
Abstract
Physical activity and sports play major roles in the overall health status of humans. It is well known that regular exercise helps to lower the risk for a broad variety of health problems, such as cardiovascular disease, type 2 diabetes, and cancer. Being physically active induces a wide variety of molecular adaptations, for example fiber type switches or other metabolic alterations, in skeletal muscle tissue. These adaptations are based on exercise-induced changes to the skeletal muscle transcriptome. Understanding their nature is crucial to improve the development of exercise-based therapeutic strategies. Recent research indicates that specifically epigenetic mechanisms, i.e., pathways that induce changes in gene expression patterns without altering the DNA base sequence, might play a major role in controlling skeletal muscle transcriptional patterns. Epigenetic mechanisms include DNA and histone modifications, as well as expression of specific microRNAs. They can be modulated by environmental factors or external stimuli, such as exercise, and eventually induce specific and fine-tuned changes to the transcriptional response. In this review, we highlight current knowledge on epigenetic changes induced in exercising skeletal muscle, their target genes, and resulting phenotypic changes. In addition, we raise the question of whether epigenetic modifications might serve as markers for the design and management of optimized and individualized training protocols, as prognostic tools to predict training adaptation, or even as targets for the design of “exercise mimics”.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Epigenetic modifications, such as DNA methylation, changes to histone proteins or microRNA (miRNA)-mediated mechanisms, can alter gene expression patterns, but do not affect the DNA base sequence. |
Recent data suggest that epigenetic mechanisms might be important in regulating skeletal muscle adaptation to exercise. |
A better understanding of these mechanisms might allow prediction of an individual’s training response and the development of optimized and individualized training strategies. |
1 Introduction
Physical exercise, when performed regularly, lowers the risk for a plethora of common health problems, such as cardiovascular disease, type 2 diabetes (T2DM), and several types of cancer. By contrast, a sedentary lifestyle is considered a major risk factor for the development of most lifestyle-related diseases (for review, see Hechanova et al. [1]). Exercise induces a broad variety of molecular changes in a multitude of target tissues, which then, in turn, bring about functional changes, such as cardiovascular or respiratory adaptations, or changes to skeletal muscle strength, metabolism and endurance (for review, see Vina et al. [2]).
Skeletal muscle adaptation to training can be achieved at various levels, such as metabolic regulation, or changes in the expression of genes encoding contractile proteins with specific functional properties, such as “fast” and “slow” myosin heavy chain isoforms. This adaptive response can be induced by both extra- and intracellular factors paralleling exercise, such as mechanical stretch, fluctuations in blood flow or temperature, an altered ATP/ADP ratio, or various hormones and systemic factors. These signals activate or repress a multitude of characteristic signaling pathways, which in turn regulate transcription and translation of specific genes. The nature of these exercise-regulated signaling pathways and the resulting changes to the skeletal muscle transcriptome are fairly well understood. Specifically, during strength training, among others, the mTORc1 (mechanistic/mammalian target of rapamycin complex 1) pathway is one of the central players that mediate transcriptional changes, eventually leading to enhanced muscle protein synthesis. By contrast, in response to endurance training, other signaling routes, such as the PGC-1α- (peroxisome proliferator-activated receptor gamma coactivator 1-alpha) pathway, play a major role. PGC-1α is a transcriptional regulator and activator of mitochondrial biogenesis, which is enriched in red (“slow”) muscle fibers. In addition, the CaMK (calcium-calmodulin-dependent kinase)-calcineurin-NFATc1 (nuclear factor of activated T cells, cytoplasmic 1) pathway, as well as various other signaling routes, are central in mediating adaptation (for review, see Hoppeler [3]).
Activation of exercise-induced signaling pathways can directly lead to induction or repression of certain genes, for example via activation of specific transcriptional regulators. However, while the development of novel methods of genome analysis, as well as of complementary DNA (cDNA) arrays, has spurred the functional analysis of exercise-relevant genes and the characterization of exercise-induced transcriptomes (for review, see Sarzynski et al. [4]), more and more evidence indicates that an individual ’s genome or “steady-state” transcriptome might not be sufficient to predict the person’s physiological response to a specific training regimen (for review, see Ntanasis-Stathopoulos et al. [5], Ehlert et al. [6], Pareja-Galeano et al. [7], Soci et al. [8]). Remarkably, recent work suggests that the analysis of epigenetic characteristics, i.e., heritable changes to the DNA that are not encoded in its base sequence and that eventually determine the transcriptome, might serve this purpose (for review, see Ntanasis-Stathopoulos et al. [5], Ehlert et al. [6], Pareja-Galeano et al. [7], Soci et al. [8]). The term “epigenetic” implies that these changes can regulate gene expression patterns “on top of genetics”, but do not alter the DNA nucleotide sequence itself. Epigenetic changes can be quite stable, might be passed on to daughter cells, and—if germ cells are affected—might become heritable. A broad spectrum of environmental factors—such as nutrition, exposure to certain chemicals, emotional challenges, or physical activity—can influence a cell’s epigenetic signature. Pre-existing epigenetic signatures can then determine how an individual reacts to a certain stimulus, i.e., a specific exercise regimen, by influencing the degree and nature of exercise-induced transcriptional changes (for review, see Ecker et al. [9]). Indeed, within the last few years, a broad variety of studies suggesting a major role of epigenetic mechanisms in skeletal muscle adaptation to exercise have been published, but, to our knowledge, to date, there is only one general review article specifically focusing on this topic (McGee and Walder [10], published in 2017).
2 Methods
2.1 Search Strategy
A PubMed search was conducted initially up to May 2018 and updated in December 2018. The following search terms were used: ‘skeletal muscle AND exercise AND epigenetics’ (34 publications retrieved), ‘skeletal muscle AND exercise AND DNA methylation’ (56 publications retrieved), ‘skeletal muscle AND exercise AND histone acetylation’ (26 publications retrieved), ‘skeletal muscle AND exercise AND histone methylation’ (22 publications retrieved), ‘skeletal muscle AND exercise AND miRNA’ (161 publications retrieved). After exclusion of duplicates and papers not relevant to our study (particularly papers with a different focus, in which the search items appeared in an unrelated context), 87 publications were selected for the current review.
3 Epigenetics and Skeletal Muscle Adaptation to Exercise
In the following sections, we discuss the three major epigenetic mechanisms, namely (1) DNA methylation, (2) acetylation and methylation of histone proteins, and (3) regulation of gene expression by microRNAs (miRNAs) (Table 1). We also sum up current knowledge on how these mechanisms are influenced by physical activity, with a specific focus on skeletal muscle, and how their modulation alters the transcriptional response in the exercising myofiber. Finally, we present data on the effects of specific exercise regimens on particular epigenetic changes in skeletal muscle tissue, and possible implications for the development of exercise-based preventive and therapeutic strategies.
3.1 DNA Methylation
Regulation of gene expression by DNA methylation is of high importance for cell function under both physiological and pathological conditions (see Meier and Recillas-Targa [11]).
DNA methylation occurs by adding a methyl group to cytosines within CpG dinucleotides to form 5-methylcytosine (5-mC) (Fig. 1). As a result, genes can be switched off. It is important to note that despite the fact that DNA methylation within the promoter region of a specific gene is associated with silencing of the latter, methylation can also occur outside of promoter regions and might then, at least in certain cases, also promote gene expression. Methylation is accomplished by specific DNA methyltransferases (DNMTs) (for review, see Meier and Recillas-Targa [11]).

