
Abstract
Transthyretin amyloidosis with polyneuropathy (ATTR-PN), a rare and progressive hereditary disorder, results from mutations in the gene coding for the transthyretin (TTR) protein that destabilize the protein’s tetrameric structure. In over 40 countries worldwide, tafamidis (Vyndaqel®) is approved for the treatment of TTR amyloidosis in adults with stage 1 symptomatic polyneuropathy, to delay peripheral neurological impairment. Tafamidis is administered orally once daily, as a soft capsule. Evidence from clinical studies, including an 18-month placebo-controlled trial and subsequent long-term, open-label extension studies (providing data from ≤ 6 years of treatment), indicate that tafamidis slowed deterioration of neurological function and maintained health-related quality of life in patients with early-stage ATTR-PN and the Val30Met mutation. TTR tetramers were stabilized in nearly all patients, and nutritional status was generally maintained or improved. Similar benefit was seen with tafamidis over 12 months in a noncomparative trial in patients with non-Val30Met ATTR-PN, although disease progression in this population (which was older and had had ATTR-PN for longer than Val30Met patients) became more notable with continued therapy in an extension study. Data for up to 10 years from large registry and referral centre studies support the long-term effectiveness and safety of tafamidis in delaying disease progression and conferring survival benefits in patients with stage 1 ATTR-PN. Tafamidis was generally well tolerated, with no new safety signals detected during the long-term trial or real-world experience. Thus, based on up to 10 years’ experience, tafamidis continues to be a valuable option in the treatment of early-stage ATTR-PN.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
First disease-modifying pharmacotherapy to be approved in this therapeutic area |
Kinetically stabilizes TTR tetramers, irrespective of amyloidogenic mutation; most patients achieve TTR tetramer stabilization |
Slows deterioration of neurological function and maintains health-related quality of life |
Generally well tolerated; suitable for long-term use |
1 Introduction
Transthyretin amyloidosis with polyneuropathy (ATTR-PN) is a rare, progressive and eventually fatal hereditary disorder that results from mutations in the gene coding for the transthyretin (TTR) protein [1,2,3]. Produced primarily by the liver, TTR is a plasma transport protein with a tetrameric structure. Amyloidogenic mutations destabilize the TTR tetramers, causing them to dissociate into monomers. These monomers partially unfold and aggregate to form insoluble fibril amyloid complexes that are preferentially deposited around the peripheral nerves, producing largely irreversible sensory, motor and autonomic neuropathy [1,2,3].
ATTR-PN, which is endemic to Portugal, Sweden and certain parts of Japan, has an estimated global prevalence of ≈ 10,000 people (and potentially as high as 40,000) [4]. While initially thought to be restricted to the aforementioned regions, diagnoses of ATTR-PN are now being made worldwide and ≈ 120 different TTR mutations (including single mutations, double mutations or deletions in the TTR gene) have been reported [1]. The most common of these mutations involves a valine being replaced with a methionine at position 30 (Val30Met) of the TTR gene [1, 3]. Disease onset varies considerably across different populations, with the mean age of onset being in the fourth decade of life [2]. Death typically occurs within 7–12 years of symptom onset [5].
Historically, orthotopic liver transplant was the standard of care for ATTR-PN [5]. Liver transplantation removes ≈ 95% of variant TTR production [1], preventing neurological progression in the majority of patients with Val30Met ATTR-PN [6]. Across mutations, it offers an overall survival rate of 55% at 20 years post-treatment [7]. Liver transplantation is associated with a number of limitations, however, including a shortage of donors, the requirement of surgery and the need for long-term immunosuppressant therapy following transplantation [3, 6, 8]. Nerve function seldom improves, and cardiac and ocular amyloid deposits may continue to accumulate [3, 5]. Furthermore, many patients with ATTR-PN are poor transplant candidates due to factors such as advanced age or disease progression [8]; those with early-onset disease (< 50 years of age), good nutritional status and shorter duration of disease have a more favourable prognosis [5, 7]. Outcomes are mutation-specific, with long-term survival rates being higher for patients with Val30Met ATTR-PN than for those with other mutations (10-year survival rate 74 vs. 44%) [1]. Due to the shortage of approved alternative therapies for ATTR-PN, physicians have repurposed drugs used to treat other conditions, based on their mechanism of action [5].
A greater understanding of TTR tetramer dissociation as the rate-limiting step in TTR-derived amyloidogenesis has more recently allowed for the development of less invasive treatment strategies for the management of ATTR-PN [1, 9]. Initially selected for clinical development due to it binding selectively and with high specificity to TTR, as well as its oral bioavailability and favourable tolerability profile [10], tafamidis (Vyndaqel®; reviewed previously in Drugs [11]) was the first disease-modifying pharmacotherapy to be approved for use in adults with early-stage symptomatic ATTR-PN to delay neurological impairment. Tafamidis is available as soft capsules containing tafamidis 20 mg as a meglumine salt (Sect. 4). Tafamidis kinetically stabilizes the quaternary structure of the TTR tetramer, precluding its dissociation into the monomers that ultimately form amyloid fibrils [10, 12, 13]. The pharmacological properties of tafamidis are provided in Table 1. This article focuses on the efficacy and safety/tolerability of tafamidis in the treatment of adults with early-stage symptomatic ATTR-PN, providing long-term data from extension studies and the clinical practice setting.
2 Therapeutic Efficacy of Tafamidis
The efficacy of oral tafamidis in adults with early-stage Val30Met ATTR-PN was evaluated in the pivotal, 18-month, double-blind, multinational, phase II/III Fx-005 registration trial [14]. Longer-term data are available from the subsequent, noncomparative phase II/III Fx-006 [15] and phase III B3461023 [16] extension studies, thus far providing efficacy data in patients treated for up to 6 years. Tafamidis was also evaluated in a small, open-label, multicentre, phase III study in Japanese patients with predominantly Val30Met ATTR-PN [17]. The efficacy of oral tafamidis is further supported by the results of the noncomparative phase II Fx1a-201 trial in adults with non-Val30Met ATTR-PN [18] (some of whom also proceeded to enter the B3461023 extension study [16]), as well as a series of post hoc analyses of the key clinical trials [19,20,21,22] and data from a number of studies in the clinical practice setting [23,24,25,26,27].
