
Abstract
Oritavancin (Orbactiv®) is a new generation lipoglycopeptide approved for use in adult patients with acute bacterial skin and skin structure infections (ABSSSI). It is administered as a single 1200 mg intravenous infusion over 3 h. In phase 3 trials in adult patients with ABSSSI, oritavancin was noninferior to vancomycin in terms of a composite outcome (cessation of spreading or reduction in the size of the baseline lesion, absence of fever and no rescue antibacterials required; primary endpoint) assessed at an US FDA-recommended early timepoint of 48–72 h after initiation of treatment. Oritavancin was also noninferior to vancomycin in terms of a ≥20 % reduction in the baseline lesion size at the early timepoint and clinical cure rate 7–14 days after the end of treatment. Oritavancin was generally well tolerated in the phase 3 trials, with most treatment-emergent adverse reactions being mild in severity. The most common adverse events occurring in oritavancin recipients were nausea, headache, vomiting, limb and subcutaneous abscesses, and diarrhoea. Oritavancin offers a number of potential advantages, including a convenient single dose treatment that may shorten or eliminate hospital stays, a reduction in healthcare resource utilization and cost, no need for dosage adjustment in patients with mild to moderate hepatic or renal impairment, no need for therapeutic drug monitoring, and elimination of compliance concerns. Therefore, oritavancin is a useful treatment option for adults with ABSSSI.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
New generation lipoglycopeptide with potent in vitro activity against Gram-positive bacteria causing ABSSSI, including methicillin-resistant Staphylococcus aureus, streptococci and enterococci |
At least three mechanisms of action, which contribute to rapid and concentration-dependent bactericidal activity |
Low potential for the emergence of oritavancin-resistant strains |
Noninferior to vancomycin for a composite clinical outcome (primary endpoint) and a ≥20 % reduction in the baseline lesion size 48–72 h after initiation of treatment, and for clinical cure rate 7–14 days after the end of treatment |
Generally well tolerated |
Convenient single dose treatment, without a need for dosage adjustment or therapeutic drug monitoring |
1 Introduction
The US FDA defines acute bacterial skin and skin structure infections (ABSSSI; previously known as complicated skin and skin structure infections) as bacterial infections of the skin with a lesion size area of ≥75 cm2 (measured by the area of redness, oedema or induration) and includes wound infection, cellulitis/erysipelas and major cutaneous abscess [1]. ABSSSI are typically caused by Gram-positive bacteria, most commonly Staphylococcus aureus [including methicillin-resistant S. aureus (MRSA)] and Streptococcus pyogenes [1]. Other Gram-positive (other Streptococcus species and Enterococcus species), Gram-negative and anaerobic bacteria, and polymicrobial infections may also be present in ABSSSI [2].
ABSSSI are associated with a substantial economic burden because of high hospitalization rates and antibacterial therapy with agents that typically require once- to thrice-daily administration for 5–14 days [3–5]. In order to achieve good clinical outcomes at minimal cost with these regimens, patient care needs to be transitioned through multiple treatment settings (often including outpatient parenteral antimicrobial therapy), using complex strategies and planning [3]. However, outpatient treatment may not overcome the shortcomings of multiple drug administrations, complex therapeutic drug monitoring, dosage adjustments, patient inconvenience caused by the use of peripherally inserted central catheters, and poor compliance [3, 6]. Longer-acting antibacterials with reduced administration frequency may mitigate some of these problems [3].
The naturally occurring glycopeptides vancomycin and teicoplanin (a first generation lipoglycopeptide; not approved in the USA) have been key parenteral antibacterials used against Gram-positive infections, particularly MRSA [7]. However, the emergence of bacterial strains resistant to these agents is associated with an increasing incidence of treatment failures and worsening clinical outcomes. The most problematic resistant strains are vancomycin-intermediate S. aureus (VISA), heterogeneous VISA (hVISA), vancomycin-resistant S. aureus (VRSA) and vancomycin-resistant enterococci (VRE). Therefore, second generation semisynthetic lipoglycopeptides (e.g. oritavancin), which have a low potential for evolution of resistance, have been developed. Furthermore, these agents are more potent and longer acting than vancomycin, and thus, permit less frequent administrations [7].
Intravenous oritavancin (Orbactiv®) is the first single-dose antibacterial therapy to be approved in the USA [8] and EU [9] for the treatment of adult patients with ABSSSI. This narrative review focuses on the clinical efficacy and tolerability of oritavancin in these patients, and provides an overview of its pharmacological properties.