DNA methylation. C5 of a cytosine base within a CpG sequence is modified by addition of a methyl group
DNA methylation patterns can be assessed in a genome-wide manner using various technologies (for review, see Yong et al. [12]). The DNA methylation pattern, however, is not entirely stable: through mechanisms involving enzymes belonging to the ten eleven translocation (TET) family, DNA demethylation can occur. These enzymes catalyze oxidation of 5-mC to generate intermediate products such as hydroxymethylcytosine, formylcytosine, or carboxylcytosine, which eventually bring about full demethylation, but which might also represent important players in epigenetic regulation of gene expression (for review, see Delatte et al. [13]). Recently, several authors demonstrated that both global and specific DNA methylation patterns are significantly altered in exercising skeletal muscle (Table 2). Initial findings have also been summarized in Ling and Rönn [14].
3.1.1 Endurance Exercise
In 2012, Barrès et al. [15] detected a decrease in global DNA methylation 20 min after one acute bout of exhaustive aerobic exercise in the skeletal muscle of sedentary, healthy, young volunteers, indicating that exercise might promote net gene activation. The respective genes might be necessary to cope with the physical stress of exercise, to regulate recovery, and/or to initiate the adaptive response, the actual “training effect”.
Moreover, changes in DNA methylation patterns can also be observed in trained versus untrained individuals. Nitert and colleagues have shown that in men aged 37.5 years on average, six months of regular endurance exercise exerted characteristic changes in DNA methylation patterns in skeletal muscle, specifically with respect to genes associated with muscle growth, differentiation, and metabolism [16]. Similar results were presented by Kanzleiter et al. [17] in skeletal muscle of trained mice. In addition, Lindholm and colleagues [18] showed that a 3-month, one-legged endurance training intervention induced changes in methylation patterns specifically of genes associated with myogenesis, muscle structure, and muscle bioenergetics. However, in contrast to other studies, the observed changes were mainly located within enhancers, gene bodies, and intergenic regions, not within CpG islands of the promoters.
Regulation of skeletal muscle metabolic and bioenergetic adaptation to exercise by differential DNA methylation could also be demonstrated by Rowlands et al. [19]. The authors showed that in middle-aged, obese, and type 2 diabetic individuals, 16 weeks of endurance exercise exerted profound changes in methylation of metabolism-associated genes: specifically, genes encoding proteins involved in oxidative, energy-releasing pathways, such as fatty acid transporter, were hypomethylated, whereas genes encoding factors associated with fat deposition, such as fatty acid synthase, were hypermethylated. Similarly, Pattamaprapanont et al. [20] demonstrated demethylation of the Nr4a3 (nuclear receptor subfamily 4 group A member 3) gene promoter in an in vitro skeletal muscle model of endurance exercise. Nr4a3 is rapidly upregulated by endurance exercise in skeletal muscle and is involved in the control of oxidative metabolism, indicating a role of DNA de(methylation) in metabolic adaptation to exercise. However, as recent data by King-Himmelreich et al. [21] indicate, metabolic control by differential DNA methylation is still not completely understood. The authors detected increased rather than decreased DNA methylation of the Ampkα2 (AMP-activated protein kinase α2) gene, which is associated with oxidative metabolism, after one single bout of treadmill running in murine skeletal muscle. However, since they also (correspondingly) observed decreased expression of this gene, the reason might lie within specific characteristics of their experimental approach, which might have been different from that of other authors. It is also very likely that differential DNA methylation plays a major role in controlling the complex crosstalk between diet- and exercise-induced effects in metabolic control. Lane et al. [22] found differential effects of carbohydrate consumption before exercise on training-induced DNA methylation patterns.
One of the best-characterized genes that is upregulated in response to endurance exercise and contributes to metabolic adaptation is the PGC-1α gene (see Sect. 1). Upon exercise, this gene appears to be demethylated within its promoter sequence in both humans [15] and mice [23, 24]. Demethylation can be observed after both one acute bout of exercise [15, 24] and a 6-week voluntary running protocol [23]. Moreover, Laker et al. [23] demonstrated that Pgc-1α hypermethylation in murine offspring, which can be induced by feeding the mother a high-fat chow 6 weeks before and during pregnancy, can be prevented by parallel voluntary wheel running of the mother, suggesting that regular exercise might be able to prevent diet-induced metabolic changes not only in the fat-consuming individual itself, but also, via epigenetic mechanisms, in the next generation. Similar results were obtained in a recent study by Kasch et al. [25]. The authors demonstrated that offspring of mothers that had been fed a high-fat diet were characterized by hypomethylation and elevated expression of the Nr4a1 (nuclear receptor subfamily 4 group A member 1) gene, a transcriptional regulator known to be associated with metabolic regulation, chronic low-grade inflammation and T2DM. After voluntary wheel running over several weeks, methylation and expression levels returned to normal levels, in contrast to sedentary controls.
Differential DNA methylation might also play a role in the control of oxidative stress associated with exercise. As Nguyen et al. [26] demonstrated, exercise-induced upregulation of the Gpx1 gene, encoding glutathione peroxidase 1, is associated with decreased methylation within exon 2 of this gene.
3.1.2 Resistance Exercise
Resistance training has also been shown to exert distinct effects on the DNA methylome. In their study of obese and type 2 diabetic individuals (see Sect. 3.1.1), Rowlands et al. [19] also found effects of a 16-week resistance exercise regimen on the DNA methylome, but which differed from those of endurance training. Similarly, very recently, Seaborne et al. [27, 28] presented a complete DNA methylome, after both acute bouts and several weeks of training, detraining, and retraining. The results are very interesting, specifically since they showed hyper- and hypomethylation of a broad variety of genes not previously implicated in adaptation to resistance exercise, and will help to elucidate the role of differential DNA methylation in muscle hypertrophy and force augmentation. Moreover, as already stated in the context of endurance exercise (see Sect. 3.1.1), recent results presented by Laker et al. [29] indicate that differential DNA methylation and demethylation might also be involved in regulation of a metabolic “crosstalk” between both diet- and exercise-induced metabolic effects.
3.1.3 Age-Dependent Effects
Interestingly, as a recent large-scale bioinformatics analysis by Brown [30] revealed, exercise-induced changes in DNA methylation patterns appear to be highly dependent on age. In this analysis, both methylation and demethylation were more pronounced in older individuals over 40 years of age when compared to younger people, and, as the author stated, might have the potential to “rewind the biological clock”. An overall increased degree of DNA methylation in aged rat skeletal muscle was recently also reported by Carter et al. [31].
3.1.4 Disuse Atrophy
Not only enhanced physical activity but also disuse atrophy induces characteristic changes in DNA methylation patterns. Fisher et al. [32] paralyzed the rat tibialis anterior muscle by tetrodotoxin (TTX) administration to the supplying nerve. As a result, they observed decreased promoter methylation of specific genes, namely “atrogenes”, such as MuRF1/Trim63 (muscle RING-finger protein-1/tripartite motif containing 63) and MAFbx/Fbxo32 (muscle atrophy F-box/F-box only protein 32), encoding factors involved in the degradation of skeletal muscle proteins, and the Chrna1 gene, encoding acetylcholine receptor subunit α1. This hypomethylation was paralleled by enhanced expression of the respective genes. After cessation of TTX application and resumption of regular physical activity, the degree of methylation of these genes rose again to control levels and a decrease in gene expression was observed.
