
Abstract
Physical exercise induces several metabolic adaptations to meet increased energy requirements. Promoter DNA methylation, histone post-translational modifications, or microRNA expression are involved in the gene expression changes implicated in metabolic adaptation after exercise. Epigenetic modifications and many epigenetic enzymes are potentially dependent on changes in the levels of metabolites, such as oxygen, tricarboxylic acid cycle intermediates, 2-oxoglutarate, 2-hydroxyglutarate, and β-hydroxybutyrate, and are therefore susceptible to the changes induced by exercise in a tissue-dependent manner. Most of these changes are regulated by important epigenetic modifiers that control DNA methylation (DNA methyl transferases, and ten–eleven-translocation proteins) and post-translational modifications in histone tails controlled by histone acetyltransferases, histone deacetylases, and histone demethylases (jumonji C proteins, lysine-specific histone demethylase, etc.), among others. Developments in mass spectrometry approaches and the comprehension of the interconnections between epigenetics and metabolism further increase our understanding of underlying epigenetic mechanisms, not only of exercise, but also of disease and aging. In this article, we describe several of these substrates and signaling molecules regulated by exercise that affect some of the most important epigenetic mechanisms, which, in turn, control the gene expression involved in metabolism.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Background
Physical exercise is one of the most commonly prescribed therapies in both health and disease [1]. Several studies have shown that persons who report increased levels of physical activity and fitness are at less risk of death [2–4]. Regular physical activity contributes to treat several chronic diseases such as pulmonary and cardiovascular diseases (chronic obstructive pulmonary disease, hypertension, intermittent claudication); metabolic disorders (type 2 diabetes, dyslipidemia, obesity, insulin resistance); muscle, bone, and joint injuries (rheumatoid arthritis, fibromyalgia, chronic fatigue syndrome, osteoporosis); cancer, and neuropsychiatric disorders [1, 5, 6]. It has been well-established that physical exercise causes alterations in the expression of several human skeletal muscle genes. However, the mechanisms by which the gene expression of important genes involved in metabolic adaptation or metabolic stress are regulated are not completely known.
Epigenetics is the study of inherited changes in the phenotype or gene expression caused by molecular mechanisms other than changes in the DNA sequence [7, 8]. However, at this point in time, the epigenetics concept has evolved and can be redefined as the mechanisms capable of regulating gene expression through changes that are independent of the nucleotide sequence of DNA. Generally, non-genetic alterations in both gene expression and the chromatin state are tightly regulated by three major mechanisms: methylation of the cytosine residues of DNA; transcriptional regulation by microRNAs (miRNAs); and chemical modifications of specific residues of histone tails (e.g. acetylation, methylation, phosphorylation, etc.) [9–11]. The classic DNA covalent modification is methylation of cytosine, which results in the addition of a methyl group at the 5′ position of the cytosine base from S-adenosylmethionine (SAM) mediated by enzymes DNMT1, 3A, and 3B [DNA (cytosine-5-)-methyltransferase] [12, 13].
Epigenetic regulation is also mediated by non-coding RNAs such as miRNAs. miRNAs are a large family of short RNA molecules of around 21 nucleotides in size [14]. These small RNA molecules regulate gene expression mainly by base-pairing to the 3′-untranslated region (UTR) or 5′-UTR of target messenger RNAs (mRNAs) [15]. miRNAs were initially described as acting mainly by downregulating gene expression (by inhibiting mRNA translation or by the degradation of mRNA molecules). However, they have also been reported as being capable of increasing the mRNA translation of some target genes [16].
Finally, histone post-translational modifications (PTMs) are involved in epigenetic regulation. The nucleosome is formed by an octamer of histone proteins in which 147 base pairs of DNA are wrapped around it. Nucleosomes contain two copies of each one of the core histone proteins H2A, H2B, H3, and H4. In addition, the H1 linker histone stabilizes the nucleosome and the linker DNA region between nucleosomes. A tail domain protrudes from histones, which is susceptible to the PTMs introduced by different enzymes and metabolic situations. These PTMs set ‘the histone code’ [11, 17].
