
Abstract
Oral once-daily, fixed-dose, ledipasvir/sofosbuvir (Harvoni®) [± ribavirin] is approved in several countries for the treatment of chronic hepatitis C (CHC) in adults and adolescents aged 12 to < 18 years, with direct-acting antiviral (DAA) regimens resulting in a paradigm shift in the treatment of the disease. In the clinical trial and/or clinical practice setting, ledipasvir/sofosbuvir (± ribavirin) was associated with high sustained virological response rates 12 weeks post-treatment (SVR12) in treatment-naive and -experienced adults and adolescents with chronic hepatitis C virus (HCV) genotype (GT) 1 infection, including in those with compensated cirrhosis or who were co-infected with HIV. SVR12 rates in real-world studies were consistent with those in trials. In other trials, ledipasvir/sofosbuvir (± ribavirin) was associated with high SVR12 rates in various CHC populations, including patients with HCV GT2, 3, 4, 5 or 6 infection, cirrhosis, pre and/or post liver or renal transplantation, inherited blood disorders or failure after prior DAA and/or interferon therapy. Thus, ledipasvir/sofosbuvir (± ribavirin) is a valuable effective and generally well tolerated option for adolescent and adult patients with HCV GT1, 4, 5 or 6 infection, including those with HIV co-infection or cirrhosis, with evidence also supporting its use in patients with chronic HCV GT2 or 3 infection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Fixed-dose tablet comprising the HCV NS5A inhibitor ledipasvir and the NS5B polymerase inhibitor sofosbuvir |
Provides high SVR12 rates and improves patient-reported outcomes in treatment-naive and -experienced adolescents (aged ≥ 12 years) and adults with chronic HCV GT1 infection |
Exhibits high SVR12 rates in other CHC populations, including in patients with HCV GT2, 3, 4, 5 or 6 infection, HIV co-infection or cirrhosis |
SVR12 rates in the real-world setting are similar to those reported in the clinical trial setting |
Generally well tolerated in the clinical trial and real-world setting |
1 Introduction
In recent years, the management of chronic hepatitis C virus (HCV) infection has been revolutionized by the introduction of interferon (IFN)-free regimens consisting of oral direct-acting antiviral agents (DAA), with DAA regimens providing markedly better antiviral efficacy [sustained virological response (SVR) of 98% with combination DAA regimens] [1] and tolerability than IFN-regimens [1,2,3]. One such DAA regimen, the fixed-dose combination of the HCV N5A inhibitor ledipasvir and the NS5B inhibitor sofosbuvir (ledipasvir/sofosbuvir; Harvoni®), is approved in numerous countries for the treatment of chronic hepatitis C (CHC), including in the EU [4]. This review, written from an EU perspective, discusses the clinical use of ledipasvir/sofosbuvir in the treatment of CHC in adults and adolescents (aged 12 to < 18 years), and summarizes its pharmacological properties.
2 Pharmacodynamic Properties
The pharmacodynamic properties of ledipasvir/sofosbuvir have been reviewed in detail [5, 6]. In brief, ledipasvir inhibits the HCV NS5A protein and sofosbuvir inhibits HCV NS5B RNA-dependent RNA polymerase, with both enzymes essential for viral replication [4,5,6]. Sofosbuvir, a nucleotide prodrug, is metabolized to the pharmacologically active metabolite GS-461203, which is incorporated into HCV RNA by NS5B polymerase where it acts as a chain terminator. In replicon assays, ledipasvir exhibits potent antiviral activity against HCV genotype (GT) 1a and 1b, with lower activity against HCV GT4a, 4b, 5a and 6a and substantially lower activity against HCV GT2a, 2b, 3a and 6c. Sofosbuvir exhibits potent pangenotypic antiviral activity against HCV GT1a, 1b, 2a, 2b, 3a, 4a, 5a and 6a in replicon assays [4,5,6].
2.1 Resistance
In vitro, the primary NS5A amino acid substitution at Y93H in HCV GT1a and 1b and also the Q30E substitution in HCV GT1a were associated with reduced susceptibility to ledipasvir [4, 7]. Based on site-directed mutagenesis of NS5A resistance-associated variants (RAVs), amino acid substitutions in GT1a conferring a > 100 to ≤ 1000 fold-change in susceptibility to ledipasvir were Q30H/R, L311/M/V, P32L and Y93T [4]. In HCV replicons, reduced susceptibility to sofosbuvir was associated with the primary NS5B substitution S28T in multiple GTs [4].
In the pivotal ION trials (Sect. 4), in patients who qualified for resistance testing, 22 of 29 patients with HCV GT1a infection had ≥ 1 NS5A RAV detected at positions K4, M28, Q30, L31, S38 and/or Y93 in post baseline isolates at the time of failure (most commonly Q30R, Y93H and L31M); no RAVs were detected in the remaining seven patients at failure [4]. Of the eight patients with HCV GT1b infection who qualified for resistance testing, seven had ≥ 1 NS5A RAV detected at positions L31 and Y93 (most commonly Y93H) in post baseline isolates at the time of failure [4]. The sofosbuvir resistance-associated NS5B S28T variant was not detected in any virological failure isolate during the ION trials [4, 8,9,10]. Amongst ledipasvir/sofosbuvir recipients, the NS5B S28T RAV (along with NS5A L31M, Y93H and Q30L substitutions) was detected in post baseline isolates at the time of treatment failure in one patient with HCV GT1 infection in a phase 2 trial [4]. At the time of virological failure, the NS5B S28T variant was also detected in isolates from one of 17 patients with HCV GT3 infection, one of three patients with HCV GT4 infection, and one of one patient with GT5 or 6 infections [4].
No cross-resistance was seen between ledipasvir and sofosbuvir, with ledipasvir fully active against the NS5B S28T variant and all ledipasvir resistance-associated NS5A substitutions fully susceptible to sofosbuvir [4]. Ledipasvir and sofosbuvir were fully active against RAVs associated with other DAAs with different mechanisms of action, including NS5B non-nucleoside inhibitors and NS3 protease inhibitors [4].
In analyses (typically pooled data [11,12,13,14]) of clinical trials in patients with HCV GT1 [11,12,13,14,15] or 2 [11] infections, ledipasvir/sofosbuvir was associated with high SVR rates, irrespective of the presence of NS5A, NS5B and/or NS3 RAVs at baseline. In the largest analysis of phase 2 and 3 trials in patients receiving ledipasvir/sofosbuvir (± ribavirin) for 6, 8, 12 or 24 weeks, 16% of patients (338/2108) had detectable NS5A RAVs at baseline and 2.5% (43/1692) had NS5B RAVs, with 93.5 and 100% of those with NS5A or NS5B RAVs achieving a SVR12 [13]. Of the 53.2% of patients (141/265) previously treated with protease inhibitors who had NS3 RAVs at baseline, 98.6% achieved a SVR12 following ledipasvir/sofosbuvir. In patients with NS5A RAVs at baseline, although high, the SVR12 rate was significantly lower than in patients who did not have NS5A RAVs at baseline (93.5 vs. 98.4%; p < 0.001), with this significant difference mainly reflecting SVR12 rates in patients with or without HCV GT1a NS5A RAVs (SVR12 92.3 vs. 98.3%; p < 0.001) rather than patients with or without HCV GT1b NS5A RAVs (SVR12 96.5 vs. 98.6%). The impact of NS5A RAVs on SVR12 rates was largely overcome by extending the duration of ledipasvir/sofosbuvir treatment and/or adding ribavirin to the regimen. After 8 weeks of ledipasvir/sofosbuvir, there was a significant reduction in the SVR12 rate in patients with NS5A RAVs exhibiting a > 100-fold resistance to ledipasvir versus that in patients without NS5A RAVs at baseline (82.8 vs. 96.4%; p = 0.011), whereas there were no between-group differences (BGDs) in SVR12 rates after 12 (95.7 vs. 99.5%) or 24 (96.2 vs. 99.4%) weeks’ treatment [13].
3 Pharmacokinetic Properties
Following oral administration of ledipasvir/sofosbuvir in patients with HCV infection, median peak plasma concentrations of ledipasvir, sofosbuvir and GS-331007 (predominant circulating metabolite; lacks activity against HCV) were reached at 4, ≈ 1 and 4 h postdose, respectively [4]. Exposure to ledipasvir was dose proportional over a range of 3–100 mg, with exposures to sofosbuvir and GS-331007 dose proportional over a range of 200–400 mg. Respective plasma protein binding of ledipasvir and sofosbuvir was > 99.8 and 61–65%; GS-331007 exhibits minimal plasma protein binding [4].
