascribed to the S100A8/A9 complex, S100A8 may function as an anti-oxidant, whereas S100A12 may chiefly act as a monocyte and mast cell chemoattractant and mast cell activator (Yang et al. 2001, 2007, 2008).
The absence of the S100A12 gene in rodents makes some studies using these in model systems difficult to interpret, particularly with neutralizing antibodies (Hofmann et al. 1999), and to relate particular functions exhibited by the human proteins to the calgranulins in these species. Moreover, the differences in expression patterns of murine and human S100A9 in activated monocytes/macrophages (Hessian et al. 1993; Santhanagopalan et al. 1995; Hu et al. 1996; Xu and Geczy 2000; Xu et al. 2001; Hsu et al. 2005) indicate important functional differences and question the roles of the S100A8/A9 complex in chronic inflammation in mice. Moreover, there are other important considerations. For example, we (McCormick et al. 2005) and others (Eue et al. 2000a, b) found no S100A8 and little S100A9 in murine models of atheroma. Interestingly, and in keeping with a protective oxidant-scavenging role, activated murine macrophages produce little MPO, making the lesion less prone to HOCl oxidation. Thus, studies of these proteins in chronic inflammation in mice should be interpreted with caution.
Roles of zinc binding
Divalent cations such as Zn2+ and Cu2+ may regulate extracellular S100 functions, particularly because Zn2+-binding causes structural changes that could influence ligand/receptor binding (Vogl et al. 2006; Moroz et al. 2009). The putative Zn2+-binding sites are structurally conserved in the calgranulins (see Fig. 2). Ca2+ and Zn2+, alone or together, induce heterotetramer formation of S100A8/A9 (Vogl et al. 2006) and oligomerization of S100A12, although in this case Zn2+ is more effective than Ca2+ (Goyette et al. 2009; Moroz et al. 2009). Evidence from chemical cross-linking, NMR, hydrophobic probe and intrinsic Tyr fluorescence experiments suggests that the structures of Zn2+-, Ca2+- and Ca2+/Zn2+-bound S100A12 complexes are subtly different (Goyette et al. 2009; Moroz et al. 2009). Zn2+ is required for S100A8/A9 to bind to a human microvascular EC line (Robinson et al. 2002) and more S100A12 is bound to the surface of human gastric carcinoma MKN74 cells in the presence of Zn2+ than in its absence (Moroz et al. 2009).
If the Zn2+-bound forms of the calgranulins indeed have different functions, this may explain some of the contradictory reports in literature, particularly as different culture media have different levels of Zn2+ (Moroz et al. 2009), and this aspect warrants clarification. Furthermore, the physiological relevance of Zn2+-binding has been unclear. Our demonstration of S100A12–Zn2+ complexes in human atheroma using an antibody that only recognizes Zn2+-dependent S100A12 complexes is the first demonstration of Zn2+-dependent calgranulin oligomers in vivo (Goyette et al. 2009). Since S100A12 inhibited matrix metalloproteinases (MMPs) by chelating Zn2+, these complexes may form as a consequence of inhibiting the large amount of MMPs, particularly MMP-9 produced in human atheroma (Goyette et al. 2009). S100A8 and S100A9 can also suppress MMP activity in a similar manner (Isaksen and Fagerhol 2001), implicating these processes in the protection of vulnerable plaque from rupture.
Zn2+ is implicated in the apoptotic activity of S100A8/A9 (Yui et al. 1995, 1997, 2002), suggesting structural changes that regulate function, although additional mechanisms probably involving receptor-mediated activation and generation of oxidants are also reported (Ghavami et al. 2004, 2008a, b). S100A8/A9 (known as calprotectin) also has broad-spectrum anti-microbial activity against numerous micro-organisms, particularly fungi, and can promote resistance to bacterial invasion (Steinbakk et al. 1990; Murthy et al. 1993; Sohnle et al. 2000; Champaiboon et al. 2009). Anti-microbial activity was proposed to be mediated by the ability of the complex to bind and control the levels of free Zn2+, an essential microbial nutrient (Steinbakk et al. 1990; Sohnle et al. 2000; Lusitani et al. 2003), although this is still contentious, particularly because S100A8/A9 may precipitate when Zn2+ is in excess (Viemann et al. 2007). The crystal structure of the S100A8/S100A9 heterotetramer indicates burial of more hydrophobic residues, particularly in S100A9, leading to a dimer of heterodimers with major contributions from the N-terminal EF hands and the C-termini. This allows the emergence of two putative Zn2+-binding sites at the subunit interface (Korndorfer et al. 2007). The C-terminal region of S100A9 contains three consecutive His residues and a His91-x-x-x-His95 motif that may mediate Zn2+-binding. An essential role for S100A9 is implicated and studies using 65Zn indicate that Zn2+-depletion is an unlikely mechanism and favor the proposal that Zn2+-dependent conformational changes may mediate activity (Murthy et al. 1993). In keeping with alternate/additional mechanisms, the anti-fungal activity of S100A8/A9 is abolished when Cys42 of S100A8 or Met63 or Met83, but not Met81, of S100A9 are mutated (Sroussi et al. 2009). Although the precise mechanisms need clarification, oxidation of these residues may contribute to their anti-microbial function.
Interestingly, S100A7 and the C-terminal domain of S100A12 have anti-microbial activity against Gram-negative bacteria (Glaser et al. 2005). These proteins do not have extended C-terminal domains containing three His residues, although both contain the His-x-x-x-His motifs. The anti-microbial activity of the C-terminal-derived S100A12 peptide is enhanced by Zn2+ (Cole et al. 2001), indicating that structural changes mediated by Zn2+ binding may modulate this activity. In contrast, Zn2+ does not mediate the antibacterial activity of S100A7 as it remains active in the presence of Zn2+, and deletion of the C-terminus had little effect on activity (Lee and Eckert 2007).
Concluding remarks
The calgranulins are highly expressed in acute and chronic inflammatory conditions, although their exact contribution to the inflammatory process remains elusive. Although these proteins are commonly labeled as ‘pro-inflammatory’, a comprehensive assessment of literature suggests that the calgranulins may play pleiotropic roles and this review attempts to highlight some of their often overlooked, protective functions. The mechanisms regulating the relative pro- or anti-inflammatory/protective roles of the calgranulins warrant more research. We propose that oxidative modifications, proteolytic cleavage to release active peptides, Zn2+-binding and complex formation may be important factors in functional diversity. Receptor-mediated functions may be governed by glycosylation of the receptor and requirements for co-receptors and/or co-stimuli. A deeper understanding of the consequences of these modifications to calgranulin function and the receptors mediating these effects may explain some of the seemingly incongruent functions proposed for these proteins.