3.1.5 “Responders” Versus “Non-responders”
It might be tempting to speculate that non-responders to a single bout of exercise or a training intervention might have hypermethylated promoter regions of adaptation-relevant genes, such as PGC-1α, or might be characterized by an inability to demethylate these regions in response to exercise-activated signaling pathways. Conversely, it is of course also possible that their adaptation defect is due to an inability to methylate and thus “silence” other genes, encoding factors that block the adaptation process. In this context, a recent study by Stephens et al. [33] is interesting. The authors demonstrated distinct DNA methylation profiles in T2DM individuals that were subsequently shown not to be responsive to an exercise regimen, in terms of phosphocreatine recovery rate, or to amelioration of insulin sensitivity and glycemic control. At least for the abovementioned PGC-1α gene, an important mechanism associated with differential DNA methylation in the context of exercise response might be nucleosome repositioning. Bajpeyi et al. [34] found that subjects who upregulate this gene in response to a single bout of endurance exercise (“high responders”) show demethylation of the − 260 nt position and nucleosome shift away from this region during exercise, whereas this is not the case in individuals without PGC-1α upregulation (“low responders”). Finally, in this context, it is interesting that Terruzzi et al. [35] could demonstrate an above-average degree of genetic polymorphism within several genes encoding enzymes involved in DNA methylation in elite endurance athletes, which might lead to reduced DNA methylation potential and higher expression of myogenic genes.
3.2 Histone Modification
The main repeat element of chromatin is the nucleosome, consisting of 146 base pairs of DNA and an octamere of histone proteins. The N-terminal tails of the latter are prone to a variety of post-translational changes such as acetylation, methylation, or phosphorylation. With respect to their function in regulating gene expression, predominantly acetylation and methylation of histones H3 and H4 have been analyzed so far (Fig. 2). Histone modifications are typically described by the number of the specific histone isoform (e.g., histone 3), the modified amino acid (e.g. K4 for lysine 4), followed by the type of modification (e.g., “ac” for acetylation or “me3” for trimethylation). Different histone modifications can exert both activating and inhibiting effects on gene expression, dependent on their specific nature and their location (promoter, enhancer, or gene body). In addition, different histone modifications can affect the effects of each other and also “cross-talk” with other types of epigenetic modifications, such as DNA methylation or miRNAs. Histone modifications are regulated bidirectionally by particular groups of enzymes, the “writers”, which deposit histone marks, for example histone acetyltransferases (HATs), or histone methyltransferases (HMTs), and the “erasers”, which remove them, for example histone deacetylases (HDACs) or histone demethylases. The mechanisms by which histone marks regulate transcription are highly diverse and include effects at all stages of transcription, namely initiation, elongation, and termination (for review, see Gates et al. [36]).

Modifications of histones H3 and H4. Schematic representation of a nucleosome. DNA is represented by a black band. Histones H3 and H4 are depicted in red and blue, respectively. Modifications known to occur at specific amino acid positions within the N-terminal ends of both histones are shown. P phosphorylation, Ac acetylation, Me methylation
Several studies indicate a major role of histone marks in skeletal muscle adaptation to physical exercise (Table 3).
3.2.1 Endurance Exercise
McGee et al. carried out a series of studies on the response to a single bout of exercise in human skeletal muscle [37]. The authors demonstrated that exercise leads to increased global acetylation of H3K36, which might be associated with enhanced transcriptional elongation of certain exercise-associated genes. In addition, the authors observed increased nuclear-to-cytosolic translocation of HDACs 4 and 5 during exercise, leading to enhanced H3 acetylation. Furthermore, dissociation of HDACs 4 and 5 from the myogenic transcription factor MEF2 (myocyte enhancer factor 2) led to enhanced DNA binding of the latter (reviewed in McGee and Hargreaves [38]). This is specifically interesting in the context of a study by Potthoff et al. [39], in which the authors claim that class II HDACs, such as HDACs 4 and 5, block oxidative adaptation of skeletal muscle cells by repressing MEF2, and that high levels of MEF2 activity are associated with enhanced running performance and decreased fatigue in mice. Similar results were presented by Smith et al. [40] and Joseph et al. [41], demonstrating hyperacetylation of histones in the area of both the MEF2 site on the Glut4 (glucose transporter type 4) gene and the NRF-1 (nuclear respiratory factor 1) binding site on the Mef2a promoter, respectively.
Association of histone acetylation with exercise tolerance is also suggested by a recent study by Hong and colleagues [42]. The authors depleted HDAC3 in mice in a skeletal muscle-specific manner and demonstrated that despite developing severe insulin resistance and reduced muscle force, these animals displayed enhanced endurance and resistance to muscle fatigue, most likely due to a specific shift in fuel oxidation from glucose to fat. Since HDAC3 association with the genome is subject to the circadian clock, these data might furthermore suggest that the exact scheduling of exercise bouts within an individual’s daily routine might warrant further investigation.
By contrast, a recent study by Ohsawa et al. [43] demonstrated reduced levels of pan-acetylated H3 in rat skeletal muscle after 4 weeks of regular treadmill running in the rat plantaris muscle. In addition, using ChIP (chromatin immunoprecipitation) analysis, the authors found significant differences with regard to gene-specific distribution of this histone modification, and further point out that not only histone modification, but also histone “variant switching”, for example replacement of a specific histone H3 isoform by another, is regulated by exercise and might eventually play a role in adaptation of skeletal muscle to training.
3.2.2 Muscle Mass and Resistance Exercise
Balanced acetylation/deacetylation might also play a role in regulation of muscle differentiation and mass by the nuclear cofactor p300, which has been shown to possess HAT activity on histone as well as on nonhistone proteins, and interacts with various transcriptional regulators (for review, see Barreiro and Sznajder [44]). This mechanism might also be central in skeletal muscle adaptation to exercise, specifically with regard to the hypertrophic response to resistance training.
Involvement of histone acetylation in the control of adaptation to resistance exercise is furthermore suggested by a study by Thalacker-Mercer and colleagues [45]. The authors analyzed muscle fiber hypertrophy in response to a 16-week progressive resistance training regimen in healthy volunteers. Subjects were classified as non-responders or responders with regard to exercise-induced myofiber hypertrophy. Interestingly, the authors found that responders displayed higher basal levels of acetylated H3K36, indicating that they were somehow “primed” to respond to the training stimulus in a more efficient manner when compared to the non-responder group. Correspondingly, responders were characterized by a basal skeletal muscle transcriptomic profile clearly different from non-responders, and showed differential expression of characteristic genes (such as the α-tubulin or p27kip gene) at the protein level after the first exercise stimulus, whereas non-responders did not. These findings are very exciting, since, based on epigenetic patterns, they might lead to the development of individualized and personalized highly specific training regimens in the future.
3.2.3 Skeletal Muscle Fiber Types
Interestingly, histone modification patterns also differ between individual muscle fiber types, namely fast and slow muscle. A very recent study by Masuzawa et al. [46] showed that in rats, enhanced H3 acetylation controls transcription from the Pgc-1α locus in response to exercise, specifically from an alternative promoter (promoter B), which appears to be very sensitive to exercise stimuli, and that there are profound differences between fast and slow skeletal muscle. In addition, Pandorf et al. [47] demonstrated that there is an association between the predominantly “activating” histone marks H3K4me3 and H3ac, and expression of the respective genes encoding fiber type-specific myosin heavy chain isoforms in “slow” and “fast” muscle types of rats. Correspondingly, fiber type switches induced by hindlimb suspension correlated with the expected changes in histone modification patterns for myosin heavy chain genes, at least in the case of myosins I, IIx, and IIb, but not IIa. These data might indicate that fiber type switches, which also occur in response to exercise, might in part, but not exclusively, be controlled by differential histone modifications. Similar results were obtained by Kawano et al. [48] in response to mechanical overload and denervation.
These data suggest that besides differential histone acetylation, differential histone methylation also appears to play a major role in exercise adaptation, particularly in metabolic control and fiber type specification.
3.2.4 Histone Methylation
The assumption that histone methylation plays a major role in exercise adaptation is supported by the data presented by Lochmann et al. [24]. The authors demonstrated that transcriptional induction of the abovementioned Pgc-1α gene with exercise is not only regulated by differential DNA methylation but also by H3K4 methylation: in one acute bout of exercise, trimethylation of lysine 4 of histone 3 (H3K4me3) within the alternative Pgc-1α promoter region (promoter B) in murine quadriceps muscle increased two- to fourfold, which might, in addition to the effects on H3 acetylation and demethylation of the “classical” Pgc-1α promoter A (see Sect. 3.2.3), also contribute to overall upregulation of this gene with exercise.