Ledipasvir undergoes slow oxidative metabolism, with systemic exposure almost exclusively due to the parent drug (accounts for > 98% of a radiolabeled dose) [4]. Sofosbuvir undergoes extensive hepatic metabolism to the active metabolite GS-461203 (not detected), which is further metabolized to the inactive metabolite GS-331007. Following administration of ledipasvir/sofosbuvir, GS-331007 accounted for ≈ 85% of total systemic exposure [4].
Primary elimination routes were biliary excretion for ledipasvir and renal clearance for GS-331007 [4]. Approximately 86% of a radiolabelled dose of ledipasvir was recovered in the faeces and, with radiolabelled sofosbuvir, ≈ 80 and 14% of the radioactive dose was recovered in the urine and faeces. The respective median terminal elimination half-lives of ledipasvir, sofosbuvir and GS-331007 were 4.7, 0.5 and 27 h [4].
The pharmacokinetics of ledipasvir, sofosbuvir and GS-331007 were not altered to a clinically relevant extent based on race, gender, age or bodyweight [4]. No dosage adjustments of ledipasvir/sofosbuvir are required in patients with mild or moderate renal impairment or mild, moderate or severe hepatic impairment [4]. Although the pharmacokinetics of ledipasvir were not altered to a clinically relevant extent in patients with severe renal impairment [estimated glomerular filtration rate (eGFR) < 30 mL/min], exposure to GS-331007 was increased in patients with severe renal impairment or end-stage renal disease [5, 16], meaning that ledipasvir/sofosbuvir dosage recommendations cannot be made in these patient groups [4].
Ledipasvir and sofosbuvir, but not GS-331007, are substrates for P-glycoprotein (P-gp) and breast cancer resistance protein (BRCP), although neither agent is a substrate for various other transporters or for CYP450 and/or uridine diphosphate glucuronyltransferase (UGT) 1A1 enzymes [4]. No clinically significant drug interactions with ledipasvir/sofosbuvir mediated by CYP50 or UGT1A1 enzymes are expected to occur. There is a potential for clinically relevant drug interactions between ledipasvir and/or sofosbuvir and potent P-gp inducers (e.g. rifampicin, rifabutin, St. John’s wort, carbamazepine, phenobarbital and phenytoin), with concomitant use of these agents contraindicated. Coadministration of ledipasvir/sofosbuvir with moderate P-gp inducers (e.g. oxcarbazepine) is not recommended [4]. Local prescribing information should be consulted for detailed information regarding potentially relevant drug interactions with ledipasvir/sofosbuvir.
4 Therapeutic Efficacy
In clinical trials and real-world studies discussed in this section, the dosage of ledipasvir/sofosbuvir was 90/400 mg once daily. When recommended, the ribavirin dosage was based on bodyweight.
4.1 In Adults with HCV Genotype 1 Infection
4.1.1 In Clinical Trials
The efficacy of ledipasvir/sofosbuvir (± ribavirin) in treatment-naive [8, 10] or -experienced [9] adults with chronic HCV GT1 infection was firmly established in the pivotal phase 3 ION-1 [8], -2 [9] and -3 [10] trials, and in a Japanese phase 3 trial [17] (Table 1), all of which have been reviewed in detail [6]. Participants in ION-2 had not had a sustained virological response with peginterferon (PegIFN)-α plus ribavirin or an NS3/4A protease inhibitor in combination with this regimen [9]. In ION-1 [8] and -2 [9], ≈ 20% of enrolled patients had compensated cirrhosis, with ION-3 enrolling patients without cirrhosis [10]. Baseline HCV RNA levels ranged from 6.3 to 6.6 log10 IU/mL in ION trials; in terms of factors traditionally considered predictive of poor outcome, 12–18% of patients were Black, 67–80% had GT1a infection and 70–88% had non-CC IL28B GT [8,9,10].
Ledipasvir/sofosbuvir was associated with high SVR12 rates (≥ 93%) in both treatment-naive and -experienced patients, irrespective of the duration of treatment (8, 12 or 24 weeks) or whether it was administered with or without ribavirin (Table 1) [8,9,10, 17]. SVR12 rates were significantly higher than specified historical response rates in all treatment groups in the ION trials [8,9,10] and, where evaluated, in treatment-naive patients without cirrhosis in the Japanese trial [17] (Table 1). In ION-3, the SVR12 rate in the 8-week ledipasvir/sofosbuvir group was noninferior to those of the 12-week ledipasvir/sofosbuvir and 8-week ledipasvir/sofosbuvir (+ ribavirin) groups (Table 1) [10]. These results support the use of a shorter duration (8 weeks) ledipasvir/sofosbuvir regimen in treatment-naive patients with HCV GT1 infection without cirrhosis (Sect. 6). In a post hoc analysis, patients with a baseline HCV RNA level of < 6 million IU/ml achieved similar SVR12 rates after 8 and 12 weeks’ ledipasvir/sofosbuvir (97 vs. 96%) [4], with no BGDs in SVR12 rates in this population based on IL28B genotype or gender [18].
Very few participants in phase 3 trials experienced virological breakthrough during or relapse after ledipasvir/sofosbuvir treatment [8,9,10, 17]. In ION-1, two patients (< 1%) experienced virological relapse by week 4 and 12 post treatment, with another patient experiencing virological breakthrough (Y93H variant detected), most likely reflecting nonadherence to treatment [8]. In ION-2, virological relapse occurred between week 4 and 12 post treatment (91% of relapses occurred by week 4) in 6 and 4% of patients in the 12-week ledipasvir/sofosbuvir and ledipasvir/sofosbuvir (+ ribavirin) groups (vs. no patients with the 24-week regimens) [9]. One patient in the 12-week ledipasvir/sofosbuvir group experienced virological breakthrough, with nonadherence to treatment suspected [9]. In ION-3, no patients in the 8-week ledipasvir/sofosbuvir, 8-week ledipasvir/sofosbuvir (+ ribavirin) or 12-week ledipasvir/sofosbuvir groups experienced virological breakthrough during treatment, with corresponding post treatment virological relapse rates of 5, 4 and 1% [10]. One patient in the Japanese trial experienced virological relapse 4 weeks post treatment, with pill counts indicating their strong adherence to treatment [17].
In treatment-naive patients, high SVR12 rates were seen across various subgroups of patients, including in patients with characteristics historically associated with a poor response, and were generally consistent with those in the overall population [8, 10]. In intent-to-treat (ITT) analyses of ION-1, SVR12 rates across treatment groups were 97–100% in patients with cirrhosis, 99–100% in patients with chronic HCV GT1a infection, 99–100% in those with non-CC IL28B and 94–100% in Black patients [8]. A retrospective pooled analysis of ION trials also showed similar SVR12 rates in Black and non-Black patients [19]. Pooled data from the ION and Japanese phase 3 trials also indicated that the SVR12 rate was similar in patients aged < 65 years to that in patients aged ≥ 65 years (97 and 98%; n = 2029 and 264) [20]. A post hoc pooled analysis of seven phase 2 and 3 randomized controlled trials (RCTs) also indicated that SVR12 rates with ledipasvir/sofosbuvir regimens were similar, irrespective of treatment duration, whether patients were treatment-naive or -experienced, or whether or not patients received ribavirin [21].
In a pooled analysis of ION trials, ledipasvir/sofosbuvir recipients (n = 1080) experienced significant (p < 0.0001) improvements from baseline in patient-reported outcomes (PROs) during 8–24 weeks’ treatment, whereas those receiving ledipasvir/sofosbuvir (+ ribavirin; n = 872) experienced significant (p < 0.0001) declines in PROs [22]. In the ledipasvir/sofosbuvir group, improvements in PROs occurred at the same time as early viral suppression, were significant (p < 0.001 vs. baseline) as early as 2 weeks after initiating treatment and were maximized at the end of treatment. In multivariate analyses, ribavirin treatment was an independent predictor of PRO impairment (p < 0.0001), with some domains of PRO measures favouring ledipasvir/sofosbuvir over ledipasvir/sofosbuvir (+ ribavirin) from 2 weeks onwards [22].
In open-label, multicentre studies (n = 51–100), high overall SVR12 rates (70–100%) were achieved after 12 or 24 weeks of ledipasvir/sofosbuvir (± ribavirin) in patients with HCV GT1 infection who had failed prior sofosbuvir-based [23,24,25] or NS5A inhibitor-based [26] regimens. In the largest study [n = 41 patients who were NS5A treatment-experienced and 59 who had failed prior sofosbuvir (± PegIFN)-based treatment; 41% of whom had cirrhosis], the overall SVR12 rate was 86% after 12 or 24 weeks of ledipasvir/sofosbuvir (± ribavirin) [24]. At baseline, 37 patients had NS5A RAVs and four had NS5B RAVs, with no patients having the S28T RAV. Virtually all (92%) of the 13 patients who experienced virological failure had NS5A RAV(s), with new treatment-emergent NS5A and NS5B RAV(s) occurring in two and six patients (three patients with NS5B RAVs had the S28T RAV) [24].