Abbreviations
- RA:
-
Rheumatoid arthritis
- IBD:
-
Inflammatory bowel disease
- ROS:
-
Reactive oxygen species
- NO:
-
Nitric oxide
- TLR:
-
Toll-like receptor
- mS100A8:
-
Murine S100A8
- mS100A9:
-
Murine S100A9
- TNFα:
-
Tumor necrosis factor α
- TGFβ:
-
Transforming growth factor β
- IFN:
-
Interferon
- LPS:
-
Lipopolysaccharide
- IL:
-
Interleukin
- COX-2:
-
Cyclo-oxygenase 2
- cAMP:
-
Cyclic adenosine monophosphate
- MAP kinase:
-
Mitogen-activated protein kinase
- EC:
-
Endothelial cells
- FGF:
-
Fibroblast growth factor
- GC:
-
Glucocorticoids
- DEX:
-
Dexamethasone
- PPAR-γ:
-
Peroxisome proliferator-activated receptor-γ
- RAGE:
-
Receptor for advanced glycation end products
- NFκB:
-
Nuclear factor κB
- EN-RAGE:
-
Extracellular newly identified RAGE-binding protein
- AGE:
-
Advanced glycation end products
- MCP-1:
-
Monocyte chemotactic protein 1
- NIF:
-
Neutrophil immobilizing factor
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- NMR:
-
Nuclear magnetic resonance
- MPO:
-
Myeloperoxidase
- MMP:
-
Matrix metalloproteinase
References
Adami C, Bianchi R, Pula G, Donato R (2004) S100B-stimulated NO production by BV-2 microglia is independent of RAGE transducing activity but dependent on RAGE extracellular domain. Biochim Biophys Acta 1742:169–177
Aguiar-Passeti T, Postol E, Sorg C, Mariano M (1997) Epithelioid cells from foreign-body granuloma selectively express the calcium-binding protein MRP-14, a novel down-regulatory molecule of macrophage activation. J Leukoc Biol 62:852–858
Akiyama H, Ikeda K, Katoh M, McGeer EG, McGeer PL (1994) Expression of MRP14, 27E10, interferon-alpha and leukocyte common antigen by reactive microglia in postmortem human brain tissue. J Neuroimmunol 50:195–201
Akpek EK, Liu SH, Thompson R, Gottsch JD (2002) Identification of paramyosin as a binding protein for calgranulin C in experimental helminthic keratitis. Invest Ophthalmol Vis Sci 43:2677–2684
Anceriz N, Vandal K, Tessier PA (2007) S100A9 mediates neutrophil adhesion to fibronectin through activation of beta2 integrins. Biochem Biophys Res Commun 354:84–89
Bai B, Yamamoto K, Sato H, Sugiura H, Tanaka T (2007) Complex regulation of S100A8 by IL-17, dexamethasone, IL-4 and IL-13 in HaCat cells (human keratinocyte cell line). J Dermatol Sci 47:259–262
Baldassarre ME, Altomare MA, Fanelli M, Carbone D, Di Bitonto G, Mautone A, Laforgia N (2007) Does calprotectin represent a regulatory factor in host defense or a drug target in inflammatory disease? Endocr Metab Immune Disord Drug Targets 7:1–5
Basso D, Greco E, Fogar P, Pucci P, Flagiello A, Baldo G, Giunco S, Valerio A, Navaglia F, Zambon CF, Falda A, Pedrazzoli S, Plebani M (2006) Pancreatic cancer-derived S-100A8 N-terminal peptide: a diabetes cause? Clin Chim Acta 372:120–128
Bausinger H, Lipsker D, Ziylan U, Manie S, Briand JP, Cazenave JP, Muller S, Haeuw JF, Ravanat C, de la Salle H, Hanau D (2002) Endotoxin-free heat-shock protein 70 fails to induce APC activation. Eur J Immunol 32:3708–3713
Berntzen HB, Fagerhol MK (1988) L1, a major granulocyte protein: antigenic properties of its subunits. Scand J Clin Lab Invest 48:647–652
Bianchi R, Adami C, Giambanco I, Donato R (2007) S100B binding to RAGE in microglia stimulates COX-2 expression. J Leukoc Biol 81:108–118
Bianchi R, Giambanco I, Donato R (2008) S100B/RAGE-dependent activation of microglia via NF-kappaB and AP-1 Co-regulation of COX-2 expression by S100B, IL-1beta and TNF-alpha. Neurobiol Aging
Bierhaus A, Humpert PM, Morcos M, Wendt T, Chavakis T, Arnold B, Stern DM, Nawroth PP (2005) Understanding RAGE, the receptor for advanced glycation end products. J Mol Med 83:876–886
Bischoff SC (2007) Role of mast cells in allergic and non-allergic immune responses: comparison of human and murine data. Nat Rev Immunol 7:93–104
Boyd JH, Kan B, Roberts H, Wang Y, Walley KR (2008) S100A8 and S100A9 mediate endotoxin-induced cardiomyocyte dysfunction via the receptor for advanced glycation end products. Circ Res 102:1239–1246
Bozinovski S, Cross M, Vlahos R, Jones JE, Hsuu K, Tessier PA, Reynolds EC, Hume DA, Hamilton JA, Geczy CL, Anderson GP (2005) S100A8 chemotactic protein is abundantly increased, but only a minor contributor to LPS-induced, steroid resistant neutrophilic lung inflammation in vivo. J Proteome Res 4:136–145
Brun JG, Haland G, Haga HJ, Fagerhol MK, Jonsson R (1995) Effects of calprotectin in avridine-induced arthritis. Apmis 103:233–240
Champaiboon C, Sappington KJ, Guenther BD, Ross KF, Herzberg MC (2009) Calprotectin S100A9 calcium-binding loops I and II are essential for keratinocyte resistance to bacterial invasion. J Biol Chem 284:7078–7090
Cole AM, Kim YH, Tahk S, Hong T, Weis P, Waring AJ, Ganz T (2001) Calcitermin, a novel antimicrobial peptide isolated from human airway secretions. FEBS Lett 504:5–10
Coleman N, Stanley MA (1994) Expression of the myelomonocytic antigens CD36 and L1 by keratinocytes in squamous intraepithelial lesions of the cervix. Hum Pathol 25:73–79
Cornish CJ, Devery JM, Poronnik P, Lackmann M, Cook DI, Geczy CL (1996) S100 protein CP-10 stimulates myeloid cell chemotaxis without activation. J Cell Physiol 166:427–437
Couper KN, Blount DG, Riley EM (2008) IL-10: the master regulator of immunity to infection. J Immunol 180:5771–5777
Dale CS, Goncalves LR, Juliano L, Juliano MA, da Silva AM, Giorgi R (2004) The C-terminus of murine S100A9 inhibits hyperalgesia and edema induced by jararhagin. Peptides 25:81–89
Dale CS, Pagano Rde L, Paccola CC, Pinotti-Guirao T, Juliano MA, Juliano L, Giorgi R (2006) Effect of the C-terminus of murine S100A9 protein on experimental nociception. Peptides 27:2794–2802
Delabie J, de Wolf-Peeters C, van den Oord JJ, Desmet VJ (1990) Differential expression of the calcium-binding proteins MRP8 and MRP14 in granulomatous conditions: an immunohistochemical study. Clin Exp Immunol 81:123–126
Devery JM, King NJ, Geczy CL (1994) Acute inflammatory activity of the S100 protein CP-10. Activation of neutrophils in vivo and in vitro. J Immunol 152:1888–1897
Donato R (2003) Intracellular and extracellular roles of S100 proteins. Microsc Res Tech 60:540–551
Eckert RL, Broome AM, Ruse M, Robinson N, Ryan D, Lee K (2004) S100 proteins in the epidermis. J Invest Dermatol 123:23–33