2.1 In Clinical Trials
2.1.1 In Patients with the Transthyretin Val30Met Mutation
2.1.1.1 Fx-005 Trial
The pivotal Fx-005 trial included adults with early-stage Val30Met ATTR-PN who had confirmed amyloid deposits, autonomic or peripheral neuropathy, and a Karnofsky performance status of ≥ 50 [14]. Eligible patients were randomized to receive tafamidis 20 mg once daily (n = 65) or placebo (n = 63) for 18 months [14]. Patients who completed Fx-005 were eligible to enter the open-label Fx-006 extension study, during which all patients received tafamidis 20 mg once daily for an additional 12 months [n = 38 having previously received tafamidis (i.e. the tafamidis–tafamidis group) and n = 33 having been switched from placebo to tafamidis (the placebo–tafamidis group)] [15]. Blinding as to which treatment was received during the initial study was maintained during Fx-006 [15]. Patients who completed both Fx-005 and Fx-006 could enrol in a second, ongoing open-label extension study (B3461023) aiming to evaluate the long-term safety and efficacy of tafamidis 20 mg once daily [16].
The co-primary endpoints in Fx-005 were the proportion of patients considered to be Neuropathy Impairment Score-Lower Limbs (NIS-LL) responders (i.e. having a < 2 point change from baseline to month 18 in NIS-LL) and the least squares mean (LSM) change from baseline in Norfolk Quality of Life Diabetic-Neuropathy Questionnaire total score (TQOL-DN) at month 18 [14]. Baseline characteristics were largely comparable between treatment arms, although the mean disease duration was 47 months in tafamidis recipients and 35 months in placebo recipients; patients were relatively young (mean ages 40 and 38 years) and 69% of all patients were on waiting lists for liver transplant at enrolment [14].
Over the course of the study, 13 patients in each group (21%) discontinued treatment (generally prior to month 12) in order to receive a liver transplant [14]. Of the 91 patients who completed Fx-005 [14], 86 enrolled in Fx-006, of whom 77 completed the study (among discontinuations, five were for liver transplantation) [15]. The B3461023 extension study enrolled 75 patients from Fx-006 with the Val30Met mutation (Table 2) and 18 non-Val30Met patients from Fx1a-201 (Sect. 2.1.2) [16]. At the time of data cut-off for the prospectively planned interim analysis (31 December 2014), 15 (20%) of the patients with Val30Met ATTR-PN were ongoing in the study, while 49 (65%) had completed the study when tafamidis became available via prescription and 11 (15%) had permanently discontinued [16].
In terms of the proportion of NIS-LL responders and the LSM change from baseline in TQOL-DN after 18 months of treatment in Fx-005, disease progression did not significantly differ between tafamidis and placebo in the intent-to-treat (ITT) population (primary analysis; inclusive of patients who discontinued due to liver transplantation), but significantly favoured tafamidis over placebo in the efficacy evaluable population (pre-specified analysis) (Table 2) [14]. Furthermore, tafamidis recipients showed no significant change from baseline in TQOL-DN, while placebo recipients significantly deteriorated (p = 0.002) [14].
Tafamidis was associated with significant benefits over placebo for a number of secondary endpoints of disease progression [small fibre function and modified body mass index (mBMI) but not large fibre function] and for TTR tetramer stabilization (defined as > 32% stabilization) [Table 2; analyzed in the ITT population] [14]. The benefits of tafamidis in terms of delaying neurological progression were further supported by various post hoc analyses, in which tafamidis was significantly (p < 0.05) more favourable than placebo with regard to LSM changes from baseline in NIS-LL at 18 months in baseline-adjusted and multiple imputation analyses and LSM changes from baseline in NIS-LL plus three small fibre nerve tests and NIS-LL plus seven nerve tests [21]. In a linear mixed-effects model for repeated measures, baseline NIS-LL was an independent predictor of neurological progression (assessed using NIS-LL; patients with lower scores at baseline showed less progression than those with higher baseline scores; p < 0.0001) during Fx-005, and neurological progression was less with tafamidis than with placebo across all levels of baseline severity (p = 0.0088) [19].
2.1.1.2 Extension Studies (Fx-006 and B3461023)
The positive effects of tafamidis on disease progression in Fx-005 were largely maintained during the initial 12-month Fx-006 extension study [15]. In patients in the tafamidis–tafamidis group, monthly rates of change in neurological function (i.e. NIS-LL, large fibre function and small fibre function) and TQOL-DN did not significantly differ between the initial double-blind trial and the open-label extension. With respect to nutritional status, the monthly mBMI change in the tafamidis–tafamidis group significantly differed between Fx-005 and Fx-006 with the previous improvement in mBMI being partially lost (1.85 vs. − 2.00/month; p = 0.0006) [15].
Patients previously receiving placebo showed a significant decline in the rate of neurological progression after being switched to tafamidis in Fx-006 (change in NIS-LL of 0.16 vs. 0.34/month in Fx-006 and Fx-005, respectively; p = 0.01), albeit with no significant changes observed in the monthly rate of deterioration in large or small fibre function [15]. After switching to tafamidis, the placebo–tafamidis group also showed significant improvements in monthly rates of change in TQOL-DN (− 0.16 vs. 0.61 in Fx-006 and Fx-005, respectively; p = 0.0003) and mBMI (5.19 vs. − 1.77; p < 0.0001), with deterioration ceasing. Benefits of early treatment initiation were seen, with significantly less deterioration in certain measures of neurological function (NIS-LL and large fibre function, but not small fibre function) at month 30 in the tafamidis–tafamidis group compared with the placebo–tafamidis group (Table 2). No significant between-group differences were seen for TQOL-DN or mBMI at month 30, and TTR tetramer stabilization was achieved in the majority of patients in each group (> 90%) (Table 2) [15].