4.1.2 In the Real-World Setting
Several large (n > 600; range 634–14,366 ledipasvir/sofosbuvir ± ribavirin recipients), real-world observational and registry studies in patients with HCV GT1 infection have confirmed the efficacy of ledipasvir/sofosbuvir regimens in the clinical practice setting, including a pooled analysis of German and US registries [27], and in US registries (ERCHIVES [28]; HCV-TARGET Study [29]; Trio Health study [30,31,32]; Veterans Affairs [33,34,35]). SVR rates reported in these studies were consistent with those in clinical trials and, in general, across a broad spectrum of patients [27, 29,30,31,32,33,34,35]. In Veteran Affairs registry studies, SVR was defined as having HCV RNA levels below the limit of quantification (LOQ) after the end of treatment, including in ≥ 1 test ≥ 10 weeks after the end of treatment [33,34,35]. In ERCHIVES (82% of 974 ledipasvir/sofosbuvir recipients had HCV GT1 infection), SVR12 rates with ledipasvir/sofosbuvir were high, irrespective of whether patient adherence (based on the percentage of doses prescribed) was 50–59% (SVR12 91%), 60–69% (89%), 70–99% (100%) or 100% (94%) [28]
Consistent with results in ION-3 [10] (Sect. 4.1.1), data from real-world registries [27, 29, 32,33,34] and a meta-analysis of six real-world cohorts [27] supported the use of a shortened regimen of ledipasvir/sofosbuvir (8 vs. 12-week) in patients with HCV GT1 infection. For example, in a real-world observational cohort study, respective SVR rates in patients completing 8 or 12 weeks’ ledipasvir/sofosbuvir or 12 weeks of ledipasvir/sofosbuvir (+ ribavirin) were 92, 95 and 92% (n = 1333, 2615 and 1120) [34].
Several factors were predictive of a lower likelihood of achieving a SVR following 8 or 12 weeks’ ledipasvir/sofosbuvir (± ribavirin) in patients with HCV GT1 infection (n = 14,366) in an observational, ITT cohort study [35]. Negative predictive factors (all p < 0.001) for achieving an SVR with ledipasvir/sofosbuvir (± ribavirin) were African-American ethnicity [odds ratio (OR) 0.80], a body mass index of < 25 (OR 0.77) or ≥ 30 kg/m2 (OR 0.77), concomitant use of a proton pump inhibitor (PPI; OR 0.81), presence of decompensated liver disease (OR 0.58) and a Fibrosis-4 (FIB-4) score of > 3.25 (OR 0.60). In the ombitasvir/paritaprevir/ritonavir plus dasabuvir (± ribavirin) cohort (n = 4688), a FIB-4 score of > 3.25 was a negative predictor for SVR (OR 0.72; p < 0.001). A 4-week on-treatment HCV RNA level of ≥ 15 IU/mL was negatively predictive (p < 0.001) of SVR post-treatment with ledipasvir/sofosbuvir (± ribavirin) [OR 0.49] or ombitasvir/paritaprevir/ritonavir plus dasabuvir (± ribavirin) [OR 0.38] [35].
Overall, ledipasvir/sofosbuvir (± ribavirin) recipients were significantly more likely to achieve an SVR than ombitasvir/paritaprevir/ritonavir plus dasabuvir (± ribavirin) recipients (OR 0.70; p < 0.001) [35]. SVR rates in the ledipasvir/sofosbuvir (n = 10,720) and ledipasvir/sofosbuvir (+ ribavirin; n = 3646) groups were 91 and 90%, and those in ombitasvir/paritaprevir/ritonavir plus dasabuvir (+ ribavirin) [n = 3832] and ombitasvir/paritaprevir/ritonavir plus dasabuvir (n = 1306) recipients were 88 and 92%.
Equivocal results regarding the impact of concomitant PPI use on the antiviral efficacy of ledipasvir/sofosbuvir were seen in two large US registry studies [31, 35]. In the larger study, albeit SVR rates were high in all ledipasvir/sofosbuvir groups (8 or 12 weeks’ treatment), these rates were significantly higher in patients who were not taking a PPI than in those taking a PPI with the pooled 8- and 12-week ledipasvir/sofosbuvir regimens (92 vs. 90%; p < 0.001; n = 7969 and 2751) and with the ledipasvir/sofosbuvir plus ribavirin regimen (91 vs. 88%; p = 0.008; n = 2468 and 1178) [35]. In the other study, the concomitant use of PPIs with ledipasvir/sofosbuvir (± ribavirin) was not associated with a reduction in SVR12 rates, irrespective of the dose or frequency of PPI use, based on a propensity-matched analysis [31].
4.2 In Adolescents with HCV Genotype 1 Infection
The efficacy of ledipasvir/sofosbuvir in treatment-naive (n = 80) and -experienced (n = 20) adolescents aged 12–17 years (median age 15 years) with HCV GT1 infection was investigated in a 12-week noncomparative, multinational, phase 2 trial [36]. At baseline, the mean HCV RNA level was 6.0 IU/mL, 84% of patients were infected via perinatal transmission, 76% had a non-CC IL28B genotype, 81% had a HCV GT1a infection, and 1% had and 42% did not have cirrhosis (cirrhosis status not known in other patients).
The overall SVR12 rate was 98% (primary endpoint), with 98 and 100% of treatment-naive and -experienced patients achieving this endpoint [36]. The two patients who did not achieve a SVR12 were lost to follow-up. No evaluable patients experienced virological failure during treatment or relapse after treatment [36].
In exploratory endpoint analyses, caregiver-assessed health-related quality of life (HRQOL) scores significantly (p < 0.05) improved from baseline after 12 weeks of ledipasvir/sofosbuvir (average improvement in the physical, emotional, social and school functioning domains of the PedsQL-4.0-SF15 measure of 5.7–8.1 points), with minimal non-significant changes over this period for patient-assessed scores (average change − 1.49 to + 2.72) [37]. At baseline, compared with the healthy adolescent population norm, adolescents with HCV infection had significantly (p < 0.05) lower total HRQOL and school functioning domain scores for both patient and caregiver assessments, with a significant difference between patients and caregivers in total scores (79.6 vs. 73.0; p = 0.0038) but not school functioning scores. Baseline scores for the physical and emotional functioning domains were also significantly (p ≤ 0.003) higher in patient’ than in caregiver’ assessments, with caregiver-assessed emotional functioning scores also significantly (p < 0.05) higher than the healthy adolescent population norm. No patients experienced a treatment-related deterioration in HRQOL during ledipasvir/sofosbuvir treatment or post treatment after up to 24 weeks follow-up, irrespective of whether these were assessed by patients or caregivers [37].
4.3 In Other Adult Populations
4.3.1 With Other HCV Genotype Infections
The efficacy of ledipasvir/sofosbuvir (± ribavirin) in patients with HCV GT2 [38], GT3 [39, 40], GT4 [41, 42], GT5 [43] and/or GT6 [40, 44] infection was evaluated in open-label, multicentre, phase 2 trials, and in a Japanese, open-label, phase 3 trial in patients with HCV GT2 infection [45]. Real-world data from the French cohort ANRS CO22 HEPATHER study (n = 1808) support the efficacy of ledipasvir/sofosbuvir across all HCV GTs, with an overall SVR12 rate of 95.6% and high SVR12 rates irrespective of the HCV GT, presence or absence of cirrhosis or whether patients were treatment-naive or -experienced [46].
In patients with chronic HCV GT2 infection, ledipasvir/sofosbuvir treatment for 12 weeks was associated with high SVR12 rates (96%) [38, 45]. In the phase 3 trial, ledipasvir/sofosbuvir was noninferior to sofosbuvir plus ribavirin treatment in terms of the primary outcome of SVR12 rates (96 vs. 95%; n = 106 and 108) [45]. In LEPTON, 25 of 26 patients (96%) achieved a SVR12 after 12 weeks’ ledipasvir/sofosbuvir, with the remaining patient withdrawing from the study after a single dose of study drug [38].