The majority of these changes are regulated by important epigenetic modifiers that control DNA methylation [DNA methyltransferases (DNMTs); ten–eleven-translocation (TET) proteins] and the PTMs in histone tails controlled by histone acetyltransferases (HATs), histone deacetylases (HDACs), and histone demethylases [jumonji C proteins (jmjC), lysine-specific histone demethylase (LSD), etc.], among others. A variety of life-based phenomena (i.e. nutrition, disease and infections, physical exercise, etc.) produce epigenetic alterations, which, in turn, affect all cells and tissues [18–20].
2 Exercise and Epigenetics Activate an Intricate Mechanism that Induces Metabolic Adaptations
Exercise training induces several adaptations at the metabolic level within muscle cells to meet increased energy requirements [21], and these adaptations require the expression of specific genes. Interestingly, epigenetics offers a new perspective to analyze changes in gene expression. It is noteworthy that global DNA hypomethylation in human skeletal muscle obtained from 14 healthy sedentary volunteers after acute exercise has been reported [22].
One of the most significant metabolic events exerted by regular exercise is the mitochondrial biogenesis pathway, in which peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), nuclear respiratory factor 1 (NRF1), and mitochondrial transcription factor A (TFAM) participate [21, 23, 24]. Upstream to PGC-1α, the metabolic sensor 5′ adenosine monophosphate-activated protein kinase (AMPK) is also involved in the activation of mitochondrial biogenesis [25–27]. Interestingly, miRNAs are involved in the regulation of this energetic sensor. In fact, miR-33 inhibition produces upregulation of the AMPK expression, leading to increased β-oxidation [28]. It has also been described that miR-29b targets PGC-1α, affects its transcription and, in turn, also affects mitochondrial biogenesis and mitochondrial adaptations [29]. Thus, these pioneering works demonstrate that metabolic sensors are regulated by epigenetic mechanisms (Fig. 1).

An overview of the described epigenetic changes induced by exercise. DNA methylation, miRNA expression, and histone PTMs are the main mechanisms related to changes in the expression of the relevant metabolic genes involved in exercise adaptations. After acute exercise, promoter methylation of PGC-1α, PDK4, and PPAR-δ occurs, thus increasing the expression of these relevant metabolic genes. Furthermore, downregulation of miR-33 induces the overexpression of AMPK which, in turn, increases the PGC-1α expression to facilitate mitochondrial adaptations. In addition, AMPK can produce the phosphorylation of class II HDACs to contribute to export HDACs from the nucleus to the cytosol. This leads to increased histone acetylation at the promoters of GLUT4 and MEF2. miRNAs microRNAs, PTMs post-translational modifications, PGC-1α peroxisome proliferator-activated receptor-gamma coactivator 1 alpha, PDK4 pyruvate dehydrogenase kinase isoenzyme 4, PPAR-δ peroxisome proliferator-activated receptor δ, AMPK 5′ adenosine monophosphate-activated protein kinase, HDACs histone deacetylases, GLUT4 glucose transporter type 4, MEF2 myocyte-specific enhancer factor-2
Another study found that exercise produces changes in the promoter methylation of crucial genes involved in body energy and glucose homeostasis, such as PGC-1α, pyruvate dehydrogenase kinase isoenzyme 4 (PDK4) and peroxisome proliferator-activated receptor δ (PPAR-δ) [22]. Surprisingly, muscle contraction is able to produce changes in promoter methylation as quickly as 45 min after contraction [22], which correlates with exercise intensity. Accordingly, other studies performed by Alibegovic et al. [30, 31] have demonstrated that 9 days of bed-rest increases PGC-1α promoter methylation (Fig. 1).
As mentioned above, histone PTMs is another epigenetic event that regulates gene expression. It is known that class II HDACs are highly expressed in human skeletal muscle and that their activity is regulated by exercise [32, 33]. For example, HDAC5 interacts with glucose transporter type 4 (GLUT4) and myocyte-specific enhancer factor-2 (MEF2) to result in the deacetylation of histones in genes GLUT4 and MEF2 to repress their transcription. However, after exercise this HDAC5 is exported from the nucleus and increases the expression of these genes. Furthermore, HDACs regulate the expression of PGC-1α in human skeletal muscle in response to a single bout of exercise. In fact, after 3 h of intense exercise in sedentary subjects, the expression of PGC-1α increases by up to 10.8 times [34]. These data indicate that epigenetic machinery is altered in skeletal muscle adaptations in response to exercise (Fig. 1).