Tafamidis effectively slowed disease progression over the long term in the interim analysis of the ongoing B3461023 extension study (Table 2) [16]. While the separation between tafamidis and placebo recipients in the rate of neurological deterioration (as measured by change from Fx-005 baseline in NIS-LL) at the end of the initial study (Fx-005) persisted at month 66 between tafamidis–tafamidis and placebo–tafamidis recipients (Table 2), the baseline-adjusted LSM between-group difference was non-significant at this latter timepoint based on the 95% CI (− 4.9; 95% CI − 10.9 to 1.2). Similarly, there was no significant baseline-adjusted LSM difference between the tafamidis–tafamidis and placebo–tafamidis groups in TQOL-DN or mBMI at month 66 (Table 2), based on 95% CIs; − 2.1 (95% CI − 13.7 to 9.5) and − 53.3 kg/m2 × g/L (95% CI − 111.0 to 4.5) [16].
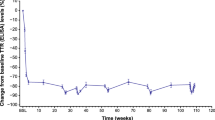
A post hoc slope analysis showed that switching from placebo to tafamidis significantly slowed disease progression (NIS-LL, NIS-LL muscle weakness and TQOL-DN; p ≤ 0.02 for months 18–66 vs. 0–18 in the placebo–tafamidis group) (Fig. 1) [16]. Although the tafamidis–tafamidis group had significantly (p ≤ 0.024) slower neurological deterioration (NIS-LL and NIS-LL muscle weakness) than the placebo–tafamidis group during the randomized trial, rates of neurological deterioration were comparable between the groups during long-term open-label treatment (months 18–66) (Fig. 1) [16].

Intent-to-treat slope analysis of a NIS-LL (total score), b NIS-LL muscle weakness, and c Norfolk QOL-DN (total score) in patients with Val30Met ATTR-PN (data from Fx-005, Fx-006 and B3461023) [14]. Slopes are adjusted at mean baseline value of the two treatment groups. Reproduced from Barroso et al. [16] with permission. NIS-LL Neuropathy Impairment Score-Lower Limbs (scale 0–88; higher scores indicate worsening function), QOL-DN TQOL Quality of Life-Diabetic Neuropathy total quality of life score (scale − 2 to 138; higher scores indicate worsening quality of life)
With respect to functional progression, two patients in the tafamidis–tafamidis group and six patients in the placebo–tafamidis group had progressed to the next ambulatory stage of the disease by year 6; estimated 6-year progression rates from the first dose of study drug were 5.5% and 34.6% in the respective groups [ambulatory data retrospectively collected; between-group difference not significant] [16]. Mean Karnofsky performance status scores were 83.8 and 80.3 in the tafamidis–tafamidis and placebo–tafamidis groups at month 30 (i.e. the start of B3461023) and were 85.9 and 78.7 in the respective groups at month 66 (reductions indicate worsening) [16].
In a post hoc subgroup analysis of patients with mild polyneuropathy at treatment start (i.e. NIS-LL ≤ 10; n = 71 analyzed) who participated in B3461023, early intervention with tafamidis was associated with minimal neurological progression and preserved nutritional status over 5.5 years [22]. Mean change from baseline in NIS-LL was 0.6 (95% CI − 0.2 to 1.4) and 5.3 (95% CI 1.6–9.1) with up to 1 and 5.5 years of treatment with tafamidis, respectively. Mean change from baseline in mBMI was 28.5 kg/m2 × g/L (95% CI 7.9–49.1) at 1 year; there was no significant deterioration in nutritional status from baseline to 5.5 years (based on 95% CI), with a mean change in mBMI of − 7.8 kg/m2 × g/L (95% CI − 44.4 to 28.8) [22].
2.1.1.3 In Japanese Patients
Tafamidis was also effective in stabilizing TTR (≥ 32% stabilization) and generally delayed disease progression in Japanese patients with ATTR-PN [n = 10; 9/10 with the Val30Met mutation (1 patient had a non-Val30Met mutation) and 7/10 with late-onset disease (i.e. diagnosed aged ≥ 50 years)] in an open-label, multicentre, phase III study [17]. Patients were treated with oral tafamidis 20 mg once daily. All patients showed TTR stabilization at weeks 8 (primary endpoint) and 26, with 9/10 and 8/10 patients maintaining TTR stabilization at weeks 52 and 78 (2/10 patients had missing data at the week 78 assessment and were not considered to be stabilized). The NIS-LL response rate (defined as an increase in NIS-LL from baseline of < 2) was 8/10, 6/10 and 4/10 at weeks 26, 52 and 78, respectively; the mean NIS-LL change from baseline to week 78 was 3.3 (17 at baseline). At week 78, the mean changes from baseline in TQOL-DN and mBMI were 10.8 and 53.7 (respective baseline values were 53 and 806), respectively, and half of the patients (4/8) had preserved ambulation status [17].
2.1.2 In Patients with Other Mutations
In the single-arm, phase II Fx1a-201 trial, patients (n = 21) with ATTR-PN mutations other than Val30Met or Val122Ile received tafamidis 20 mg once daily for 6 weeks (part 1), with those patients demonstrating TTR stabilization (defined as > 32% stabilization) at week 6 continuing treatment for ≤ 12 months in total (part 2) [18]. At baseline, their mean duration of ATTR-PN-related symptoms was 65 months and their mean age at onset was 59 years; neurological function and disease-specific quality of life (QOL) were considerably impaired (mean NIS and TQOL-DN values were 49 and 48, respectively) [18]. All patients who completed Fx1a-201 (n = 18) proceeded to enter the B3461023 extension study and were included in the efficacy and safety analyses (Sect. 2.1.1) [16]. At the time of data cut-off for the B3461023 interim analysis, two patients were ongoing in the study, while seven patients had completed the study (when tafamidis became available via prescription) and nine had permanently discontinued [16].
Tafamidis effectively stabilized TTR tetramers in patients with non-Val30Met ATTR-PN [18]. TTR tetramer stabilization was achieved by 94.7% of patients with evaluable data at week 6 (18/19 patients; primary outcome) and in all patients with evaluable data at months 6 (n = 18) and 12 (n = 17) [secondary endpoints]. Exploratory efficacy outcomes indicated minimal deterioration in neurological function from baseline to month 12, as indicated by the respective mean change from baseline in NIS, NIS-LL and NIS-Upper Limbs (5.3, 2.7 and 2.5; respective baseline values were 49, 28, 21) and the proportion of patients with a change in NIS of ≤ 4 points (12/18 patients). Moreover, TQOL-DN and mBMI did not change to any clinically relevant degree [18].
