
Abstract
Exercise training elicits acute and adaptive long term changes in human physiology that mediate the improvement of performance and health state. The responses are integrative and orchestrated by several mechanisms, as gene expression. Gene expression is essential to construct the adaptation of the biological system to exercise training, since there are molecular processes mediating oxidative and non-oxidative metabolism, angiogenesis, cardiac and skeletal myofiber hypertrophy, and other processes that leads to a greater physiological status. Epigenetic is the field that studies about gene expression changes heritable by meiosis and mitosis, by changes in chromatin and DNA conformation, but not in DNA sequence, that studies the regulation on gene expression that is independent of genotype. The field approaches mechanisms of DNA and chromatin conformational changes that inhibit or increase gene expression and determine tissue specific pattern. The three major studied epigenetic mechanisms are DNA methylation, Histone modification, and regulation of noncoding RNA-associated genes. This review elucidates these mechanisms, focusing on the relationship between them and their relationship with exercise training, physical performance and the enhancement of health status. On this chapter, we clarified the relationship of epigenetic modulations and their intimal relationship with acute and chronic effect of exercise training, concentrating our effort on skeletal muscle, heart and vascular responses, that are the most responsive systems against to exercise training and play crucial role on physical performance and improvement of health state.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Exercise is an ambient stimulus that elicits acute and adaptive long term changes in locomotor and physiological system. The changes challenge the whole-body homeostasis and mediate the improvement of performance and health state. These responses are integrative and occur both to cellular and systemic level in several tissues, and organs, as the endothelial/vascular, cardiomyocyte/heart, myocyte/skeletal muscle due or by the increase of metabolic demand of contracting skeletal muscle [1].
There is an intricate network of mechanisms that orchestrate the acute and adaptive exercise-induced response and there is much to be clarified on this issue despite the ongoing investigation. These mechanisms can be considered at several levels: related to the stimulus that triggers them, as well as the signaling pathways involved, structural level, metabolic level, and finally the mechanisms of regulation of gene expression [2].
Gene expression can be regarded as “bricks” that build the exercise-induced effects. There are molecular basis to all mechanisms involved in adaptive response exercise-induced, as muscle contraction, increased mitochondrial mass, increase of oxidative and non-oxidative metabolism, enhanced angiogenesis, cardiac and skeletal myofiber hypertrophy.
All the processes involved are mediated by several signaling events, pre- and post-transcriptional events and regulation, translation and protein processing and there are a complex molecular spatial and temporal interactions between the phases, elements and mechanisms that orchestrate the integrated response to exercise practice, also accounting genomic, ambient and exercise stimuli as intensity, duration and frequency [3].
One aspect of this spatial and temporal relationship of gene expression is the epigenetic regulation. Epigenetic is the study of changes of the gene expression independently of genotype that is heritable or dynamically modifiable. These field approaches the several DNA and chromatin conformational changes, that inhibit, increase and finally determines the developmental or tissue specific pattern of gene expression and the phenotype [4,5,6]. From the epigenetic point of view, the cell nucleus can be considered a chemical reactor of infinite complexity and high turnover.
The three major regulatory groups for epigenetic mechanisms are DNA methylation, histone modification, and the regulation of noncoding RNA-associated genes. The following section explains these modifications and their relationship with exercise training, performance and health status. Additionally, the following sections review on their relationship with the acute and chronic effect of exercise training focusing on skeletal muscle, heart and vascular system, that are highly responsive to exercise and crucial to performance and health state.
2 Epigenetics: History and Concepts
The term “epigenetics” was first used by Conrad Waddington in 1946 when he defined it as “branch of biology that studies the casual interaction between genes and their products”. Despite its breadth, this definition opened perspective about epigenetics as modulator of gene transcription and the increase of evidence by studies resulted in currently most accepted approach, postulating that “epigenetic is the study about gene expression changes heritable by meiosis and mitosis, by changes in chromatin and DNA conformation, but not in DNA sequence” [7]. Also, these chemical modifications in DNA occur and are reversed constantly during the life span of the individual, except to those that are constitutive, that are frequently induced in an individual during the life span and can be heritable. They perform role in nucleosome assembly, mitosis, meiosis, cell cycle, transcription, recombination and repair of DNA [8,9,10].
Different characteristics in monozygotic twins, progressive changes in chromatin function over development and aging, are some examples of inherited phenotypic differences of immobilized DNA sequences not mutated [11, 12]. Due the epigenetic code has a highly dynamic character it is also understood that it may influence the susceptibility to diseases, especially those related to metabolic dysfunctions, cancer and cardiovascular diseases, from random environmental influences, exposure to chemical reagents, and also behavioral patterns by previous generations. This influence is also attributed to gene expression changes due epigenetic variations in coding or non-genome regions.
The exercise training induces an adaptive pattern of gene expression that improves the performance and contributes to health state [2, 7]. Epigenetic regulation regulates in part this pattern and is interesting to know how these events and their consequences can be the usable to develop exercise and therapeutic methods of intervention, capable to prevent harmful changes to current and future generation, and also minimize harmful to current generation. Also, exercise is a viable form to study gene expression patterns that antagonizes disease, elucidating about mechanisms that intermediate both situations [11,12,13,14].
3 Epigenetic Mechanisms
3.1 DNA Methylation
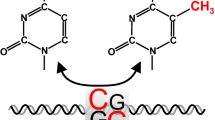
DNA methylation is a highly conserved to bacteria and eukaryotes and affects stability of genome, gene expression and development. These change involves covalent addition of methyl groups (alkyl derived from methane, containing one carbon atom bonded to three hydrogen atoms — CH3) to the fifth carbon of cytosine residues (5-mC) in DNA, usually at the cytosine–phosphate-guanine dinucleotides (also called CpGs islands) sites. The CpG islands correspond to genomic regions with more than 500 base pairs in length, 55% of those are located within promoter regions in 40% of mammals genes [12, 15].
The methylation in non CPG sites is less common and regarded as aberrant, also involved in tumorigenesis and abnormal cell functions [16].
The effect that DNA methylation has on gene expression is dependent on the site, within the genome in that occur. If methyl is added to CpG islands of gene promoters the transcription is repressed, since prevents transcription factors from binding to the promoter or by inducing binding of methyl CpG binding proteins to methylated DNA [17]. Thus methylation condenses the chromatin making transcriptional machinery inaccessible. The process of methylation/demethylation exerts important role in the embryonic development and cell differentiation, to establish properties of tissue identity (Fig. 16.1).

DNA Methylation. This mechanism is due the addition of a methyl group (CH3) to the CpGs island sites of DNA. Once CH3 is added to promoter genes, transcription is repressed by the condensation of chromatin. DNA methyltransferases (DNMTs) are enzymes responsible for adding a methyl group to the cytosine, making transcriptional machinery inaccessible. On the other hand, TET enzymes (ten-eleven translocation) are responsible for the removal of CH3 group, which brings the chromatin to its optimal condensation, making transcription possible once again [239]
Methylation at the promoter and enhancer regions of genes is associated with transcriptional repression, whereas the unmethylated state is regarded as permissive to transcription [18]. Inversely, there is evidence of active transcription as nucleotide within body of gene is methylated [19].
The frequency of 5-mC corresponds to less than 1% of the total number of nucleotides in the genome, smaller amount than the expected. Nevertheless, a striking feature, of eukaryotic genomes is the presence of regions methylated interposed in unmethylated regions. However, the distributions of dinucleotides CpG (cytosine-phosphate guanine) and 5-methylcytidines are non-random [20, 21]. CpG dinucleotides, generally are methylated in normal cells, but an exception is hypomethylation of adjacent CpG regions to active genes [22].
The CPG islands also are strongly acetylated and often acetylation occurs without histone lysine 9 residue H3, leaving a chromatin in its active configuration [21, 23]. They kept unmethylated, except in genomic imprinting or located on the inactive X chromosome [24, 25], which allow a connection of proteins and enzymes that triggers transcription. In contrast, methylated CpG islands are related to transcriptional silencing [8, 26]. Around of 70% of the CpG islands at human genome are constitutively methylated, in contrast the majority of CPG island are unmethylated. Most of methylated CpG dinucleotides exist in genomic regions of transposable elements and prevent the initiation of transcription of these elements [27].
The modification of cytosine to 5-methylcytidine, prevents binding of transcription factors such as AP-2, cMYC/MYN, CREB, E2F, and NF-kB in promoter regions of transcription initiation. Although, also other proteins as MeCP-2, MDB1, 2, 3, 4 can bind these sites, that stimulate chromatin condensation and inactivate the gene [21, 23].
The methyl-binding proteins can also mediate histone modifications and direct DNA methylation cross-talk with histone modifications contribute to the DNA methylation-related transcriptional silencing [20, 28, 29].
Enzymes that catalyze group addition methyl to the cytosine molecules belong to the family of DNA methyltransferases (DNMTs), including DNMT1, DNMT3A, DNMT3B and their isoforms, and DNMT3L. DNMT1 is the main responsible for maintaining the standards of DNA methylation during mitosis. The DNMTs3, are responsible for the de novo methylation of DNA molecules synthesized, being particularly important during early stages of embryonic development, since copies pre-existing methylation patterns onto the new DNA strand during DNA replication. DNMT3L is an alternative splicing expressed DNMT-related protein that does not contain intrinsic DNA methyltransferase activity, but physically interacts with DNMT3a and DNMT3b modulating their catalytic activity. In combination, these de novo and maintenance methyltransferases seem to constitute the core enzymatic components of the DNA methylation system in mammals [30, 31].
A fourth DNA methyltransferase, DNMT2, shows weak DNA methyltransferase activity in vitro, and deletion of the DNMT2 gene in embryonic stem cells causes no detectable effect on global DNA methylation, suggesting that this enzyme has little involvement in setting DNA methylation patterns. The role of DNMT2 is poorly knew and studies suggest that possible that both DNMT1 and DNMT2 can functionally compensate each other [24, 32].
Another enzyme that have role in regulation of the methylation pattern in DNA is the TET (ten-eleven translocation) that oxidize 5-mC to 5-hydroxymethylcytosine (5hmC) and thus starts active removal of DNA methyl. There are three isoforms: TET1, TET2 and TET3 [33].
Studies using ESCs and iPSCs culture (embryonic and induced pluripotent stem cells) suggests that TET proteins and 5hmC abundance are involved in regulating pluripotency and differentiation in potential. Inversely of observed during ESC differentiation, reprogramming of differentiated cells into iPSCs is associated with the activation of Tet1 and Tet2 and accumulation of 5hmC [34].
Additionally, 5hmC and pluripotency is further highlighted by the existence of a cluster of binding sites for pluripotency related transcription factors upstream of TET1 and TET2: Knockdown of TET1 and TET2 causes downregulation of a group of genes that includes pluripotency-related genes including: Esrrb, Prdm14, Dppa3, Klf2, Tcl1 and Zfp42 and a concomitant increase in methylation of their promoters, together with an increased propensity of ESC cells for differentiation [35]. The Factor transcription, OCT4 and SOX2 directly control the levels of both TET1 and TET2 [34]. Importantly, TET1 depletion in ESCs downregulates pluripotency-associated genes [35, 36]. Inversely, promoters of genes that are silenced during embryo body formation lose gain 5mC at their promoters [35].
Furthermore, differentiation markers as Cdx2, Gata4 and Gata6 are induced in TET1-depleted cells. This derepression induces the potential to ESC generate extra-embryonic tissues [34, 37]. Inversely, the depletion of TET2 does not increase the differentiation potential of ESCs, and simultaneous knockdown of TET1 and TET2 appears to be less efficient in increasing the trans differentiation potential when compared to TET1 knockdown alone [34].
TET2-null mices increase their quantity of hematopoietic stem cell numbers, and myeloid progenitor cells. TET2−/− and TET2+/− stem cells have an increase in self-renewal ability in culture than wild-type cells do, suggesting that TET2 expression promotes hematopoietic differentiation. The highest levels of 5hmC of any tissue are found in the adult brain, particularly in the hypothalamus and in the cerebral cortex, but they are also high in other parts. Interestingly, as neuronal cells in the adult brain normally ceased to divide mitotically, this profile is similar that of 5-mC/5hmC that is located in gene bodies was found to be associated with higher levels of transcription, as in ESCs [38, 39]. It is possible that 5hmC in gene bodies is a more general epigenetic feature, whereas its presence in promoters may particularly be a feature of pluripotent cell types [34, 40,41,42].
The demethylation mediated by TET3 seems has a role in oocytes activity to reprogram somatic nuclei during cloning (somatic cell nuclear transfer) and finally, loss of 5-mC from the paternal genome in the fertilized egg correlates with an increase in 5hmC in the male pronucleus at a time when the female pronucleus remains methylated and contains low levels of 5hmC. This suggests an involvement of 5hmC in demethylation of the paternal genome. Indeed zygotes that lack TET3 increases developmental failure since fail demethylate male pronucleus and promoter regions [43,44,45].
The CPG islands are strongly acetylated and often acetylation occurs without histone lysine 9 residue H3 (H3K9), leaving a chromatin in its active configuration [21, 23]. They kept unmethylated, except in genomic imprinting or located on the inactive X chromosome [24, 25], which allow a connection of proteins and enzymes that start a transcript. In contrast, methylated CpG islands are related to transcriptional silencing [8, 26]. Around of 70% of the CpG islands at human genome are constitutively methylated, in contrast the majority of CPG island are unmethylated. Most of methylated CpG dinucleotides exist in genomic regions of transposable elements and prevent the initiation of transcription of these elements [19].
The studies show evidence that the methylation/demethylation state is a very important epigenetic process and that correlates with activation or gene silencing and interacts with other epigenetic regulation to orchestrate the pattern of gene expression.
3.2 Histone Modifications
Acetylation was the first posttranslational modification of histones, reported by Vincent Alfrey in 1964 [46]. After that, in later 1990s high resolution X-ray suggested that these modification was from N-terminal tails that protrude from their own nucleosome and make contact with adjacent nucleosomes [47]. In present times, is established that the N-terminal modification of histones can change the link between nucleosomes and also chromatin conformation, and consequently expose DNA sites and influence transcription, as other DNA processes as repair, replication and recombination. The changes regulate chromatin structure and also enzymatically induce remodeling from ATP to reorganize nucleosomes [4]. This section will explain about these changes and how structurally affects the chromatin and consequently the gene expression (Fig. 16.2).