Extreme exercise capacity is not completely understood. The angiotensin-converting enzyme (ACE), a dipeptidyl carboxypeptidase of the M2-metalloprotease family, has been proposed to play an important role in physical endurance [35]. Nonetheless, inconsistencies for polymorphism association with ACE activity exist depending on the subjects’ ethnicity. There is evidence for the epigenetic regulation of the ACE gene [36]. However, very little information on this issue is available for the time being. Rivière et al. [37], using human cell lines and mice tissue, demonstrated that the ACE gene expression is regulated by epigenetic phenomena. Accordingly, treatments with 5-azacytidine (a DNA methylation inhibitor) and trichostatin A (an HDAC inhibitor) bring about increased ACE expression. In addition, the authors demonstrated that the promoter methylation of the ACE gene is cell-type specific and correlates with ACE gene transcription, thus supporting the influence of epigenetic mechanisms on ACE expression.
Furthermore, a single bout of exercise is enough to increase the tricarboxylic acid (TCA) cycle flux that correlates positively with the increase in TCA cycle intermediates, such as citrate, isocitrate, succinate, and fumarate [38]. Increases in the number, size, and activity of mitochondria lead to higher metabolism with the subsequent increased capability of oxygen utilization and a resultant drop in intracellular oxygen [39]. Therefore, this effect leads to very relevant programs that are able to control distinct cellular pathways.
Under hypoxic conditions, hypoxia-inducible factor 1 (HIF-1) initiates a gene expression program that facilitates increased oxygen supply to overcome the initial hypoxic insult. HIF-1α is continuously targeted for ubiquitination and degradation dependently on the hydroxylation of two proline residues in the oxygen-dependent degradation domain through prolyl hydroxylases (PHDs) [40, 41]. PHDs require, as co-substrates, oxygen, ferrous iron [Fe(II)], and 2-oxoglutarate (2-OG) for the activity of this reaction. Consequently, exercise-induced hypoxia is blocked and HIF-1 is stabilized. Moreover, PGC-1α is coupled to HIF signaling in regulating intracellular oxygen availability and in allowing cells to match the increased oxygen demand after mitochondrial biogenesis with increased oxygen supply [39].
Yet despite PHD transcription and activity being modulated mostly by oxygen availability, adaptation to physical exercise can also be regulated by other different mechanisms in a more complex manner and by a wide range of dynamic microenvironments. Some of these relevant systems involved in adaptation to physical exercise are specific epigenetic mechanisms that use metabolic intermediates and oxygen to regulate epigenetic signals [19, 22].
As previously described, the majority of these changes are regulated by important epigenetic modifiers that control methylation of the DNA, miRNAs, or PTMs introduced or erased by HATs and HDACs, respectively. However, other epigenetic machineries are involved in the regulation of the epigenetic signature as, for example, TET proteins and histone demethylases (jmjC and LSD, etc.), among others. Recent evidence has revealed the interdependence between PHDs, histone demethylases (i.e. jmjC), and TET proteins. These groups of proteins contain the same structural catalytic domain and belong to the same 2-OG and Fe(II)-dependent dioxygenase superfamily. Overall, they are activated or inhibited by similar mechanisms [42].
3 Important Epigenetic Mechanisms are Regulated by the Oxygen and Metabolites Produced During Exercise
Histone demethylases are essential enzymes involved in epigenetic regulation by demethylation of histone lysines. Histone demethylase jmjC proteins present 2-OG and Fe(II)-dependent demethylase activity; specifically, jmjC can demethylate histone H3 lysine 36 dimethylation (H3K36me2) [43] (Fig. 2). Furthermore, TET proteins (TET1, TET2, and TET3) are codified for the TET genes and display methylcytosine dioxygenase activity.