In post hoc analyses of data from Fx-005 (Sect. 2.1.1) and Fx1a-201, tafamidis delayed neurological progression comparably in patients with Val30Met and non-Val30Met ATTR-PN during the first year of treatment, with the baseline-adjusted LSM change in NIS-LL being 1.60 and 1.62 in the respective populations at 12 months [20]. In contrast, the change in NIS-LL was 4.72 in Val30Met placebo recipients; the Val30Met tafamidis group had a significantly more favorable change in NIS-LL than the placebo group (p = 0.0055), while the difference between the non-Val30Met tafamidis group and Val30Met placebo group did not reach significance (p = 0.0592). The magnitude of predicted LSM change in NIS-LL was comparable between the Val30Met and non-Val30Met tafamidis-treated cohorts across a range of baseline NIS-LL values, and was consistently numerically lower than that observed with placebo [20].
Disease progression was observed in tafamidis recipients with non-Val30Met ATTR-PN during the long-term B3461023 extension study [16]. From Fx1a-201 baseline to month 48, there were LSM changes of 14.2 (95% CI 8.0–20.4), 8.9 (95% CI 4.1–13.7) and 2.6 (95% CI 0.9–4.2) in the respective NIS-LL total score, muscle weakness subscale score and sensation subscale score (31, 17 and 8.9 at baseline), 24.8 (95% CI 7.0–42.7) in TQOL-DN (54 at baseline), − 13.4 (95% CI − 20.4 to − 6.4) in Karnofsky performance status score (72 at baseline) and − 1 kg/m2 (95% CI − 2.2 to 0.2) in BMI (25 kg/m2 at baseline) [16].
2.2 In the Clinical Practice Setting
The efficacy of tafamidis is generally supported by experience in the clinical practice setting, which includes a number of fully published analyses [23,24,25,26,27], the two largest of which [23, 25] are the focus here.
Tafamidis delayed neurological progression relative to no treatment for up to 2 years from baseline in a non-randomized, longitudinal, matched cohort analysis of stage 1 ATTR-PN patients from the international Transthyretin Amyloidosis Outcomes Survey (THAOS), an ongoing, global, non-interventional disease registry for ATTR amyloidosis [25]. Patients were matched within mutation groups, defined as Val30Met and non-Val30Met. In the matched treated sample, the majority of patients had the Val30Met mutation (92.5%), were relatively young (mean age 40.4 years) and were born in Portugal (80.2%). Dosage recommendations for tafamidis were based on clinical judgement and the local product label, or open-label study protocol; the indicated dosage is 20 mg once daily (Sect. 4). After 24 months of treatment, tafamidis recipients (n = 252) had significantly lower rates of neurological deterioration [as measured by NIS-LL and Neurologic Composite Score (NCS) total scores and NCS sensory, motor and reflex subscale scores] than the untreated controls (n = 252) [p ≤ 0.023 for all]. Relative to no treatment, tafamidis was also associated with a significant improvement in QOL (as measured by TQOL-DN; p < 0.001). There were no significant between-group differences in Karnofsky performance status score or mBMI. The treated and untreated patients did not significantly differ in time to disease progression, with the majority of patients in both groups remaining in stage 1 at 24 months (98.6 vs. 97.3%). As no deaths were reported in tafamidis recipients, the primary survival analysis could not estimate a hazard ratio (HR) versus matched untreated patients; a secondary analysis without regard to matching indicated a significantly greater risk of death in untreated patients than in tafamidis recipients (HR 3.95; 95% CI 1.54–10.14; p = 0.0042) [25].
Tafamidis also conferred survival benefits relative to no treatment in a multi-institutional, hospital-based cohort study in Portugal [23]. Symptomatic adults with stage 1 ATTR-PN and the Val30Met mutation were prospectively followed until December 2016. Patients received tafamidis (n = 432) or a liver transplant (n = 957), or were untreated (n = 1771). Median overall survival was not reached in tafamidis recipients (maximum follow-up 10 years), 24.73 years since disease onset in liver transplant recipients and 11.61 years in untreated patients. In tafamidis recipients, the observed survival rate was 92.9% at 10 years after treatment initiation. Each year that treatment with tafamidis was delayed by increased the mortality risk by 39% (HR 1.39; 95% CI 1.05–1.83; p = 0.022), while sex and disease onset (early vs. late) had no impact. In patients with early-onset disease, tafamidis (n = 347) reduced mortality risk by 91% relative to no treatment (n = 755) [HR 0.09; 95% CI 0.03–0.25; p < 0.0001] and by 63% relative to liver transplant (n = 855) [HR 0.37; 95% CI 0.14–1.00; p = 0.050]; the probabilities of survival at 10 years after ATTR-PN onset were 96, 73 and 72% in tafamidis recipients, liver transplant recipients and untreated patients, respectively. Prior tafamidis treatment had no impact on mortality risk after liver transplantation. In patients with late-onset disease, tafamidis (n = 85) reduced the risk of mortality by 82% relative to no treatment (n = 116) [HR 0.18; 95% CI 0.06–0.49; p = 0.001]; the 10-year survival probabilities were 92 and 49% in tafamidis recipients and untreated patients, respectively [23].
3 Tolerability of Tafamidis
Tafamidis was generally well tolerated in adults with ATTR-PN in clinical trials [14,15,16,17,18] and real-world studies [24, 26], with most adverse events (AEs) being mild or moderate in severity where specified [16, 17, 28]. The focus of this section is on tolerability and safety data from the placebo-controlled Fx-005 trial in patients with Val30Met ATTR-PN (Sect. 2.1.1) [14] and its ongoing long-term extension (B3461023) [which also included patients with non-Val30Met ATTR-PN previously enrolled in the open-label Fx1a-201 trial; Sect. 2.1.2] [16]. In the long-term extension study, mean cumulative tafamidis exposure was 5.1 and 3.5 years in patients with Val30Met ATTR-PN who had initially received tafamidis (n = 38) or placebo (n = 37), respectively, and 3.6 years in patients with non-Val30Met ATTR-PN (n = 18) [16].