Histones Acetylation. This mechanism consists of the addiction of an acetyl groupo (CH3CO) in the lysine and arginine residues of histones. The balance between the deacetylated and acetylated states of histones is controlled by the antagonistic actions of two types of enzymes: histone deacetylases (HDACs) and histone acetyltransferases (HATs). HATs generally are seen as transcriptional coactivators, while HDACs are considered transcriptional repressors [239]
3.3 Chromatin Structure, Nucleosomes and Histones
First, in this section, we will give a brief overview of the assembly of Chromatin to better elucidate about histone modifications and their mechanism and functions in gene expression.
Chromatin assembly of packaged units of DNA wrapped around histones isoform (H2, H3, H4) protein structures called nucleosomes. The histone core is positively charged and forms a tight structure with negatively charged DNA backbone, thereby restricting access of transcriptional factors to DNA and suppressing gene expression [3].
The nucleosome particle contains one H3-H4 tetramer and two H2A-H2B dimers and wraps two times 147 pb of DNA [48]. The histones are unable to self-assemble into nucleosomes under physiological conditions and tend to interact with DNA nonspecifically [49]. The regulation between histones and DNA are performed by histone chaperones [50] that regulate interactions between chaperones assist in assembly and disassembly by regulation of interactions. Assembly factors in complex with histone dimers are then recruited to sites of chromatin assembly by chromatin-associated proteins, where they are deposited. After the nucleosomes formation, chromatin organization is restored by chromatin remodeling complexes, which promote nucleosome spacing [48].
Summarizing the stepwise in assembly of histone proteins onto chromatin: First the Nucleosomes form from H3-H4 dimer precursors, and associate with histone chaperones that prevent nonspecific histone–DNA interactions. These chaperones hand off histone dimers to assembly factors, which are specific to the histone that recognize. Second the H3-H4 assembly factor is recruited to target chromatin regions by DNA-bound recruiter proteins, allowing the deposition of (H3-H4) 2 tetramers. Third, upon the incorporation of H2A-H2B dimers, the new nucleosome forms. Last, the spacing creates disorganization of new and old nucleosomes, that is restored by the chromatin organizers proteins [48].
Modifications to the amino acids residues in the histone proteins, notably H3 and H4 induce major changes on chromatin structure and consequently in gene expression, since alter the histone/DNA association and is more permissive to transcriptional factors binding to DNA [3]. Such modification include lysine, arginine acetylation and methylation, lysine ubiquination, serine phosphorylation, and Poly-ADP-ribosilation of histones and will be approached in next sections.
3.4 Acetylation
Acetylation consists in reversible addiction of acetyl group (CHCO3) in lysine and arginine residues in histones. Histone acetylation is a process closely linked to gene transcriptional activation, while histone deacetylation consistently results in gene transcriptional repression. Acetylation/deacetylation patterns control the nucleosome assembly, the folding of chromatin and gene transcription, since that overcomes repressive effect of nucleosomes, and allows dynamic changes in gene transcription [51].
The balance between the acetylated and deacetylated states of histones is controlled by the antagonistic actions of two types of enzymes: histone acetyltransferases (HATs) and histone deacetylases (HDACs). As such, HATs are generally seen as transcriptional coactivators, while HDACs are considered transcriptional corepressors [51].
The HATs neutralize the lysine’s positive charge and weaken the interactions between histones and DNA, using acetyl-coA as cofactor. They catalyze the transfer of an acetyl group to the ε-amino group of lysine side chains. HATs are divided into major classes: type-A (HATsA) and type-B (HATsB). HATsB are predominantly cytoplasmic, acetylating free histones that were not deposited into chromatin. HATsB acetylate newly synthesized histone H4 at lysine 5 and lysine 12 (and some sites within H3). This pattern of acetylation is important for deposition of the histones, after which the marks are removed [52].
The HATsA are a more broad family of enzymes than the HATsB and they can be classified into three separated groups (GNAT, MYST and CBP/p300) families depending on amino-acid sequence homology and conformational structure [53]. HATsA are associated in large multiprotein complexes that play important roles in controlling enzyme recruitment, activity and substrate specificity. Each of these enzymes modifies multiple sites within the histone N-terminal tails but not only in histone tails but also in sites within globular histone core: as side chain of lysine 56 (H3), in human by hGCN5 [54]. This class functions in several transcriptional coactivators, and also has ability to neutralize positive charges, thereby disrupting the stabilizing influence of electrostatic interaction [55].
Inversely to HATs, HDACs reverse lysine acetylation, thus restoring the positive charge and repress transcription. HDACs are divided into four class and acetylate histones and non-histones protein [55].
The histones deacetylation by HDACs leads to chromatin condensation, and therefore, predominantly represses gene transcription [51]. Classified in accord to the expression patterns, cellular localization, enzymatic activity, and protein structure, mammalian genomes encode 11 members of the HDAC protein family, divides into four classes (class I, IIa, IIb, III and IV HDAC). Class I, II, and IV HDAC contain a Zn2 + − dependent deacetylase domain [55, 56].
Class I are expressed ubiquitously, localized in the nucleus and presents high enzymatic activity. Their structures are simple, consisting of the conserved deacetylase domain and short amino- and carboxy-terminal extensions. HDAC1, 3 5 and 8 are members of this class.
Class II has large N-terminal extensions with conserved binding sites for the transcription factor myocyte enhancer factor 2 (MEF2) and the chaperone protein 14–3-3, which increase their responsive signal. Consecutively, occur phosphorylation by kinases, such as calcium/calmodulin-dependent protein kinase (CamK) and protein kinase D (PKD), and these HDACs bind and shuttle from the nucleus to the cytoplasm [4]. This class show restricted expression patterns. HDAC5 and HDAC9 are highly expressed in skeletal muscle, heart and brain [57]. HDAC4 is highly expressed in the brain and growth plates of the bones [58], and HDAC7 is enriched in endothelial cells and in T-cell precursors derived from the thymus. The class IIa HDACs recruits class I HDACs through their C-terminal HDAC domain, which probably accounts for a portion of their repressive activity [59]. In addition, the regulatory domains of class IIa HDACs interact with transcriptional repressors, such as C-terminal-binding protein (CTBP) and heterochromatin protein 1 (HP1) 37, 38, 40 performing role as adaptors to nucleate multiple types of transcriptional regulators and to confer signal responsiveness to downstream target genes [40].
Class IIb HDACs are constituted by HDAC6 and HDAC10. HDAC6 is the main cytoplasmic deacetylase in mammalian cells. The proteins directly affected by HDAC6 are cytoskeletal proteins such as α-tubulin and cortactin, transmembrane proteins such as the interferon receptor IFNαR, and chaperones This isoform has two catalytic domains and a C-terminal zinc finger [60,61,62,63]. HDAC10 are enriched in cytoplasm and highly expressed in mammalian tissue and interacts with HDAC3 but the role of this interaction remains to be clarified [10, 60].
The Class IV has only one isoform: HDAC11. Little is known about its role, although it is enriched in several tissues: as brain, heart, skeletal muscle, kidney and testis. HDAC11 has a catalytic domain homologous to HDACI and II classes with short N- and C-terminal extensions [64].
In contrast, class III HDAC, also called sirtruins, characterized by NAD + −dependent deacetylase domains [65, 66]. The sirtruins are divides into five subclasses (I–IV and U) based on the phylogenetic conservation of a core domain of 250 amino acids [67]. The enzymatic activity of this class is linked to the energy status of the cell due its dependence of NAD+ [68]. Thus this class in mammals have an important role in regulation of metabolic functions, especially the class I Sirtruin I. Sirtruins have roles in metabolic homeostasis during fasting and caloric restriction by acetylation level and activity of key metabolic targets PGC-1a, FOXO1, FOXO3, NF-kB, MEF2, p53, 300 e MyoD [69].
The activities of HATs and HDACs are very important for gene transcription, since the dynamics of active genes presents high levels of acetylation turnover. It is thought that this dynamics is orchestrated by a first recruitment of HATs by transcriptional activators leading to high levels of acetylation and that HDACs are also recruited to the active gene and act as part of their global function. Also, is possible that due to the activity of HATs being more stronger then HDACs, the balance of acetylation and deacetylation changes is to more acetylated state. Also can occur overlapping specificity of HATs and HDACs, as most lysines are acetylated by HATs, but some lysines are stronger targets of deacetylation by HDACs. Thus, a combination of two mechanisms determines the functional outcome of histone acetylation and deacetylation. Also the modification exerts functions by two ways: the acetylation/deacetylation is coordinated or redundant and dictates function and second, acetylation/deacetylation of specific residues has alone functional effects [51]. Concluding, the functional state of acetylation and protein binding at a gene requires a complex analysis of HAT and HDAC recruitment, histone acetylation. Transcription profile is highly dynamic state, very sensitive to methylation/acetylation relation and other epigenetic modifications during gene activation and repression. In this way, is possible to have a comprehensive view of the various layers in which acetylation, deacetylation, and other epigenetic and histones modifications regulate gene activity (Table 16.1).
3.5 Other Histones Modification
Although acetylation is the best characterized histone modification, This section approaches other modification that influences chromatin structure and thus gene expression: Phosphorylation, Deamination, β-N-acetil-glucosamine, Poly-ADP-ribosylation, ubiquination, sumoylation, histone tail clipping, isomerization.
Phosphorylation
This change is highly dynamic, and occurs on serine, threonine and tyrosine, and not exclusively in the N-terminal histone tails but in core, as the phosphorylation of H3Y41 exerted by JAK2 [70]. This reaction is catalyzed by histone kinases of four enzyme families with ATPase activity (switching defective/sucrose no fermenting – SWI/SNF, imitation switch (ISWI), chromo domain helicase DNA binding (CHD) inositol requiring 80 (INO 80), that transfer a phosphate group from ATP to the hydroxyl group of the target amino-acid side chain. The effect is addition of negative charge to the histone that influences the chromatin structure. For major part of these enzymes remain unclear the site which the kinases exerts their effects and is possible that chromatin bound factor binds to DNA to assist the interaction. An exception is the mammalian MAPK1, that has an DNA-binding domain with which it is tethered to the DNA [71]. The phosphatases that dephosphorylate the sites in histones are less known, although is thought that there are high phosphatase activity within the nucleus due rapid histone phosphorylation [72, 73].
Deimination
This irreversible reaction is catalyzed by the peptidyl deiminase PADI4 and consists the conversion of an arginine to a citrulline, neutralizing the positive charge of the arginine and thus exposing sites in DNA to transcription [74, 75].
β-N-acetyl glucosamine
This histones modification occur in serine and threonine side chains with single β-N-acetyl glucosamine (O-GlcNAc) sugar residues and is catalyzed by O-GlcNAc transferase, consisting in the transfer of the sugar from the donor substrate, UDP-GlcNAc, to the residue. The modification occur in H2A, H2B and H4, is highly dynamic and rapidly reversible by β-N-acetylglucosaminidase (O-GlcNAcase) [76].
ADP-ribosylation
This modification is reversible and consists in addiction of ADP molecules on glutamate and arginine, that correlates with relaxation of chromatin [77]. These reaction is catalyzed by poly-ADPribose polymerase (PARP) and reversed by the poly-ADP-ribose-glycohydrolase family of enzymes. PARP-1 activity induces to high acetylation core histone and its ribosylation in the HK4me3 inhibits demethylation and excludes H1 making promoters of genes accessible [78, 79].
Addiction of only one ADP molecule is catalyzed by mono-ADP-ribosyltransferases on H2a, H2b, H3 and H4, core histones, and also in the linker H1. These modification are related with the pathway of DNA damage [77].
Ubiquination and Sumoylation
This covalent modification is larger than others and highly dynamic, consisting in the attachment of Ubiquitin, a polypeptide, covalent to histone lysine via the sequential action of three enzymes: E1, activator of Ubiquitin, E2, conjugator of Ubiquitin and E3-ligation enzymes. The modification is removed via the action of isopeptidases [80]. Although the sites H2A and H2B. H2AK119ub1 are related with gene silencing, and H2BK123ub1 with transcriptional initiation and elongation little is known about this modification [75, 81]. Sumoylation is addiction of small molecules like-ubiquitin lysine in the H2A, H2B, H3 and H4. This modification is considered be repressive since, antagonizes acetylation and ubiquination in the side chain of lysine [82,83,84].
Histone Tail Clipping
This modification consists in remotion of several amino acids residues of histone N-terminal tail in Histone that they reside [85,86,87]. In mammals the mouse enzyme was identified as Cathepsin L, and cleaves the N-terminus of H3 during ES cell differentiation Histone tail clipping is also related with DNA damage and show a consequent modification in acetylation and methylation patterns, but direct relation with transcription and gene expression in human is not stablished [87].
Isomerization
This modification is not covalent and consists in the interconversion of proline between the cis and trans conformations, being catalyzed by isomerases [88, 89]. Recently, this modification is related to aspartate residues in brain mices, being D-asparagyl related to activation of chromatin and increased transcription activity.
3.6 Non Coding RNAs
Nowadays is clear that Non-coding RNAs (ncRNAs) have different regulatory functions in Eukaryotes, including in mammalians. Recently, in last decade, ncRNAs have been implicated also in epigenetic mechanisms, as transposon activity and silencing, paramutation, variegation and X-chromossome inactivation. The ncRNAs are also able to direct the DNA methylation and histone modifications and thus related to gene expression control in complexes organisms [90].