In two trials in treatment-naive [39, 40] and/or -experienced [40] patients with HCV GT3 infection, 12 weeks of ledipasvir/sofosbuvir (± ribavirin) provided effective antiviral efficacy in the majority of patients. SVR12 rates (primary endpoint) were 89% (99/111 patients) [39] and 100% (26/26) [40] in treatment-naive patients after ledipasvir/sofosbuvir (+ ribavirin) therapy, and 64% (16/25) after ledipasvir/sofosbuvir alone [40]. In treatment-experienced patients, 82% of patients (41/50) achieved a SVR12 after ledipasvir/sofosbuvir (+ ribavirin) therapy [40]. In patients with or without cirrhosis, respective SVR12 rates were 73 and 89% in treatment-experienced patients. At baseline, cirrhosis was present in 44% of treatment-experienced patients and ≈ 20% of treatment-naive patients (all of whom achieved SVR12) [40]. These data are supported by real-world evidence from the German DHC-R study [47] and a Georgia registry database [48], with SVR12 rates after ledipasvir/sofosbuvir (± ribavirin) of 97.6% (244/250 patients) in the Georgia study [48] and 74% (61/82 ITT patients) in the DHC-R study [47].
In treatment-naive and -experienced patients with HCV GT4 infection, high SVR12 rates (93 [41] and 95% [42]) were seen after 12 weeks of ledipasvir/sofosbuvir therapy in two open-label, phase 2 trials. In the larger trial, SVR12 rates were similar in treatment-naive and -experienced patients (SVR12 91 and 95%; n = 22/group; 23% of patients had compensated cirrhosis) [41]. The three patients who did not achieve SVR12 experienced virological relapse within 4 weeks of completing treatment; these patients had a baseline HCV RNA level of ≥ 800,000 IU/mL, a non-CC IL28B GT and pre-treatment NS5A RAVs [41]. These data are supported by evidence from clinical trials [49,50,51] (Sect. 4.3.2) and real world studies (Sect. 4.3.3) in which some participants had HCV GT4 infection.
Ledipasvir/sofosbuvir for 12 weeks provided effective antiviral efficacy in treatment-naive (n = 21) and -experienced (n = 20) patients with HCV GT5 infection, with SVR12 rates of 95% in both groups (primary endpoint) [43]. SVR12 rates in patients with (n = 9) or without cirrhosis (n = 32) were 89 and 97% [43].
In patients with HCV GT6 infection, SVR12 was achieved by 95% of patients after 8 weeks’ ledipasvir/sofosbuvir in treatment-naive patients without cirrhosis (n = 20) or 12 weeks’ ledipasvir/sofosbuvir (n = 40) in treatment-naive patients with cirrhosis or treatment-experienced patients (± cirrhosis) [44]. A SVR was maintained at 24 weeks’ follow-up in all patients who achieved SVR12. At baseline in the 12-week group, two patients had decompensated cirrhosis, three had hepatocellular carcinoma (HCC) and 14 were treatment experienced; four patients in this group did not have prior treatment-failure or cirrhosis [44]. These data are supported by evidence from a trial in treatment-naive patients with HCV GT6 infection in which the SVR12 rate was 96% (24/25 patients) after 12 weeks’ ledipasvir/sofosbuvir [40]. The patient who did not achieve SVR12 experienced virological relapse 4 weeks after discontinuing treatment early at week 8 [40].
4.3.2 Patients with HIV Co-Infection
The efficacy of ledipasvir/sofosbuvir in treatment-naive and -experienced patients co-infected with HCV GT1 or 4 and HIV-1 was investigated in the 12-week, noncomparative, multicentre, phase 3 ION-4 trial (n = 335) [51]. Patients were receiving stable antiretroviral therapy with emtricitabine/tenofovir disoproxil fumarate (+ efavirenz, rilpivirine or raltegravir). At baseline, 75, 23 and 2% of patients had chronic HCV GT1a, 1b or 4 infections, respectively, 20% had compensated cirrhosis and 55% had not responded to prior anti-HCV treatment. The primary endpoint was the HCV SVR12 rate [51].
HCV SVR12 rates were high in the overall population (96%) and in all subgroups of patients (SVR12 85–100%), including in treatment-naive (SVR12 95%) and -experienced patients (97%), patients without (97%) or with cirrhosis (94%), Black (90%) and non-Black (99.5%) patients, and in patients with HCV GT1a (96%), 1b (96%) and 4 (100%) infections [51]. Of the 13 patients in the overall population who failed to achieve SVR12, one patient died after 4 weeks’ treatment, two had HCV breakthrough suspected to be due to nonadherence to study drug and ten had HCV relapse. All patients who experienced HCV relapse were of Black ethnicity, with seven patients having the TT allele in the IL28B gene and eight previously treated with efavirenz. No patients had confirmed HIV-1 virological rebound [51].
In a retreatment study that enrolled 9 of 10 patients who experienced HCV relapse in ION-4, 89% of patients achieved SVR12 after retreatment (within 60 days of relapse) with ledipasvir/sofosbuvir (+ ribavirin) for 12 weeks [52]. The other patient experienced HCV relapse 4 weeks after retreatment [52].
In ION-4, ledipasvir/sofosbuvir recipients experienced significant improvements in PROs during treatment, based on FACIT-F, SF-36 physical component and CLDQ-HCV scores [53]. These benefits in PROs during ledipasvir/sofosbuvir treatment were maintained in patients achieving a SVR12. Conversely, based on historical data from the PHOTON-1 trial, sofosbuvir plus ribavirin treatment was associated with a significant (p < 0.001) deterioration in PRO scores for FACIT-F and SF-36 physical functioning [53].
4.3.3 Patients with Cirrhosis and/or Pre-Transplant or Post-Liver Transplant
In adults with chronic HCV GT1 infection [49, 50, 54] (or GT4 [49, 50]) and cirrhosis [49, 50, 54] or who had undergone a liver transplant [49, 50], ledipasvir/sofosbuvir (± ribavirin) treatment for 12 or 24 weeks resulted in high SVR12 rates (primary outcome) in open-label (SOLAR-1 [50] and SOLAR-2 [49]) or double-blind (SIRIUS [54]), multicentre, phase 2 trials. These data are supported by evidence from studies in the real-world setting in patients with HCV infection and cirrhosis (n = 91 [55] and 424 [56]). Further evidence comes from a pairwise meta-analysis of seven RCTs in adults with HCV GT1 infection with cirrhosis (n = 2560–2566) in which there were no significant differences in rapid virological response, SVR4 and SVR12 rates or the rate of virological relapse between ledipasvir/sofosbuvir and ledipasvir/sofosbuvir (+ ribavirin) treatment (I2 = 0 for all endpoints) [57].
Eligible patients in SIRIUS had compensated cirrhosis and were randomized to 12 weeks of placebo and then 12 weeks of ledipasvir/sofosbuvir plus ribavirin (n = 77) or to 24 weeks of ledipasvir/sofosbuvir plus placebo (n =78; placebo group) [54]. SVR12 rates in the ledipasvir/sofosbuvir (+ ribavirin) and placebo groups were 96 and 97% [54]. Patients in both groups who achieved an SVR12 experienced significant (p < 0.05) improvements in PROs, including in general health, social functioning, emotional well-being and most domains of CLDQ-HCV [58]. Significant (p < 0.05) improvements in most PROs were evident after 12 weeks of ledipasvir/sofosbuvir (+ ribavirin), whereas placebo recipients only experienced a significant (p < 0.05) improvement in the worry domain of CLDQ-HCV at this timepoint. At the end of treatment (week 24), compared with ledipasvir/sofosbuvir (+ ribavirin) therapy, patients receiving ledipasvir/sofosbuvir (+ placebo) experienced significantly (p < 0.05) greater improvements in emotional and systemic CLDQ-HCV domains and in physical functioning and mental component summary scores [58].
In SOLAR-2, eligible patients were pre-transplant and had decompensated cirrhosis with Child-Turcotte-Pugh Class (CTC) B or C hepatic impairment (n = 56 and 51) [cohort A] or were post-liver transplant patients who had no cirrhosis (n = 101), CTC A, B or C hepatic impairment (n = 67, 45 and 8, respectively) or fibrosing cholestatic hepatitis (n = 5) [cohort B] [49]. Participants were randomized to 12 or 24 weeks’ ledipasvir/sofosbuvir (+ ribavirin). In the overall population, 89 and 11% of patients had HCV GT1 and GT4 infection. In patients with GT1 infection, SVR12 rates ranged from 50% in a group with two patients to 78–100% in groups with 2–45 patients. In those with GT4 infection, the number of patients/group was small, with SVR12 rates ranging from 0% (one patient/group) to 50–100% (1–7 patients/group) [49]. These data are supported by evidence from the similarly designed SOLAR-1 trial [50].