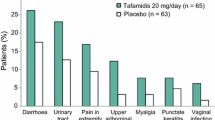
In Fx-005, AEs were reported in the majority of patients (92.3% of tafamidis recipients and 96.8% of placebo recipients) [14]. The most common AEs (occurring in ≥ 5% of tafamidis recipients and with a ≥ 2% higher incidence than with placebo) were diarrhoea, urinary tract infection, pain in extremity, upper abdominal pain, myalgia, punctate keratitis and vaginal infection (Fig. 2). AEs infrequently led to treatment discontinuation, doing so in only four (6.2%) tafamidis recipients and three (4.8%) placebo recipients. Serious AEs (SAEs) occurred in 9.2% of tafamidis recipients and 7.9% of placebo recipients. Urinary tract infection (reported in two tafamidis recipients) was the only SAE to occur in more than one patient. Complications arising from liver transplantation resulted in two deaths in the tafamidis group and three deaths in the placebo group [14].

During long-term administration (≤ 6 years) in patients with Val30Met or non-Val30Met ATTR-PN in B3461023, tafamidis had a comparable tolerability profile to that observed during the placebo-controlled period of Fx-005; no new safety signals were detected [16]. One-quarter of patients (24.7%; 23 and 33% of patients with Val30Met and non-Val30Met ATTR-PN, respectively) experienced ≥ 1 treatment-related AE (TRAE). The most common TRAEs were headache, oedema peripheral and urinary tract infection, each reported in two patients (2.2%). The rate of discontinuations resulting from AEs was 4.3% (3 and 11% of patients with Val30Met and non-Val30Met ATTR-PN); two of the AEs that led to treatment discontinuation were considered to be treatment-related (faecal incontinence and renal impairment). An additional 2.2% of patients temporarily discontinued tafamidis due to TRAEs [16].
Severe AEs and SAEs reported during the long-term extension study were seldom considered to be treatment-related [16]. One treatment-related severe AE was reported (pericardial effusion), and treatment-related SAEs occurred in one patient (pericardial effusion, cardiac disorder and aggravated renal function). There were seven deaths during treatment or within 30 days of study completion or discontinuation, and one post-therapy death of unknown cause that occurred 2 years after study discontinuation due to an AE (faecal incontinence); none of these eight deaths (three patients with Val30Met ATTR-PN and five patients with non-Val30Met ATTR-PN) were considered to be treatment related [16].
Laboratory parameters (including measures of thyroid function) showed no clinically relevant changes during the placebo-controlled Fx-005 trial [14]. During long-term treatment, the most common laboratory abnormalities in patients with Val30Met ATTR-PN were neutrophil levels > 1.2 × upper limit of normal (ULN) [14/75 patients; 18.7%], lymphocyte levels < 0.8 × lower limit of normal (LLN) [12/75 patients; 16.0%], gamma glutamyl transferase levels > 3.0 × ULN (8/75 patients; 10.7%) and thyrotropin levels < 0.8 × LLN (6/75 patients; 8.0%), while the most common in patients with non-Val30Met ATTR-PN were prothrombin time > 1.1 × ULN (6/11 patients; 54.5%) and blood urea nitrogen levels > 1.3 × ULN (3/18 patients; 16.7%) [16].
No safety concerns relating to cardiac function were observed in the Fx1a-201 trial in patients with non-Val30Met and non-Val122Ile ATTR-PN (n = 21) [29]. Tafamidis had no clinically relevant effects on mean echocardiographic and electrocardiographic parameters, and cardiac biomarkers were stable during therapy. There was a significant increase from baseline to month 12 in the proportion of patients with normal heart rate variability (21 vs. 42%; p < 0.05). While septal wall thickness was typically stable, ≥ 2 mm increases in interventricular septal thickness were observed in four patients. At baseline, all patients were in New York Heart Association class I or II (47.6 and 52.4%) and four patients (19.0%) had cardiac pacemakers [29]. In the long-term extension study (B3461023), a Fridericia-corrected QT interval > 500 ms was noted in four patients (10.5%) with Val30Met ATTR-PN in the tafamidis–tafamidis group, one patient (2.7%) with Val30Met ATTR-PN in the placebo–tafamidis group and four tafamidis recipients (22.2%) with non-Val30Met ATTR-PN [16].
Tolerability in other trials [15, 17, 18] and the clinical practice setting [24, 26] was generally consistent with that observed in the pivotal trial and long-term extension. In a small trial in Japanese patients (Sect. 2.1.1), one SAE (sudden death) was assessed as being related to tafamidis [17].
4 Dosage and Administration of Tafamidis
In numerous countries including those of the EU [13], tafamidis is approved for the treatment of TTR amyloidosis in adults with stage 1 symptomatic polyneuropathy, to delay peripheral neurological impairment. In several other countries including Japan [30], tafamidis is approved for inhibiting the progression of peripheral neuropathy in patients with ATTR-PN. Tafamidis is available in soft capsules, with each capsule containing 20 mg of micronized tafamidis meglumine (equivalent to 12.2 mg of tafamidis) [13, 30]. The recommended dose of tafamidis meglumine is 20 mg orally, once per day [13, 30]. Capsules should be swallowed whole, and may be taken with or without food [13]. As the capsule shell contains sorbitol (E420), tafamidis should not be administered in patients with rare hereditary problems of fructose intolerance. Tafamidis should be added to the standard of care for the treatment of ATTR-PN and should be discontinued if the patient has a liver transplant [13]. Local prescribing information should be consulted for detailed information concerning contraindications, special warnings and precautions for use, drug interactions and use in specific patient populations.
5 Place of Tafamidis in the Management of Transthyretin Amyloidosis with Polyneuropathy
Access to approved treatments has considerably improved the long-term prognosis for patients with ATTR-PN [23]. Historically, the excess mortality rate in untreated patients with ATTR-PN was 10-fold relative to the general population; in patients with ATTR-PN who receive tafamidis or liver transplant, the excess mortality rate decreases to 4-fold that of the general population [23]. Approved in the EU in 2011 and several other countries thereafter for the delay of peripheral neurological progression in this setting (Sect. 4), tafamidis was the first pharmacological alternative to liver transplantation in patients with stage 1 ATTR-PN [11, 31]. Tafamidis inhibits neurological progression through targeting TTR tetramer dissociation (the rate-limited step in amyloidogenesis), kinetically stabilizing wild-type and mutant TTR (Sect. 1).