Mechanisms mediated by epigenetic 2-OG-Fe(II)-oxygen-dependent enzymes TET and jmjC. a The 5-methylcytosine formation mechanism by DNMTs and the demethylation mechanism mediated by TET proteins, in which the first step generates 5-hmeC. b The histone lysine demethylase mechanism by which the jmjC enzyme, using 2-OG and oxygen as co-substrates, induces the hydroxylation of methyllysine to release succinate and CO2. Afterward, formaldehyde is eliminated after the decomposition of the N-hydroxymethyl group. 2-OG 2-oxoglutarate, Fe(II) ferrous iron, TET ten–eleven-translocation, jmjC jumonji C, DNMTs DNA methyltransferases, 5-hmeC 5-hydroxymethylcytosine
The founder of the TET family, TET1, was originally identified as a fusion partner of MLL in acute myeloid leukemia with t(10;11)(q21;q32) translocation. TET proteins produce 5-methylcytosine hydroxylation, resulting in the generation of 5-hydroxymethylcytosine (5-hmeC) [Fig. 2]. Although the role of 5-hmeC is being revealed, it has been found to be enriched in actively transcribed genes and in the promoters of polycomb-repressed elements that are activated normally during the development of mouse embryonic stem cells [44]. 5-hmeC formation has been proposed as an oxidative process that leads to remove methyl groups from DNA [45]. In addition, high 5-hmeC levels have also been found in specific neuronal populations [46]. Tahiliani et al. [44] demonstrated that TET1 is a 2-OG and Fe(II)-dependent dioxygenase that oxidizes 5-methylcytosine (5-meC) to 5-hmeC. Thus, TET1 activity can be modulated by similar mechanisms to those observed for PHDs.
It is well-known that exercise induces gene expression changes that trigger metabolic adaptations in skeletal muscle. Furthermore, metabolic changes during exercise may also affect gene expression through epigenetic mechanisms. In fact, Barrès et al. [22] suggested that demethylation at the promoter of PGC-1α, PDK4, and PPAR-δ did not occur through the hydroxylation mediated by TETs. One possible explanation is that the regulation of the methylation of these specific genes is highly controlled by mechanisms independently of TET activity. However, one would assume that the resultant drop in intracellular oxygen after acute exercise would have an effect on whole-genome methylation levels.
Moreover, the availability of other metabolites produced during exercise may affect epigenetic machinery activity in the aforementioned specific genes and involved in mitochondrial biogenesis (Fig. 3). Thus, 2-hydroxyglutarate produced from 2-OG, may alter the activity of 2-OG and Fe(II)-dependent dioxygenases, including PHDs and jmjC histone demethylases, as well as TET proteins. The reduction of 2-OG lowers the level of this important substrate used by jmjC histone demethylases and TET proteins, thus producing low histone demethylation and also low 5-hmeC levels [47]. Furthermore, 2-OG and Fe(II)-dependent dioxygenases can be inhibited by succinate accumulation. The effect of succinate on jmjC histone demethylases and PHDs has been confirmed by the observations made by Cervera et al. [48], who treated human cells with succinate dehydrogenase (SDH) inhibitors which induce succinate accumulation and, in turn, produce enhanced histone methylation; other authors [49–51] also reported evidence that tumors with SDH mutations show enhanced HIF stability.

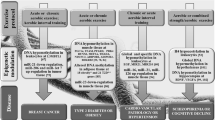
Flowchart representing the key metabolic events after exercise which control epigenetic mechanisms. Exercise induces energy requirements, which are met by increased mitochondrial biogenesis by the PGC-1α, NRF1, and TFAM pathway. Increases in the number, size, and activity of mitochondria lead to a subsequent increase in the TCA cycle metabolites pool and β-hydroxybutyrate. Moreover, this increased capability of oxygen utilization leads to a drop in intracellular oxygen that blocks the 2-OG-Fe(II)-oxygen-dependent enzymes to, in turn, block PHDs, jmjC, and TET proteins. All these pathways lead to the complex regulation of several epigenetic pathways through histone demethylases, acetyltransferases, and deacetylases. PGC-1α peroxisome proliferator-activated receptor-gamma coactivator 1 alpha, NRF1 nuclear respiratory factor 1, TFAM mitochondrial transcription factor A, TCA tricarboxylic acid, 2-OG 2-oxoglutarate, Fe(II) ferrous iron, PHDs prolyl hydroxylases, jmjC jumonji C, TET ten–eleven-translocation, HDACs histone deacetylases
In addition, reduced 2-OG levels, due to physical exercise [38], and deregulation or mutations in isocitrate dehydrogenase (IDH), among other mechanisms, can limit 2-OG generation upstream in the TCA cycle [47].