SVR12 rates were also high in pre-transplant recipients with cirrhosis [59] and/or post-liver transplant recipients [59, 60] treated with ledipasvir/sofosbuvir (± ribavirin) in prospective [59] or retrospective [60], observational, multicentre, real-world studies. In the retrospective study, most patients had HCV GT1 (96% of patients) [60] and, in the prospective study, all patients had HCV GT4 infection [59]. In patients with chronic HCV GT4 infection, after 12 or 24 weeks’ ledipasvir/sofosbuvir (± ribavirin), SVR12 rates in pre- and post-transplant patients (n = 61 and 50) were 92 and 86% [59].
The longitudinal HCV-TARGET real-world study provides further support for the efficacy of ledipasvir/sofosbuvir (± ribavirin) in liver (n = 347), kidney (n = 60) and dual liver kidney transplant (n = 36) recipients [61]. The majority (87%) of patients had HCV GT1 infection, 42% had cirrhosis, 54% had failed prior antiviral therapy, and 85% received ledipasvir/sofosbuvir (± ribavirin), 9% sofosbuvir plus daclatasvir (± ribavirin) and 6% ombitasvir/paritaprevir/ritonavir plus dasabuvir (± ribavirin). The overall SVR12 rate across all treatment regimens was 96%, with respective SVR12 rates in liver, kidney and dual transplant recipients of 97, 95 and 91% [61].
4.3.4 Post Kidney Transplant
The efficacy of ledipasvir/sofosbuvir (12 or 24 weeks’ treatment) in kidney transplant recipients with chronic HCV GT1 (91% of patients) or GT4 (9%) infection was evaluated in an open-label, multinational, phase 2 trial (n = 114) [62]. At baseline, 31% of patients were treatment-experienced, 85% had no cirrhosis, 15% had compensated cirrhosis and the median eGFR was 56 mL/min. All patients in the 12- and 24-week groups achieved SVR12 (primary outcome), including a patient in the 12-week group who discontinued study drug at week 4. With the exception of one patient who discontinued the study prior to the post-treatment week 24 visit for reasons not considered to be treatment related, all patients achieved SVR24 [62].
4.3.5 Patients with Inherited Blood Disorders
Based on results from the 12-week, exploratory, multicentre, phase 2 ELECTRON-1 trial in patients with chronic HCV GT1 infection and transfusion-dependent thalassaemia (all 14 patients achieved SVR12) [63], the efficacy of 12 or 24 weeks’ ledipasvir/sofosbuvir was evaluated in patients with chronic HCV GT1 or 4 infection with an inherited blood disorder in an open-label, multicentre, phase 2b trial [64]. Consequent to emerging results from ION-2, the protocol was amended to extend ledipasvir/sofosbuvir therapy from 12 to 24 weeks in treatment-experienced patients with HCV GT1 infection and cirrhosis who had not yet completed treatment. At baseline, 99% of patients had HCV GT1 infection and one patient had HCV GT4 infection, 63% had haemophilia A, 28% had haemophilia B, 10% had von Willebrand disease, 42% were treatment-experienced and 32% had cirrhosis. The SVR12 rates after 12 (n = 99) and 24 (n = 5) weeks’ ledipasvir/sofosbuvir were 99 and 100% (primary outcome), with the remaining patient in the 12-week group lost to follow-up; 8 weeks after initiating treatment, this patient had a HCV RNA level below the LOQ [64].
These data are supported by a 12-week, open-label, historically-controlled, multicentre trial in patients with HCV GT1 or 4 infection and transfusion-dependent thalassaemia (n = 100); at baseline, 16% of patients had cirrhosis and 37% were treatment-naive [65]. High SVR12 rates were achieved (98% in the overall population) after 12 weeks of ledipasvir/sofosbuvir, irrespective of whether patients had cirrhosis at baseline or had experienced treatment failure on prior antiviral therapy [65].
5 Safety and Tolerability
Ledipasvir/sofosbuvir (± ribavirin) was generally well tolerated in the clinical trial and real-world settings in treatment-naive and -experienced adult patients with HCV infections, based on studies discussed in Sect. 4. Discussion focuses on pooled analyses of the ION trials in patients with HCV GT1 infections [4, 66, 67]. The tolerability profile of patients with other HCV GT infections was generally similar to that in patients with HCV GT1 infection. Adverse reactions seen in the phase 2 trial in adolescents (aged 12 to < 18 years) (Sect. 4.2) were consistent with those seen in clinical trials of ledipasvir/sofosbuvir in adults [4].
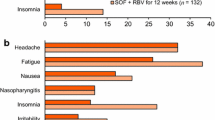
In a pooled analysis of ION trials (8–24 weeks’ treatment), treatment-emergent adverse events (TEAEs) occurred in 74% of 1080 ledipasvir/sofosbuvir and 85% of 872 ledipasvir/sofosbuvir (+ ribavirin) recipients, with most TEAEs of mild to moderate severity [66]. Very few patients receiving ledipasvir/sofosbuvir for 8, 12 or 24 weeks (n = 215, 539 and 326, respectively) permanently discontinued treatment because of TEAEs (0, < 1 and 1%, respectively), with corresponding rates of discontinuation in the 8-, 12- and 24-week ledipasvir/sofosbuvir (+ ribavirin) groups of < 1, 0 and 2% (n = 216, 328 and 328) [4]. The most common treatment-related adverse events (TRAEs) occurring in ≥ 5% of patients in the 8-, 12- and 24-week ledipasvir/sofosbuvir groups were fatigue (16, 13 and 18%, respectively), headache (11, 14 and 17%), nausea (6, 7 and 9%), diarrhoea (4, 3 and 7%) and insomnia (3, 5 and 6%), most of which were of grade 1 severity [67]. Treatment-emergent serious adverse events (SAEs) occurred in 2% of ledipasvir/sofosbuvir recipients and 3% of ledipasvir/sofosbuvir (+ ribavirin) recipients; of which, ≤ 0.4% were considered treatment-related [66].
The most frequent TRAEs occurring during ledipasvir/sofosbuvir (+ ribavirin) were consistent with the known safety profile of ribavirin, with no increase in the frequency or severity of these expected adverse drug reactions [4]. In a pooled analysis, the addition of concomitant ribavirin to ledipasvir/sofosbuvir therapy was associated with an increased incidence of TRAEs (71 vs. 45% with ledipasvir/sofosbuvir alone) and laboratory abnormalities (e.g. anaemia, increased total bilirubin levels and decreased lymphocytes, reflecting ribavirin-induced haemolysis), irrespective of the presence or absence of cirrhosis [66]. Concomitant ribavirin was associated with more frequent adverse events leading to dose modifications/discontinuations than ledipasvir/sofosbuvir alone, irrespective of the presence (20 vs. < 1% of patients; n = 113 and 111) or absence (13 vs. < 1%; n = 759 and 969) of cirrhosis. Medical intervention was required by approximately twice as many ledipasvir/sofosbuvir (+ ribavirin) as ledipasvir/sofosbuvir recipients, irrespective of whether patients were treated for 8 (22 vs. 11%), 12 (30 vs. 16%) or 24 weeks (36 vs. 22%) [66]. Pairwise meta-analyses of RCTs were consistent with these results, with the addition of ribavirin to ledipasvir/sofosbuvir associated with a worsening safety profile compared with ledipasvir/sofosbuvir alone, including an increased risk of any adverse event [risk ratio (RR) 0.89; 95% CI 0.85–0.94; p < 0.00001; I2 19%; n = 7 RCTs], anaemia (RR 0.09; 95% CI 0.05–0.19; p < 0.00001; I26%; n = 6 RCTs) and rash events (RR 0.38; 95% CI 0.21–0.68; p = 0.001; I263%; n = 4 RCTs) [57].
In trials in adults with decompensated cirrhosis and/or who were awaiting liver transplant or post liver transplant (Sect. 4.3.3), ledipasvir/sofosbuvir (± ribavirin) was generally well tolerated. During ledipasvir/sofosbuvir (± ribavirin) treatment, no new adverse drug reactions were detected, with adverse events typical of those expected as clinical sequelae of advanced liver disease and/or transplantation or consistent with the known safety profile of ribavirin [4]. In SOLAR-1 and SOLAR-2, reductions in haemoglobin of < 10 g/dL and < 8.5 g/dL occurred in 39 and 13% of ledipasvir/sofosbuvir (+ ribavirin) recipients; ribavirin was discontinued in 15% of patients and 7% of liver transplant recipients required modification of their immunosuppressive regimens [4].