The most recent best practice guidelines issued by the European Network for ATTR-PN (ATTReuNET) recommend tafamidis as the first-line treatment for patients with stage 1 ATTR-PN (i.e. symptomatic patients who can walk unaided outside), with tafamidis thus replacing liver transplantation as the standard of care in these patients [5]. If disease progression occurs during tafamidis therapy, liver transplantation is recommended in younger patients (aged < 50 years) with no contraindications for this surgery. In Japan, liver transplantation remains the first-line treatment for ATTR-PN; tafamidis should be administered in patients who are unsuitable candidates for this surgery [3]. Recent Brazilian consensus guidelines acknowledge liver transplantation and tafamidis as the only disease-modifying treatments for ATTR-PN currently available [32]. Clinicians should take a multidisciplinary approach to the management of ATTR-PN [5, 32], including a neurologist, cardiologist and potentially an ophthalmologist in initially assessing and later reviewing the patient; during treatment, continuous monitoring is essential [5]. It should be noted that following the ATTReuNET guidelines being produced, two additional pharmacological treatments (inotersen and patisiran) have received approval for use in stage 1 and 2 ATTR-PN in the EU [33, 34]. These agents are also approved for ATTR-PN in the USA [35, 36]. Inotersen, an antisense oligonucleotide, and patisiran, a small interfering RNA, inhibit hepatic TTR production (wild-type and mutant) via post-transcriptional gene silencing [33, 34, 37].
Approval of tafamidis was based largely on the findings of the well-designed phase II/III Fx-005 registration trial (Sect. 2.1.1.1). While the effects of the drug on the neurological and health-related QOL co-primary endpoints did not reach statistical significance in the ITT population, this may reflect the proportion of patients who discontinued due to liver transplantation being higher than anticipated (21% vs. an estimated 10%) [14]. Indeed, in prespecified analyses in the efficacy evaluable population (which excluded liver transplant patients rather than classing them as nonresponders), tafamidis recipients showed significant benefits relative to placebo recipients for both co-primary endpoints (Table 2). Tafamidis also improved nutritional status relative to placebo. Consistent with the positive effects of tafamidis on disease progression, TTR tetramer stabilization was achieved in nearly all tafamidis recipients (Sect. 2.1.1). Most efficacy data for tafamidis come from patients with early-onset, Val30Met ATTR-PN, which appears to have a slower progression rate than disease that is late-onset or caused by non-Val30Met variants [38]. While tafamidis has not been evaluated in non-Val30Met ATTR-PN in a randomized, placebo-controlled trial, results from a small open-label trial in patients with ATTR-PN and a range of amyloidogenic TTR mutations suggest the effects of tafamidis are generally comparable to those observed in Val30Met-positive patients (Sect. 2.1.2); the majority of patients with non-Val30Met ATTR-PN achieved TTR tetramer stabilization from week 6 onwards. This was as expected, given the activity of tafamidis in stabilizing TTR tetramers with various mutations in vitro (Table 1). Although tafamidis has been evaluated in a small number of patients with stage 1 late-onset Val30Met ATTR-PN [39], efficacy has not been established in this patient population.
The long-term efficacy and safety of tafamidis is being evaluated in an ongoing open-label extension study (B3461023; Sects. 2 and 3), which currently constitutes the longest longitudinal evaluation of a disease-modifying pharmacological treatment for ATTR-PN (≤ 6 years of data) [7]. At the time of the interim analysis (after ≤ 66 months of treatment), tafamidis recipients with Val30Met ATTR-PN showed minimal deterioration in neurological function and health-related QOL, with nutritional status being generally maintained or improved (Sect. 2.1.1). It should be noted that while neurological progression is slowed by tafamidis, it is not stopped completely. At 6 years, the proportion of patients who had progressed to the next ambulatory stage of the disease was six times lower among those who had received tafamidis for the full duration of the studies than in those who commenced tafamidis 18 months later (i.e. those initially randomized to placebo) (Sect. 2.1.1). With respect to patients with non-Val30Met ATTR-PN, some deterioration in neurological function, Karnofsky performance status score and health-related QOL was observed over 4 years of treatment with tafamidis in B3461023 (Sect. 2.1.2). While the small sample size and absence of a comparator hampers the interpretation of these results, the patients with non-Val30Met disease were older, with more severe disease and a longer disease duration at baseline than those with Val30Met ATTR-PN (Sect. 2.1). As with other interventional therapies for ATTR-PN, clinical trials and experience in the real-world setting suggest that the benefits of tafamidis are greatest in patients with shorter disease duration and less severe disease (Sect. 2), supporting the desirability of early diagnosis and treatment initiation in ATTR-PN [5, 32].
Data from the largest international rare disease registry for patients with ATTR amyloidosis (THAOS) supports the real-world effectiveness of tafamidis in delaying neurological progression in patients with stage 1 disease (Sect. 2.2) [25]. Most patients in the matched-cohort analysis were relatively young, with Val30Met disease (Sect. 2.2). While survival data in tafamidis recipients are currently limited, a multi-institutional hospital-based study in Portugal found that tafamidis significantly reduced the risk of mortality relative to no treatment in both early-onset and late-onset Val30Met ATTR-PN (Sect. 2.2). Tafamidis and liver transplantation (for which there are survival data at ≥ 20 years’ post-treatment [11, 25]) have not been compared in controlled trials. Although tafamidis appeared to reduce mortality risk relative to liver transplantation in patients with early-onset ATTR-PN in the Portuguese study (Sect. 2.2), this result should be interpreted with caution due to the relatively short follow-up in tafamidis recipients (maximum follow-up of 10 years, with median overall survival not reached) [23]. Nevertheless, tafamidis offers a number of practical benefits over liver transplantation, including suitability for use in patients with late-onset disease (Sect. 1) and a favourable safety record (Sect. 3). Liver transplantation (for which there is a shortage of donors) is a surgical procedure that carries considerable risk, as well as the requirement for long-term immunosuppressant therapy (Sect. 1).