Edgeworth J, Gorman M, Bennett R, Freemont P, Hogg N (1991) Identification of p8, 14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. J Biol Chem 266:7706–7713
Ehlermann P, Eggers K, Bierhaus A, Most P, Weichenhan D, Greten J, Nawroth PP, Katus HA, Remppis A (2006) Increased proinflammatory endothelial response to S100A8/A9 after preactivation through advanced glycation end products. Cardiovasc Diabetol 5:6
Endoh Y, Chung YM, Clark IA, Geczy CL, Hsu K (2009) IL-10-dependent S100A8 gene induction in monocytes/macrophages by double-stranded RNA. J Immunol 182:2258–2268
Eue I, Langer C, Eckardstein A, Sorg C (2000a) Myeloid related protein (MRP) 14 expressing monocytes infiltrate atherosclerotic lesions of ApoE null mice. Atherosclerosis 151:593–597
Eue I, Pietz B, Storck J, Klempt M, Sorg C (2000b) Transendothelial migration of 27E10 + human monocytes. Int Immunol 12:1593–1604
Eversole LR, Miyasaki KT, Christensen RE (1992) The distribution of the antimicrobial protein, calprotectin, in normal oral keratinocytes. Arch Oral Biol 37:963–968
Eversole LR, Miyasaki KT, Christensen RE (1993) Keratinocyte expression of calprotectin in oral inflammatory mucosal diseases. J Oral Pathol Med 22:303–307
Foell D, Kucharzik T, Kraft M, Vogl T, Sorg C, Domschke W, Roth J (2003) Neutrophil derived human S100A12 (EN-RAGE) is strongly expressed during chronic active inflammatory bowel disease. Gut 52:847–853
Foell D, Frosch M, Sorg C, Roth J (2004a) Phagocyte-specific calcium-binding S100 proteins as clinical laboratory markers of inflammation. Clin Chim Acta 344:37–51
Foell D, Hernandez-Rodriguez J, Sanchez M, Vogl T, Cid MC, Roth J (2004b) Early recruitment of phagocytes contributes to the vascular inflammation of giant cell arteritis. J Pathol 204:311–316
Foell D, Wittkowski H, Vogl T, Roth J (2007) S100 proteins expressed in phagocytes: a novel group of damage-associated molecular pattern molecules. J Leukoc Biol 81:28–37
Foell D, Wittkowski H, Roth J (2009) Monitoring disease activity by stool analyses: from occult blood to molecular markers of intestinal inflammation and damage. Gut 58:859–868
Freemont P, Hogg N, Edgeworth J (1989) Sequence identity. Nature 339:516
Frosch M, Strey A, Vogl T, Wulffraat NM, Kuis W, Sunderkotter C, Harms E, Sorg C, Roth J (2000) Myeloid-related proteins 8 and 14 are specifically secreted during interaction of phagocytes and activated endothelium and are useful markers for monitoring disease activity in pauciarticular-onset juvenile rheumatoid arthritis. Arthritis Rheum 43:628–637
Fu X, Mueller DM, Heinecke JW (2002) Generation of intramolecular and intermolecular sulfenamides, sulfinamides, and sulfonamides by hypochlorous acid: a potential pathway for oxidative cross-linking of low-density lipoprotein by myeloperoxidase. Biochemistry 41:1293–1301
Gabrielsen TO, Dale I, Brandtzaeg P, Hoel PS, Fagerhol MK, Larsen TE, Thune PO (1986) Epidermal and dermal distribution of a myelomonocytic antigen (L1) shared by epithelial cells in various inflammatory skin diseases. J Am Acad Dermatol 15:173–179
Galli SJ, Nakae S, Tsai M (2005) Mast cells in the development of adaptive immune responses. Nat Immunol 6:135–142
Gao B, Tsan MF (2003a) Endotoxin contamination in recombinant human heat shock protein 70 (Hsp70) preparation is responsible for the induction of tumor necrosis factor alpha release by murine macrophages. J Biol Chem 278:174–179
Gao B, Tsan MF (2003b) Recombinant human heat shock protein 60 does not induce the release of tumor necrosis factor alpha from murine macrophages. J Biol Chem 278:22523–22529
Gebhardt C, Breitenbach U, Tuckermann JP, Dittrich BT, Richter KH, Angel P (2002) Calgranulins S100A8 and S100A9 are negatively regulated by glucocorticoids in a c-Fos-dependent manner and overexpressed throughout skin carcinogenesis. Oncogene 21:4266–4276
Gebhardt C, Nemeth J, Angel P, Hess J (2006) S100A8 and S100A9 in inflammation and cancer. Biochem Pharmacol 72:1622–1631
Geczy C (1996) Regulation and proinflammatory properties of the chemotactic protein, CP-10. Biochim Biophys Acta 1313:246–252
Ghavami S, Kerkhoff C, Los M, Hashemi M, Sorg C, Karami-Tehrani F (2004) Mechanism of apoptosis induced by S100A8/A9 in colon cancer cell lines: the role of ROS and the effect of metal ions. J Leukoc Biol 76:169–175
Ghavami S, Kerkhoff C, Chazin WJ, Kadkhoda K, Xiao W, Zuse A, Hashemi M, Eshraghi M, Schulze-Osthoff K, Klonisch T, Los M (2008a) S100A8/9 induces cell death via a novel, RAGE-independent pathway that involves selective release of Smac/DIABLO and Omi/HtrA2. Biochim Biophys Acta 1783:297–311
Ghavami S, Rashedi I, Dattilo BM, Eshraghi M, Chazin WJ, Hashemi M, Wesselborg S, Kerkhoff C, Los M (2008b) S100A8/A9 at low concentration promotes tumor cell growth via RAGE ligation and MAP kinase-dependent pathway. J Leukoc Biol 83:1484–1492
Glaser R, Harder J, Lange H, Bartels J, Christophers E, Schroder JM (2005) Antimicrobial psoriasin (S100A7) protects human skin from Escherichia coli infection. Nat Immunol 6:57–64
Goebeler M, Roth J, Burwinkel F, Vollmer E, Bocker W, Sorg C (1994) Expression and complex formation of S100-like proteins MRP8 and MRP14 by macrophages during renal allograft rejection. Transplantation 58:355–361
Goetzl EJ, Austen KF (1972) A neutrophil-immobilizing factor derived from human leukocytes, I: generation and partial characterization. J Exp Med 136:1564–1580
Goetzl EJ, Gigli I, Wasserman S, Austen KF (1973) A neutrophil immobilizing factor derived from human leukocytes, II: specificity of action on polymorphonuclear leukocyte mobility. J Immunol 111:938–945
Gordon S (2003) Alternative activation of macrophages. Nat Rev Immunol 3:23–35
Gottsch JD, Liu SH, Minkovitz JB, Goodman DF, Srinivasan M, Stark WJ (1995) Autoimmunity to a cornea-associated stromal antigen in patients with Mooren’s ulcer. Invest Ophthalmol Vis Sci 36:1541–1547
Gottsch JD, Eisinger SW, Liu SH, Scott AL (1999a) Calgranulin C has filariacidal and filariastatic activity. Infect Immun 67:6631–6636
Gottsch JD, Li Q, Ashraf F, O’Brien TP, Stark WJ, Liu SH (1999b) Cytokine-induced calgranulin C expression in keratocytes. Clin Immunol 91:34–40