The ncRNAs are RNAs molecules that do not encode proteins, but have regulatory functions. According to their size they can be divides into small and long ncRNAs [91]. These small ncRNAs, includes single strand endogenous microRNAs, siRNAs (small interfering RNAs) e PiRNAs (Piwi-interacting RNAs) (17–29), that differentiates in enzymatic process and Argonaut recruitment in their biogenesis. Since all are negative regulators of translation step in protein synthesis, they play important roles in numerous cellular and tissue processes, as apoptosis, proliferation, differentiation, growth, DNA damage, and pluripotency [92]. Currently there are 2603 mature sequences of small ncRNAs discovered stored at data base airbase 21 (mirbase.org). They act by the coupling in the messenger RNA (mRNA) transcript from a gene and initially was believed that only coupled in 3′ UTR (untranslated region) regions. Today we know that there are several functional sites to small non coding in cell as 5′ UTR region and that there are miRNAs interactions with others microRNAs and proteins. The small ncRNAs are “promiscuous” molecules, since they have hundreds to thousands of targets and several can act in one target simultaneously to effective their function. Small non coding RNAs have multiple pairing possibilities in sites within their target gene. To approach exactly their functional target genes and “wave” effects remains an important challenge to researchers [93, 94].
Small Non Coding RNAs
In mammalian, the small ncRNAs are not directly related with epigenetic regulation, although in disease, aberrant expression of microRNAs and their cluster can alter the global DNA or chromatin state by negatively inhibition of DMNTs 3 and 1 and these regulation [95,96,97].
The loss of a crucial component in biogenesis of small ncRNAs, Dicer, that processes long RNAs, was able to induce aberrant accumulation of long non-coding RNAs in centromeres and consequently there was loss of histone H3 lysine 9 methylation (H3K9me), which was detrimental to the functions of the centromere [98, 99].
There are silencing protein complexes (RITS, RISC), guided to small ncRNA that acts in pairing between centromeres, complementary DNA sequence and in negative regulation in targets messenger RNA (mRNA) [92, 100, 101]. These complexes also have methyltransferase activity to repetitive regions to methylate histone H3 at lysine 9 (H3K9me). This modification is able in turn recruits chromodomain-containing proteins, such as Chp1, Chp2, and Swi6, to initiate the spreading and establishment of heterochromatin domains. RITS is able to interact with another complex called the RDRC (RNA-dependent RNA polymerase complex), that Rdp1 and Dicer activity and have RNA processing capacity, showing interaction between small ncRNAs biogenesis and epigenetic modification that formats heterochromatin structure at the centromere [102].
The Argonaut 4, other protein involved in maturation and function of small ncRNAs, has the role direct siRNA to the targets and simultaneously histone and DNA methylation [103].
The piRNAs recruit repressor factors HP1a and Su(vary)3–9 to specific genomic loci to and consequently repress RNA polymerase II transcription [104, 105]. PiRNA binds to Piwi protein complexes to the promoter region of the repressor factor CREB2 in nervous cells and mediate its memory-related DNA methylation [106].
These findings show evidence that small ncRNA molecules and machinery have roles in epigenetic regulation. Further mechanistic studies of small ncRNAs involved in this process may show others roles that remain unclear and elucidate about epigenome interactions [91].
Long Non Coding RNAs
The lncRNAs can range their length from few hundred to 100 kilobase species. Evidence now suggests that large non-coding regions of the human genome are transcribed during normal and diseased cellular function. lncRNAs can express from introns, exons or intergenic regions [28]. They can be divided into five categories: (1) Sense, (2) Antisense lncRNAs that are transcribed in the opposite direction of protein-coding genes and have partial overlap with genes. (3) bidirectional when its expression and of neighbor coding transcript is initiated in close genomic proximity, (4) Intronic when is completely within an intron of a second transcript (can be precursors by smalls ncRNAs) (5) Intergenic lncRNAs reside in intergenic regions without overlapping gene [107].
The versatile lncRNAs are implicated in epigenetic gene regulation by several forms that are only recently clarified. Initially implicated in two heritable epigenetic processes, Inactivation of X Chromosome and Imprinting, nuclear roles, lncRNAs also has been implicated in gene regulation as CERNA (Competitive Endogenous RNA). There are lncRNAs that exerts roles in nucleus and cytosol. In the nucleus, lncRNAs are able to recruit and bind complexes (including chromatin regulation and transcriptional machinery proteins as RNA polymerase) that regulate directly gene expression in promoters and gene loci and also directly bind in DNA to promote cis and trans activation. Additionally, the lncRNA binding at DNA sites influenciates acetylation and methylation pattern in adjacent sites [108, 109]. In cytosol lncRNAs are able to bind mature sncRNAs and inhibit their function and also “coating” tertiary structure of transcription factors and splicing factors [108, 109].
Concluding this section, there is evidence that the several mechanisms of epigenome “cross talk” to regulate gene expression and consequently the adaptive/compensatory phenotype required in life situation, as health/disease, development/growth. The adaptive response to training includes several signaling pathways to induce hypertrophy, angiogenesis, and metabolic gains. The signaling pathways recruit activator and repressor factors that influence the pattern of gene expression and effective the response. Thus, is not amazing that epigenetic regulation is a multilevel regulation that may orchestrate the exercise-induced response. In the next sections we will approach about the epigenetic regulation involved in exercise response in the skeletal muscle, heart and endothelial tissue.
3.7 Epigenetic Regulation, Exercise and Heart
Epigenetic events such as DNA methylation, histone modifications and microRNA regulation play an important role in the programming gene expression, in cardiogenesis and are crucial in correctly development of the heart after birth [110, 111].
Epigenetic changes are strongly influenced by lifestyle. It is known that the type of diet and physical exercise can lead to epigenetic alterations [112]. Recent studies have shown that physiological stress, promoted by physical exercise, can be determinant in epigenetic variations in myocardium [113]. In view of this, numerous researchers have focused on studies epigenetic related to the increase in phenotype variability in individuals who practice aerobic and resistance training, including epigenetic changes that can be transmitted during fetal development due to physical exercise [114,115,116]. It is believed that, especially, the exercise habits of pregnant women during the gestational period, can significantly impact the cardiac metabolism of the child [117]. This impact results from the binding of compounds in certain genes altering their expression which could influence future adaptations to exercise in adult life [118]. This may explain why many studies show different adaptations in the heart of individuals undergoing the same physical training [119]. These studies show evidence that the heterogeneity are not only linked to gene patterns but also linked to epigenetics alterations [120].
There is a strong evidence to epigenetic impact in a set of morphological and functional adaptations with exercise training in cardiac adult life [121, 122]. Historically, the study of the changes in the base sequence of DNA, cardiac transcriptional and translation processes and intracellular pathways signaling has been very informative in understanding the array of the events in myocardium to exercise training [123, 124]. Recently the rapidly evolving field of cardiac microRNAs has further broadened our understanding of cardiac response to exercise, however, there is still little understanding of other epigenetic alterations [111, 125]. In this section, we will describe epigenetic cardiac and their role in controlling cardiac response to aerobic and resistance training.
3.8 Aerobic Exercise
Aerobic training is well known to promote beneficial adaptations in the myocardium that includes cardiac hypertrophy, cardiac metabolism control, improvement in contraction and relaxation, formations of new blood vessels and decreased collagen content, improved myocardial antioxidant capacity, and decreased mitochondrial dysfunction and has been shown to prevent cardiomyocyte apoptosis [126, 127].
In recent years it has been demonstrated that the development of these hypertrophic phenotypes is closely linked to changes in regulatory regions of DNA. Epigenetic processes responsible for controlling the methylation and acetylation of chromatin through histones have been demonstrated in the heart. In addition, a class of non-coding RNAs has been shown to play an important role in the control of hypertrophic processes of the heart [105, 128].
The eccentric hypertrophy due to volume overload is characterized by addition of sarcomeres in series and longitudinal cardiomyocyte growth. The phenotype of this remodeling is typically related to exercises, such as running where greater volumetric overload is required. The left ventricle remodeling induced by physiological stimuli leads to preserved or even enhanced left ventricle function [114, 129].
On the other hand, cardiovascular diseases associated with the pathological cardiac hypertrophy phenotype generate exponential searches for pharmacological and non-pharmacological therapies for the prevention and treatment of this phenotype. Some studies in the last decades have suggested that the manipulation of HDACs may be interesting therapies, since its interaction with transcription factors and histones seems to regulate this phenotype of cardiac hypertrophy [130,131,132,133]. This gene regulation given by epigenetic mechanisms seems to play a key role in the development of the heart and its adaptation to stimuli generated in the myocardium in the postnatal period [134]. As already described, histones are modified by several mechanisms, which may result in the activation or suppression of gene expression.
3.8.1 DNA Methylation
To heart, physical exercise seems offers an epigenetic regulation that holds benefits with several health domains [115, 127, 135]. The exercise promotes consistent cardiovascular benefits but yet the involvement of methylation mechanisms remains to be completely elucidated. There are several studies involving physical exercise and cardiovascular health improvement but few implicating methylation [136,137,138].
Denham et al. applied an exercise training consisting of sprint interval training, showed that the increase of cardiorespiratory fitness of 12 healthy young men participants and the improvement of their maximal running performance was concomitant with a decrease of low density lipoprotein concentration and genome-wide DNA methylation changes in their sperm. Several CpG island and gene promoter regions were demethylated after exercise, indicating increased genome-wide transcriptional changes, including epidermal growth factor (EGF) that presented reduced gene expression. MicroRNAs miR-21 and miR-210 locus (MIR21 gene) changed DNA methylation, which induced their expression in other genes involved in improvement of cardiovascular function [139]. Concluding, exercise training is able to change the gene expression to promote health and prevent disease and there is evidence that methylation is involved in this adaptive process, that provides contributions to respiratory and cardiovascular health and regeneration. Exercise, as epigenetic regulator, implies the potential to counteract pathophysiological processes, and health-related changes in skeletal muscle, cardiovascular cells and other [7]. The underlying molecular mechanisms, as methylation regulation remains to be completely clarified, as their relationship with populations, dose-response, modality, intensity of exercise.
3.8.2 Histones Acetylation
HDACs are the most studied class of regulatory enzymes in relation to the epigenetic mechanisms related to the phenotype of cardiac hypertrophy. Studies with animals such as that of Zhang et al. (2002) show the participation of these epigenetic mechanisms related to cardiac disorders [133]. In 2002 this group published a striking article in the journal Cell where they demonstrated that two class II HDACs are responsible for controlling the phenotype of cardiac hypertrophy by interacting with myocyte enhancer factor 2 (MEF2), a transcription factor that activates several genes of the genetic load fetal heart disease [133]. In this study mutant mice were created for the HDAC9 gene, being refractory to the phosphorylation of HDAC kinase, thus preventing the action of HDAC9 as a suppressor of pro-hypertrophic transcription factors [133]. Still, these young animals, in the absence of stress, did not show any difference in cardiac function and morphology in relation to the controls, however, with aging or pressure overload or stimulation of calcineurin (an important activator of pro-inflammatory pathways and pro-thrombotic); these animals had a pronounced phenotype of pathological cardiac hypertrophy. The authors correlated this hypertrophy with an over activation of MEF2. Thus, they conclude in this study that class II HDAC may act in the adult heart as suppressor of fetal load genes and its manipulation may serve as a potential therapeutic target [133].
Later in 2004, this same research group, published a new study showing once again the role of these class II HDACs as suppressors of pathological cardiac hypertrophy [130]. In this study they created a mutant animal for HDAC5, which as well as the HDAC9 deficient also spontaneously with aging had a pathological hypertrophic phenotype. To further substantiate the role of these HDACs as controllers of the cartilage remodeling process, the authors created a double mutant animal for both HDACs (HDAC5 and HDAC9) and a high percentage of these animals died during the embryogenesis period [130].
One of the most powerful weapons for the prevention and regression of this phenotype of pathological cardiac hypertrophy is physical training [140, 141]. Physical training has been used as an effective non-pharmacological tool, however little is known about its role in epigenetic mechanisms in the heart. In a recent study by Soci et al. (2016), animals were submitted to an aerobic swimming training protocol that mimics volume and intensity of training equated to that of an athlete [135]. The group of animals trained in this protocol showed a significant decrease of HDAC4, being a class II HDAC that is also associated with the development of pathological cardiac hypertrophy [135]. This HDAC4 is phosphorylated by calcium/calmodulin-dependent protein kinases I and II (CAMKI and CAMKII) which is an enzyme strongly correlated with pathological cardiac hypertrophy. With this study, the authors show for the first time that aerobic physical training may be able to modulate the expression of class II HDAC by regulating the gene expression of the fetal genetic load, thus functioning as a therapeutic alternative for cardiac disorders. Furthermore, in this study the exercise was able to modulate the expression of HP1β (heterochromatin protein 1β) which is a MEF2 corepressor, contributing to suppression of the expression of fetal reprogramming genes [135].
In 2007, Montgomery and colleagues published a study where they demonstrated the importance of class I histone (HDAC1 and HDAC2) for heart development and growth as well as cardiac function [132]. The authors conclude that these two HDACs perform a redundant work, since when the specific deletion of each was made, there were no differences in the development or function of the myocardium; however, when they were deleted simultaneously, there was an increase in lethality in these animals [132]. Other recent study showed that aerobic swimming training increased HDAC1 expression and decreased HDAC3 expression [135]. This data is in agreement with another study where treadmill training was used for 4 weeks, the authors also found an increase in the expression of HDAC1 with aerobic exercise [142]. In this study, the authors conclude that diabetic mice (db/db) have a compensatory mechanism between the two HDACs (HDAC1 and HDAC2), and there is a decrease in HDAC2 after the fourth week, when there is cardiac hypertrophy, showing so possibly HDAC2 is more closely linked to cardiac hypertrophy than HDAC1 [142].
3.8.3 MicroRNAs
Many miRNAs have emerged as promising therapeutic targets for cardiac disorders [128, 129, 143, 144]. In 2006, Van Rooij and colleagues published in the journal PNAS a signature pattern of miRNAs that are modified in the heart in response to transverse aortic constriction or expression of activated calcineurin and may play an important role for pathologic cardiac hypertrophy and for the phenotype of heart failure. In this study, of the 16 miRNAs that were selected because they were up- or down regulated in response to CT or Calcineurin activation [128]. One of the most prominent microRNAs was miRNA-195, thus they created a lineage of animals with overexpression of this miRNA. These animals showed spontaneous increase of myocytes with cardiac dysfunction from the sixth week after birth [128]. Therefore, the identification of miRNAs differentially expressed in conditions of myocardial stress as done in the Van Rooij study opens new possibilities in the search for therapeutic approaches to cardiac disorders.