In a retrospective cohort study, patients with HCV infection and pre-existing renal dysfunction [i.e. creatinine clearance (CLCR) < 60 mL/min; n = 17] were more likely to experience worsening of kidney disease during 8 (n = 15), 12 (n = 67) or 24 (n = 8) weeks’ ledipasvir/sofosbuvir than patients with CLCR of ≥ 60 mL/min [68]. Of the patients with pre-existing renal dysfunction, 42% experienced a decline in CLCR during treatment, with univariate (p = 0.016) and multivariate (p = 0.04) analyses indicating that a baseline CLCR of < 60 mL/min was significantly associated with a worsening of renal function during ledipasvir/sofosbuvir therapy [68].
Cases of severe bradycardia and heart block have occurred in patients receiving concomitant ledipasvir/sofosbuvir plus amiodarone and/or other drugs that lower the heart rate [4]. Coadministration of amiodarone with ledipasvir/sofosbuvir is not recommended; in patients without alternative, viable treatment options, cardiac monitoring is recommended [4]. In thorough QT studies, the maximum recommended dose of sofosbuvir (400 mg) and supratherapeutic doses of ledipasvir (120 mg) and sofosbuvir (1200 mg) did not prolong the QTc interval to a clinically relevant extent [4, 5, 69], and were not associated with clinically significant adverse events, ECG abnormalities or changes in physical examinations or vital signs [5].
6 Dosage and Administration
In the EU [4], ledipasvir/sofosbuvir is indicated for the treatment of CHC in adults and in adolescents aged 12 to < 18 years. The recommended dosage of ledipasvir/sofosbuvir is 90/400 mg once daily taken without regard to food, with concomitant ribavirin recommended in specified patient groups [4]. The duration of ledipasvir/sofosbuvir treatment varies in specific HCV patient populations (typically 12 or 24 weeks), based on several factors such as the HCV GT and presence or absence of liver cirrhosis; in treatment-naive patients with GT1, 4, 5 or 6 CHC without cirrhosis, 8 weeks’ treatment may be considered [4]. Local prescribing information should be consulted for detailed information, including duration of treatment, weight-based ribavirin dosages, contraindications, warnings, precautions and use in specific populations.
7 Place of Ledipasvir/Sofosbuvir in the Management of Chronic Hepatitis C
Globally, CHC affects an estimated 71 million individuals and remains a leading causes of chronic liver disease and HCC [70]. CHC treatment targets attaining a cure (i.e. achievement of an SVR12 or SVR24; > 99% of patients achieving an SVR are cured) to prevent the long-term complications of HCV-related liver and extrahepatic diseases, including hepatic necroinflammation, fibrosis, cirrhosis, decompensation of cirrhosis, HCC, extrahepatic manifestations and death [71]. Given their efficacy, ease of use and safety profiles, DAA regimens are considered valuable treatment options for individual HCV GTs in recent 2016 EASL guidelines, with ledipasvir/sofosbuvir (± ribavirin) considered valuable for treating HCV GT1, 4, 5 and 6, and sofosbuvir/velpatasvir (± ribavirin) and sofosbuvir plus daclatasvir (± ribavirin) regimens considered valuable for treating all HCV GTs (i.e. GT1-6) [71]. The approval of the pangenotypic sofosbuvir/velpatasvir/voxilaprevir DAA regimen in the EU [72] (and elsewhere) is too recent for it to have been considered in EASL guidelines [71], a regimen that may be particularly beneficial in patients with HCV GT3 infection. Ultimately, the choice of DAA regimen needs to take several factors into account, including viral and host factors (e.g. HCV genotype; the presence of RAVs), patient characteristics (e.g. previous treatment history; comorbidities such as cirrhosis, liver disease) and the potential for drug-drug interactions, with numerous drug-drug interactions possible with HCV DAAs [71, 73]. Despite the significant advantages of DAA regimens over IFN-regimens, a major barrier to their potential to effectively eradicate CHC worldwide is their high acquisition costs [1, 73].
In general, HCV NS5B polymerase inhibitors have a high genetic barrier to resistance, whereas HCV NS5A inhibitors have a low-resistance barrier [3]. SVR12 rates were generally high after 12 or 24 weeks of ledipasvir/sofosbuvir treatment in adults with HCV GT1 infection who had failed prior sofosbuvir- or NS5A-based regimens (Sect. 4.1.1) and irrespective of the presence of NS5A, NS5B and/or NS3 RAVs at baseline (based on meta-analyses of RCTs, Sect. 2.1). No cross-resistance was seen between ledipasvir and sofosbuvir, with these drugs fully active against RAVs associated with other DAAs with different mechanisms of action such as NS5B non-nucleoside inhibitors and NS3 protease inhibitors (Sect. 2.1).
Extensive experience in the clinical trial and real-world settings has firmly established the antiviral efficacy (in terms of SVR12 rates) of ledipasvir/sofosbuvir (± ribavirin) in treatment-naive and -experienced adults with chronic HCV GT1 infection (Sect. 4.1), including in those with and without compensated cirrhosis (Sect. 4.1.1) and in patients co-infected with HIV (Sect. 4.3.2). In adults with HCV GT1 infection, SVR12 rates in the real world setting (Sect. 4.1.2) were consistent with those seen in clinical trials (Sect. 4.1.1), including the efficacy of a shorter duration of treatment (i.e. 8 weeks vs. typical duration of 12 or 24 weeks) in adults with chronic HCV GT1 infection without cirrhosis. Ledipasvir/sofosbuvir (± ribavirin) also provides high antiviral efficacy in treatment-naive and -experienced adolescents (aged ≥ 12 years) with chronic HCV GT1 infection (Sect. 4.2) and in adults with chronic HCV infections of other genotypes (GT 2–6) (Sect. 4.3.1), pre- and/or post-liver transplantation (Sect. 4.3.3), post kidney transplantation (Sect. 4.3.4), inherited blood disorders (Sect. 4.3.5), or failure after prior DAA and/or interferon therapy (Sect. 4.1.1).
Ledipasvir/sofosbuvir was generally well tolerated in treatment-naive and-experienced adult and adolescent patients with CHC in the clinical trial and real world settings, with most TEAEs of mild to moderate severity and very few patients discontinuing treatment because of these events (Sect. 5). As expected, the addition of ribavirin to ledipasvir/sofosbuvir treatment was associated with a worsening safety profile, reflecting ribavirin-related adverse events such as anaemia.
Thus, ledipasvir/sofosbuvir (± ribavirin) is a valuable effective and generally well tolerated treatment option in adolescent and adult patients with chronic HCV GT1, 4, 5 or 6 infection, including those with HIV co-infection or cirrhosis, with evidence also supporting its use in patients with chronic HCV GT2 or 3 infection.
Data Selection Ledipasvir/Sofosbuvir: 458 records
Duplicates removed | 71 |
Excluded during initial screening (e.g. press releases; news reports; not relevant drug/indication; preclinical study; reviews; case reports; nonrandomized trial) | 249 |
Excluded during writing (e.g. reviews; duplicate data; small patient number; nonrandomized/phase I/II trials) | 64 |
Cited efficacy/tolerability articles | 61 |
Cited articles not efficacy/tolerability | 12 |
Search Strategy: EMBASE, MEDLINE and PubMed from 2015 to present. Previous Adis Drug Evaluation published in 2015 was hand-searched for relevant data. Clinical trial registries/databases and websites were also searched for relevant data. Key words were Harvoni, Ledipasvir/sofosbuvir, LDV/SOF, GS-5885/GS-7977, Hepatitis C. Records were limited to those in English. Searches last updated 4 Jan 2018 | |
References
Younossi Z, Birerdinc A, Henry L. Hepatitis C infection: a multi-faceted systemic disease with clinical, patient-reported and economic consequences. J Hepatol. 2016;65:S109–19.
Asselah T, Boyer N, Saadoun D, et al. Direct-acting antivirals for the treatment of hepatitis C virus infection: optimizing current IFN-free treatment and future perspectives. Liver Int. 2016;36(Suppl S1):47–7.
Majumdar A, Kitson MT, Roberts SK. Treatment of hepatitis C in patients with cirrhosis: remaining challenges for direct-acting antiviral therapy. Drugs. 2015;75:823–34.
European Medicines Agency. Harvoni 90 mg/400 mg film-coated tablets: summary of product characteristics. 2017. http://www.ema.europa.eu. Accessed 2 Nov 2017.
German P, Mathias A, Brainard D, et al. Clinical pharmacokinetics and pharmacodynamics of ledipasvir/sofosbuvir, a fixed-dose combination tablet for the treatment of hepatitis C. Clin Pharmacokinet. 2016;55(11):1337–51.
Keating GM. Ledipasvir/sofosbuvir: a review of its use in chronic hepatitis C. Drugs. 2015;75(6):675–85.