Goyette J, Yan WX, Yamen E, Chung YM, Lim SY, Hsu K, Rahimi F, di Girolamo N, Song C, Jessup W, Kockx M, Bobryshev YV, Freedman SB, Geczy C (2009) Pleiotropic roles of S100A12 in coronary atherosclerotic plaque formation and rupture. J Immunol 183(1):593–603
Greenlee KJ, Corry DB, Engler DA, Matsunami RK, Tessier P, Cook RG, Werb Z, Kheradmand F (2006) Proteomic identification of in vivo substrates for matrix metalloproteinases 2 and 9 reveals a mechanism for resolution of inflammation. J Immunol 177:7312–7321
Gribenko AV, Makhatadze GI (1998) Oligomerization and divalent ion binding properties of the S100P protein: a Ca2 +/Mg2 + -switch model. J Mol Biol 283:679–694
Grimbaldeston MA, Geczy CL, Tedla N, Finlay-Jones JJ, Hart PH (2003) S100A8 induction in keratinocytes by ultraviolet A irradiation is dependent on reactive oxygen intermediates. J Invest Dermatol 121:1168–1174
Guignard F, Mauel J, Markert M (1995) Identification and characterization of a novel human neutrophil protein related to the S100 family. Biochem J 309(Pt 2):395–401
Gurish MF, Austen KF (2001) The diverse roles of mast cells. J Exp Med 194:F1–F5
Harrison CA, Raftery MJ, Walsh J, Alewood P, Iismaa SE, Thliveris S, Geczy CL (1999) Oxidation regulates the inflammatory properties of the murine S100 protein S100A8. J Biol Chem 274:8561–8569
Hasegawa T, Kosaki A, Kimura T, Matsubara H, Mori Y, Okigaki M, Masaki H, Toyoda N, Inoue-Shibata M, Kimura Y, Nishikawa M, Iwasaka T (2003) The regulation of EN-RAGE (S100A12) gene expression in human THP-1 macrophages. Atherosclerosis 171:211–218
Hayashi N, Kido J, Kido R, Wada C, Kataoka M, Shinohara Y, Nagata T (2007) Regulation of calprotectin expression by interleukin-1alpha and transforming growth factor-beta in human gingival keratinocytes. J Periodontal Res 42:1–7
Hazell LJ, Stocker R (1993) Oxidation of low-density lipoprotein with hypochlorite causes transformation of the lipoprotein into a high-uptake form for macrophages. Biochem J 290:165–172
Hazell LJ, van den Berg JJ, Stocker R (1994) Oxidation of low-density lipoprotein by hypochlorite causes aggregation that is mediated by modification of lysine residues rather than lipid oxidation. Biochem J 302:297–304
Healy AM, Pickard MD, Pradhan AD, Wang Y, Chen Z, Croce K, Sakuma M, Shi C, Zago AC, Garasic J, Damokosh AI, Dowie TL, Poisson L, Lillie J, Libby P, Ridker PM, Simon DI (2006) Platelet expression profiling and clinical validation of myeloid-related protein-14 as a novel determinant of cardiovascular events. Circulation 113:2278–2284
Heizmann CW, Cox JA (1998) New perspectives on S100 proteins: a multi-functional Ca(2 +)-, Zn(2 +)- and Cu(2 +)-binding protein family. Biometals 11:383–397
Heizmann CW, Fritz G, Schafer BW (2002) S100 proteins: structure, functions and pathology. Front Biosci 7:d1356–d1368
Hermani A, De Servi B, Medunjanin S, Tessier PA, Mayer D (2006) S100A8 and S100A9 activate MAP kinase and NF-kappaB signaling pathways and trigger translocation of RAGE in human prostate cancer cells. Exp Cell Res 312:184–197
Hess DT, Matsumoto A, Kim S-O, Marshall HE, Stamler JS (2005) Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol 6:150–166
Hessian PA, Edgeworth J, Hogg N (1993) MRP-8 and MRP-14, two abundant Ca(2 +)-binding proteins of neutrophils and monocytes. J Leukoc Biol 53:197–204
Hessian PA, Wilkinson L, Hogg N (1995) The S100 family protein MRP-14 (S100A9) has homology with the contact domain of high molecular weight kininogen. FEBS Lett 371:271–275
Hetland G, Talgo GJ, Fagerhol MK (1998) Chemotaxins C5a and fMLP induce release of calprotectin (leucocyte L1 protein) from polymorphonuclear cells in vitro. Mol Pathol 51:143–148
Hiratsuka S, Watanabe A, Aburatani H, Maru Y (2006) Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat Cell Biol 8:1369–1375
Hitomi J, Yamaguchi K, Kikuchi Y, Kimura T, Maruyama K, Nagasaki K (1996) A novel calcium-binding protein in amniotic fluid, CAAF1: its molecular cloning and tissue distribution. J Cell Sci 109(Pt 4):805–815
Hitomi J, Kimura T, Kusumi E, Nakagawa S, Kuwabara S, Hatakeyama K, Yamaguchi K (1998) Novel S100 proteins in human esophageal epithelial cells: CAAF1 expression is associated with cell growth arrest. Arch Histol Cytol 61:163–178
Hobbs JA, May R, Tanousis K, McNeill E, Mathies M, Gebhardt C, Henderson R, Robinson MJ, Hogg N (2003) Myeloid cell function in MRP-14 (S100A9) null mice. Mol Cell Biol 23:2564–2576
Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM (1999) RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 97:889–901
Hogg N, Allen C, Edgeworth J (1989) Monoclonal antibody 5.5 reacts with p8, 14, a myeloid molecule associated with some vascular endothelium. Eur J Immunol 19:1053–1061
Hsu K, Passey RJ, Endoh Y, Rahimi F, Youssef P, Yen T, Geczy CL (2005) Regulation of S100A8 by glucocorticoids. J Immunol 174:2318–2326
Hu SP, Harrison C, Xu K, Cornish CJ, Geczy CL (1996) Induction of the chemotactic S100 protein, CP-10, in monocyte/macrophages by lipopolysaccharide. Blood 87:3919–3928
Hunter MJ, Chazin WJ (1998) High level expression and dimer characterization of the S100 EF-hand proteins, migration inhibitory factor-related proteins 8 and 14. J Biol Chem 273:12427–12435
Ikemoto M, Murayama H, Itoh H, Totani M, Fujita M (2007) Intrinsic function of S100A8/A9 complex as an anti-inflammatory protein in liver injury induced by lipopolysaccharide in rats. Clin Chim Acta 376:197–204
Isaksen B, Fagerhol MK (2001) Calprotectin inhibits matrix metalloproteinases by sequestration of zinc. Mol Pathol 54:289–292
Ishikawa K, Nakagawa A, Tanaka I, Suzuki M, Nishihira J (2000) The structure of human MRP8, a member of the S100 calcium-binding protein family, by MAD phasing at 1.9 A resolution. Acta Crystallogr D Biol Crystallogr 56:559–566
Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH (2001) Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol 3:193–197
Jinquan T, Vorum H, Larsen CG, Madsen P, Rasmussen HH, Gesser B, Etzerodt M, Honore B, Celis JE, Thestrup-Pedersen K (1996) Psoriasin: a novel chemotactic protein. J Invest Dermatol 107:5–10
Kerkhoff C, Sorg C, Tandon NN, Nacken W (2001) Interaction of S100A8/S100A9-arachidonic acid complexes with the scavenger receptor CD36 may facilitate fatty acid uptake by endothelial cells. Biochemistry 40:241–248