Tafamidis was generally well tolerated in clinical trials and real-world studies, with safety data available in patients with Val30Met and non-Val30Met ATTR-PN treated for ≤ 6 years (Sect. 3). During long-term treatment, the most common TRAEs were headache, oedema peripheral and urinary tract infection, which all occurred at low rates (2.2% each); TRAEs were seldom severe or serious, and no deaths in the long-term extension study were considered to be treatment related (which may be contrasted with a mortality rate of 14% observed in the first year following liver transplantation [23]). Its long-term tolerability was consistent with that initially observed during the placebo-controlled registration study, with no new safety signals identified in the extension trial or clinical practice setting (Sect. 3). With a lack of NSAID activity (Table 1) and no apparent safety concerns relating to cardiovascular function (Sect. 3), tafamidis is suitable for use in patients with ATTR-PN who also have cardiomyopathy (confirmed by results from a recent phase III trial [40]).
Tafamidis is administered orally once daily (Sect. 4), which some patients might find more convenient than the subcutaneous and intravenous injection routes offered by inotersen (administered once weekly) [33] and patisiran (administered once every 3 weeks) [34], respectively. As is the case with tafamidis and liver transplantation, no clinical studies have directly compared the efficacy of tafamidis with that of inotersen or patisiran; however, these gene silencing drugs slow or reverse disease progression in ATTR-PN [41]. An indirect treatment comparison using the standard pairwise Bucher method for endpoints used in both the tafamidis and patisiran clinical trials has suggested that patisiran may be more effective than tafamidis in preventing deterioration in neurological function and health-related QOL (assessed using NIS-LL and TQOL-DN), although there are limitations to such a design (e.g. differences between study groups in baseline characteristics, in this case including age, disease severity and mutation type) [42]. To date, no such analyses have compared tafamidis and inotersen, or all three drugs, and these would be of interest. Given that multiple pharmaceutical options are now available, there is a need for investigation into the patient-specific factors that will determine the most appropriate therapy for each patient [41]. Robust cost-benefit analyses comparing tafamidis with the silencing drugs will be important in further elucidating their respective roles in the treatment of stage 1 ATTR-PN [41].
In conclusion, tafamidis is an effective and generally well tolerated pharmacological option for delaying peripheral neurological progression in adults with stage 1 symptomatic ATTR-PN. Based on up to 10 years’ experience in clinical trials and the real-world setting, tafamidis continues to be a valuable option in the treatment of early-stage ATTR-PN.
Data Selection Tafamidis: 342 records identified
Duplicates removed | 106 |
Excluded during initial screening (e.g. press releases; news reports; not relevant drug/indication; preclinical study; reviews; case reports; not randomized trial) | 96 |
Excluded during writing (e.g. reviews; duplicate data; small patient number; nonrandomized/phase I/II trials) | 96 |
Cited efficacy/tolerability articles | 17 |
Cited articles not efficacy/tolerability | 27 |
Search Strategy: EMBASE, MEDLINE and PubMed from 2012 to present. Previous Adis Drug Evaluation published in 2012 was hand searched for relevant data. Clinical trial registries/databases and websites were also searched for relevant data. Key words were tafamidis, Vyndaqel, FX1006A, PF6291826, familial amyloid polyneuropathy, FAP, TTR-FAP, hATTR. Records were limited to those in English language. Searches last updated 2 May 2019 | |
References
Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31.
Hou X, Aguilar M-I, Small DH. Transthyretin and familial amyloidotic polyneuropathy: recent progress in understanding the molecular mechanism of neurodegeneration. FEBS J. 2007;274(7):1637–50.
Sekijima Y, Ueda M, Koike H, et al. Diagnosis and management of transthyretin familial amyloid polyneuropathy in Japan: red-flag symptom clusters and treatment algorithm. Orphanet J Rare Dis. 2018;13(1):6.
Schmidt HH, Waddington-Cruz M, Botteman MF, et al. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2018;57(5):829–37.
Adams D, Suhr OB, Hund E, et al. First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Curr Opin Neurol. 2016;29(Suppl 1):S14–26.
Hawkins PN, Ando Y, Dispenzeri A, et al. Evolving landscape in the management of transthyretin amyloidosis. Ann Med. 2015;47(8):625–38.
Ericzon BG, Wilczek HE, Larsson M, et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation. 2015;99(9):1847–54.
Sekijima Y. Recent progress in the understanding and treatment of transthyretin amyloidosis. J Clin Pharm Ther. 2014;39(3):225–33.
Plante-Bordenave V. Transthyretin familial amyloid polyneuropathy: an update. J Neurol. 2018;265:976–83.
Coelho T, Merlini G, Bulawa CE, et al. Mechanism of action and clinical application of tafamidis in hereditary transthyretin amyloidosis. Neurol Ther. 2016;5(1):1–25.
Scott LJ. Tafamidis: a review of its use in familial amyloid polyneuropathy. Drugs. 2014;74(12):1371–8.
Bulawa CE, Connelly S, DeVit M, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci USA. 2012;109(24):9629–34.
European Medicines Agency. Vyndaqel (tafamidis) 20 mg soft capsules: summary of product characteristics. 2016. http://www.ema.europa.eu/. Accessed 2 May 2019.
Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785–92.
Coelho T, Maia LF, Martins da Silva A, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260(11):2802–14.
Barroso FA, Judge DP, Ebede B, et al. Long-term safety and efficacy of tafamidis for the treatment of hereditary transthyretin amyloid polyneuropathy: results up to 6 years. Amyloid. 2017;24(3):194–204.
Ando Y, Sekijima Y, Obayashi K, et al. Effects of tafamidis treatment on transthyretin (TTR) stabilization, efficacy, and safety in Japanese patients with familial amyloid polyneuropathy (TTR-FAP) with Val30Met and non-Val30Met: a phase III, open-label study. J Neurol Sci. 2016;362:266–71.
Merlini G, Planté-Bordeneuve V, Judge DP, et al. Effects of tafamidis on transthyretin stabilization and clinical outcomes in patients with non-Val30Met transthyretin amyloidosis. J Cardiovasc Transl Res. 2013;6(6):1011–20.