By contrast, Willkomm et al. [49] demonstrated that after metabolically demanding high-intensity resistance exercise, H3K4 trimethylation was decreased. Based on parallel in vitro studies with cultured C2C12 myoblasts, the authors reason that high local lactate accumulation might be triggering this decrease. Since H3K4me3 appears to promote expression of muscle-specific genes, the physiological function of this mechanism is currently enigmatic, since decreased expression of myogenic genes should rather block exercise adaptation. Future studies might aim at determining the exact target genes of the H3K4me3 mark in this setting, as well as the kinetics of its decrease/increase within more extended time frames, and in response to regular exercise as well as different exercise regimens. For instance, it is very possible that this reaction is rather short-lived and serves the purpose of temporarily blocking adaptation, especially after unaccustomed exercise, as a means to allow other pathways to proceed, which might bring about metabolic recovery or regeneration.
Finally, recent data by Ohsawa et al. [43] suggest that not only modification of H3 but also of other histones might play an important role in skeletal muscle adaptation to exercise. In rat plantaris muscle, the authors demonstrated that regular treadmill running over several weeks had significant effects on trimethylation of histone 4 at lysine 20 (H4K20me3). Using ChIP analysis, the authors also showed significant gene-specific effects of training on this type of modification.
3.2.5 Histone-modifying Enzymes as Potential Targets for “Exercise Mimics”
Overall, these data indicate that histone modifications and histone-modifying enzymes in general, might play a major, and probably complex, role in skeletal muscle adaptation to exercise, not only in fiber-type determination, metabolism, and exercise tolerance, but also in adaptation of muscle force and hypertrophy. Given the background that histone-modifying enzymes can already be targeted by various pharmacological agents, of which more and more are being developed and some are highly specific (for review, see Ribich et al. [50]), future research is warranted to carefully disentangle these pathways and their interaction, which might lead to the development of “exercise mimics” in the future. As stated by several authors, novel HDAC inhibitors in particular might become a major focus within the next few years (for review, see Penna and Costelli [51]).
3.3 miRNAs
miRNAs are small, trans-acting RNA molecules that have been highly conserved throughout evolution. They are non-protein-coding. miRNA molecules silence their target messenger RNA(s) (mRNA(s)) in a post-transcriptional manner, eventually regulating abundance and thus function of their target protein(s). In specific cases, however, miRNAs can also stabilize and activate their targets (Vasudevan et al. [52]). Around 50% of protein-coding genes are regulated by miRNAs. miRNAs play important roles in the control of cell growth and differentiation, as well as metabolic pathways and many other cellular functions (for review, see Polakovičová et al. [53], Silva et al. [54], Wei et al. [55]).
Within the last few years, an increasing number of miRNA species have been detected in the human genome, and most of them have been associated with cardiovascular disease, cancer, diabetes, or other pathologies (for review, see Paul et al. [56]). In general, miRNAs appear to be particularly important under non-homeostatic conditions, for example after trauma (for review, see Daskalakis et al. [57]).
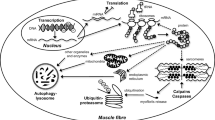
DNA sequences encoding miRNAs can be located within protein-coding genes, both within introns and exons, and in intergenic regions. Their transcription yields so-called primary miRNAs (pri-miRNAs), which are double-stranded and can encode one single or several miRNAs. pri-miRNAs are recognized and processed by the endoribonuclease Drosha, a member of the ribonuclease III superfamily that cleaves double-stranded RNA, and its cofactor Pasha (in humans also known as DGCR8: DiGeorge syndrome critical region 8). The resulting precursor miRNAs (pre-miRNAs), which are 60–70 nucleotides (nt) in length and display a hairpin-like structure, are exported from the nucleus via the exportin-5 transporter. In the cytosol, through action of another endoribonuclease, the so-called Dicer enzyme, they are then again processed into double-stranded RNA molecules of approximately 20 nt. After unwinding and degradation of the inactive, “passenger” strand, the active, “guide” strand is transferred to the RNA-induced silencing complex (RISC), which contains another endoribonuclease, Argonaute-2, where it binds to the 5’ or 3’ untranslated region (UTR) of its target mRNA, causing regulation of gene expression via multiple mechanisms, among them degradation of the target mRNA, as well as inhibition of its transcription or translation (Fig. 3; for review, see Polakovičová et al. [53], Silva et al. [54], Wei et al. [55]).

Pathways of miRNA processing and miRNA-driven epigenetic regulation of gene expression. Transcription from a specific sequence with activating histone marks (shown as green circles) by RNA polymerase II (Pol II) yields primary miRNA (pri-miRNA), which is cleaved by the Drosha-DiGeorge syndrome critical region 8 (DGCR8/Pasha) complex, resulting in a hairpin-structure pre-miRNA. The latter is again cleaved by the Dicer enzyme to yield a short miRNA duplex, which is then incorporated into the RNA-induced silencing complex (RISC), containing the Argonaute-2 protein (AGO). Degradation of the passenger strand leads to formation of the mature RISC complex, containing the mature miRNA. The latter can influence gene expression via three major mechanisms: (1) degradation via Argonaute proteins (top), (2) inhibition of ribosomal translation, specifically by blocking translational initiation or elongation or promote premature termination of translation (middle), and (3) recruiting epigenetic modifiers, such as histone methyltransferases (HMTs) and histone deacetylases (HDACs), to the promoter of the respective gene, thereby inducing inhibiting histone marks (shown as red circles), thus preventing or slowing down translation (bottom)
There is ample evidence suggesting that miRNAs might play an important role in regulating skeletal muscle plasticity, such as skeletal muscle hypertrophy, or functional characteristics of the muscle cell, such as metabolic aspects or fiber-type specificity. Furthermore, several (cardiac and/or skeletal) muscle-specific miRNAs have been identified, specifically miR-1, miR-133a, miR-206 (skeletal muscle-specific), miR-208a, miR-499, and sometimes also miR-486, which is not specific for but which is enriched in muscle cells, for which the term “myomiRNAs” has been coined. In addition, a broad variety of other miRNAs with more ubiquitous expression patterns have been shown to be important players in skeletal muscle development and plasticity (for review, see McCarthy [58], Luo et al. [59], Horak et al. [60]).
Against this background, it is not surprising that several studies have already shown that miRNA expression patterns in skeletal muscle tissue can be modulated by exercise. In the following sections, we briefly sum up these papers. For more details on specific issues, the reader may refer to the literature cited.
3.3.1 Endurance Exercise
Initial analyses were carried out in the mouse model system, with Safdar et al. [61] in 2009 demonstrating differential expression of several miRNAs after one single 90-min bout of treadmill running in murine quadriceps femoris muscle. In particular, whereas a moderate increase in miR-1 expression could be detected in this study, miR-133 remained unaltered. Remarkably, however, the authors demonstrated decreased miR-23 expression after exercise, which is particularly interesting, since this miRNA is a negative regulator of the PGC-1α mRNA. Consistent with these findings, Aoi et al. [62] found a profound downregulation of miR-696, which might also target the PGC-1α mRNA, after 4 weeks of progressive running exercise. Another important miRNA in this context might be miR-494, which was found to be decreased after 7 days of extensive swimming exercise, thus potentially allowing increased expression of mitochondrial transcription factor A (Tfam) and forkhead box j3 (Foxj3), encoding activators of mitochondrial biogenesis [63]. Keller et al. [64] carried out miRNA profiling after 6 weeks of supervised endurance training in young, sedentary males, and primarily found downregulation of miRNAs targeting the transcription factors RUNX1 (runt-related transcription factor 1), SOX9 (SRY (sex-determining region Y)-box 9), and PAX3 (paired-box transcription factor 3), indicating a function of miRNAs in regulating these factors in response to training.