In the search for these therapeutic approaches many studies have demonstrated the efficacy of physical training in modulating the expression of several miRNAs in the heart, thereby regulating its several target genes in several hypertrophic processes against pathological and physiological stimuli, under this circunstances, physical exercise is an important non-pharmacological therapy for the prevention and reversal of pathological processes [115, 116, 129, 140].
In 2015, Liu et al. presented in an elegant study that miRNA-222 is required to induce physiological cardiac hypertrophy caused in response to aerobic physical training, both swimming and treadmill. Furthermore, this miRNA seems to exert cardioprotective effect against pathological cardiac remodeling [145]. In this study two animal training models, aerobic treadmill and aerobic swimming were used to identify which miRNAs would present a pattern of expression that would induce physiological cardiac hypertrophy in both models [145]. The miRNA-222 was increased 2.1- and 2.8-fold in the swimming and running exercise models, respectively (p < 0.003 and 0.02), so it was selected with particular interest.
After selection of miRNA-222, the authors created a transgenic animal with overexpression of this miRNA and induced an ischemic injury in these animals. As results, they obtained that the overexpression of this miRNA caused cardioprotective effect reducing fibrotic content in relation to the control group [145].
Aerobic physical training has already been described as capable of modulating the expression of several miRNAs responsible for cardiac remodeling [115, 127, 135, 146, 147]. In 2011, Soci et al. showed that miRNA-29c expression was increased with swimming training and was negatively correlated with the expression of collagen content, improving myocardial compliance [115]. Fernandes et al. (2011) demonstrated that the same protocol of aerobic physical training of swimming was responsible for increasing the cardiac expression of miRNAs-27a and -27b and followed by a decrease of miRNA-143, which presented as direct targets components of the renin angiotensin system (RAS) Strictly related to cardiac remodeling [127]. The miRNA-126 was also correlated with physical swimming training inducing cardiac remodeling, Da Silva et al. Demonstrated that training induced physiological cardiac remodeling followed by increased angiogenesis, which correlated with a decrease in Spred-1, which favored an increase in pathway signaling Pro-angiogenic reactions such as Raf-1/ERK ½ [148]. In addition, the miRNA-126 also targets the PI3KR2 protein, was decreased in the trained groups, causing an increase in PI3K/Akt/eNOS pathway signaling in these groups [148] (Fig. 16.3).

Schematic summary of the epigenetic cardiac regulation in the cardiac remodeling induced by aerobic and resistance exercise in animal model. MiRNAs involved in aerobic training adaptive response, resistance training adaptive response and both training response. Methylation and Acetylation are known to regulate aerobic training responses, but not resistance training response. All epigenetic regulation interacts to orchestrate the physiological cardiac remodeling. MiRNAs (microRNA), HDAC. (Histone deacetylases) [148, 154, 155, 239]
3.8.4 Resistance Exercise
Several studies suggest that resistance training has beneficial effects on the cardiac morphological and contractility and can potentially be an effective treatment for various clinical conditions as such heart disease [149, 150]. It is well established, that cardiac after-load due to intermittent increases in blood pressure during resistance training induces pressure overload to the left ventricle. This intermittent pressure stimulus to the heart increases cardiomyocyte cell width and consequently increase left ventricular wall thickness. This cardiac hypertrophy features observed in weight-lifting athletes are defined as concentric and physiological [127]. On the contrary, the cardiac hypertrophy occurs in response to situations such as hypertension and valve diseases are defined as concentric and pathological [151]. Although the cardiac hypertrophy in cardiovascular system already have been established with resistance training [152], little is known about the molecular mechanisms responsible for mediating the different forms of cardiac hypertrophy (Fig. 16.4).

Schematic summary of the epigenetic skeletal-muscle regulation in biological process related to exercise training. DNA Methylation, Histone acetylation and MicroRNAs interacts to regulate protein synthesis and thus skeletal muscle myogenesis, hypertrophy and angiogenesis. HDAC (Histona deacetilases) [239]
Resistance training-induced beneficial adaptation to the cardiovascular system was neglected by many years, thus many of the mechanisms of resistance training induced cardiovascular adaptations are still uncovered [149, 153]. One of the main problems is still the lack of models animals with similar protocols of the resistance training performed for humans. Our group and others has used an animal model of resistance training that mimic the exercise performed for bodybuilding in rats and today it is the most used model by many laboratories [154].
In 2005, our group using this animal model of the resistance training showed left ventricular hypertrophy in rats in response to resistance training [155]. The extent of left ventricular hypertrophy found in the trained group (12%) was similar to that reported in some human studies involving weight lifters training for no longer than 3 months, but smaller than that seen in other studies investigating athletes engaged in this type of exercise for more than 1 year [156, 157]. In addition, our group also investigated the morphology of single left ventricular myocytes induced after 8 weeks of resistance training. In this study confirmed that resistance training model promoted an increase the width and volume of left ventricular myocytes when compared to sedentary control animals [158].
Many researchers have focused on miRNA studies predominate in the field of cardiovascular system [159], however little is known about their expression patterns or role of the miRNA in physiological cardiac hypertrophy conditions, especially with resistance training [127]. On the another hand, in studies to analyze the cardiac miRNA expression signature of miRNA with use of the microarray platforms indicated that miRNAs are aberrantly expressed in pathological cardiac hypertrophic mouse [128]. In this regard, miRNA expression profile under either experimental or clinical conditions of cardiac hypertrophy has been revealed [160]. Studies have identified anti-hypertrophic miRNAs (miRNA-1, -133, -26, -9, -98, -29, -378, and -145) and pro-hypertrophic miRNAs (miRNA-143, -103, -130a, -146a, -21, -210, -221, -222, -27a/b, -199a/b, -208, -195, -499, -34a/b/c, -497, -23a, and -15a/b) in the heart and miRNAs that are expressed only in cardiac tissue (miRNA-1, -133a/b, -208a/b, and -499) [127]. Interestingly, in experiments with transverse aortic constriction that promotes concentric cardiac hypertrophy occurred an downregulation of the miRNA-1 and -208a while overexpression of miRNAs-208a and -499 were involved with pathological cardiac hypertrophy [161,162,163,164].
Additionally, Melo et al. observed in animals subjected the same protocol to resistance training that the isolated cardiomyocytes had an improvement in contraction and relaxation [158] and Pinter et al. showed that the improvement in cardiomyocyte contractility was due to an increase in myosin ATPase activity, the papillary muscles developing more isometric force and an enhanced Ca2+ influx and in the trained group [165]. These results shown that there is not agreement with results from other studies that cardiac function is not altered in resistance training individuals [157]. Although, Melo et al. did not test all mechanisms, the increase in the expression of calcium regulatory proteins such as Serca 2a which, show in cells of the training group in this study, that is responsible in rat ventricular cells for 92% of Ca2+ reuptake may partly explain the improvement the time to peak and time to half relaxation [158, 166]. From these data our group was the first to identify Serca-2a target miRNA differently expressed in cardiac left ventricular induced by resistance training. Through in silico analysis of predicted targets for miRNA, we verified a possible relationship between Serca2a and miRNA-214. Our results showed that decreased miRNA-214 levels in the trained group may explain the increased expression of Serca2a. This relationship becomes of great interest because our results show that the regulation of Serca2a by miRNA-214 occurs by resistance training [158]. However, other microRNAs may also be acting in the regulation of Serca 2a. Gurha et al., also showed that genetic ablation of miRNA-22 regulates target proteins that function as transcription factors for Serca2a expression, Wahlquist et al., showed that miRNA-25 regulates Serca2a and contributes to declining cardiac function during heart failure [167, 168]. On the other hand, the miRNA-214 may also be acting in the regulation of other contractile proteins as well as Aurora et al., reported that miRNA-214 targets both sodium/calcium exchanger 1 (NCX) and pro-apoptotic effectors of Ca2 + signaling pathways like CaMKII and cyclophilin D [169]. Although we and others have used an animal model of resistance training to study cardiovascular adaptations data are still scarce with regard to role of the miRNA in contraction and relaxation function.
It is well-known that cardiomyocyte contractility depends on the expression of α- and β- myosin heavy chain (MHC), since MHC is the major contractile protein of the heart, and is crucial to the efficiency of cardiac performance. Studies have shown a shift from α- toward β-MHC composition of the adult heart under pathological conditions accompanied by higher expression of fetal gene reprogramming, which correlates with impaired cardiac performance [163]. Using this animal model of resistance training, Barauna et al. shown that none of the pathological cardiac hypertrophy molecular markers factor as such atrial natriuretic or α-MHC-to-β-MHC ratio, were changed in resistance training rats [155]. Interestingly, miRNA-208 encoded by the α-MHC gene, has been shown to be involved in pathological cardiac growth and up-regulation of β-MHC expression [163]. Recently, Soci et al., observed that high-volume swim training, improved cardiac diastolic function, induced cardiac hypertrophy and decreased the expression of miRNA-208a and 208b [135]. On the other hand, Van Rooij et al. (2006) showed that overexpression of miRNA-208a is required for expression of β-MHC in response to pathological stimuli [128]. Although the data indicate that different phenotypical changes observed in response to pathological state can be regulated by miRNA alterations, little is known about the mechanisms involved cardiac epigenetic with resistance training.
3.9 Epigenetic Regulation, Exercise and Skeletal Muscle
The muscular mass corresponds to more than one third of the weight of a healthy adult individual, thus the skeletal muscle is one of the most abundant tissues of the human body. The main functions of this tissue are sustentation, joint movement, heat production and caloric control [170]. The skeletal muscle is composed of cylindrical and long cells, organized in bundles and with the presence of many nuclei. This multinuclear characteristic confers to this tissue a great plasticity and that generates a rapid response to diverse stimuli such as nutritional, mechanical, hormonal and humoral [171]. These responses also generate adaptations in skeletal muscle that may be beneficial or harmful and which in turn are closely related to epigenetic factors [171].
Currently it is very clear that a number of diseases, especially metabolic chronic diseases, generate stimuli that induce harmful responses in skeletal muscle [172,173,174]. Muscle cachexia [175], sarcopenia [176], Inflammatory processes in the skeletal muscle [177] and microvascular rarefaction [178], are just a few examples of the responses that skeletal muscle can present as a result of pathological stimuli. All of these processes are responses that occur through innumerable molecular changes and in turn can be orchestrated without changes in DNA, for example by stimulating decompensated expression of non-coding RNAs as miRNAs, by inducing acetylation or deacetylation of chromatin and also through methylation processes [3, 171, 179, 180].
On the other hand, there are stimuli that cause changes in skeletal muscle, but these changes are beneficial. In the case of physical exercise, the muscular contractions induce the increase of the muscular mass [181], increased angiogenesis [182] and increased mitochondrial biogenesis [183]. These processes can also occur without changes in DNA [3]. Physical exercise, in turn, is also able to counteract the responses generated by some diseases in skeletal muscle, acting as a therapeutic tool.
With all that we have said so far we can understand that skeletal muscle adapts to the stimuli that individuals pass through life. Responses to these stimuli, beneficial or harmful, are linked to various signaling pathways, for example, the PI3K/AKT/mTOR that contributes to protein synthesis and hypertrophy [184], and pathways of degradation such as the PTEN and FOXO pathway [185].
The phosphorylation and activation of AKT depends on a variety of signals, such as cytokine and hormone growth factors, this activation is conditioned by the phosphorylation of PI3K [184]. Knockout animals for the AKT1 gene (AKT1 -/-) show deficiency in muscle growth [186] and mice that overexpress AKT1 result in a hypertrophic phenotype characterized by increased tissue size [187].
Another important function of AKT in skeletal muscle trophism is the regulation of gene transcription through the inactivation of FOXO, a transcription factor responsible for the transactivation of genes involved with components of the proteolytic system coordinated by the ubiquitin-proteasome system [188].
The FOXO isoforms are predominantly located in the nuclear compartment where they are expressed in the active form, but when phosphorylated, mainly by AKT, FOXO proteins are sequestered to the cytosol, where they are unable to transcribe genes involved in the process of muscular atrophy. Therefore, in muscle atrophy situations, the decreased AKT signaling pathway allows for the transcription of atrogin-1 and MuRF1, two-component skeletal muscle component of the E3 ubiquitin ligases [184, 185, 188].
Another important protein is PGC-1α, a transcriptional coactivator that regulates genes involved in energy metabolism and mitochondrial biogenesis. This protein interacts with several transcription factors from binding to the nuclear receptors [189].
The good functioning of the contractile muscular machinery is associated with the adequate expression of some protein pathways, changes in these pathways contribute to the great plasticity of the skeletal muscle and there are epigenetic mechanisms to control them. Thus, in this part of the chapter we will talk about how skeletal muscle is influenced by epigenetic mechanisms and how harmful stimuli such as chronic diseases and beneficial stimuli such as physical exercise also influence the epigenetic responses in skeletal muscle.
3.9.1 Methylation
The vitamins B6 and B12, obtained from nutrition, regulate the metabolism of homocysteine, an epigenetic product of DNA/RNA/protein methylation [190]. Hyperhomocysteinemia (HHCy) is implicated in elderly frailty and linked to vitamin deficiency, being a risk factor for cardiovascular and neurodegenerative diseases, as well as osteoporotic fractures and complications during pregnancy. A study applied an exercise schedule to reverse HHCy-induced changes in CBS+/− mice showing greater fatigability, due to reduced ATP levels, with a lesser generation of contractile force. Molecular changes, elevated during HHcy were reversed after exercise: amount of NRF- 1, a transcriptional regulator of mitochondrial transcription factor A (mtTFA), was decreased together with mtTFA protein quantity in homocysteine treated cells, concomitant with an increase in DNMT3a and DNMT3b proteins and global DNA methylation levels in skeletal muscle [191].
Other study identified imprinted genes in skeletal muscle gene networks and observed exercise-associated DNA methylation alterations. The bioinformatics meta-analyzed only imprinted exercise-related genes, and showed that overall methylation pattern appears to be predictive to population selection and quantification of exercise. Some genes that were differentially methylated in response to exercise-activity (RB1, MEG3, UBE3A, PLAGL1, SGCE, INS) were important for muscle gene networks [192].