Kerkhoff C, Hofmann HA, Vormoor J, Melkonyan H, Roth J, Sorg C, Klempt M (2002) Binding of two nuclear complexes to a novel regulatory element within the human S100A9 promoter drives the S100A9 gene expression. J Biol Chem 277:41879–41887
Kerkhoff C, Nacken W, Benedyk M, Dagher MC, Sopalla C, Doussiere J (2005) The arachidonic acid-binding protein S100A8/A9 promotes NADPH oxidase activation by interaction with p67phox and Rac-2. Faseb J 19:467–469
Kido J, Hayashi N, Kataoka M, Nagata T (2005) Calprotectin expression in human monocytes: induction by porphyromonas gingivalis lipopolysaccharide, tumor necrosis factor-alpha, and interleukin-1beta. J Periodontol 76:437–442
Klebanoff SJ (2005) Myeloperoxidase: friend and foe. J Leukoc Biol 77:598–625
Kligman D, Hilt DC (1988) The S100 protein family. Trends Biochem Sci 13:437–443
Koike T, Harada N, Yoshida T, Morikawa M (1992) Regulation of myeloid-specific calcium binding protein synthesis by cytosolic protein kinase C. J Biochem (Tokyo) 112:624–630
Komada T, Araki R, Nakatani K, Yada I, Naka M, Tanaka T (1996) Novel specific chemtactic receptor for S100L protein on guinea pig eosinophils. Biochem Biophys Res Commun 220:871–874
Korndorfer IP, Brueckner F, Skerra A (2007) The crystal structure of the human (S100A8/S100A9)2 heterotetramer, calprotectin, illustrates how conformational changes of interacting alpha-helices can determine specific association of two EF-hand proteins. J Mol Biol 370:887–898
Kube E, Becker T, Weber K, Gerke V (1992) Protein–protein interaction studied by site-directed mutagenesis: characterization of the annexin II-binding site on p11, a member of the S100 protein family. J Biol Chem 267:14175–14182
Kumar A, Steinkasserer A, Berchtold S (2003) Interleukin-10 influences the expression of MRP8 and MRP14 in human dendritic cells. Int Arch Allergy Immunol 132:40–47
Lackmann M, Cornish CJ, Simpson RJ, Moritz RL, Geczy CL (1992) Purification and structural analysis of a murine chemotactic cytokine (CP-10) with sequence homology to S100 proteins. J Biol Chem 267:7499–7504
Lackmann M, Rajasekariah P, Iismaa SE, Jones G, Cornish CJ, Hu S, Simpson RJ, Moritz RL, Geczy CL (1993) Identification of a chemotactic domain of the pro-inflammatory S100 protein CP-10. J Immunol 150:2981–2991
Lagasse E, Clerc RG (1988) Cloning and expression of two human genes encoding calcium-binding proteins that are regulated during myeloid differentiation. Mol Cell Biol 8:2402–2410
Lau D, Baldus S (2006) Myeloperoxidase and its contributory role in inflammatory vascular disease. Pharmacol Ther 111:16–26
Lau W, Devery JM, Geczy CL (1995) A chemotactic S100 peptide enhances scavenger receptor and Mac-1 expression and cholesteryl ester accumulation in murine peritoneal macrophages in vivo. J Clin Invest 95:1957–1965
Lazoura E, McLeish MJ, Aguilar MI (2000) Studies on the conformational properties of CP-10(42–55), the hinge region of CP-10, using circular dichroism and RP-HPLC. J Pept Res 55:411–418
Leach ST, Yang Z, Messina I, Song C, Geczy CL, Cunningham AM, Day AS (2007) Serum and mucosal S100 proteins, calprotectin (S100A8/S100A9) and S100A12, are elevated at diagnosis in children with inflammatory bowel disease. Scand J Gastroenterol 42:1321–1331
Leclerc E, Fritz G, Weibel M, Heizmann CW, Galichet A (2007) S100B and S100A6 differentially modulate cell survival by interacting with distinct RAGE (receptor for advanced glycation end products) immunoglobulin domains. J Biol Chem 282:31317–31331
Lee KC, Eckert RL (2007) S100A7 (Psoriasin)–mechanism of antibacterial action in wounds. J Invest Dermatol 127:945–957
Li J, Schmidt AM (1997) Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J Biol Chem 272:16498–16506
Lim SY, Raftery M, Cai H, Hsu K, Yan WX, Hseih HL, Watts RN, Richardson D, Thomas S, Perry M, Geczy CL (2008) S-nitrosylated S100A8: novel anti-inflammatory properties. J Immunol 181:5627–5636
Lim SY, Raftery MJ, Goyette J, Hsu K and Geczy CL (2009). Oxidative modifications of S100 proteins: functional regulation by redox. J Leukoc Biol
Lusitani D, Malawista SE, Montgomery RR (2003) Calprotectin, an abundant cytosolic protein from human polymorphonuclear leukocytes, inhibits the growth of Borrelia burgdorferi. Infect Immun 71:4711–4716
Manitz MP, Horst B, Seeliger S, Strey A, Skryabin BV, Gunzer M, Frings W, Schonlau F, Roth J, Sorg C, Nacken W (2003) Loss of S100A9 (MRP14) results in reduced interleukin-8-induced CD11b surface expression, a polarized microfilament system, and diminished responsiveness to chemoattractants in vitro. Mol Cell Biol 23:1034–1043
McClintock KA, Shaw GS (2003) A novel S100 target conformation is revealed by the solution structure of the Ca2 + –S100B–TRTK-12 complex. J Biol Chem 278:6251–6257
McCormick MM, Rahimi F, Bobryshev YV, Gaus K, Zreiqat H, Cai H, Lord RS, Geczy CL (2005) S100A8 and S100A9 in human arterial wall. Implications for atherogenesis. J Biol Chem 280:41521–41529
McNeill E, Conway SJ, Roderick HL, Bootman MD, Hogg N (2007) Defective chemoattractant-induced calcium signalling in S100A9 null neutrophils. Cell Calcium 41:107–121
Mikkelsen SE, Novitskaya V, Kriajevska M, Berezin V, Bock E, Norrild B, Lukanidin E (2001) S100A12 protein is a strong inducer of neurite outgrowth from primary hippocampal neurons. J Neurochem 79:767–776
Miranda LP, Tao T, Jones A, Chernushevich I, Standing KG, Geczy CL, Alewood PF (2001) Total chemical synthesis and chemotactic activity of human S100A12 (EN-RAGE). FEBS Lett 488:85–90
Mirmohammadsadegh A, Tschakarjan E, Ljoljic A, Bohner K, Michel G, Ruzicka T, Goos M, Hengge UR (2000) Calgranulin C is overexpressed in lesional psoriasis. J Invest Dermatol 114:1207–1208
Mogensen TH (2009). Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22:240–273 (table of contents)
Mork G, Schjerven H, Mangschau L, Soyland E, Brandtzaeg P (2003) Proinflammatory cytokines upregulate expression of calprotectin (L1 protein, MRP-8/MRP-14) in cultured human keratinocytes. Br J Dermatol 149:484–491
Moroz OV, Burkitt W, Wittkowski H, He W, Ianoul A, Novitskaya V, Xie J, Polyakova O, Lednev IK, Shekhtman A, Derrick PJ, Bjoerk P, Foell D, Bronstein IB (2009) Both Ca2 + and Zn2 + are essential for S100A12 protein -oligomerization and function. BMC Biochem 10:11