The accumulation of succinate and fumarate, as a result of SDH mutations [52], but also due to exercise [38], inhibits 2-OG-dependent dioxygenases, such as PHD [53, 54]. Moreover, free radicals production by mitochondria under hypoxic conditions diminishes the availability of Fe(II), which reduces the activity of PHDs [55]. Therefore, any stress capable of inducing a persistent boost of free radicals should affect the regulation of 2-OG-Fe(II)-oxygen-dependent enzymes such as PHDs, jmjC, and TET [54], as in the case of physical exercise.
One of the most interesting metabolites that can exert effects on epigenetic regulation is β-hydroxybutyrate [56, 57]. This molecule is produced at millimolar levels after prolonged exercise (2–3 days of strenuous exercise) [58]. It derives from acetoacetate, which is catabolyzed by β-hydroxybutyrate dehydrogenase in an NAD+-dependent reaction. Chemically, β-hydroxybutyrate is similar to butyrate, an inhibitor of class I and II HDACs; in fact, β-hydroxybutyrate has proved to inhibit HDAC1, HDAC3, HDAC4 in a dose-dependent manner in human embryonic kidney 293 (HEK293) cells, and also in mice models during fasting and calorie restriction during which significantly increased levels of histone H3 lysine 9 acetylation (H3K9ac) and histone h3 lysine 14 acetylation (H3K14ac) have been observed [56]. These results indicate that β-hydroxybutyrate is able to regulate the epigenetic state of chromatin by inhibiting the activity of class I and II HDACs.
The mechanisms described in this article clearly suggest that exercise regulates several epigenetic mechanisms. Furthermore, the metabolites generated by continuous, acute, moderate, or strenuous exercise control the activity of some highly relevant epigenetic enzymes that regulate gene expression.
4 Mass Spectrometry as a Tool for Identifying Metabolites that Regulate Epigenetic Phenomena During Exercise
The combination of accurate mass data for a large collection of metabolites, and the identification of the metabolites achieved by searching metabolomic databases [for example, the Human Metabolome Database (HMDB)], and the mass spectrometry (MS) technologies applied to analyze histone PTMs and DNA methylation in specific genes (i.e. Sequenom) will increase our knowledge in the near future on the metabolic pathways involved in epigenetic control.
Sequenom is a MassARRAY platform used for single nucleotide polymorphism (SNP) genotyping, DNA methylation, and quantitative gene expression studies based on matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) MS. For DNA methylation analysis, the EpiTYPER tool by Sequenom allows the discovery and quantification of DNA methylation. This assay is based on the bisulfite conversion of genomic DNA. Basically, cytosine (C) is deaminated by sodium bisulfite to uracil (U), although 5-methylcytosine remains unchanged. After polymerase chain reaction amplification in the non-methylated DNA fragment, U changes to thymine (T) and a T7-promoter tag, required for the next step, are introduced. Afterward, in vitro transcription on the reverse strand using specific T7 polymerase, generates strands with an AC (adenine-cytosine) sequence in the corresponding positions where non-methylated CG (cytosine-guanine) initially existed. These variations are a replacement of A instead of G in non-methylated CpG, which results in a mass difference of 16 Da per CG site that is easily detected by the MassARRAY of Sequenom. For the generation of restriction fragments, U-specific cleavage mediated by RNase A is included. This enzyme cleaves the transcription products at specific U or C bases in separate reactions. In the C-cleavage reactions, methylated regions are cleaved at every C to create fragments that contain at least one CpG site each. However in the U-cleavage reaction, both methylated and non-methylated CpG sequences are cleaved at every U. It is important to note that for both the U- and C-reactions, the cleavage products resulting from the methylated and non-methylated DNA are the same in length, but differs in nucleotide composition terms. Generally speaking, a U-cleavage reaction captures most information (Fig. 4). This property is used to analyze differences in mass by MALDI-TOF for the identification of fragments and the assignation of methylation at specific CpGs. This platform is indicated especially for the DNA methylation analysis of several amplicons of specific genes [59, 60].