Amass L, Li H, Gundapaneni B, et al. Influence of baseline neurologic severity on disease progression and the associated disease-modifying effects of tafamidis in patients with transthyretin amyloid polyneuropathy. Orphanet J Rare Dis. 2018;13:225.
Gundapaneni BK, Sultan MB, Keohane DJ, et al. Tafamidis delays neurological progression comparably across Val30Met and non-Val30Met genotypes in transthyretin familial amyloid polyneuropathy. Eur J Neurol. 2018;25(3):464–8.
Keohane D, Schwartz J, Gundapaneni B, et al. Tafamidis delays disease progression in patients with early stage transthyretin familial amyloid polyneuropathy: additional supportive analyses from the pivotal trial. Amyloid. 2017;24(1):30–6.
Waddington Cruz M, Amass L, Keohane D, et al. Early intervention with tafamidis provides long-term (5.5-year) delay of neurologic progression in transthyretin hereditary amyloid polyneuropathy. Amyloid. 2016;23(3):178–83.
Coelho T, Inês M, Conceição I, et al. Natural history and survival in stage I Val30Met transthyretin familial amyloid polyneuropathy. Neurology. 2018;91(21):e1999–2009.
Cortese A, Vita G, Luigetti M, et al. Monitoring effectiveness and safety of tafamidis in transthyretin amyloidosis in Italy: a longitudinal multicenter study in a non-endemic area. J Neurol. 2016;263(5):916–24.
Mundayat R, Stewart M, Alvir J, et al. Positive effectiveness of tafamidis in delaying disease progression in transthyretin familial amyloid polyneuropathy up to 2 years: an analysis from the Transthyretin Amyloidosis Outcomes Survey (THAOS). Neurol Ther. 2018;7(1):87–101.
Plante-Bordeneuve V, Gorram F, Salhi H, et al. Long-term treatment of transthyretin familial amyloid polyneuropathy with tafamidis: a clinical and neurophysiological study. J Neurol. 2017;264(2):268–76.
Sousa S, Valdrez K, Fonseca I, et al. Prediction of nerve conduction studies outcomes in patients with familial amyloidotic polyneuropathy receiving tafamidis therapy. J Neuromuscul Dis. 2016;3(Suppl 1):S77–8.
European Medicines Agency. Tafamidis: CHMP assessment report. 2011. http://www.ema.europa.eu/. Accessed 2 May 2019.
Damy T, Judge DP, Kristen AV, et al. Cardiac findings and events observed in an open-label clinical trial of tafamidis in patients with non-Val30Met and non-Val122Ile hereditary transthyretin amyloidosis. J Cardiovasc Transl Res. 2015;8(2):117–27.
Pfizer Inc. Vyndaqel® capsules 20 mg: Japanese prescribing information. 2016. http://www.pmda.go.jp/. Accessed 2 May 2019.
McKeage K, Lyseng-Williamson KA, Scott LJ. Tafamidis in transthyretin amyloidosis: a guide to its use in delaying peripheral neurological impairment in patients with stage 1 polyneuropathy. Drug Ther Perspect. 2017;33(2):47–53.
Pinto MV, Barreira AA, Bulle AS, et al. Brazilian consensus for diagnosis, management and treatment of transthyretin familial amyloid polyneuropathy. Arq Neuropsiquiatr. 2018;76(9):609–21.
Keam SJ. Inotersen: first global approval. Drugs. 2018;78:1–6.
Hoy S. Patisiran: first global approval. Drugs. 2018;78:1625–31.
Alnylam Pharmaceuticals Inc. ONPATTRO (patisiran) lipid complex injection, for intravenous use. 2018. http://www.alnylam.com/. Accessed 2 May 2019.
Ionis Pharmaceuticals Inc. TEGSEDI (inotersen) injection, for subcutaneous use. 2018. http://tegsedi.com/. Accessed 2 May 2019.
Adams D, Théaudin M, Cauquil C, et al. FAP neuropathy and emerging treatments. Curr Neurol Neurosci Rep. 2014;14(3):435.
Mariani L-L, Lozeron P, Théaudin M, et al. Genotype-phenotype correlation and course of transthyretin familial amyoid polyneuropathies in France. Ann Neurol. 2015;78(6):901–16.
Lozeron P, Theaudin M, Mincheva Z, et al. Effect on disability and safety of tafamidis in late onset of Met30 transthyretin familial amyloid polyneuropathy. Eur J Neurol. 2013;20(12):1539–45.
Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;27:1007–16.
Vita G, Vita GL, Stancanelli C, et al. Genetic neuromuscular disorders: living the era of a therapeutic revolution. Part 1: peripheral neuropathies. Neurol Sci. 2019;40(4):661–9.
Plante-Bordeneuve V, Lin H, Gollob J, et al. An indirect treatment comparison of the efficacy of patisiran and tafamidis for the treatment of hereditary transthyretin-mediated amyloidosis with polyneuropathy. Expert Opin Pharmacother. 2018;20(4):473–81.
Klamerus KJ, Watsky E, Moller R, et al. The effect of tafamidis on the QTc interval in healthy subjects. Br J Clin Pharmacol. 2015;79(6):918–25.
Suhr OB, Conceicao IM, Karayal ON, et al. Post hoc analysis of nutritional status in patients with transthyretin familial amyloid polyneuropathy: impact of tafamidis. Neurol Ther. 2014;3(2):101–12.
Acknowledgements
During the peer review process, the manufacturer of tafamidis was also offered an opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Conflict of interest
Yvette Lamb and Emma Deeks are salaried employees of Adis/Springer, responsible for the article content and declare no relevant conflicts of interest.
Additional information
The manuscript was reviewed by:F. Barroso, Department of Neurology, Institute for Neurological Research Raul Carrea (FLENI), Buenos Aires, Argentina; M. Waddington Cruz, National Amyloidosis Referral Center (CEPARM), University Hospital, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil.
Rights and permissions
About this article

Cite this article
Lamb, Y.N., Deeks, E.D. Tafamidis: A Review in Transthyretin Amyloidosis with Polyneuropathy. Drugs 79, 863–874 (2019). https://doi.org/10.1007/s40265-019-01129-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-019-01129-6