Similarly, Nielsen and colleagues [65] showed that in response to acute endurance exercise, expression of miRs-1, -133a, -133b, and -206 increased in vastus lateralis muscle of healthy, young, untrained men, indicating that these miRNA species might play a major role in regulating skeletal muscle adaptation to physical activity. Interestingly, this induction could not be observed any more after a 12-week endurance training regimen. Moreover, trained subjects displayed decreased resting expression of these myomiRs [65], which increased back to baseline within 2 weeks after cessation of training, suggesting that myomiRs might be lower in trained versus untrained muscle.
Consistent with these findings, Russell et al. [66] found that a single bout of moderate-intensity endurance cycling led to upregulation of several miRNAs, among them miRs-1, -133a, and -133b, in skeletal muscle of young, untrained, subjects, whereas others were downregulated. Furthermore, expression of genes encoding components of the miRNA processing machinery, such as Drosha, Dicer, or exportin-5, were all increased, suggesting an overall enhanced miRNA biosynthesis rate. Remarkably, this is in contrast with the abovementioned mouse study by Safdar et al. [61], which could not detect enhanced expression of the Drosha or Dicer genes in their model system. In this context, it is interesting that in Dicer knockout mice, 2 weeks of voluntary wheel running induced skeletal muscle metabolic adaptation similarly to controls, paralleled by only moderately reduced miRNA levels in skeletal muscle, suggesting that there might be Dicer-independent pathways of miRNA biogenesis (Oikawa et al. [67]). After 10 days of endurance training, Russell et al. [66] found further characteristic alterations in miRNA expression patterns, which might reflect muscle adaptation to the training stimulus.
3.3.2 Resistance Exercise
Compared with endurance exercise, miRNA expression kinetics in skeletal muscle in response to resistance or eccentric exercise might be even more dependent on additional variables, such as age, a specific exercise regimen, or exercise history. In a mouse model of muscle hypertrophy induced by functional overload, McCarthy and Esser [68] demonstrated reduced expression of miR-1 and miR-133a, paralleled by enhanced expression of the genes encoding Drosha and exportin-5, but not that of the Dicer gene. Similarly, Mueller et al. [69] detected a decrease in miR-1 expression in response to both a 12-week regimen of resistance or eccentric exercise in elderly human subjects with a mean age of 80 years, whereas Davidsen et al. [70] did not observe a decrease in a comparable setting with young men.
Similar to endurance exercise, expression of miRNAs might already be changed after a single bout of resistance or eccentric exercise. In response to a single bout of leg extension resistance exercise paralleled by essential amino acid ingestion, Drummond et al. [71] demonstrated downregulation of miR-1 in skeletal muscle of young but not older subjects. By contrast, Ringholm et al. [72] found that one 45-min bout of knee extensor exercise did not influence miR-1 or miR-133 expression in the vastus lateralis muscle of young, healthy men after 1 week of bedrest.
miRNAs might also serve as prognostic biomarkers for adaptation to strength training. Davidsen et al. [70] demonstrated that expression patterns of certain miRNAs in human vastus lateralis muscle in response to a 12-week resistance training regimen are characteristic for “high” and “low” responders. Specifically, the authors demonstrated that low responders were characterized by downregulation of miR-378, miR-29a, and miR-26a, as well as upregulation of miR-451. The authors speculated that the differential expression of these miRNAs in low responders might be a “compensatory” phenomenon, “reflecting a failure to “activate” growth and remodeling genes”.
3.3.3 Concurrent Training and Different Training Modes
So far, little is known about the effects of concurrent training, i.e., regimens with combined endurance and resistance exercise blocks, and also different exercise modes, such as HIIT (high-intensity interval training) versus MICT (moderate-intensity continuous training), on miRNA expression patterns. Rowlands et al. [19] found profound differences with regard to miRNA expression (and also DNA methylation) patterns after 16 weeks of endurance versus resistance exercise regimens in a cohort ofT2DM, obese, middle-aged subjects. Furthermore, in a study of healthy young males, Fyfe et al. [73] observed that prior HIIT, but not MICT cycling, compromised resistance exercise-induced changes in miRNA expression patterns.
3.3.4 Circulating miRNAs
miRNAs can also be extracted from plasma and serum, specifically in response to stress (such as exercise) and trauma, with their origin and mode of secretion or release still being largely enigmatic. Since in comparison to skeletal muscle specimens, blood is more easily accessible, determining miRNA patterns in plasma and serum is particularly interesting, especially when considering their use as biomarkers for training monitoring, control, and management (for review, see Mooren et al. [74], Makarova et al. [75]). The most important findings in this area are summarized in the following paragraphs. Detailed meta-analyses can be found in two recent reviews by Polakovičová et al. [53] and Silva et al. [54].
Baggish et al. [76] analyzed circulating miRNAs in human plasma before and after 90 days of intensified, mainly endurance-based rowing training in young, active rowers. Interestingly, the authors found a correlation between peak levels of miR-146a and maximal volumes of relative oxygen consumption per kg bodyweight (VO2max), both at baseline and after the training period. Upregulation of circulating miR-146a after endurance exercise has also been demonstrated by Wardle et al. [77]. By contrast, resistance training, both acute and over a period of several weeks, had the opposite effect [77, 78]. Interestingly, Baggish et al. [76] could furthermore detect a direct correlation between changes in miR-20a expression pre- and post-training and the relative gain in maximal O2 consumption induced by the training, indicating that miRNA concentration changes might indeed serve as markers for training adaptation. Similarly, there seems to be a correlation between high levels of circulating miRNAs-21 and 210 and low VO2max [79]. However, regulation of miRNA-21 appears to be quite context-dependent [76, 77, 80]. A single study [81] analyzed miR-486 concentrations in serum after both acute and 4 weeks of thrice-weekly chronic cycling exercise and found decreases in both cases, indicating that this miRNA might also serve as a robust marker for exercise impact.
Over the last few years, a broad variety of studies have specifically demonstrated upregulation of circulating miR-1 and miR-133a in response to single bouts of exercise and long-term training, irrespective of the type of exercise (endurance- or resistance-based) (for review, see Polakovičová et al. [53], Silva et al. [54]). Since these two miRNAs are muscle cell-specific and appear to be central players in these cells (for review, see Polakovičová et al. [53], Silva et al. [54]), it is likely that they might be released at least in part from the vigorously exercising skeletal muscle.
However, despite the fact that these two miRNAs are upregulated in response to most exercise regimens, Uhlemann et al. [82] showed that different modes of exercise evoke different patterns of circulating miR-133. Concentrations of this species increased after resistance training and after marathon running, but not after a maximum ergometer test or 4 h of cycling at 70% VO2max. Similarly, Wardle et al. [77] observed characteristic miRNA patterns in plasma samples of endurance- and resistance-trained elite male athletes, both in comparison to each other and in relation to untrained controls.
Based on these initial exploratory experiments, a series of further studies analyzed circulating miRNA patterns, mostly in plasma, but sometimes also in serum, in distinct exercise settings (for review, see Polakovičová et al. [53], Silva et al. [54]). Remarkably, a central finding of several studies was again a distinct upregulation of circulating miR-1 and miR-133a immediately after a marathon run (Mooren et al. [75], for review, see Polakovičová et al. [53]). Furthermore, in a recent study, Horak et al. [83] compared the effects of 8 weeks of explosive or hypertrophic strength training versus high-intensity interval sprint training, and found characteristic patterns for each training mode, and Håkansson et al. [84] have recently presented data suggesting that there are profound differences with regard to circulating miRNA patterns when the effects of exhaustive and non-exhaustive endurance exercise were compared.