Cheng G, Tian Y, Doehle B, et al. In vitro antiviral activity and resistance profile characterization of the hepatitis C virus NS5A inhibitor ledipasvir. Antimicrob Agents Chemother. 2016;60(3):1847–53.
Afdhal N, Zeuzem S, Kwo P, et al. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med. 2014;370(20):1889–98.
Afdhal N, Reddy KR, Nelson DR, et al. Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med. 2014;370(16):1483–93.
Kowdley KV, Gordon SC, Reddy KR, et al. Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N Engl J Med. 2014;370(20):1879–88.
Mizokami M, Dvory-Sobol H, Izumi N, et al. Resistance analyses of Japanese hepatitis co-infected patients receiving sofosbuvir or ledipasvir/sofosbuvir containing regimens in phase 3 studies. J Viral Hepat. 2016;23(10):780–8.
Wyles D, Dvory-Sobol H, Svarovskaia ES, et al. Post-treatment resistance analysis of hepatitis C virus from phase II and III clinical trials of ledipasvir/sofosbuvir. J Hepatol. 2017;66(4):703–10.
Sarrazin C, Dvory-Sobol H, Svarovskaia ES, et al. Prevalence of resistance-associated substitutions in HCV NS5A, NS5B, or NS3 and outcomes of treatment with ledipasvir and sofosbuvir. Gastroenterology. 2016;151(3):501-12.e1.
Zeuzem S, Mizokami M, Pianko S, et al. NS5A resistance-associated substitutions in patients with genotype 1 hepatitis C virus: prevalence and effect on treatment outcome. J Hepatol. 2017;66:910–8.
Kan H, Imamura M, Kawakami Y, et al. Emergence of drug resistance-associated variants and changes in serum lipid profiles in sofosbuvir plus ledipasvir-treated chronic hepatitis C patients. J Med Virol. 2017;89(11):1963–72.
Kirby BJ, Symonds WT, Kearney BP, et al. Pharmacokinetic, pharmacodynamic, and drug-interaction profile of the hepatitis C virus NS5B polymerase inhibitor sofosbuvir. Clin Pharmacokinet. 2015;54:677–90.
Mizokami M, Yokosuka O, Takehara T, et al. Ledipasvir and sofosbuvir fixed-dose combination with or without ribavirin for 12 weeks in treatment-naive and previously treated Japanese patients with genotype 1 hepatitis C: an open-label, randomised, phase 3 trial. Lancet Infect Dis. 2015;15(6):645–53.
Kowdley KV, An D, Pang PS, et al. Analysis of subgroup differences in the ION-3 trial of ledipasvir-sofosbuvir in chronic hepatitis C infection. Open Forum Infect Dis. 2015;2(2):1–2.
Wilder JM, Jeffers LJ, Ravendhran N, et al. Safety and efficacy of ledipasvir-sofosbuvir in black patients with hepatitis C virus infection: a retrospective analysis of phase 3 data. Hepatology. 2016;63(2):437–44.
Saab S, Park SH, Mizokami M, et al. Safety and efficacy of ledipasvir/sofosbuvir for the treatment of genotype 1 hepatitis C in subjects aged 65 years or older. Hepatology. 2016;63(4):1112–9.
Reddy KR, Bourliere M, Sulkowski M, et al. Ledipasvir and sofosbuvir in patients with genotype 1 hepatitis C virus infection and compensated cirrhosis: an integrated safety and efficacy analysis. Hepatology. 2015;62(1):79–86.
Younossi ZM, Stepanova M, Marcellin P, et al. Treatment with ledipasvir and sofosbuvir improves patient-reported outcomes: results from the ION-1, -2, and -3 clinical trials. Hepatol. 2015;61(6):1798–808.
Wyles D, Pockros P, Morelli G, et al. Ledipasvir-sofosbuvir plus ribavirin for patients with genotype 1 hepatitis C virus previously treated in clinical trials of sofosbuvir regimens. Hepatology. 2015;61(6):1793–7.
Lawitz E, Pockros P, Yang JC, et al. Ledipasvir/sofosbuvir regimens for the retreatment of patients who failed sofosbuvir-based regimens [abstract no. O-011]. Hepatol Int. 2016;10(Suppl 1):S9.
Tam E, Luetkeymer AF, Mantry PS, et al. Ledipasvir/sofosbuvir for treatment of hepatitis C virus in sofosbuvir-experienced, NS5A treatment-naive patients: findings from two randomized trials. Liver Int. 2017. https://doi.org/10.1111/liv.13616.
Akuta N, Sezaki H, Suzuki F, et al. Ledipasvir plus sofosbuvir as salvage therapy for HCV genotype 1 failures to prior NS5A inhibitors regimens. J Med Virol. 2017;89(7):1248–54.
Kowdley KV, Sundaram V, Jeon CY, et al. Eight weeks of ledipasvir/sofosbuvir is effective for selected patients with genotype 1 hepatitis C virus infection. Hepatology. 2017;65(4):1094–103.
Butt AA, Yan P, Shaikh OS, et al. Treatment adherence and virological response rates in hepatitis C virus infected persons treated with sofosbuvir-based regimens: results from ERCHIVES. Liver Int. 2016;36(9):1275–83.
Terrault NA, Zeuzem S, Di Bisceglie AM, et al. Effectiveness of ledipasvir-sofosbuvir combination in patients with hepatitis C virus infection and factors associated with sustained virologic response. Gastroenterology. 2016;151(6):1131-40.e5.
Tapper EB, Bacon BR, Curry MP, et al. Real-world effectiveness for 12 weeks of ledipasvir-sofosbuvir for genotype 1 hepatitis C: the Trio Health study. J Viral Hepat. 2017;24(1):22–7.
Tapper EB, Bacon BR, Curry MP, et al. Evaluation of proton pump inhibitor use on treatment outcomes with ledipasvir and sofosbuvir in a real-world cohort study. Hepatology. 2016;64(6):1893–9.
Curry MP, Tapper EB, Bacon B, et al. Effectiveness of 8- or 12-weeks of ledipasvir and sofosbuvir in real-world treatment-naive, genotype 1 hepatitis C infected patients. Aliment Pharmacol Ther. 2017:1–9.
Backus LI, Belperio PS, Shahoumian TA, et al. Real-world effectiveness of ledipasvir/sofosbuvir in 4,365 treatment-naive, genotype 1 hepatitis C-infected patients. Hepatology. 2016;64(2):405–14.
Backus LI, Belperio PS, Shahoumian TA, et al. Comparative effectiveness of ledipasvir/sofosbuvir ± ribavirin vs. ombitasvir/paritaprevir/ritonavir + dasabuvir ± ribavirin in 6961 genotype 1 patients treated in routine clinical practice. Aliment Pharmacol Ther. 2016;44:400–10.
Backus LI, Belperio PS, Shahoumian TA, et al. Real-world effectiveness and predictors of sustained virological response with all-oral therapy in 21,242 hepatitis C genotype-1 patients. Antivir Ther. 2017. https://doi.org/10.3851/IMP3117.
Balistreri WF, Murray KF, Rosenthal P, et al. The safety and effectiveness of ledipasvir-sofosbuvir in adolescents 12-17 years old with hepatitis C virus genotype 1 infection. Hepatology. 2017;66(2):371–8.
Younossi ZM, Stepanova M, Balistreri W, et al. Health-related quality of life in adolescent patients with hepatitis C genotype 1 treated with sofosbuvir and ledipasvir. J Ped Gastroenterol Nutr. 2018;66(1):1112–6.
Gane EJ, Hyland RH, Yang Y, et al. Efficacy of ledipasvir plus sofosbuvir for 8 or 12 weeks in patients with hepatitis C virus genotype 2 infection. Gastroenterology. 2017;152(6):1366–71.
Feld JJ, Ramji A, Shafran SD, et al. Ledipasvir-sofosbuvir plus ribavirin in treatment-naive patients with hepatitis C virus genotype 3 infection: an open-label study. Clin Infect Dis. 2017;65(1):13–9.
Gane EJ, Hyland RH, An D, et al. Efficacy of ledipasvir and sofosbuvir, with or without ribavirin, for 12 weeks in patients with HCV genotype 3 or 6 infection. Gastroenterology. 2015;149(6):1454–61.
Abergel A, Metivier S, Samuel D, et al. Ledipasvir plus sofosbuvir for 12 weeks in patients with hepatitis C genotype 4 infection. Hepatology. 2016;64(4):1049–56.
Kohli A, Kapoor R, Sims Z, et al. Ledipasvir and sofosbuvir for hepatitis C genotype 4: a proof-of-concept phase 2a cohort study. Lancet Infect Dis. 2015;15(9):1049–54.