Voisin et al. also showed that DNA methylation decreased with exercise (60% of loci), suggesting increased gene transcription. Exercise-associated DNA methylation was stronger among older people (age accounted approximately for 30% of variation). Among older people, genes exhibiting DNA methylation decreases were in part of a miRNA-19b regulation that is tumor suppressor. Controlled exercise interventions could help the aging epigenome, especially among older patients that normally presents a several disease phenotype, including cancer propensity and cardiovascular [193].
Exercise intensity benefits for positive epigenetic changes in terms of mitochondrial biogenesis were shown by Edgett et al. Also, healthy human male subjects that perfomed interval cycling at 73, 100 or 133% presented peak power output (PPO) and post-exercise changes in gene expression of PGC-1α (peroxisome proliferator-activated receptor gamma coactivator 1 alpha, a protein encoded by the PPARGC1A gene) and its regulators were estimated in skeletal muscle biopsies. Notably, increases in the mRNA levels of the regulators Sirt-1, PDK4 and RIP140 (metabolic genes) also occurred [194].
3.9.2 MicroRNAs
MiRNAs are part of the class of non-coding RNAs and there are an abundance of studies with these molecules in the skeletal muscle. MiRNAs control much of the expression of the encoded proteins in the human body; therefore, many biological processes are controlled by miRNAs in a post-transcriptional mode. Skeletal muscle together with cardiac tissue have their own set of miRNAs, such as miRNA-1, miRNA -133a / b, miRNA -206, miRNA -208a/b, miRNA-486 and miRNA-499 which are referred to as myomiRs [195], But it is to be noted that miRNA-206 is expressed only in skeletal muscle and miRNA-208a is expressed only in the heart. These miRNAs control the biogenesis, regeneration and maintenance of skeletal muscle tissue [196]. In cases of chronic diseases it is common to find abnormal expression of these accompanied by injury to skeletal and cardiac muscle.
MicroRNA-1 and microRNA-206 are very similar. Their hairpin differ in only 3 base pairs and have the same seed region, so they share many targets and have similar functions, so they will be quoted together in this section.
These miRNAs are strongly related to muscle development, specifically there is a remarkable increase in the expression of these during the differentiation of myoblast. The high expression of these miRNAs in this period is related to the interruption of cell proliferation, an effect present in the two miRNAs. Studies inhibiting their expression show the importance of these in decreasing the proliferative phase and allowing the development of myoblasts to start [197].
Skeletal muscle satellite cells are a cell type that have an important function in regeneration and muscular hypertrophy. These are in the quiescent state until required by cellular signals that are given to the muscle after some injury. If muscle damage occurs, these cells enter the cell cycle and increase their proliferation, fuse the remaining muscle cells, differentiate and this process is one of the main ways to occur muscle regeneration; This phenomenon in turn is also orchestrated by the expression of some miRNAs among them miRNAs-1 and 206.
After an injury to the skeletal muscle occurs a remarkable decrease in the expression of the miRNAs 1/206 which is succeeded by a great increase in the expression; This phenomenon is related to the cycle of cell proliferation and differentiation. MiRNAs-1 and 206 are involved in the process of myogenesis and many of their targets have a role in proliferation and differentiation pathways, the main targets of these miRNAs regulating these processes are the proteins HDAC4, PAX3 and PAX7 [196].
PAX3 and PAX7 are important cell proliferation factors, mainly satellite cells, and these are both targets of the miRNAs-1 and 206, these data are supported by studies that have performed the superexpression of these miRNAs at very early stages of the satellites skeletal muscle cell culture and observed the premature proliferation blockade and the onset of differentiation [197].
The importance of these miRNAs during the muscle regeneration process can be observed in a study by Liu et al. that promoted the deletion of microRNA-206 and subjected the animals to muscle injuries and muscle regeneration was significantly decreased [198].
Another study by Li et al. shows the involvement of miRNAs-1 and 206 in the regulation of the cell cycle through inhibition of the CCND2 (cyclin D2) and CCND1 (cyclin D1) proteins indispensable factors for cell cycle progression [40]. The suppression of these proteins has an anti-proliferative effect and thus leading to inhibition of muscle growth and suggests a specific role of these miRNAs in decreasing the cell cycle during the differentiation process. Muscle cells with dicreased expression of miRNAs-1 and 206 result in increased anti-apoptotic factors inhibiting cell death [196].
MiRNAs 133a and 133b are extremely similar and divide many targets and function; they are related to the regulation of myogenesis, tropism and muscle regeneration.
On myogenesis, the miRNAs-133a and 133b have an ambiguous role; There is evidence that these miRNAs promote the increase of myoblast proliferation, however there is other evidence showing these miRNAs are related to suppression of proliferation and increased differentiation of myoblast. These results suggest that these miRNAs take a role in both processes, varying according to the context and phenotype. The suppression of myoblasts proliferation and the increase of differentiation occur by MAPK protein regulation. MiRNAs-133a/b, which in turn indirectly control the MAPK pathway, this miRNAs targets the FGFR1 and PP2AC proteins thus preventing the activation of MAPK. The inhibition of the MAPK pathway leads to the formation of extremely small myotubes. In this way MAPK is important in the preliminary stages of myogenesis, this protein allows the accumulation of myoblast enough to fuse and form functional myotubules [196].
However, a study by Luo et al. showed that overexpression of miRNA 133a in C2C12 muscle cells generates a significant increase in myotubes formation; one of the possible mechanisms for this phenomenon to occur is the binding of the miRNA to the FOXL2 protein, this target acts on the negative control of the protein MyoG protein responsible for the cell differentiation, that is, the miRNA 133a decreases the expression of FOXL2 leading to the indirect increase of MyoG [199].
The literature also shows that knockout animals for miRNA-133a present severe myopathies, mitochondrial dysfunction, myofibroblast morphology and cell death [198]. On the other hand, super-expression of miRNA-133a added to microRNAs-1 and 206, injected into the skeletal muscle of mice after suffering muscle damage, led to the indirect increase of MyoG and MyoD1 proteins, increasing muscle regeneration and preventing fibrosis [200].
Studies indicate that physical exercise is able to modulate the expression of several miRNAs. The work of Baggish et al. shows that exercise is able to increase the expression of several miRNAs, which are involved in the decrease of inflammatory factors, such as miRNAs-21 and 146a, and miRNAs that are involved with trophism and cardiac muscle contractility and skeletal muscle, such as miRNA-133a [201]. Moore et al. (2014) also points out that physical exercise alters the expression of miRNAs in the circulation, and some miRNAs such as -206, 1 and 21 can be used as biomarkers of aerobic training [202].
Further studies show that skeletal muscle trophism is regulated by the action of certain miRNAs, and exercise is able to control the expression of these miRNAs, for example myomiRs. The main target of miRNA-133 is the IGF-I receptor, IGF-IR; Studies performed with cells show that IGF-IR over-expression or knockdown results in the modulation of PI3K-AKT pathway phosphorylation. MiRNA-133 decreases the expression of IGF-IR and, as such, it directly contributes to the reduction of the cascade of reactions that lead to the phosphorylation of AKT, leading to decreases in the development of skeletal muscle [203]. Furthermore, a decrease of this miRNA by chronic exercise is suggested [204].
Accumulated evidence shows that physical exercise is able to modulate even the biogenesis of miRNAs by influencing the expression of proteins related to this process, for example Drosha, Dicer, and Exportin-5. The study by Russel et al. evaluated muscle biopsies of nine healthy subjects 3 hours after an acute moderate intensity cycling session. A miRNAs large scale analysis was performed on the samples and it was shown that the Drosha, Dicer and Exportin-5 proteins had their expression increased, as well as the expression of the miRNAs-1, 133a, 133b, and 181, on the other MiRNAs-9, 23a, 23b and 31 had decreased expression [205].
3.9.3 Histone Acetylation
Histone deacetylases (HDACs) remove the acetyl groups from histones; removal of the acetyl group increases the condensation of the chromatin, which in turn leads to decreased transcriptional activity. HDACs are classified as class I (HDACs 1, 2, 3 and 8), class IIa (HDACs 4, 5, 7 and 9), class IIb (HDACs 6 and 10), class III HDACs (sirtuins- sirt), and class IV (HDAC 11) [206].
The literature points out the involvement of several of these HDACs and sirtuins in the regulation of cellular trophism. Some are closely related to the process of muscular atrophy through various stimuli. The Beharry & Judge study shows that the levels of p300, Cbp, Pcaf, HDAC2, HDAC4, HDAC4, HDAC6 and Sirt1 mRNA expression increase in the process of muscle atrophy, whereas HDAC7 proteins and mRNA decrease in this condition [180]. HDAC4 proteins have already been shown to be an important trophic regulation factor, only the overexpression of this protein is capable of generating atrophy of the muscle fibers and the knockout of HDAC4 is able to attenuate the muscular atrophy promoted by denervation [207].
Epigenetic processes do not necessarily occur separately, miRNAs, for example, often present as HDAC targets [208]. From the inhibition of the HDAC4 protein through the action of the miRNAs-1 and 206 there is an increase in cell differentiation and decrease in proliferation as mentioned above [198].
Physical exercise in turn also plays a role in the modulation of HDACS in skeletal muscle. McGee et al. conducted a study to examine the effect of physical exercise on the overall histone changes in skeletal muscle in humans. The study shows that physical exercise does not generate proteosomal degradation of class IIa of HDACs, however HDAC4 and 5 are exported from the nucleus during exercise, thus removing its function of transcriptional repression. It has also been shown that there is a greater activation of the AMPK and CaMKII proteins in response to physical exercise, that are kinases that induce the nuclear export of HDAC class IIa dependent phosphorylation [206].
To conclude, physical exercise has been gaining a prominent role over the years as our society has been raising awareness through scientific studies that physical activity practice is a practical, cheap and effective therapy for the control of various diseases and to promote health. Physical exercise causes important physiological stimuli that are able to counter pathological stimuli. Skeletal muscle is one of the most important of the tissues for the practice of physical exercise, the continuous mechanical contraction of skeletal muscle during physical activity triggers a series of signaling pathways that lead to improvement of the structure and function of muscle tissue through gene expression.
Epigenetic mechanisms that lead to beneficial adaptations of exercise in skeletal muscle have not yet been fully elucidated. MicroRNAs and HDACs are the most studied molecules, but there is still a vast field to be explored. The array technologies as “omics”, certainly will propitiate more discoveries and increase the knowledge about epigenetics and physical exercise, in several other tissues and cells.
3.10 Blood Vessels, Exercise Training and Epigenetics
The vascular system has as its main function the distribution of blood flow throughout the other organs of the human body through the fine regulation of vascular resistance and blood pressure. This characteristic allows fundamental processes to the homeostasis of the organism, such as gas exchanges, the supply of nutrients, as well as the removal of metabolic residues [209]. According to its function, the vascular system is divided into conductance arteries, resistance arteries, exchange vessels, and capacitance vessels. In its structure, this system has three distinct and extremely interconnected layers. The most internal layer is called tunica intima; The central, middle layer, is called tunica media; And the outer one, called tunica adventitia, finally, there is the extracellular matrix that has its composition varied according to each type of vascular segment [210].
Due complexity on structure and function and intimate relationship with the other systems, the vascular system can undergo modulations against various stimuli, either pathological or physiological, resulting in the process known as vascular remodeling. Restenosis is an important pathological remodeling process, and consists in the accumulation of vascular smooth muscle cells (VSMC) or hypertrophy of the tunica media of the vessel in arterial hypertension. As a physiological response, we have the processes known as angiogenesis or embryonic arteriogenesis, and angiogenesis triggered by physical training. Through this process, the physical training is able to increase the blood supply, as well as to reestablish the microvascular network, decreasing cellular apoptosis through the increase of oxygen supply, a mechanism impaired by capillary rarefaction triggered by a series of pathologies such as arterial hypertension [211]. In the biochemical and molecular sphere, the role of nitric oxide (NO) and vascular endothelial growth factor (VEGF), as well as the balance of apoptosis stimulators and inhibitors, can be cited as the main factors responsible for the correction of endothelial dysfunction [212,213,214,215].
Over the last few decades, the understanding of physical training as one of the most important non-pharmacological measures for disease prevention and treatment has been intensified. In the same sense, the search for intrinsic responses to endothelial cells and VSMC induced by physical training has aroused the interest of the scientific community in the epigenetic study of the vascular modulations resulting from physical training. The following will be shown studies that relate epigenetic regulation and exercise in vascular tissue.
3.10.1 DNA Methylation
Cardiorespiratory fitness is associated with improvement of endothelial and VSMC function. The ageing induces failure to balance reactive oxygen species (ROS) levels, and oxidative stress (OS). The OS is able to damage endothelial cell functions and leads to senescence. The endogenous antioxidant enzyme manganese superoxide dismutase (SOD) maintains low levels of ROS. Evidence that methylation is related with exercise effect is very recent. A study comparing aged mice sedentary, exercised and treated with the antioxidant catechin for 12 months showed that only methylation patterns in SOD promoter was lower in sedentary aged mice [216].
Hyperhomocysteinemia(HHCy), as cited in skeletal muscle section, is prevalent in aged and also hypertensive subjects [217, 218]. In blood vessels, HHCy causes imbalance between matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) [219]. Thus methylation is involved in these pathogenic process. A study, on aorta arteries of HHCy mice, showed that 5-aza-2′-deoxycytidine (Aza), a DNMT1 inhibitor, was able to reduce extra cellular matrix remodeling and consequently blood pressure by reduction in resistive index and wall-to-lumen ratio. Vascular response to phenylephrine, acetylcholine, and sodium nitroprusside also improved after Aza [191]. Exercise training promotes physiological vascular remodeling, arteriogenesis and angiogenesis by a set of pathways [148, 220]. Thus is not surprisingly that exercise could also alter expression and or activity of DNMTs also in blood vessel, and that methylation process is involved in beneficial exercise-induced vascular remodeling.
Exercise also increases the expression of endothelial growth factors, as VEGF, NO production by shear stress and induces endothelial progenitor cells (EPCs) in to circulation [221]. Thus, although there are few evidences that methylation is involved in vascular exercise-induced effect, further investigation is needed and relevant to be performed.