However, as already mentioned at the beginning of this section, the origins, triggers, and modes of secretion of circulating miRNAs are still largely enigmatic. Ramos et al. [85] recently carried out an interesting study, addressing the issue of potential functions of exercise-induced changes in circulating miRNAs. The authors demonstrated increased levels of circulating, but decreased amounts of muscular, miR-133 after 4 weeks of high-intensity treadmill running in mice. They hypothesized that this miRNA species might be actively secreted into the circulation upon exercise, thus lowering its concentration in muscle cells, which might be important for exercise-induced adaptation. Furthermore, secreted miRNAs might also exert paracrine or endocrine functions in other tissues and organs.
Some authors also analyzed miRNA expression patterns in different types of white blood cells. As in plasma, defined changes were found in these cells, but they did not always reflect those found in plasma (for review, see Denham et al. [86]).
It is currently unclear if miRNA expression patterns at rest or after a single bout of exercise might also serve as prospective “predictors” for training outcome, or, even more, for the efficacy of a specific training regimen. If so, this would open the door to novel and possibly more efficient strategies in training control and management.
Taken together, miRNAs can act as modulators of skeletal muscle cell proliferation, differentiation, and hypertrophy. In addition, they might be important regulators of skeletal muscle metabolism. All these features might render them important candidates for determining an individual’s response to a specific exercise regimen.
4 Conclusions
Regular exercise undoubtedly yields considerable health benefits, associated with adaptation of the body to the physical workload. Within the last few years, it has become clear that an individual’s adaptation to a specific training regimen depends not only on his or her genetic background, but also on so-called epigenetic mechanisms. These include processes that influence gene expression without altering the DNA base sequence, specifically: (1) DNA methylation, (2) post-translational modifications of histone proteins, and (3) regulation of gene expression via specific miRNAs. In contrast to the DNA base sequence, an individual’s epigenetic “signature” is subject to environmental influences, such as diet or prior training experience. Recently, considerable progress has been made towards a better understanding of the “epigenetic response” to specific training stimuli. It has become more and more clear that both single bouts of exercise as well as long-term training regimens alter epigenetic characteristics. However, whereas exercise-induced changes in miRNA patterns have already been analyzed quite thoroughly in a broad variety of contexts, little is still known about differential DNA methylation and histone modification patterns in response to physical activity, specifically in skeletal muscle tissue. Finally, a major aim of future studies will be the development of epigenetic marker panels that allow prediction of an individual’s reaction to a specific training stimulus. It is very likely that a person’s individual “history”, specifically his or her lifestyle, but also probably biographical and emotional experiences, have shaped his or her epigenome—and that this epigenetic “memory” determines this particular person’s reaction to a specific training stimulus in the present (Sharples et al. [87]). Better knowledge of these connections would allow the development of individualized training regimens, which would allow a person to reach his or her training goals with maximum efficiency. Specifically, it will be interesting to elucidate the role of epigenetics in regulating adaptation to concurrent exercise, i.e., combined endurance-resistance training regimens, as well as different training modes, such as HIIT versus MICT.
References
Hechanova RL, Wegler JL, Forest CP. Exercise: a vitally important prescription. JAAPA. 2017;30:17–22.
Vina J, Sanchis-Gomar F, Martinez-Bello V, Gomez-Cabrera MC. Exercise acts as a drug; the pharmacological benefits of exercise. Br J Pharmacol. 2012;167:1–12.
Hoppeler H. Molecular networks in skeletal muscle plasticity. J Exp Biol. 2016;219:205–13.
Sarzynski MA, Ghosh S, Bouchard C. Genomic and transcriptomic predictors of response levels to endurance exercise training. J Physiol. 2017;595:2931–9.
Ntanasis-Stathopoulos J, Tzanninis JG, Philippou A, Koutsilieris M. Epigenetic regulation on gene expression induced by physical exercise. J Musculoskelet Neuronal Interact. 2013;13:133–46.
Ehlert T, Simon P, Moser DA. Epigenetics in sports. Sports Med. 2013;4:93–110.
Pareja-Galeano H, Sanchis-Gomar F, García-Giménez JL. Physical exercise and epigenetic modulation: elucidating intricate mechanisms. Sports Med. 2014;44:429–36.
Soci UPR, Melo SFS, Gomes JLP, Silveira AC, Nóbrega C, de Oliveira EM. Exercise training and epigenetic regulation: multilevel modification and regulation of gene expression. Adv Exp Med Biol. 2017;1000:281–322.
Ecker S, Pancaldi V, Valencia A, Beck S, Paul DS. Epigenetic and transcriptional variability shape phenotypic plasticity. Bioessays. 2018;40(2):1700148.
McGee SL, Walder KR. Exercise and the skeletal muscle epigenome. Cold Spring Harb Perspect Med. 2017;7(9):a029876. https://doi.org/10.1101/cshperspect.a029876.
Meier K, Recillas-Targa F. New insights on the role of DNA methylation from a global view. Front Biosci (Landmark Ed). 2017;22:644–68.
Yong W-S, Hsu F-M, Chen P-Y. Profiling genome-wide DNA methylation. Epigenetics Chromatin. 2016;9:26.
Delatte B, Deplus R, Fuks F. Playing TETris with DNA modifications. EMBO J. 2014;33:1198–211.
Ling C, Rönn T. Epigenetic adaptation to regular exercise in humans. Drug Discov Today. 2014;19:1015–8.
Barrès R, Yan J, Egan B, Treebak JT, Rasmussen M, Fritz T, et al. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012;15:405–11.
Nitert MD, Dayeh T, Volkov P, Elgzyri T, Hall E, Nilsson E, et al. Impact of an exercise intervention on DNA methylation in skeletal muscle from first-degree relatives of patients with type 2 diabetes. Diabetes. 2012;61:3322–32.
Kanzleiter T, Jähnert M, Schulze G, Selbig J, Hallahan N, Schwenk RW, et al. Exercise training alters DNA methylation patterns in genes related to muscle growth and differentiation in mice. Am J Physiol Endocrinol Metab. 2015;308(10):E912–20.
Lindholm ME, Marabita F, Gomez-Cabrero D, Rundqvist H, Ekström TJ, Tegnér J, et al. An integrative analysis reveals coordinated reprogramming of the epigenome and the transcriptome in human skeletal muscle after training. Epigenetics. 2014;9:1557–69.
Rowlands DS, Page RA, Sukala WR, Giri M, Ghimbovschi SD, Hayat I, et al. Multi-omic integrated networks connect DNA methylation and miRNA with skeletal muscle plasticity to chronic exercise in type 2 diabetic obesity. Physiol Genom. 2014;46:747–65.
Pattamaprapanont P, Garde C, Fabre O, Barrès R. Muscle contraction induces acute hydroxymethylation of the exercise-responsive gene Nr4a3. Front Endocrinol (Lausanne). 2016;7:165. https://doi.org/10.3389/fendo.2016.00165 (eCollection 2016).
King-Himmelreich TS, Schramm S, Wolters MC, Schmetzer J, Möser CV, Knothe C, et al. The impact of endurance exercise on global and AMPK gene-specific DNA methylation. Biochem Biophys Res Commun. 2016;474:284–90.
Lane SC, Camera DM, Lassiter DG, Areta JL, Bird SR, Yeo WK, et al. Effects of sleeping with reduced carbohydrate availability on acute training responses. J Appl Physiol (1985). 2015;119(6):643–55.