Abergel A, Asselah T, Metivier S, et al. Ledipasvir-sofosbuvir in patients with hepatitis C virus genotype 5 infection: an open-label, multicentre, single-arm, phase 2 study. Lancet Infect Dis. 2016;16(4):459–64.
Nguyen MH, Trinh H, Do S, et al. Open label study of 8 vs. 12 weeks of ledipasvir/sofosbuvir in genotype 6 treatment naive or experienced patients. Am J Gastroenterol. 2017;112(12):1824–31.
Asahina Y, Itoh Y, Ueno Y, et al. Ledipasvir/sofosbuvir in the treatment of Japanese patients with chronic HCV genotype 2 infection [abstract no. 1184]. Hepatology. 2017;66(1 Suppl):638A.
Pol S, ANRS/AFEF HEPATHER study group. Safety and efficacy of the combination sofosbuvir-ledipasvir in HCV mono-infected patients from the French Cohort ANRS CO22 HEPATHER [abstract no. 1087]. Hepatology. 2017;66(1 Suppl):582A.
Cornberg M, Petersen J, Schober A, et al. Real-world use, effectiveness and safety of anti-viral treatment in chronic hepatitis C genotype 3 infection. Aliment Pharmacol Ther. 2017;45(5):688–700.
Tsertsvadze T, Chkhartishvilli N, Abutidze A, et al. Real-world effectiveness of sofosbuvir and ledipasvir/sofosbuvir based regimens in hepatitis C virus genotype 3 infection within national hepatitis C elimination program in the country of Georgia [abstract no.1167]. Hepatology. 2017;66(1 Suppl):627-8A.
Manns M, Samuel D, Gane EJ, et al. Ledipasvir and sofosbuvir plus ribavirin in patients with genotype 1 or 4 hepatitis C virus infection and advanced liver disease: a multicentre, open-label, randomised, phase 2 trial. Lancet Infect Dis. 2016;16(6):685–97.
Charlton M, Everson GT, Flamm SL, et al. Ledipasvir and sofosbuvir plus ribavirin for treatment of HCV infection in patients with advanced liver disease. Gastroenterology. 2015;149(3):649–59.
Naggie S, Cooper C, Saag M, et al. Ledipasvir and sofosbuvir for HCV in patients coinfected with HIV-1. N Engl J Med. 2015;373(8):705–13.
Cooper CL, Naggie S, Saag MS, et al. Successful retreatment of HCV/HIV-coinfected patients who failed 12 weeks of LDV/SOF. Clin Infect Dis. 2016;63:528–31.
Younossi ZM, Stepanova M, Sulkowski M, et al. Sofosbuvir and ledipasvir improve patient-reported outcomes in patients co-infected with hepatitis C and human immunodeficiency virus. J Viral Hepat. 2016;23(11):857–65.
Bourliere M, Bronowicki JP, de Ledinghen V, et al. Ledipasvir-sofosbuvir with or without ribavirin to treat patients with HCV genotype 1 infection and cirrhosis non-responsive to previous protease-inhibitor therapy: a randomised, double-blind, phase 2 trial (SIRIUS). Lancet Infect Dis. 2015;15(4):397–404.
Hagiwara S, Nishida N, Watanabe T, et al. Outcome of combination therapy with sofosbuvir and ledipasvir for chronic type C liver disease. Oncology. 2017;92(Suppl 1):3–9.
Barone M, Iannone A, Shahini E, et al. A different perspective on sofosbuvir-ledipasvir treatment of patients with HCV genotype 1b cirrhosis: the ital-c network. J Viral Hepatitis. 2017. https://doi.org/10.1111/jvh.12765.
Ferreira VL, Assis Jarek NA, Tonin FS, et al. Ledipasvir/sofosbuvir with or without ribavirin for the treatment of chronic hepatitis C genotype 1: a pairwise meta-analysis. J Gastroenterol Hepatol. 2017;32(4):749–55.
Younossi ZM, Stepanova M, Pol S, et al. The impact of ledipasvir/sofosbuvir on patient-reported outcomes in cirrhotic patients with chronic hepatitis C: the SIRIUS study. Liver Int. 2016;36(1):42–8.
Abbalkhail F, Elsiesy H, Elbeshbeshy H, et al. Treatment of patients with hepatitis C virus infection (genotype 4) with ledipasvir-sofosbuvir in the liver transplant setting. Transplant Proc. 2017. https://doi.org/10.1097/TP.0000000000001907.
Kwok RM, Ahn J, Schiano TD, et al. Sofosbuvir plus ledispasvir for recurrent hepatitis C in liver transplant recipients. Liver Transpl. 2016;22(11):1536–43.
Saxena V, Khungar V, Verna EC, et al. Safety and efficacy of current direct-acting antiviral regimens in kidney and liver transplant recipients with hepatitis C: results from the HCV-TARGET study. Hepatology. 2017;66(4):1090–101.
Colombo M, Aghemo A, Liu H, et al. Treatment with ledipasvir-sofosbuvir for 12 or 24 weeks in kidney transplant recipients with chronic hepatitis C virus genotype 1 or 4 infection: a randomized trial. Ann Intern Med. 2017;166(2):109–17.
Stedman CAM, Hyland RH, Ding X, et al. Once daily ledipasvir/sofosbuvir fixed-dose combination with ribavirin in patients with inherited bleeding disorders and hepatitis C genotype 1 infection. Haemophilia. 2016;22(2):214–7.
Walsh CE, Workowski K, Terrault NA, et al. Ledipasvir-sofosbuvir and sofosbuvir plus ribavirin in patients with chronic hepatitis C and bleeding disorders. Haemophilia. 2017;23(2):198–206.
Mangia A, Sarli R, Gamberini R, et al. Randomised clinical trial: sofosbuvir and ledipasvir in patients with transfusion-dependent thalassaemia and HCV genotype 1 or 4 infection. Aliment Pharmacol Ther. 2017;46(4):424–31.
Alqahtani SA, Afdhal N, Zeuzem S, et al. Safety and tolerability of ledipasvir/sofosbuvir with and without ribavirin in patients with chronic hepatitis C virus genotype 1 infection: analysis of phase III ION trials. Hepatology. 2015;62(1):25–30.
Gilead Sciences. HARVONI® (ledipasvir and sofosbuvir): US prescribing information. 2017. http://www.gilead.com. Accessed 1 Nov 2017
Rosenblatt R, Kumar S, Mehta A, et al. Baseline creatinine clearance is a predictor of worsening renal function while on treatment with sofosbuvir-ledipasvir [abstract no. Su1464]. Gastroenterology. 2016;150(4 Suppl. 1):S1107.
German P, Mathias A, Brainard D, et al. A thorough QT study to evaluate the effects of supratherapeutic doses of ledipasvir on the QTc interval in healthy subjects. Clin Pharmacol Drug Dev. 2017. https://doi.org/10.1002/cpdd.390.
WHO. Global hepatitis report 2017. 2017. http://www.who.int/hepatitis/. Accessed 30 Nov 2017.
European Association for the Study of the Liver. EASL recommendations on treatment of hepatitis C 2016. J Hepatol. 2016;61(1):153–94.
European Medicines Agency. Vosevi (sofosbuvir, velpatasvir and voxilaprevir) 400 mg/100 mg/10 mg film coated tablets: summary of product characteristics. 2017. http://www.ema.europa.eu. Accessed 4 Nov 2017.
Carter W, Connelly S, Struble K. Reinventing HCV treatment: past and future perspectives. J Clin Pharmacol. 2017;57(3):287–96.
Acknowledgements
During the peer review process, the manufacturer of tocilizumab was also offered an opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Conflict of interest
Lesley Scott is a salaried employee of Adis/Springer, is responsible for the article content and declares no relevant conflicts of interest.
Additional information about this Adis Drug Review can be found at http://www.medengine.com/Redeem/F3DCF0600DB0B722.
Additional information
The manuscript was reviewed by: A. Ascione, Fatebefratelli Hospital, Centre for Liver Disease, Napoli, Italy; S. Karatapanis, General Hospital of Rhodes, First Department of Internal Medicine, Rhodes, Greece; R. Pisapia, Epidemiology of Emerging and Re-emerging Infections and AIDs Reference Center, Istituto Nazionale per la Malattie Infettive “L. Spallanzani”, Rome, Italy; R. E. Stauber, Medical University of Graz, Department of Internal Medicine, Graz, Austria.
Rights and permissions
About this article

Cite this article
Scott, L.J. Ledipasvir/Sofosbuvir: A Review in Chronic Hepatitis C. Drugs 78, 245–256 (2018). https://doi.org/10.1007/s40265-018-0864-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-018-0864-z