3.10.2 Histone Acetylation
It is well known that a bout of exercise increases the vascular shear stress, that can increase transcription of eNOS by chromatin remodeling on histone H3 and H4. Illi et al. (2003) established for first time the relationship between shear stress (SS) and chromatin remodeling. HUVECs were exposed to SS, and chromatin within c-fos and c-jun promoters was specifically immunoprecipitated by an antibody against acetylated histone H3 on K14. SS plus trichostatin A (HDAC inhibitor) induced histone H3 serine phosphorylation at position 10 (S10) and lysine acetylation at position 14 (K14). The results indicate that SS induces modifications of histones and show evidence of a blood flow-dependent regulation of gene expression [222].
Exercising is known to reduce blood pressure and improves wall properties by expression of eNOS and elastic fiber genes, improving cardiorespiratory fitness and SMC function, increases vascular growth factors and mobilizes EPCs, benefits MMP activity, decreasing cardiovascular risk [223,224,225]. Although it is well known that endothelial cell function, angiogenesis, vasculogenesis, and endothelial stem/progenitor cells are strongly affected by exercise, a direct link between histone acetylation and other histones modification in vascular tissues remains to be established.
3.10.3 MicroRNAS and Long non Coding RNAS
RNAs derived from non-coding sequences may exert an important function of regulating gene expression through small interfering RNAs, long non-coding RNAs (lncRNA) or miRNAs. The lncRNA have, in their majority, more than 200 nucleotides, being able to interact in diverse levels of the cell as in the structure, the conformation and in mechanisms of activation and cellular repression [226]. In the vascular system, lncRNA expressed in the endothelium and in the smooth muscle were recently demonstrated as regulators of both growth and endothelial function processes and the contractile phenotype of VSMC, respectively [227].
Despite the recent progress in studies on lncRNAs, the major class of RNA modulated by physical training and capable of promoting important function in the vascular system are miRNAs. These small RNA molecules are able to control about two-thirds of the protein-encoding genes, according to bioinformatics studies [213], bringing no surprises as regards their important and fundamental role in the vascular system [228].
Researchers has been demonstrating a series of miRNAs involved in physiological cardiac remodeling induced by aerobic physical training, as reviewed by Fernandes et al. [114], when the authors found evidence that physiological cardiac hypertrophy involves the regulation of miRNAs-27a and b, in addition to miRNA-143, through induction to the positive balance in the formation pathway of Angiotensin (1–7), a vasodilator, in detriment to the vasoconstriction pathway of this peptide. We also observed increased expression of miRNA-29 compared to two protocols of aerobic physical training, one of moderate and one of high intensity, in female rats. This miRNA targets the collagen gene. Thus, increased miRNA-29 expression triggers a reduction in the expression of collagen I and III, relevant for increased compliance and left ventricular function [115]. Melo et al. [116] found that aerobic swimming training was also able to promote increased expression of miRNAs 29a and 29b, with consequent prevention of increased expression of collagen I and III on the border and remote region of myocardial infarction, Suggesting positive effects of aerobic physical training in reducing the negative effects of cardiovascular diseases.
In vascular diseases, such as atherosclerosis, there are a variety of miRNAs influenced by aerobic physical training. In this pathology, physical training, as well as statin therapy, may lead to the increase of miRNA-146a, which targets the Toll-like receptor 4 (TLR4) and tumor necrosis factor receptor 6 (TRAF6), triggering, for example, a reduction in the inflammatory in vascular injury [229].
Also, in this perspective, we have that in arterial hypertension; there is a reduction in miRNA-126 microvascular expression, in addition to an increase in miRNAs-16 and miRNA-21 expressions. Considering that miRNA-126 is specifically expressed in endothelial cells, it has been described as a regulator of the migration of inflammatory cells, in addition to the formation of the capillary network and cellular survival, and is related to vascular dysfunction, inflammation and rarefaction in pathologies [148, 212, 230, 231]. One of its targets, PI3KR2 (regulatory subunit 2 of phosphatidylinositol 3,4,5-triphosphate), negatively regulates the signaling of VEGF (vascular endothelial growth factor) [148, 230, 232]. Interestingly, VEGF and Bcl-2 anti-apoptotic protein were validated as targets of miRNA-16 in endothelial cells [233,234,235,236]. The upturn in miRNA expression was also associated with decreased proliferation, migration, and formation of endothelial cell tubes in vitro [237]. In accordance with these data, miRNA-21 is also shown to be an apoptotic modulator of Bcl-2, thus suggesting a role in the regulation of angiogenic activity [233,234,235, 238]. In animals with arterial hypertension, aerobic physical training was able to reestablish the expression of miRNAs-126, 16 and 21, in parallel with the correction of capillary rarefaction, suggesting that angiogenesis may depend on the balance between pro and anti-angiogenesis factors through the miRNAs action [148, 182]. Thus, the role of physical training as a treatment for several cardiovascular diseases, through the modulation of miRNAs, is emphasized, presenting its important therapeutic potential.
4 Final Considerations
Epigenetics and its relationship with exercise-induced gene expression is a promising and emerging research field. Although the number of studies is increasing, much remains to be investigated and discovered about. Studies approaching about methylation, acetylation and miRNAs are more common to be found, nevertheless, the other modifications remain practically unexplored. Physical activity contributes to improve health and prevent disease, inducing skeletal muscle and cardiovascular adaption. The results show that exercise is a potent ambient stimulus to induce epigenetic regulation and also that induces a gene expression pattern that counteracts in several points of disease. These candidate genes and their forms of epigenetic regulation have potential to elucidate the several mechanisms about the exercise effect, and in long term, effective therapeutic approach, nutritional and training methods to preserve and improve health.
References
Hawley JA, Hargreaves M, Joyner MJ et al (2014) Integrative biology of exercise. Cell 159(4):738–749
Bernardo BC, Weeks KL, Pretorius L et al (2010) Molecular distinction between physiological and pathological cardiac hypertrophy: experimental findings and therapeutic strategies. Pharmacol Ther 128(1):191–227
Hargreaves M (2015) Exercise and gene expression. Prog Mol Biol Transl Sci 135:457–469
Bannister AJ, Kouzarides T (2011) Regulation of chromatin by histone modifications. Cell Res 21(3):381–395
Varley KE, Gertz J, Bowling KM et al (2013) Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res 23(3):555–567
Denham J, Marques FZ, O'Brien BJ et al (2014) Exercise: putting action into our epigenome. Sports Med 44(2):189–209
Zimmer P, Bloch W (2015) Physical exercise and epigenetic adaptations of the cardiovascular system. Herz 40(3):353–360
Howell PM, Liu SH, Ren SP et al (2009) Epigenetics in human melanoma. Cancer Control 16(3):200–218
Jirtle RL, Skinner MK (2007) Environmental epigenomics and disease susceptibility. EMBO Rep 12(7):620
Tong JJ, Liu J, Bertos NR et al (2002) Identification of HDAC10, a novel class II human histone deacetylase containing a leucine-rich domain. Nucleic Acids Res 30(5):1114–1123
Webster AL, Yan MS, Marsden PA (2013) Epigenetics and cardiovascular disease. Can J Cardiol 29(1):46–57
Jiang YZ, Manduchi E, Jimenez JM et al (2015) Endothelial epigenetics in biomechanical stress: disturbed flow-mediated epigenomic plasticity in vivo and in vitro. Arterioscler Thromb Vasc Biol 35(6):1317–1326
Bird A (2007) Perceptions of epigenetics. Nature 447(7143):396–398
Turgeon PJ, Sukumar AN, Marsden PA (2014) Epigenetics of cardiovascular disease – a new “beat” in coronary artery disease. Med Epigenet 2(1):37–52
Nuhrenberg T, Gilsbach R, Preissl S et al (2014) Epigenetics in cardiac development, function, and disease. Cell Tissue Res 356(3):585–600
Han H, Cortez CC, Yang X et al (2011) DNA methylation directly silences genes with non-CpG island promoters and establishes a nucleosome occupied promoter. Hum Mol Genet 20(22):4299–4310
Nan X, Ng HH, Johnson CA et al (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393(6683):386–389
Deaton AM, Bird A (2011) CpG islands and the regulation of transcription. Genes Dev 25(10):1010–1022
Jones PA (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13(7):484–492
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16(1):6–21
Caiafa P, Zampieri M (2005) DNA methylation and chromatin structure: the puzzling CpG islands. J Cell Biochem 94(2):257–265
Lund AH, van Lohuizen M (2004) Epigenetics and cancer. Genes Dev 18(19):2315–2335
Wilson AS, Power BE, Molloy PL (2007) DNA hypomethylation and human diseases. Biochim Biophys Acta 1775(1):138–162
Hermann A, Schmitt S, Jeltsch A (2003) The human Dnmt2 has residual DNA-(Cytosine-C5) methyltransferase activity. J Biol Chem 278(34):31717–31721
Issa JP (2004) CpG island methylator phenotype in cancer. Nat Rev Cancer 4(12):988–993
Egger G, Liang G, Aparicio A et al (2004) Epigenetics in human disease and prospects for epigenetic therapy. Nature 429(6990):457–463
Wang H, Wang L, Erdjument-Bromage H et al (2004) Role of histone H2A ubiquitination in Polycomb silencing. Nature 431(7010):873–878
Cao J (2014) The functional role of long non-coding RNAs and epigenetics. Biol Proced Online 16:11
Klose RJ, Zhang Y (2007) Regulation of histone methylation by demethylimination and demethylation. Nat Rev Mol Cell Biol 8(4):307–318
Suetake I, Shinozaki F, Miyagawa J et al (2004) DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J Biol Chem 279(26):27816–27823
Nimura K, Ishida C, Koriyama H et al (2006) Dnmt3a2 targets endogenous Dnmt3L to ES cell chromatin and induces regional DNA methylation. Genes Cells 11(10):1225–1237
Okano M, Xie S, Li E (1998) Dnmt2 is not required for de novo and maintenance methylation of viral DNA in embryonic stem cells. Nucleic Acids Res 26(11):2536–2540
Branco MR, Ficz G, Reik W (2011) Uncovering the role of 5-hydroxymethylcytosine in the epigenome. Nat Rev Genet 13(1):7–13
Koh KP, Yabuuchi A, Rao S et al (2011) Tet1 and Tet2 regulate 5-hydroxymethylcytosine production and cell lineage specification in mouse embryonic stem cells. Cell Stem Cell 8(2):200–213
Ficz G, Branco MR, Seisenberger S et al (2011) Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 473(7347):398–402
Wu H, D'Alessio AC, Ito S et al (2011) Dual functions of Tet1 in transcriptional regulation in mouse embryonic stem cells. Nature 473(7347):389–393
Dawlaty MM, Ganz K, Powell BE et al (2011) Tet1 is dispensable for maintaining pluripotency and its loss is compatible with embryonic and postnatal development. Cell Stem Cell 9(2):166–175
Szwagierczak A, Bultmann S, Schmidt CS et al (2010) Sensitive enzymatic quantification of 5-hydroxymethylcytosine in genomic DNA. Nucleic Acids Res 38(19):e181
Munzel M, Globisch D, Carell T (2011) 5-Hydroxymethylcytosine, the sixth base of the genome. Angew Chem Int Ed Engl 50(29):6460–6468
Li Z, Cai X, Cai CL et al (2011) Deletion of Tet2 in mice leads to dysregulated hematopoietic stem cells and subsequent development of myeloid malignancies. Blood 118(17):4509–4518
Quivoron C, Couronne L, Della Valle V et al (2011) TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20(1):25–38
Moran-Crusio K, Reavie L, Shih A et al (2011) Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20(1):11–24
Wossidlo M, Nakamura T, Lepikhov K et al (2011) 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat Commun 2:241
Iqbal K, Jin SG, Pfeifer GP et al (2011) Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. P Natl Acad Sci USA 108(9):3642–3647
TP G, Guo F, Yang H et al (2011) The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477(7366):606–610
Allfrey VG, Faulkner R, Mirsky AE (1964) Acetylation and methylation of histones and their possible role in the regulation of Rna synthesis. Proc Natl Acad Sci U S A 51:786–794
Bentley GA, Lewit-Bentley A, Finch JT et al (1984) Crystal structure of the nucleosome core particle at 16 a resolution. J Mol Biol 176(1):55
Chen CC, Mellone BG (2016) Chromatin assembly: journey to the CENter of the chromosome. J Cell Biol 214(1):13–24
Wilhelm FX, Wilhelm ML, Erard M et al (1978) Reconstitution of chromatin: assembly of the nucleosome. Nucleic Acids Res 5(2):505–521
Haushalter KA, Kadonaga JT (2003) Chromatin assembly by DNA-translocating motors. Nat Rev Mol Cell Biol 4(8):613–620
Shahbazian MD, Grunstein M (2007) Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem 76:75–100
Parthun MR (2007) Hat1: the emerging cellular roles of a type B histone acetyltransferase. Oncogene 26(37):5319–5328
Hodawadekar SC, Marmorstein R (2007) Chemistry of acetyl transfer by histone modifying enzymes: structure, mechanism and implications for effector design. Oncogene 26(37):5528–5540