Murthy AR, Lehrer RI, Harwig SS, Miyasaki KT (1993) In vitro candidastatic properties of the human neutrophil calprotectin complex. J Immunol 151:6291–6301
Nacken W, Roth J, Sorg C, Kerkhoff C (2003) S100A9/S100A8: myeloid representatives of the S100 protein family as prominent players in innate immunity. Microsc Res Tech 60:569–580
Nacken W, Mooren FC, Manitz MP, Bode G, Sorg C, Kerkhoff C (2005) S100A9 deficiency alters adenosine-5′******-triphosphate induced calcium signalling but does not generally interfere with calcium and zinc homeostasis in murine neutrophils. Int J Biochem Cell Biol 37:1241–1253
Newton RA, Hogg N (1998) The human S100 protein MRP-14 is a novel activator of the beta 2 integrin Mac-1 on neutrophils. J Immunol 160:1427–1435
Nicholls SJ, Hazen SL (2005) Myeloperoxidase and cardiovascular disease. Arterioscler Thromb Vasc Biol 25:1102–1111
Nishimura F, Terranova VP, Sawa T, Murayama Y (1999) Migration inhibitory factor related protein-8 (MRP-8) is an autocrine chemotactic factor for periodontal ligament cells. J Dent Res 78:1251–1255
Odink K, Cerletti N, Bruggen J, Clerc RG, Tarcsay L, Zwadlo G, Gerhards G, Schlegel R, Sorg C (1987) Two calcium-binding proteins in infiltrate macrophages of rheumatoid arthritis. Nature 330:80–82
Orlova VV, Choi EY, Xie C, Chavakis E, Bierhaus A, Ihanus E, Ballantyne CM, Gahmberg CG, Bianchi ME, Nawroth PP, Chavakis T (2007) A novel pathway of HMGB1-mediated inflammatory cell recruitment that requires Mac-1-integrin. EMBO J 26:1129–1139
Otsuka K, Terasaki F, Ikemoto M, Fujita S, Tsukada B, Katashima T, Kanzaki Y, Sohmiya K, Kono T, Toko H, Fujita M, Kitaura Y (2009) Suppression of inflammation in rat autoimmune myocarditis by S100A8/A9 through modulation of the proinflammatory cytokine network. Eur J Heart Fail 11:229–237
Paccola CC, Gutierrez VP, Longo I, Juliano L, Juliano MA, Giorgi R (2008) Antinociceptive effect of the C-terminus of murine S100A9 protein on experimental neuropathic pain. Peptides 29:1806–1814
Pagano RL, Dias MA, Dale CS, Giorgi R (2002) Neutrophils and the calcium-binding protein MRP-14 mediate carrageenan-induced antinociception in mice. Mediators Inflamm 11:203–210
Pagano RL, Sampaio SC, Juliano L, Juliano MA, Giorgi R (2005) The C-terminus of murine S100A9 inhibits spreading and phagocytic activity of adherent peritoneal cells. Inflamm Res 54:204–210
Pagano RL, Mariano M, Giorgi R (2006) Neutrophilic cell-free exudate induces antinociception mediated by the protein S100A9. Mediators Inflamm 2006:36765
Pascual G, Glass CK (2006) Nuclear receptors versus inflammation: mechanisms of transrepression. Trends Endocrinol Metab 17:321–327
Passey RJ, Williams E, Lichanska AM, Wells C, Hu S, Geczy CL, Little MH, Hume DA (1999) A null mutation in the inflammation-associated S100 protein S100A8 causes early resorption of the mouse embryo. J Immunol 163:2209–2216
Perez-Mato I, Castro C, Ruiz FA, Corrales FJ, Mato JM (1999) Methionine adenosyltransferase S-nitrosylation is regulated by the basic and acidic amino acids surrounding the target thiol. J Biol Chem 274:17075–17079
Petri B, Bixel MG (2006) Molecular events during leukocyte diapedesis. FEBS J 273:4399–4407
Petri B, Phillipson M, Kubes P (2008) The physiology of leukocyte recruitment: an in vivo perspective. J Immunol 180:6439–6446
Petty HR, Kindzelskii AL, Adachi Y, Todd RF 3rd (1997) Ectodomain interactions of leukocyte integrins and pro-inflammatory GPI-linked membrane proteins. J Pharm Biomed Anal 15:1405–1416
Petty HR, Worth RG, Todd RF 3rd (2002) Interactions of integrins with their partner proteins in leukocyte membranes. Immunol Res 25:75–95
Propper C, Huang X, Roth J, Sorg C, Nacken W (1999) Analysis of the MRP8-MRP14 protein–protein interaction by the two-hybrid system suggests a prominent role of the C-terminal domain of S100 proteins in dimer formation. J Biol Chem 274:183–188
Raftery MJ, Geczy CL (2002) Electrospray low energy CID and MALDI PSD fragmentations of protonated sulfinamide cross-linked peptides. J Am Soc Mass Spectrom 13:709–718
Raftery MJ, Yang Z, Valenzuela SM, Geczy CL (2001) Novel intra- and inter-molecular sulfinamide bonds in S100A8 produced by hypochlorite oxidation. J Biol Chem 276:33393–33401
Rahimi F, Hsu K, Endoh Y, Geczy CL (2005) FGF-2, IL-1beta and TGF-beta regulate fibroblast expression of S100A8. Febs J 272:2811–2827
Rammes A, Roth J, Goebeler M, Klempt M, Hartmann M, Sorg C (1997) Myeloid-related protein (MRP) 8 and MRP14, calcium-binding proteins of the S100 family, are secreted by activated monocytes via a novel, tubulin-dependent pathway. J Biol Chem 272:9496–9502
Raptis SZ, Pham CT (2005) Neutrophil-derived serine proteases in immune complex-mediated diseases. Immunol Res 32:211–215
Ravasi T, Hsu K, Goyette J, Schroder K, Yang Z, Rahimi F, Miranda LP, Alewood PF, Hume DA, Geczy C (2004) Probing the S100 protein family through genomic and functional analysis. Genomics 84:10–22
Reed RC, Berwin B, Baker JP, Nicchitta CV (2003) GRP94/gp96 elicits ERK activation in murine macrophages. A role for endotoxin contamination in NF-kappa B activation and nitric oxide production. J Biol Chem 278:31853–31860
Robinson MJ, Hogg N (2000) A comparison of human S100A12 with MRP-14 (S100A9). Biochem Biophys Res Commun 275:865–870
Robinson MJ, Tessier P, Poulsom R, Hogg N (2002) The S100 family heterodimer, MRP-8/14, binds with high affinity to heparin and heparan sulfate glycosaminoglycans on endothelial cells. J Biol Chem 277:3658–3665
Roth J, Vogl T, Sorg C, Sunderkotter C (2003) Phagocyte-specific S100 proteins: a novel group of proinflammatory molecules. Trends Immunol 24:155–158
Rouleau P, Vandal K, Ryckman C, Poubelle PE, Boivin A, Talbot M, Tessier PA (2003) The calcium-binding protein S100A12 induces neutrophil adhesion, migration, and release from bone marrow in mouse at concentrations similar to those found in human inflammatory arthritis. Clin Immunol 107:46–54
Ryckman C, McColl SR, Vandal K, de Medicis R, Lussier A, Poubelle PE, Tessier PA (2003a) Role of S100A8 and S100A9 in neutrophil recruitment in response to monosodium urate monohydrate crystals in the air-pouch model of acute gouty arthritis. Arthritis Rheum 48:2310–2320
Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA (2003b) Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol 170:3233–3242
Ryckman C, Gilbert C, de Medicis R, Lussier A, Vandal K, Tessier PA (2004) Monosodium urate monohydrate crystals induce the release of the proinflammatory protein S100A8/A9 from neutrophils. J Leukoc Biol 76:433–440
Sakaguchi T, Yan SF, Yan SD, Belov D, Rong LL, Sousa M, Andrassy M, Marso SP, Duda S, Arnold B, Liliensiek B, Nawroth PP, Stern DM, Schmidt AM, Naka Y (2003) Central role of RAGE-dependent neointimal expansion in arterial restenosis. J Clin Invest 111:959–972
Salama I, Malone PS, Mihaimeed F, Jones JL (2008) A review of the S100 proteins in cancer. Eur J Surg Oncol 34:357–364
Santhanagopalan V, Hahn BL, Sohnle PG (1995) Resistance of zinc-supplemented Candida albicans cells to the growth inhibitory effect of calprotectin. J Infect Dis 171:1289–1294
Schafer BW, Heizmann CW (1996) The S100 family of EF-hand calcium-binding proteins: functions and pathology. Trends Biochem Sci 21:134–140
Schmidt AM, Yan SD, Yan SF, Stern DM (2001) The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest 108:949–955
Schnekenburger J, Schick V, Kruger B, Manitz MP, Sorg C, Nacken W, Kerkhoff C, Kahlert A, Mayerle J, Domschke W, Lerch MM (2008) The calcium binding protein S100A9 is essential for pancreatic leukocyte infiltration and induces disruption of cell–cell contacts. J Cell Physiol 216:558–567
Seemann J, Weber K, Gerke V (1996) Structural requirements for annexin I-S100C complex-formation. Biochem J 319(Pt 1):123–129
Sellmayer A, Krane SM, Ouellette AJ, Bonventre JV (1992) 1 alpha, 25-(OH)2 vitamin D3 enhances expression of the genes encoding Ca(2 +)-binding proteins MRP-8 and MRP-14. Am J Physiol 262:C235–C242
Shepherd CE, Goyette J, Utter V, Rahimi F, Yang Z, Geczy CL, Halliday GM (2006) Inflammatory S100A9 and S100A12 proteins in Alzheimer’s disease. Neurobiol Aging 27:1554–1563
Shibata F, Miyama K, Shinoda F, Mizumoto J, Takano K, Nakagawa H (2004) Fibroblast growth-stimulating activity of S100A9 (MRP-14). Eur J Biochem 271:2137–2143
Shibata F, Ito A, Ohkuma Y, Mitsui K (2005) Mitogenic activity of S100A9 (MRP-14). Biol Pharm Bull 28:2312–2314
Smith SP, Shaw GS (1998) A change-in-hand mechanism for S100 signalling. Biochem Cell Biol 76:324–333
Sohnle PG, Hunter MJ, Hahn B, Chazin WJ (2000) Zinc-reversible antimicrobial activity of recombinant calprotectin (migration inhibitory factor-related proteins 8 and 14). J Infect Dis 182:1272–1275
Sorci G, Riuzzi F, Agneletti AL, Marchetti C, Donato R (2003) S100B inhibits myogenic differentiation and myotube formation in a RAGE-independent manner. Mol Cell Biol 23:4870–4881
Sorci G, Riuzzi F, Agneletti AL, Marchetti C, Donato R (2004) S100B causes apoptosis in a myoblast cell line in a RAGE-independent manner. J Cell Physiol 199:274–283
Sperandio M (2006) Selectins and glycosyltransferases in leukocyte rolling in vivo. FEBS J 273:4377–4389
Srikrishna G, Panneerselvam K, Westphal V, Abraham V, Varki A, Freeze HH (2001) Two proteins modulating transendothelial migration of leukocytes recognize novel carboxylated glycans on endothelial cells. J Immunol 166:4678–4688
Sroussi HY, Berline J, Dazin P, Green P, Palefsky JM (2006) S100A8 triggers oxidation-sensitive repulsion of neutrophils. J Dent Res 85:829–833
Sroussi HY, Berline J, Palefsky JM (2007) Oxidation of methionine 63 and 83 regulates the effect of S100A9 on the migration of neutrophils in vitro. J Leukoc Biol 81:818–824
Sroussi HY, Kohler GA, Agabian N, Villines D, Palefsky JM (2009) Substitution of methionine 63 or 83 in S100A9 and cysteine 42 in S100A8 abrogate the antifungal activities of S100A8/A9: potential role for oxidative regulation. FEMS Immunol Med Microbiol 55:55–61
Steinbakk M, Naess-Andresen CF, Lingaas E, Dale I, Brandtzaeg P, Fagerhol MK (1990) Antimicrobial actions of calcium binding leucocyte L1 protein, calprotectin. Lancet 336:763–765
Sunahori K, Yamamura M, Yamana J, Takasugi K, Kawashima M, Yamamoto H, Chazin WJ, Nakatani Y, Yui S, Makino H (2006) The S100A8/A9 heterodimer amplifies proinflammatory cytokine production by macrophages via activation of nuclear factor kappa B and p38 mitogen-activated protein kinase in rheumatoid arthritis. Arthritis Res Ther 8:R69
Suryono Kido J, Hayashi N, Kataoka M, Shinohara Y, Nagata T (2006) Norepinephrine stimulates calprotectin expression in human monocytic cells. J Periodontal Res 41:159–164
Terasaki F, Fujita M, Shimomura H, Tsukada B, Otsuka K, Katashima T, Ikemoto M, Kitaura Y (2007) Enhanced expression of myeloid-related protein complex (MRP8/14) in macrophages and multinucleated giant cells in granulomas of patients with active cardiac sarcoidosis. Circ J 71:1545–1550
Test S, Weiss SJ (1986) The generation and utilization of chlorinated oxidants by human neutrophils. Adv Free Radic Biol Med 2:91–116
Thorey IS, Roth J, Regenbogen J, Halle JP, Bittner M, Vogl T, Kaesler S, Bugnon P, Reitmaier B, Durka S, Graf A, Wockner M, Rieger N, Konstantinow A, Wolf E, Goppelt A, Werner S (2001) The Ca2 + -binding proteins S100A8 and S100A9 are encoded by novel injury-regulated genes. J Biol Chem 276:35818–35825
Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, Kiener PA, Coyle AJ (2007) Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol 8:487–496
Todd RF 3rd, Petty HR (1997) Beta 2 (CD11/CD18) integrins can serve as signaling partners for other leukocyte receptors. J Lab Clin Med 129:492–498
Tsan MF, Gao B (2004) Endogenous ligands of Toll-like receptors. J Leukoc Biol 76:514–519
Turovskaya O, Foell D, Sinha P, Vogl T, Newlin R, Nayak J, Nguyen M, Olsson A, Nawroth PP, Bierhaus A, Varki N, Kronenberg M, Freeze HH, Srikrishna G (2008) RAGE, carboxylated glycans and S100A8/A9 play essential roles in colitis-associated carcinogenesis. Carcinogenesis 29:2035–2043
van Lent PL, Grevers L, Blom AB, Sloetjes A, Mort JS, Vogl T, Nacken W, van den Berg WB and Roth J (2007) Myeloid related proteins S100A8/S100A9 regulate joint inflammation and cartilage destruction during antigen-induced arthritis. Ann Rheum Dis