Laker RC, Lillard TS, Okutsu M, Zhang M, Hoehn KL, Connelly JJ, et al. Exercise prevents maternal high-fat diet-induced hypermethylation of the Pgc-1α gene and age-dependent metabolic dysfunction in the offspring. Diabetes. 2014;63:1605–11.
Lochmann TL, Thomas RR, Bennett JP Jr, Taylor SM. Epigenetic modifications of the PGC-1α promoter during exercise induced expression in mice. PLoS One. 2015;10(6):e0129647.
Kasch J, Kanzleiter I, Saussenthaler S, Schürmann A, Keijer J, van Schothorst E, et al. Insulin sensitivity linked skeletal muscle Nr4a1 DNA methylation is programmed by the maternal diet and modulated by voluntary exercise in mice. J Nutr Biochem. 2018;57:86–92.
Nguyen A, Duquette N, Mamarbachi M, Thorin E. Epigenetic regulatory effect of exercise on glutathione peroxidase 1 expression in the skeletal muscle of severely dyslipidemic mice. PLoS One. 2016;11(3):e0151526.
Seaborne RA, Strauss J, Cocks M, Shepherd S, O’Brien TD, Someren KAV, et al. Methylome of human skeletal muscle after acute and chronic resistance exercise training, detraining and retraining. Sci Data. 2018;5:180213.
Seaborne RA, Strauss J, Cocks M, Shepherd S, O’Brien TD, van Someren KA, et al. Human skeletal muscle possesses an epigenetic memory of hypertrophy. Sci Rep. 2018;8:1898.
Laker RC, Garde C, Camera DM, Smiles WJ, Zierath JR, Hawley JA, et al. Transcriptomic and epigenetic responses to short-term nutrient-exercise stress in humans. Sci Rep. 2017;7:15134.
Brown WM. Exercise-associated DNA methylation change in skeletal muscle and the importance of imprinted genes: a bioinformatics meta-analysis. Br J Sports Med. 2015;49:1567–78.
Carter HN, Pauly M, Tryon LD, Hood DA. Effect of contractile activity on PGC-1α transcription in young and aged skeletal muscle. J Appl Physiol. 1985;2018(124):1605–15.
Fisher AG, Seaborne RA, Hughes TM, Gutteridge A, Stewart C, Coulson JM, et al. Transcriptomic and epigenetic regulation of disuse atrophy and the return to activity in skeletal muscle. FASEB J. 2017;31:5268–82.
Stephens NA, Brouwers B, Eroshkin AM, Yi F, Cornnell HH, Meyer C, et al. Exercise response variations in skeletal muscle PCr recovery rate and insulin sensitivity relate to muscle epigenomic profiles in individuals with type 2 diabetes. Diabetes Care. 2018;41:2245–54.
Bajpeyi S, Covington JD, Taylor EM, Stewart LK, Galgani JE, Henagan TM. Skeletal muscle PGC1α -1 nucleosome position and -260 nt DNA methylation determine exercise response and prevent ectopic lipid accumulation in men. Endocrinology. 2017;158(7):2190–9.
Terruzzi I, Senesi P, Montesano A, La Torre A, Alberti G, Benedini S, et al. Genetic polymorphisms of the enzymes involved in DNA methylation and synthesis in elite athletes. Physiol Genom. 2011;43:965–73.
Gates LA, Foulds CE, O’Malley BW. Histone marks in the ‘driver’s seat’: functional roles in steering the transcription cycle. Trends Biochem Sci. 2017;42:977–89.
McGee SL, Fairlie E, Garnham AP, Hargreaves M. Exercise-induced histone modifications in human skeletal muscle. J Physiol. 2009;587:5951–8.
McGee SL, Hargreaves M. Histone modifications and exercise adaptations. J Appl Physiol. 1985;2011(110):258–63.
Potthoff MJ, Wu H, Arnold MA, Shelton JM, Backs J, McAnally J, et al. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J Clin Investig. 2007;117:2459–67.
Smith JA, Kohn TA, Chetty AK, Ojuka EO. CaMK activation during exercise is required for histone hyperacetylation and MEF2A binding at the MEF2 site on the Glut4 gene. Am J Physiol Endocrinol Metab. 2008;295:E698–704.
Joseph JS, Ayeleso AO, Mukwevho E. Exercise increases hyper-acetylation of histones on the Cis-element of NRF-1 binding to the Mef2a promoter: implications on type 2 diabetes. Biochem Biophys Res Commun. 2017;486:83–7.
Hong S, Zhou W, Fang B, Lu W, Loro E, Damle M, et al. Dissociation of muscle insulin sensitivity from exercise endurance in mice by HDAC3 depletion. Nat Med. 2017;23:223–34.
Ohsawa I, Konno R, Masuzawa R, Kawano F. Amount of daily exercise is an essential stimulation to alter the epigenome of skeletal muscle in rats. J Appl Physiol (1985). 2018;125(4):1097–104.
Barreiro E, Sznajder JI. Epigenetic regulation of muscle phenotype and adaptation: a potential role in COPD muscle dysfunction. J Appl Physiol. 1985;2013(114):1263–72.
Thalacker-Mercer A, Stec M, Cui X, Cross J, Windham S, Bamman M. Cluster analysis reveals differential transcript profiles associated with resistance training-induced human skeletal muscle hypertrophy. Physiol Genom. 2013;45:499–507.
Masuzawa R, Konno R, Ohsawa I, Watanabe A, Kawano F. Muscle type-specific RNA polymerase II recruitment during PGC-1α gene transcription after acute exercise in adult rats. J Appl Physiol (1985). 2018. https://doi.org/10.1152/japplphysiol.00202.2018.
Pandorf CE, Haddad F, Wright C, Bodell PW, Baldwin KM. Differential epigenetic modifications of histones at the myosin heavy chain genes in fast and slow skeletal muscle fibers and in response to muscle unloading. Am J Physiol Cell Physiol. 2009;297:C6–16.
Kawano F, Nimura K, Ishino S, Nakai N, Nakata K, Ohira Y. Differences in histone modifications between slow- and fast-twitch muscle of adult rats and following overload, denervation, or valproic acid administration. J Appl Physiol. 1985;2015(119):1042–52.
Willkomm L, Gehlert S, Jacko D, Schiffer T, Bloch W. p38 MAPK activation and H3K4 trimethylation is decreased by lactate in vitro and high intensity resistance training in human skeletal muscle. PLoS One. 2017;12(5):e0176609.
Ribich S, Harvey D, Copeland RA. Drug discovery and chemical biology of cancer epigenetics. Cell Chem Biol. 2017;24:1120–47.
Penna F, Costelli P. New developments in investigational HDAC inhibitors for the potential multimodal treatment of cachexia. Expert Opin Investig Drugs. 2018. https://doi.org/10.1080/13543784.2019.1557634 (Epub ahead of print).
Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science. 2007;318(5858):1931–4.
Polakovičová M, Musil P, Laczo E, Hamar D, Kyselovič J. Circulating microRNAs as potential biomarkers of exercise response. Int J Mol Sci. 2016;17(10):E1553.
Silva GJJ, Bye A, El Azzouzi H, Wisløff U. MicroRNAs as important regulators of exercise adaptation. Prog Cardiovasc Dis. 2017;2017(60):130–51.
Wei JW, Huang K, Yang C, Kang CS. Non-coding RNAs as regulators in epigenetics. Oncol Rep. 2017;37:3–9.
Paul P, Chakraborty A, Sarkar D, Langthasa M, Rahman M, Bari M, et al. Interplay between miRNAs and human diseases. J Cell Physiol. 2018;233:2007–18.
Daskalakis NP, Provost AC, Hunter RG, Guffanti G. Noncoding RNAs: stress, glucocorticoids, and posttraumatic stress disorder. Biol Psychol. 2018;83:849–65.