Tjeertes JV, Miller KM, Jackson SP (2009) Screen for DNA-damage-responsive histone modifications identifies H3K9Ac and H3K56Ac in human cells. EMBO J 28(13):1878–1889
Yang XJ, Seto E (2007) HATs and HDACs: from structure, function and regulation to novel strategies for therapy and prevention. Oncogene 26(37):5310–5318
Haberland M, Montgomery RL, Olson EN (2009) The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 10(1):32–42
Clayton AL, Hazzalin CA, Mahadevan LC (2006) Enhanced histone acetylation and transcription: a dynamic perspective. Mol Cell 23(3):289–296
Vega RB, Matsuda K, Oh J et al (2004) Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 119(4):555–566
Fischle W, Dequiedt F, Hendzel MJ et al (2002) Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell 9(1):45–57
Tang X, Gao JS, Guan YJ et al (2007) Acetylation-dependent signal transduction for type I interferon receptor. Cell 131(1):93–105
Zhang X, Yuan Z, Zhang Y et al (2007) HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell 27(2):197–213
Kovacs JJ, Murphy PJ, Gaillard S et al (2005) HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell 18(5):601–607
Matsuyama A, Shimazu T, Sumida Y et al (2002) In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J 21(24):6820–6831
Liu H, Hu Q, Kaufman A et al (2008) Developmental expression of histone deacetylase 11 in the murine brain. J Neurosci Res 86(3):537–543
Haigis MC, Mostoslavsky R, Haigis KM et al (2006) SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell 126(5):941–954
North BJ, Verdin E (2004) Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome Biol 5(5):224
Frye RA (2000) Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem Biophys Res Commun 273(2):793–798
Anderson RM, Bitterman KJ, Wood JG et al (2003) Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423(6936):181–185
Schwer B, Verdin E (2008) Conserved metabolic regulatory functions of sirtuins. Cell Metab 7(2):104–112
Dawson MA, Bannister AJ, Gottgens B et al (2009) JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 461(7265):819–822
Hu S, Xie Z, Onishi A et al (2009) Profiling the human protein-DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell 139(3):610–622
Sugiyama K, Sugiura K, Hara T et al (2002) Aurora-B associated protein phosphatases as negative regulators of kinase activation. Oncogene 21(20):3103–3111
Goto H, Yasui Y, Nigg EA et al (2002) Aurora-B phosphorylates histone H3 at serine28 with regard to the mitotic chromosome condensation. Genes Cells 7(1):11–17
Cuthbert GL, Daujat S, Snowden AW et al (2004) Histone deimination antagonizes arginine methylation. Cell 118(5):545–553
Wang Y, Wysocka J, Sayegh J et al (2004) Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 306(5694):279–283
Sakabe K, Wang ZH, Hart GW (2010) Beta-N-acetylglucosamine (O-GlcNAc) is part of the histone code. P Natl Acad Sci USA 107(46):19915–19920
Hassa PO, Haenni SS, Elser M et al (2006) Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol Mol Biol R 70(3):789–829
Cohen-Armon M, Visochek L, Rozensal D et al (2007) DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: a link to histone acetylation. Mol Cell 25(2):297–308
Krishnakumar R, Kraus WL (2010) PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol Cell 39(5):736–749
Hershko A, Ciechanover A (1998) The ubiquitin system. Annu Rev Biochem 67:425–479
Lee JS, Shukla A, Schneider J et al (2007) Histone crosstalk between H2B monoubiquitination and H3 methylation mediated by COMPASS. Cell 131(6):1084–1096
Seeler JS, Dejean A (2003) Nuclear and unclear functions of SUMO. Nat Rev Mol Cell Biol 4(9):690–699
Shilo Y, Eisenman RN (2003) Histone sumoylation is associated with transcriptional repression. P Natl Acad Sci USA 100(23):13225–13230
Nathan D, Ingvarsdottir K, Sterner DE et al (2006) Histone sumoylation is a negative regulator in Saccharomyces Cerevisiae and shows dynamic interplay with positive-acting histone modifications. Genes Dev 20(8):966–976
Allis CD, Wiggins JC (1984) Proteolytic processing of micronuclear H3 and histone phosphorylation during conjugation in Tetrahymena Thermophila. Exp Cell Res 153(2):287–298
Santos-Rosa H, Kirmizis A, Nelson C et al (2009) Histone H3 tail clipping regulates gene expression. Nat Struct Mol Biol 16(1):17–22
Duncan EM, Muratore-Schroeder TL, Cook RG et al (2008) Cathepsin L proteolytically processes histone H3 during mouse embryonic stem cell differentiation. Cell 135(2):284–294
Chen ZZ, Zang JY, Whetstine J et al (2006) Structural insights into histone demethylation by JMJD2 family members. Cell 125(4):691–702
Nelson CJ, Santos-Rosa H, Kouzarides T (2006) Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell 126(5):905–916
Costa FF (2008) Non-coding RNAs, epigenetics and complexity. Gene 410(1):9–17
Chen J, Xue Y (2016) Emerging roles of non-coding RNAs in epigenetic regulation. Sci China Life Sci 59(3):227–235
He L, Hannon GJ (2004) MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5(7):522–531
Chi SW, Zang JB, Mele A et al (2009) Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature 460(7254):479–486
Xue Y, Ouyang K, Huang J et al (2013) Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell 152(1-2):82–96
Benetti R, Gonzalo S, Jaco I et al (2008) A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases. Nat Struct Mol Biol 15(9):998
Denis H, Ndlovu MN, Fuks F (2011) Regulation of mammalian DNA methyltransferases: a route to new mechanisms. EMBO Rep 12(7):647–656
Garzon R, Liu S, Fabbri M et al (2009) MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 113(25):6411–6418
Hall IM, Shankaranarayana GD, Noma K et al (2002) Establishment and maintenance of a heterochromatin domain. Science 297(5590):2232–2237
Volpe TA, Kidner C, Hall IM et al (2002) Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 297(5588):1833–1837
Noma K, Sugiyama T, Cam H et al (2004) RITS acts in cis to promote RNA interference-mediated transcriptional and post-transcriptional silencing. Nat Genet 36(11):1174–1180
Verdel A, Jia S, Gerber S et al (2004) RNAi-mediated targeting of heterochromatin by the RITS complex. Science 303(5658):672–676
Motamedi MR, Verdel A, Colmenares SU et al (2004) Two RNAi complexes, RITS and RDRC, physically interact and localize to noncoding centromeric RNAs. Cell 119(6):789–802
Zilberman D, Cao X, Jacobsen SE (2003) ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science 299(5607):716–719
Huang XA, Yin H, Sweeney S et al (2013) A major epigenetic programming mechanism guided by piRNAs. Dev Cell 24(5):502–516
Huang ZP, Chen J, Seok HY et al (2013) MicroRNA-22 regulates cardiac hypertrophy and remodeling in response to stress. Circ Res 112(9):1234–1243
Rajasethupathy P, Antonov I, Sheridan R et al (2012) A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell 149(3):693–707
Ponting CP, Oliver PL, Reik W (2009) Evolution and functions of long noncoding RNAs. Cell 136(4):629–641
Salmena L, Poliseno L, Tay Y et al (2011) A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 146(3):353–358
Engreitz JM, Haines JE, Perez EM et al (2016) Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 539(7629):452–455
van Weerd JH, Koshiba-Takeuchi K, Kwon C et al (2011) Epigenetic factors and cardiac development. Cardiovasc Res 91(2):203–211
Martinez SR, Gay MS, Zhang L (2015) Epigenetic mechanisms in heart development and disease. Drug Discov Today 20(7):799–811
Kaliman P, Parrizas M, Lalanza JF et al (2011) Neurophysiological and epigenetic effects of physical exercise on the aging process. Ageing Res Rev 10(4):475–486
van Rooij E, Sutherland LB, Qi X et al (2007) Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 316(5824):575–579
Fernandes T, Barauna VG, Negrao CE et al (2015) Aerobic exercise training promotes physiological cardiac remodeling involving a set of microRNAs. Am J Physiol-Heart C 309(4):H543–H552
Soci UP, Fernandes T, Hashimoto NY et al (2011) MicroRNAs 29 are involved in the improvement of ventricular compliance promoted by aerobic exercise training in rats. Physiol Genomics 43(11):665–673
Melo SF, Fernandes T, Barauna VG et al (2014) Expression of MicroRNA-29 and collagen in cardiac muscle after swimming training in myocardial-infarcted rats. Cell Physiol Biochem 33(3):657–669
D'Urso A, Brickner JH (2014) Mechanisms of epigenetic memory. Trends Genet 30(6):230–236
Papait R, Cattaneo P, Kunderfranco P et al (2013) Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc Natl Acad Sci U S A 110(50):20164–20169
George K, Spence A, Naylor LH et al (2011) Cardiac adaptation to acute and chronic participation in endurance sports. Heart 97(24):1999–2004
Ling C, Ronn T (2014) Epigenetic adaptation to regular exercise in humans. Drug Discov Today 19(7):1015–1018
Scharhag J, George K, Shave R et al (2008) Exercise-associated increases in cardiac biomarkers. Med Sci Sports Exerc 40(8):1408–1415
Weiner RB, Baggish AL (2012) Exercise-induced cardiac remodeling. Prog Cardiovasc Dis 54(5):380–386
Diffee GM (2004) Adaptation of cardiac myocyte contractile properties to exercise training. Exerc Sport Sci Rev 32(3):112–119
Green DJ, Naylor LH, George K (2006) Cardiac and vascular adaptations to exercise. Curr Opin Clin Nutr Metab Care 9(6):677–684
Matkovich SJ, Hu Y, Dorn GW 2nd (2013) Regulation of cardiac microRNAs by cardiac microRNAs. Circ Res 113(1):62–71
Bye A, Langaas M, Hoydal MA et al (2008) Aerobic capacity-dependent differences in cardiac gene expression. Physiol Genomics 33(1):100–109
Fernandes T, Hashimoto NY, Magalhaes FC et al (2011) Aerobic exercise training-induced left ventricular hypertrophy involves regulatory MicroRNAs, decreased angiotensin-converting enzyme-angiotensin ii, and synergistic regulation of angiotensin-converting enzyme 2-angiotensin (1-7). Hypertension 58(2):182–189
van Rooij E, Sutherland LB, Liu N et al (2006) A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. P Natl Acad Sci USA 103(48):18255–18260
Fernandes T, Soci UP, Oliveira EM (2011) Eccentric and concentric cardiac hypertrophy induced by exercise training: microRNAs and molecular determinants. Braz J Med Biol Res 44(9):836–847
Chang S, McKinsey TA, Zhang CL et al (2004) Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol 24(19):8467–8476
McGee SL, Swinton C, Morrison S et al (2014) Compensatory regulation of HDAC5 in muscle maintains metabolic adaptive responses and metabolism in response to energetic stress. FASEB J 28(8):3384–3395
Montgomery RL, Davis CA, Potthoff MJ et al (2007) Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev 21(14):1790–1802
Zhang CL, McKinsey TA, Chang SR et al (2002) Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110(4):479–488
Recchioni R, Marcheselli F, Antonicelli R et al (2016) Physical activity and progenitor cell-mediated endothelial repair in chronic heart failure: is there a role for epigenetics? Mech Ageing Dev 159:71–80
Soci UP, Fernandes T, Barauna VG et al (2016) Epigenetic control of exercise training-induced cardiac hypertrophy by miR-208. Clin Sci (Lond) pii:CS20160480
Samitz G, Egger M, Zwahlen M (2011) Domains of physical activity and all-cause mortality: systematic review and dose-response meta-analysis of cohort studies. Int J Epidemiol 40(5):1382–1400
Agarwal SK (2012) Cardiovascular benefits of exercise. Int J Gen Med 5:541–545
Cooney JK, Law R-J, Matschke V et al (2011) Benefits of exercise in rheumatoid arthritis. J Aging Res 2011:1–14
Denham J, O'Brien BJ, Harvey JT et al (2015) Genome-wide sperm DNA methylation changes after 3 months of exercise training in humans. Epigenomics 7(5):717–731
McMullen JR, Jennings GL (2007) Differences between pathological and physiological cardiac hypertrophy: novel therapeutic strategies to treat heart failure. Clin Exp Pharmacol P 34(4):255–262
Hill JA, Olson EN (2008) Cardiac plasticity. N Engl J Med 358(13):1370–1380
Cox EJ, Marsh SA (2013) Exercise and diabetes have opposite effects on the assembly and O-GlcNAc modification of the mSin3A/HDAC1/2 complex in the heart. Cardiovasc Diabetol 12:101
Abdellatif M (2012) Differential expression of microRNAs in different disease states. Circ Res 110(4):638–650
van Rooij E (2012) Introduction to the series on MicroRNAs in the cardiovascular system. Circ Res 110(3):481–482
Liu X, Xiao J, Zhu H et al (2015) miR-222 is necessary for exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell Metab 21(4):584
Ramasamy S, Velmurugan G, Shanmugha Rajan K et al (2015) MiRNAs with apoptosis regulating potential are differentially expressed in chronic exercise-induced physiologically hypertrophied hearts. PLoS One 10(3):e0121401
Ellison GM, Waring CD, Vicinanza C et al (2012) Physiological cardiac remodelling in response to endurance exercise training: cellular and molecular mechanisms. Heart 98(1):5–10
Silva DA, ND J, Fernandes T, Soci UP et al (2012) Swimming training in rats increases cardiac MicroRNA-126 expression and angiogenesis. Med Sci Sports Exerc 44(8):1453–1462
Grans CF, Feriani DJ, Abssamra ME et al (2014) Resistance training after myocardial infarction in rats: its role on cardiac and autonomic function. Arq Bras Cardiol 103(1):60–68
Wise FM, Patrick JM (2011) Resistance exercise in cardiac rehabilitation. Clin Rehabil 25(12):1059–1065
Petriz BA, Franco OL (2014) Effects of hypertension and exercise on cardiac proteome remodelling. Biomed Res Int 2014:634132
Shimizu I, Minamino T (2016) Physiological and pathological cardiac hypertrophy. J Mol Cell Cardiol 97:245–262