Flow diagram for the DNA methylation analysis using EpiTyper by Sequenom. The quantitative DNA methylation analysis is based on the ability of bisulfite to introduce methylation-dependent sequence variations of C to T into PCR amplification products. These post-PCR C/T variations appear as G/A variations after in vitro transcription, which results in a mass difference of 16 Da per CpG site, as detected by MALDI-TOF MS, and based on the overview scheme of the quantitative methylation analysis process by Sequenom. C cytosine, T thymine, PCR polymerase chain reaction, MALDI-TOF matrix-assisted laser desorption/ionization-time of flight, MS mass spectrometry
In addition, the PTMs of purified histones can be analyzed for their specific PTMs by MALDI-TOF and nanoelectrospray ionization tandem MS [60, 61], which are powerful tools to analyze the histone code in specific genes regulated by exercise.
Another main approach to completely decipher the regulation of epigenetic machinery by the metabolites produced during exercise can be attained by MS. To date, the quantification of TCA cycle metabolites is based on studying the distribution of carbon fluxes and their regulation using 13C isotopic substrates [62]. The labeled 13C atoms incorporated into intracellular metabolites [63] are quantified by MS and/or nuclear magnetic resonance (NMR) [64] [65]. However, it is difficult to measure several key TCA cycle metabolites directly because of their instability and low concentration [62].
Recently, Koubaa et al. developed a robust technique based on liquid chromatography-tandem MS (LC–MS/MS) and gas chromatography-MS (GC–MS) synergy to measure 13C-labeling TCA cycle intermediates (succinate, fumarate, malate, 2-OG, citrate, and isocitrate) in a more accurate and validated manner. As the authors suggested, this approach may be applicable to aerobic organisms and tissues [62]. Finally, Dahl et al. [66] were able to measure β-hydroxybutyrate in body fluids by ultra-high pressure liquid chromatography (UPLC)-MSMS. A direct application of this approach to analyze β-hydroxybutyrate in tissues will be most useful for evaluating the levels of this inhibitor of class I and II HDACs.
5 Conclusions and Future Perspectives
Epigenetic modifications and many epigenetic enzymes are potentially dependent on changes in the levels of metabolites such as oxygen, 2-OG, 2-hydroxyglutarate, succinate, fumarate, β-hydroxybutyrate. Therefore, they are susceptible to changes induced by exercise in a tissue-dependent manner.
We surmise that increasingly more sophisticated approaches (i.e. new MS methodologies) will eventually resolve many pending issues. Hopefully, these combined approaches will provide a detailed understanding of the epigenetic mechanisms underlying metabolic regulation by lifestyle to help biomedicine in future interventions based on exercise in order to improve the human health span and longevity.
References
Vina J, Sanchis-Gomar F, Martinez-Bello V, et al. Exercise acts as a drug; the pharmacological benefits of exercise. Br J Pharmacol. 2012;167(1):1–12.
Blair SN, Kohl HW 3rd, Paffenbarger RS Jr, et al. Physical fitness and all-cause mortality: a prospective study of healthy men and women. JAMA. 1989;262(17):2395–401.
Macera CA, Hootman JM, Sniezek JE. Major public health benefits of physical activity. Arthritis Rheum. 2003;49(1):122–8.
Myers J, Kaykha A, George S, et al. Fitness versus physical activity patterns in predicting mortality in men. Am J Med. 2004;117(12):912–8.
Pedersen BK, Saltin B. Evidence for prescribing exercise as therapy in chronic disease. Scand J Med Sci Sports. 2006;16(Suppl 1):3–63.
Warburton DE, Nicol CW, Bredin SS. Health benefits of physical activity: the evidence. CMAJ. 2006;174(6):801–9.
Allis CD, Jenuwein T, Reinberg D. Epigenetics. New York: Cold Spring Harbor Laboratory Press; 2007. p. 502.
Bird A. Perceptions of epigenetics. Nature. 2007;447(7143):396–8.
Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128(4):635–8.
Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128(4):669–81.
Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–80.
Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–92.
Irizarry RA, Ladd-Acosta C, Wen B, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41(2):178–86.
He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5(7):522–31.
Lytle JR, Yario TA, Steitz JA. Target mRNAs are repressed as efficiently by microRNA-binding sites in the 5′ UTR as in the 3′ UTR. Proc Natl Acad Sci U S A. 2007;104(23):9667–72.
Vasudevan S, Tong Y, Steitz JA. Switching from repression to activation: microRNAs can up-regulate translation. Science. 2007;318(5858):1931–4.
Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403(6765):41–5.
Alegria-Torres JA, Baccarelli A, Bollati V. Epigenetics and lifestyle. Epigenomics. 2011;3(3):267–77.
Sanchis-Gomar F, Garcia-Gimenez JL, Perez-Quilis C, et al. Physical exercise as an epigenetic modulator: Eustress, the “positive stress” as an effector of gene expression. J Strength Cond Res. 2012;26(12):3469–72.
Talens RP, Christensen K, Putter H, et al. Epigenetic variation during the adult lifespan: cross-sectional and longitudinal data on monozygotic twin pairs. Aging Cell. 2012;11(4):694–703.
Holloszy JO, Coyle EF. Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J Appl Physiol. 1984;56(4):831–8.
Barrès R, Yan J, Egan B, et al. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012;15(3):405–11.
Vina J, Gomez-Cabrera MC, Borras C, et al. Mitochondrial biogenesis in exercise and in ageing. Adv Drug Deliv Rev. 2009;61(14):1369–74.
Holloszy JO. Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem. 1967;242(9):2278–82.
Bergeron R, Ren JM, Cadman KS, et al. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab. 2001;281(6):E1340–6.
Hardie DG. AMP-activated protein kinase: a key system mediating metabolic responses to exercise. Med Sci Sports Exerc. 2004;36(1):28–34.
Hardie DG, Sakamoto K. AMPK: a key sensor of fuel and energy status in skeletal muscle. Physiology (Bethesda). 2006;21:48–60.
Davalos A, Goedeke L, Smibert P, et al. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc Natl Acad Sci U S A. 2011;108(22):9232–7.
Wang H, Garzon R, Sun H, et al. NF-kappaB-YY1-miR-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell. 2008;14(5):369–81.
Alibegovic AC, Sonne MP, Hojbjerre L, et al. Insulin resistance induced by physical inactivity is associated with multiple transcriptional changes in skeletal muscle in young men. Am J Physiol Endocrinol Metab. 2010;299(5):E752–63.
Alibegovic AC, Hojbjerre L, Sonne MP, et al. Impact of 9 days of bed rest on hepatic and peripheral insulin action, insulin secretion, and whole-body lipolysis in healthy young male offspring of patients with type 2 diabetes. Diabetes. 2009;58(12):2749–56.
McKinsey TA, Zhang CL, Olson EN. Control of muscle development by dueling HATs and HDACs. Curr Opin Genet Dev. 2001;11(5):497–504.
McGee SL, Hargreaves M. Histone modifications and exercise adaptations. J Appl Physiol (1985). 2011;110(1):258–63.
Egan B, Carson BP, Garcia-Roves PM, et al. Exercise intensity-dependent regulation of peroxisome proliferator-activated receptor coactivator-1 mRNA abundance is associated with differential activation of upstream signalling kinases in human skeletal muscle. J Physiol. 2010;588(Pt 10):1779–90.
Puthucheary Z, Skipworth JR, Rawal J, et al. The ACE gene and human performance: 12 years on. Sports Med. 2011;41(6):433–48.
Raleigh SM. Epigenetic regulation of the ACE gene might be more relevant to endurance physiology than the I/D polymorphism. J Appl Physiol. (1985). 2012;112(6):1082–3.
Rivière G, Lienhard D, Andrieu T, et al. Epigenetic regulation of somatic angiotensin-converting enzyme by DNA methylation and histone acetylation. Epigenetics. 2011;6(4):478–89.
Gibala MJ, MacLean DA, Graham TE, et al. Tricarboxylic acid cycle intermediate pool size and estimated cycle flux in human muscle during exercise. Am J Physiol. 1998;275(2 Pt 1):E235–42.
O’Hagan KA, Cocchiglia S, Zhdanov AV, et al. PGC-1alpha is coupled to HIF-1alpha-dependent gene expression by increasing mitochondrial oxygen consumption in skeletal muscle cells. Proc Natl Acad Sci U S A. 2009;106(7):2188–93.
Ivan M, Kondo K, Yang H, et al. HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292(5516):464–8.
Bruick RK, McKnight SL. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 2001;294(5545):1337–40.