Vandal K, Rouleau P, Boivin A, Ryckman C, Talbot M, Tessier PA (2003) Blockade of S100A8 and S100A9 suppresses neutrophil migration in response to lipopolysaccharide. J Immunol 171:2602–2609
Viemann D, Strey A, Janning A, Jurk K, Klimmek K, Vogl T, Hirono K, Ichida F, Foell D, Kehrel B, Gerke V, Sorg C, Roth J (2005) Myeloid-related proteins 8 and 14 induce a specific inflammatory response in human microvascular endothelial cells. Blood 105:2955–2962
Viemann D, Barczyk K, Vogl T, Fischer U, Sunderkotter C, Schulze-Osthoff K, Roth J (2007) MRP8/MRP14 impairs endothelial integrity and induces a caspase-dependent and -independent cell death program. Blood 109:2453–2460
Voganatsi A, Panyutich A, Miyasaki KT, Murthy RK (2001) Mechanism of extracellular release of human neutrophil calprotectin complex. J Leukoc Biol 70:130–134
Vogl T, Propper C, Hartmann M, Strey A, Strupat K, van den Bos C, Sorg C, Roth J (1999a) S100A12 is expressed exclusively by granulocytes and acts independently from MRP8 and MRP14. J Biol Chem 274:25291–25296
Vogl T, Roth J, Sorg C, Hillenkamp F, Strupat K (1999b) Calcium-induced noncovalently linked tetramers of MRP8 and MRP14 detected by ultraviolet matrix-assisted laser desorption/ionization mass spectrometry. J Am Soc Mass Spectrom 10:1124–1130
Vogl T, Ludwig S, Goebeler M, Strey A, Thorey IS, Reichelt R, Foell D, Gerke V, Manitz MP, Nacken W, Werner S, Sorg C, Roth J (2004) MRP8 and MRP14 control microtubule reorganization during transendothelial migration of phagocytes. Blood 104:4260–4268
Vogl T, Leukert N, Barczyk K, Strupat K, Roth J (2006) Biophysical characterization of S100A8 and S100A9 in the absence and presence of bivalent cations. Biochim Biophys Acta 1763:1298–1306
Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, Nacken W, Foell D, van der Poll T, Sorg C, Roth J (2007) Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med 13:1042–1049
Wilckens T (1995) Glucocorticoids and immune function: physiological relevance and pathogenic potential of hormonal dysfunction. Trends Pharmacol Sci 16:193–197
Wolf R, Howard OM, Dong HF, Voscopoulos C, Boeshans K, Winston J, Divi R, Gunsior M, Goldsmith P, Ahvazi B, Chavakis T, Oppenheim JJ, Yuspa SH (2008) Chemotactic activity of S100A7 (Psoriasin) is mediated by the receptor for advanced glycation end products and potentiates inflammation with highly homologous but functionally distinct S100A15. J Immunol 181:1499–1506
Wu N, Davidson JM (2004) Migration inhibitory factor-related protein (MRP)8 and MRP14 are differentially expressed in free-electron laser and scalpel incisions. Wound Repair Regen 12:327–336
Xie J, Burz DS, He W, Bronstein IB, Lednev I, Shekhtman A (2007) Hexameric calgranulin C (S100A12) binds to the receptor for advanced glycated end products (RAGE) using symmetric hydrophobic target-binding patches. J Biol Chem 282:4218–4231
Xu K, Geczy CL (2000) IFN-gamma and TNF regulate macrophage expression of the chemotactic S100 protein S100A8. J Immunol 164:4916–4923
Xu K, Yen T, Geczy CL (2001) Il-10 up-regulates macrophage expression of the S100 protein S100A8. J Immunol 166:6358–6366
Yan WX, Armishaw C, Goyette J, Yang Z, Cai H, Alewood P, Geczy CL (2008) Mast cell and monocyte recruitment by S100A12 and its hinge domain. J Biol Chem 283:13035–13043
Yanamandra K, Alexeyev O, Zamotin V, Srivastava V, Shchukarev A, Brorsson AC, Tartaglia GG, Vogl T, Kayed R, Wingsle G, Olsson J, Dobson CM, Bergh A, Elgh F, Morozova-Roche LA (2009) Amyloid formation by the pro-inflammatory S100A8/A9 proteins in the ageing prostate. PLoS ONE 4:e5562
Yang Z, Tao T, Raftery MJ, Youssef P, Di Girolamo N, Geczy CL (2001) Proinflammatory properties of the human S100 protein S100A12. J Leukoc Biol 69:986–994
Yang Z, Yan WX, Cai H, Tedla N, Armishaw C, Di Girolamo N, Wang HW, Hampartzoumian T, Simpson JL, Gibson PG, Hunt J, Hart P, Hughes JM, Perry MA, Alewood PF, Geczy CL (2007) S100A12 provokes mast cell activation: a potential amplification pathway in asthma and innate immunity. J Allergy Clin Immunol 119:106–114
Yen T, Harrison CA, Devery JM, Leong S, Iismaa SE, Yoshimura T, Geczy CL (1997) Induction of the S100 chemotactic protein, CP-10, in murine microvascular endothelial cells by proinflammatory stimuli. Blood 90:4812–4821
Yong HY, Moon A (2007) Roles of calcium-binding proteins, S100A8 and S100A9, in invasive phenotype of human gastric cancer cells. Arch Pharm Res 30:75–81
Yui S, Mikami M, Yamazaki M (1995) Purification and characterization of the cytotoxic factor in rat peritoneal exudate cells: its identification as the calcium binding protein complex, calprotectin. J Leukoc Biol 58:307–316
Yui S, Mikami M, Tsurumaki K, Yamazaki M (1997) Growth-inhibitory and apoptosis-inducing activities of calprotectin derived from inflammatory exudate cells on normal fibroblasts: regulation by metal ions. J Leukoc Biol 61:50–57
Yui S, Nakatani Y, Hunter MJ, Chazin WJ, Yamazaki M (2002) Implication of extracellular zinc exclusion by recombinant human calprotectin (MRP8 and MRP14) from target cells in its apoptosis-inducing activity. Mediators Inflamm 11:165–172
Zimmer DB, Wright Sadosky P, Weber DJ (2003) Molecular mechanisms of S100–target protein interactions. Microsc Res Tech 60:552–559
Zreiqat H, Howlett CR, Gronthos S, Hume D, Geczy CL (2007) S100A8/S100A9 and their association with cartilage and bone. J Mol Histol 38:381–391
Zwadlo G, Bruggen J, Gerhards G, Schlegel R, Sorg C (1988) Two calcium-binding proteins associated with specific stages of myeloid cell differentiation are expressed by subsets of macrophages in inflammatory tissues. Clin Exp Immunol 72:510–515
Acknowledgments
The authors acknowledge the National Health and Medical Research Council of Australia for funding and members of the laboratory who contributed to the research discussed in this review, particularly Dr. Kenneth Hsu, Ms. Su Yin Lim, Dr. Zheng Yang, Dr. Weixing Yan and Dr. Mark Raftery and our long-term collaborator, Professor Paul Alewood.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Goyette, J., Geczy, C.L. Inflammation-associated S100 proteins: new mechanisms that regulate function. Amino Acids 41, 821–842 (2011). https://doi.org/10.1007/s00726-010-0528-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00726-010-0528-0