Haykowsky MJ, Dressendorfer R, Taylor D et al (2002) Resistance training and cardiac hypertrophy: unravelling the training effect. Sports Med 32(13):837–849
Tamaki T, Uchiyama S, Nakano S (1992) A weight-lifting exercise model for inducing hypertrophy in the hindlimb muscles of rats. Med Sci Sports Exerc 24(8):881–886
Barauna VG, Batista ML Jr, Costa Rosa LF et al (2005) Cardiovascular adaptations in rats submitted to a resistance-training model. Clin Exp Pharmacol Physiol 32(4):249–254
Fagard RH (1996) Athlete’s heart: a meta-analysis of the echocardiographic experience. Int J Sports Med 17(S 3):S140–S144
Pluim BM, Zwinderman AH, van der Laarse A et al (2000) The athlete’s heart. A meta-analysis of cardiac structure and function. Circulation 101(3):336–344
Melo SFS, Barauna VG, Carneiro MA et al (2015) Resistance training regulates cardiac function through modulation of miRNA-214. Int J Mol Sci 16(4):6855–6867
van Rooij E, Olson EN (2012) MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat Rev Drug Discov 11(11):860–872
Seok HY, Chen J, Kataoka M et al (2014) Loss of MicroRNA-155 protects the heart from pathological cardiac hypertrophy. Circ Res 114(10):1585–1595
Sayed D, Hong C, Chen IY et al (2007) MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res 100(3):416–424
Care A, Catalucci D, Felicetti F et al (2007) MicroRNA-133 controls cardiac hypertrophy. Nat Med 13(5):613–618
van Rooij E, Quiat D, Johnson BA et al (2009) A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev Cell 17(5):662–673
Shieh JT, Huang Y, Gilmore J et al (2011) Elevated miR-499 levels blunt the cardiac stress response. PLoS One 6(5):e19481
De CCEPR, Padilha AS, de Oliveira EM et al (2008) Cardiovascular adaptive responses in rats submitted to moderate resistance training. Eur J Appl Physiol 103(5):605–613
Bers DM (2002) Cardiac excitation-contraction coupling. Nature 415(6868):198–205
Gurha P, Abreu-Goodger C, Wang T et al (2012) Targeted deletion of microRNA-22 promotes stress-induced cardiac dilation and contractile dysfunction. Circulation 125(22):2751–2761
Wahlquist C, Jeong D, Rojas-Munoz A et al (2014) Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature 508(7497):531–535
Aurora AB, Mahmoud AI, Luo X et al (2012) MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca(2)(+) overload and cell death. J Clin Invest 122(4):1222–1232
Ahima RS, Park HK (2015) Connecting myokines and metabolism. Endocrinol Metab (Seoul) 30(3):235–245
Bassel-Duby R, Olson EN (2006) Signaling pathways in skeletal muscle remodeling. Annu Rev Biochem 75:19–37
Tisdale MJ (2009) Mechanisms of cancer cachexia. Physiol Rev 89(2):381–410
Nicoletti I, Cicoira M, Zanolla L et al (2003) Skeletal muscle abnormalities in chronic heart failure patients: relation to exercise capacity and therapeutic implications. Congest Heart Fail 9(3):148–154
Groarke JD, Cheng S, Jones LW, et al (2013) Cancer cachexia: getting to the heart of the matter. Eur Heart J. doi:10.1093/eurheartj/eht424
Sandri M (2016) Protein breakdown in cancer cachexia. Semin Cell Dev Biol 54:11–19
Morley JE, Malmstrom TK (2013) Frailty, sarcopenia, and hormones. Endocrinol Metab Clin N Am 42(2):391–405
Lim JP, Leung BP, Ding YY et al (2015) Monocyte chemoattractant protein-1: a proinflammatory cytokine elevated in sarcopenic obesity. Clin Interv Aging 10:605–609
Frisbee JC, Goodwill AG, Frisbee SJ et al (2014) Distinct temporal phases of microvascular rarefaction in skeletal muscle of obese Zucker rats. Am J Physiol Heart Circ Physiol 307(12):H1714–H1728
Walsh ME, Van Remmen H (2016) Emerging roles for histone deacetylases in age-related muscle atrophy. Nutr Healthy Aging 4(1):17–30
Beharry AW, Judge AR (2015) Differential expression of HDAC and HAT genes in atrophying skeletal muscle. Muscle Nerve 52(6):1098–1101
Ruas JL, White JP, Rao RR et al (2012) A PGC-1 alpha isoform induced by resistance training regulates skeletal muscle hypertrophy. Cell 151(6):1319–1331
Fernandes T, Magalhaes FC, Roque FR et al (2012) Exercise training prevents the microvascular rarefaction in hypertension balancing angiogenic and apoptotic factors: role of microRNAs-16, −21, and −126. Hypertension 59(2):513–520
Egan B, Carson BP, Garcia-Roves PM et al (2010) Exercise intensity-dependent regulation of peroxisome proliferator-activated receptor. Coactivator-1 alpha mRNA abundance is associated with differential activation of upstream signalling kinases in human skeletal muscle. J Physiol-London 588(10):1779–1790
Latres E, Amini AR, Amini AA et al (2005) Insulin-like growth factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin (PI3K/Akt/mTOR) pathway. J Biol Chem 280(4):2737–2744
Goodman CA, McNally RM, Hoffmann FM et al (2013) Smad3 induces atrogin-1, inhibits mTOR and protein synthesis, and promotes muscle atrophy in vivo. Mol Endocrinol 27(11):1946–1957
Cho H, Thorvaldsen JL, Chu QW et al (2001) Akt1/PKB alpha is required for normal growth but dispensable for maintenance of glucose homeostasis in mice. J Biol Chem 276(42):38349–38352
Bodine SC, Stitt TN, Gonzalez M et al (2001) Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3(11):1014–1019
Van Der Heide LP, Hoekman MF, Smidt MP (2004) The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem J 380(2):297–309
Schnyder S, Handschin C (2015) Skeletal muscle as an endocrine organ: PGC-1alpha, myokines and exercise. Bone 80:115–125
Tyagi SC, Joshua IG (2014) Exercise and nutrition in myocardial matrix metabolism, remodeling, regeneration, epigenetics, microcirculation, and muscle. Can J Physiol Pharmacol 92(7):521–523
Veeranki S, Winchester LJ, Tyagi SC (2015) Hyperhomocysteinemia associated skeletal muscle weakness involves mitochondrial dysfunction and epigenetic modifications. Biochim Biophys Acta 1852(5):732–741
Brown WM (2015) Exercise-associated DNA methylation change in skeletal muscle and the importance of imprinted genes: a bioinformatics meta-analysis. Br J Sports Med 49(24):1567–1578
Voisin S, Eynon N, Yan X et al (2015) Exercise training and DNA methylation in humans. Acta Physiol (Oxf) 213(1):39–59
Edgett BA, Foster WS, Hankinson PB et al (2013) Dissociation of increases in PGC-1alpha and its regulators from exercise intensity and muscle activation following acute exercise. PLoS One 8(8):e71623
Brown DM, Goljanek-Whysall K (2015) microRNAs: modulators of the underlying pathophysiology of sarcopenia? Ageing Res Rev 24(Pt B):263–273
Horak M, Novak J, Bienertova-Vasku J (2016) Muscle-specific microRNAs in skeletal muscle development. Dev Biol 410(1):1–13
Goljanekwhysall K, Sweetman D, Abuelmagd M et al (2011) MicroRNA regulation of the paired-box transcription factor Pax3 confers robustness to developmental timing of myogenesis. Proc Natl Acad Sci 108(29):11936
Liu N, Williams AH, Maxeiner JM et al (2012) microRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice. J Clin Investig 122(6):2054
Luo Y, Wu X, Ling Z et al (2015) microRNA133a targets Foxl2 and promotes differentiation of C2C12 into myogenic progenitor cells. Dna & Cell Biol 34(1):29
Nakasa T, Ishikawa M, Shi M et al (2010) Acceleration of muscle regeneration by local injection of muscle-specific microRNAs in rat skeletal muscle injury model. Journal of cellular. Mol Med 14(10):2495–2505
Baggish AL, Park J, Min PK et al (2014) Rapid upregulation and clearance of distinct circulating microRNAs after prolonged aerobic exercise. J Appl Physiol (1985) 116(5):522–531
Mooren FC, Viereck J, Kruger K et al (2014) Circulating micrornas as potential biomarkers of aerobic exercise capacity. Am J Physiol-Heart C 306(4):H557–H563
Yu H, Lu Y, Li Z et al (2014) microRNA-133: expression, function and therapeutic potential in muscle diseases and cancer. Curr Drug Targets 15(9):817–828
Safdar A, Saleem A, Tarnopolsky MA (2016) The potential of endurance exercise-derived exosomes to treat metabolic diseases. Nat Rev Endocrinol 12(9):504
Russell AP, Lamon S, Boon H et al (2013) Regulation of miRNAs in human skeletal muscle following acute endurance exercise and short-term endurance training. J Physiol 591(18):4637–4653
McGee SL, Fairlie E, Garnham AP et al (2009) Exercise-induced histone modifications in human skeletal muscle. J Physiol 587(24):5951–5958
Bongers KS, Fox DK, Ebert SM et al (2013) Skeletal muscle denervation causes skeletal muscle atrophy through a pathway that involves both Gadd45a and HDAC4. Am J Physiol-Endoc M 305(7):E907–E915
Winbanks CE, Beyer C, Hagg A et al (2013) miR-206 represses hypertrophy of myogenic cells but not muscle fibers via inhibition of HDAC4. PLoS One 8(9):e73589
Herbert SP, Stainier DY (2011) Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol 12(9):551–564
Tennant M, McGeachie JK (1990) Blood vessel structure and function: a brief update on recent advances. Aust N Z J Surg 60(10):747–753
Melo RM, Martinho E Jr, Michelini LC (2003) Training-induced, pressure-lowering effect in SHR: wide effects on circulatory profile of exercised and nonexercised muscles. Hypertension 42(4):851–857
Nazari-Jahantigh M, Wei Y, Schober A (2012) The role of microRNAs in arterial remodelling. Thromb Haemost 107(4):611–618
Hartmann D, Thum T (2011) MicroRNAs and vascular (dys)function. Vasc Pharmacol 55(4):92–105
Hammond SM (2015) An overview of microRNAs. Adv Drug Deliv Rev 87:3–14
Neth P, Nazari-Jahantigh M, Schober A et al (2013) MicroRNAs in flow-dependent vascular remodelling. Cardiovasc Res 99(2):294–303
Nguyen A, Leblond F, Mamarbachi M et al (2016) Age-dependent demethylation of Sod2 promoter in the mouse femoral artery. Oxidative Med Cell Longev 2016:8627384
Moroz P, Le MT, Norman PE (2007) Homocysteine and abdominal aortic aneurysms. ANZ J Surg 77(5):329–332
Guthikonda S, Haynes WG (2006) Homocysteine: role and implications in atherosclerosis. Curr Atheroscler Rep 8(2):100–106
Ovechkin AV, Tyagi N, Sen U et al (2006) 3-Deazaadenosine mitigates arterial remodeling and hypertension in hyperhomocysteinemic mice. Am J Physiol Lung Cell Mol Physiol 291(5):L905–L911
Fernandes T, Barauna VG, Negrao CE et al (2015) Aerobic exercise training promotes physiological cardiac remodeling involving a set of microRNAs. Am J Physiol Heart Circ Physiol 309(4):H543–H552
Koutroumpi M, Dimopoulos S, Psarra K et al (2012) Circulating endothelial and progenitor cells: evidence from acute and long-term exercise effects. World J Cardiol 4(12):312–326
Illi B, Nanni S, Scopece A et al (2003) Shear stress-mediated chromatin remodeling provides molecular basis for flow-dependent regulation of gene expression. Circ Res 93(2):155–161
Nualnim N, Barnes JN, Tarumi T et al (2011) Comparison of central artery elasticity in swimmers, runners, and the sedentary. Am J Cardiol 107(5):783–787
Maeda S, Tanabe T, Otsuki T et al (2004) Moderate regular exercise increases basal production of nitric oxide in elderly women. Hypertens Res 27(12):947–953
Silva JF, Rocha NG, Nobrega AC (2012) Mobilization of endothelial progenitor cells with exercise in healthy individuals: a systematic review. Arq Bras Cardiol 98(2):182–191
Heo JB, Lee YS, Sung S (2013) Epigenetic regulation by long noncoding RNAs in plants. Chromosom Res 338(6):1435–1439
Uchida S, Dimmeler S (2015) Long noncoding RNAs in cardiovascular diseases. Circ Res 116(4):737–750
Small EM, Olson EN (2011) Pervasive roles of microRNAs in cardiovascular biology. Nature 469(7330):336–342
XD W, Zeng K, Liu WL et al (2014) Effect of aerobic exercise on miRNA-TLR4 signaling in atherosclerosis. Int J Sports Med 35(4):344–350
Fish JE, Santoro MM, Morton SU et al (2008) miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell 15(2):272–284
Staszel T, Zapala B, Polus A et al (2011) Role of microRNAs in endothelial cell pathophysiology. Pol Arch Med Wewn 121(10):361–366
Wang S, Aurora AB, Johnson BA et al (2008) The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell 15(2):261–271
Urbich C, Kuehbacher A, Dimmeler S (2008) Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc Res 79(4):581–588
Suarez Y, Sessa WC (2009) MicroRNAs as novel regulators of angiogenesis. Circ Res 104(4):442–454
Quintavalle C, Garofalo M, Croce CM et al (2011) “ApoptomiRs” in vascular cells: their role in physiological and pathological angiogenesis. Vasc Pharmacol 55(4):87–91
Chamorro-Jorganes A, Araldi E, Penalva LO et al (2011) MicroRNA-16 and microRNA-424 regulate cell-autonomous angiogenic functions in endothelial cells via targeting vascular endothelial growth factor receptor-2 and fibroblast growth factor receptor-1. Arterioscler Thromb Vasc Biol 31(11):2595–2606
Cimmino A, Calin GA, Fabbri M et al (2005) miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci U S A 102(39):13944–13949
Sen CK, Gordillo GM, Khanna S et al (2009) Micromanaging vascular biology: tiny microRNAs play big band. J Vasc Res 46(6):527–540
Les Laboratoires Servier (2017) Servier. http://www.servier.com/Powerpoint-image-bank. Accessed 14 Feb 2017
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Soci, U.P.R., Melo, S.F.S., Gomes, J.L.P., Silveira, A.C., Nóbrega, C., de Oliveira, E.M. (2017). Exercise Training and Epigenetic Regulation: Multilevel Modification and Regulation of Gene Expression. In: Xiao, J. (eds) Exercise for Cardiovascular Disease Prevention and Treatment. Advances in Experimental Medicine and Biology, vol 1000. Springer, Singapore. https://doi.org/10.1007/978-981-10-4304-8_16
Download citation
DOI: https://doi.org/10.1007/978-981-10-4304-8_16
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-4303-1
Online ISBN: 978-981-10-4304-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)