McCarthy JJ. The MyomiR network in skeletal muscle plasticity. Exerc Sport Sci Rev. 2011;39:150–4.
Luo W, Nie Q, Zhang X. MicroRNAs involved in skeletal muscle differentiation. J Genet Genom. 2013;40:107–16.
Horak M, Novak J, Bienertova-Vasku J. Muscle-specific microRNAs in skeletal muscle development. Dev Biol. 2016;410(1):1–13.
Safdar A, Abadi A, Akhtar M, Hettinga BP, Tarnopolsky MA. miRNA in the regulation of skeletal muscle adaptation to acute endurance exercise in C57Bl/6J male mice. PLoS One. 2009;4(5):e5610.
Aoi W, Naito Y, Mizushima K, Takanami Y, Kawai Y, Ichikawa H, et al. The microRNA miR-696 regulates PGC-1 alpha in mouse skeletal muscle in response to physical activity. Am J Physiol Endocrinol Metab. 2010;298:E799–806.
Yamamoto H, Morino K, Nishio Y, Ugi S, Yoshizaki T, Kashiwagi A, et al. MicroRNA-494 regulates mitochondrial biogenesis in skeletal muscle through mitochondrial transcription factor A and Forkhead box j3. Am J Physiol Endocrinol Metab. 2012;303:E1419–27.
Keller P, Vollaard NB, Gustafsson T, Gallagher IJ, Sundberg CJ, Rankinen T, et al. A transcriptional map of the impact of endurance exercise training on skeletal muscle phenotype. J Appl Physiol. 1985;110:46–59.
Nielsen S, Scheele C, Yfanti C, Akerström T, Nielsen AR, Pedersen BK, et al. Muscle specific microRNAs are regulated by endurance exercise in human skeletal muscle. J Physiol. 2010;588:4029–37.
Russell AP, Lamon S, Boon H, Wada S, Güller I, Brown EL, et al. Regulation of miRNAs in human skeletal muscle following acute endurance exercise and short-term endurance training. J Physiol. 2013;591:4637–53.
Oikawa S, Lee M, Motohashi N, Maeda S, Akimoto T. An inducible knockout of Dicer in adult mice does not affect endurance exercise-induced muscle adaptation. Am J Physiol Cell Physiol. 2018. https://doi.org/10.1152/ajpcell.00278.2018.
McCarthy JJ, Esser KA. MicroRNA-1 and microRNA-133a expression are decreased during skeletal muscle hypertrophy. J Appl Physiol. 1985;2007(102):306–13.
Mueller M, Breil FA, Lurman G, Klossner S, Flück M, Billeter R, et al. Different molecular and structural adaptations with eccentric and conventional strength training in elderly men and women. Gerontology. 2011;57:528–38.
Davidsen PK, Gallagher IJ, Hartman JW, Tarnopolsky MA, Dela F, Helge JW, et al. High responders to resistance exercise training demonstrate differential regulation of skeletal muscle microRNA expression. J Appl Physiol. 1985;2011(110):309–17.
Drummond MJ, McCarthy JJ, Fry CS, Esser KA, Rasmussen BB. Aging differentially affects human skeletal muscle microRNA expression at rest and after an anabolic stimulus of resistance exercise and essential amino acids. Am J Physiol Endocrinol Metab. 2008;295:E1333–40.
Ringholm S, Biensø RS, Kiilerich K, Guadalupe-Grau A, Aachmann-Andersen NJ, Saltin B, et al. Bed rest reduces metabolic protein content and abolishes exercise-induced mRNA responses in human skeletal muscle. Am J Physiol Endocrinol Metab. 2011;301:E649–58.
Fyfe JJ, Bishop DJ, Zacharewicz E, Russell AP, Stepto NK. Concurrent exercise incorporating high-intensity interval or continuous training modulates mTORC1 signaling and microRNA expression in human skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 2016;31:R1297–311.
Makarova JA, Maltseva DV, Galatenko VV, Abbasi A, Maximenko DG, Grigoriev AI, et al. Exercise immunology meets miRNAs. Exerc Immunol Rev. 2014;20:135–64.
Mooren FC, Viereck J, Krüger K, Thum T. Circulating microRNAs as potential biomarkers of aerobic exercise capacity. Am J Physiol Heart Circ Physiol. 2014;306(4):H557–63.
Baggish AL, Hale A, Weiner RB, Lewis GD, Systrom D, Wang F, et al. Dynamic regulation of circulating microRNA during acute exhaustive exercise and sustained aerobic exercise training. J Physiol. 2011;589:3983–94.
Wardle SL, Bailey ME, Kilikevicius A, Malkova D, Wilson RH, Venckunas T, et al. Plasma microRNA levels differ between endurance and strength athletes. PLoS One. 2015;10(4):e0122107.
Sawada S, Kon M, Wada S, Ushida T, Suzuki K, Akimoto T. Profiling of circulating microRNAs after a bout of acute resistance exercise in humans. PLoS One. 2013;8(7):e70823.
Bye A, Røsjø H, Aspenes ST, Condorelli G, Omland T, Wisløff U. Circulating microRNAs and aerobic fitness—the HUNT-Study. PLoS One. 2013;8(2):e57496.
Nielsen S, Åkerström T, Rinnov A, Yfanti C, Scheele C, Pedersen BK, et al. The miRNA plasma signature in response to acute aerobic exercise and endurance training. PLoS One. 2014;9(2):e87308.
Aoi W, Ichikawa H, Mune K, Tanimura Y, Mizushima K, Naito Y, et al. Muscle-enriched microRNA miR-486 decreases in circulation in response to exercise in young men. Front Physiol. 2013;4:80.
Uhlemann M, Möbius-Winkler S, Fikenzer S, Adam J, Redlich M, Möhlenkamp S, et al. Circulating microRNA-126 increases after different forms of endurance exercise in healthy adults. Eur J Prev Cardiol. 2014;21:484–91.
Horak M, Zlamal F, Iliev R, Kucera J, Cacek J, Svobodova L, et al. Exercise-induced circulating microRNA changes in athletes in various training scenarios. PLoS One. 2018;13(1):e0191060. https://doi.org/10.1371/journal.pone.0191060 eCollection 2018.
Håkansson KEJ, Sollie O, Simons KH, Quax PHA, Jensen J, Nossent AY. Circulating Small non-coding RNAs as biomarkers for recovery after exhaustive or repetitive exercise. Front Physiol. 2018;9:1136.
Ramos AE, Lo C, Estephan LE, Tai YY, Tang Y, Zhao J, et al. Specific circulating microRNAs display dose-dependent responses to variable intensity and duration of endurance exercise. Am J Physiol Heart Circ Physiol. 2018;315(2):H273–83.
Denham J, Marques FZ, O’Brien BJ, Charchar FJ. Exercise: putting action into our epigenome. Sports Med. 2014;44:189–209.
Sharples AP, Stewart CE, Seaborne RA. Does skeletal muscle have an ‘epi’-memory? The role of epigenetics in nutritional programming, metabolic disease, aging and exercise. Aging Cell. 2016;15:603–16.
Acknowledgements
The authors thank Thomas Beiter for help with preparation of the figures.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This review was supported by a grant from the intramural graduate school “iReAct” of the University and the University Hospital Tübingen.
Conflict of interest
Manuel Widmann, Andreas Nieß, and Barbara Munz declare that they have no conflicts of interest relevant to the content of this review.
Rights and permissions
About this article

Cite this article
Widmann, M., Nieß, A.M. & Munz, B. Physical Exercise and Epigenetic Modifications in Skeletal Muscle. Sports Med 49, 509–523 (2019). https://doi.org/10.1007/s40279-019-01070-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40279-019-01070-4