Cyr AR, Domann FE. The redox basis of epigenetic modifications: from mechanisms to functional consequences. Antioxid Redox Signal. 2011;15(2):551–89.
Tsukada Y, Fang J, Erdjument-Bromage H, et al. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 2006;439(7078):811–6.
Tahiliani M, Koh KP, Shen Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–5.
Hirsila M, Koivunen P, Gunzler V, et al. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J Biol Chem. 2003;278(33):30772–80.
Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324(5929):929–30.
Zhao S, Lin Y, Xu W, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324(5924):261–5.
Cervera AM, Bayley JP, Devilee P, et al. Inhibition of succinate dehydrogenase dysregulates histone modification in mammalian cells. Mol Cancer. 2009;8:89.
Gimenez-Roqueplo AP, Favier J, Rustin P, et al. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. Am J Hum Genet. 2001;69(6):1186–97.
Gimenez-Roqueplo AP, Favier J, Rustin P, et al. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003;63(17):5615–21.
Selak MA, Armour SM, MacKenzie ED, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7(1):77–85.
Brugnara L, Vinaixa M, Murillo S, et al. Metabolomics approach for analyzing the effects of exercise in subjects with type 1 diabetes mellitus. PLoS One. 2012;7(7):e40600.
Xiao M, Yang H, Xu W, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26(12):1326–38.
Berra E, Ginouves A, Pouyssegur J. The hypoxia-inducible-factor hydroxylases bring fresh air into hypoxia signalling. EMBO Rep. 2006;7(1):41–5.
Gerald D, Berra E, Frapart YM, et al. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118(6):781–94.
Shimazu T, Hirschey MD, Newman J, et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339(6116):211–4.
Zinker BA, Britz K, Brooks GA. Effects of a 36-hour fast on human endurance and substrate utilization. J Appl Physiol. 1990;69(5):1849–55.
Koeslag JH, Noakes TD, Sloan AW. Post-exercise ketosis. J Physiol. 1980;301:79–90.
Gupta R, Nagarajan A, Wajapeyee N. Advances in genome-wide DNA methylation analysis. Biotechniques. 2010;49(4):iii–xi.
Garcia-Gimenez JL, Sanchis-Gomar F, Lippi G, et al. Epigenetic biomarkers: a new perspective in laboratory diagnostics. Clin Chim Acta. 2012;413(19–20):1576–82.
Cocklin RR, Wang M. Identification of methylation and acetylation sites on mouse histone H3 using matrix-assisted laser desorption/ionization time-of-flight and nanoelectrospray ionization tandem mass spectrometry. J Protein Chem. 2003;22(4):327–34.
Koubaa M, Cocuron JC, Thomasset B, et al. Highlighting the tricarboxylic acid cycle: liquid and gas chromatography-mass spectrometry analyses of (13)C-labeled organic acids. Anal Biochem. 2013;436(2):151–9.
Choi J, Grossbach MT, Antoniewicz MR. Measuring complete isotopomer distribution of aspartate using gas chromatography/tandem mass spectrometry. Anal Chem. 2012;84(10):4628–32.
Koubaa M, Mghaieth S, Thomasset B, et al. Gas chromatography-mass spectrometry analysis of 13C labeling in sugars for metabolic flux analysis. Anal Biochem. 2012;425(2):183–8.
O’Grady J, Schwender J, Shachar-Hill Y, et al. Metabolic cartography: experimental quantification of metabolic fluxes from isotopic labelling studies. J Exp Bot. 2012;63(6):2293–308.
Dahl SR, Olsen KM, Strand DH. Determination of gamma-hydroxybutyrate (GHB), beta-hydroxybutyrate (BHB), pregabalin, 1,4-butane-diol (1,4BD) and gamma-butyrolactone (GBL) in whole blood and urine samples by UPLC-MSMS. J Chromatogr B Analyt Technol Biomed Life Sci. 2012;885–886:37–42.
Acknowledgments
No sources of funding were used to help prepare this article. The authors have no potential conflicts of interest that are directly relevant to the content of this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pareja-Galeano, H., Sanchis-Gomar, F. & García-Giménez, J.L. Physical Exercise and Epigenetic Modulation: Elucidating Intricate Mechanisms. Sports Med 44, 429–436 (2014). https://doi.org/10.1007/s40279-013-0138-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40279-013-0138-6