2 Pharmacodynamic Properties of Oritavancin
2.1 Mechanism of Action
Oritavancin is a vancomycin analogue derived from chloroeremomycin, from which it differs by the addition of a lipophilic N-4-(4-chlorophenyl)benzyl side chain [10]. It has multiple mechanisms of action which contribute to its concentration-dependent bactericidal activity. Oritavancin inhibits cell wall synthesis by inhibiting the transglycosylation (polymerization) and transpeptidation (crosslinking) steps by binding to the carboxyl terminal acyl-d-alanyl-d-alanine residues of the stem pentapeptide in nascent peptidoglycan chain and peptidic crosslinking segments, respectively. Unlike other glycopeptides, oritavancin is able to bind to depsipeptides, including d-alanyl-d-lactate residues, which are present in organisms exhibiting VanA-type resistance [10]. Additionally, oritavancin disrupts cell membrane integrity, resulting in depolarization, increased permeability and rapid cell death [10, 11]; an ultrastructural study revealed that oritavancin caused septal distortions in MRSA and VRE [12]. The lipophilic side chain anchors the drug to the cell membrane and thus, enhances its affinity for the target site [10].
2.2 Antibacterial Activity
2.2.1 In Vitro Activity
This section mainly focuses on the antibacterial activity of oritavancin against Gram-positive bacteria causing ABSSSI as specified in the US manufacturer’s prescribing information (Table 1) [8]. In surveillance studies discussed, clinical isolates were collected between 2005 and 2014 in the USA and/or Europe [13–20], USA, Europe and Asia [21–26], Canada [27] or worldwide [28]. Most studies were conducted as part of the SENTRY Antimicrobial Surveillance programme [14, 15, 17, 18, 20–25, 28]. In all studies, the minimum inhibitory concentration (MIC) required to inhibit the growth of 90 % of isolates (MIC90) was determined using broth microdilution techniques, with the addition of polysorbate 80 to testing media. Susceptibility to oritavancin was based on the US FDA breakpoints (Table 1). As with glycopeptide antibacterials in general, oritavancin has no intrinsic activity against Gram-negative bacteria [9].
Oritavancin showed potent in vitro activity against the target pathogens, with MIC90 values several-fold lower than those of vancomycin, and where reported, ≥98 % of clinical isolates were susceptible to oritavancin and all isolates were susceptible to vancomycin (Table 1). At least 90 % of vancomycin-susceptible Enterococcus faecium demonstrated an in vitro MIC of ≤0.12 µg/mL (the susceptibility breakpoint for vancomycin-susceptible Enterococcus faecalis) for oritavancin; however, the efficacy and safety of oritavancin in treating clinical infections caused by this species is not established in adequate well-controlled clinical trials [8]. Oritavancin MIC90 values for MRSA did not differ between clinical isolates collected in the USA and Europe, with the differences within twofold for Streptococcus agalactiae, Streptococcus dysgalactiae, S. pyogenes and Streptococcus anginosus group [15, 19, 20]. In phase 3 trials (Sect. 4), oritavancin MIC90 values against MRSA and methicillin-susceptible S. aureus (MSSA) isolates (0.12 µg/mL for both; n = 404 and 535) [13] were consistent with those in the surveillance studies (Table 1).
The in vitro activity of oritavancin has been tested against clinical isolates with reduced susceptibility to vancomycin and other drugs. The MIC90 value for oritavancin was ≤0.12 μg/mL against multidrug-resistant (MDR) S. aureus strains (susceptibility ≥98 %; n = 337–1345) [20, 22, 25, 27], MRSA strains with a vancomycin MIC of 2 µg/mL (n = 124) [17] and S. aureus strains with a vancomycin MIC of 2 µg/mL (n = 205) or a daptomycin MIC of 1–4 µg/mL (n = 100) [20]. MIC90 values for oritavancin against VRSA (n = 10), hVISA (n = 11) and VISA (n = 14) were 0.5, 1 and 2 µg/mL, respectively [29]. Oritavancin MIC90 values did not change between erythromycin-susceptible and -nonsusceptible strains of S. agalactiae and S. pyogenes [26] or between MDR and non-MDR strains of β-haemolytic and viridans streptococci [30]. Against vancomycin-resistant E. faecalis, oritavancin exhibited good in vitro activity against VanB-type (MIC90 0.03 or 0.06 µg/mL; n = 19 and 17) as well as VanA-type (MIC90 0.5–1 µg/mL; n = 20–65) strains [17, 18, 21, 24, 26]. Of note, oritavancin was more potent than dalbavancin against E. faecalis (n = 14) and E. faecium (n = 15) strains exhibiting VanA-type resistance (MIC90 ≤0.5 vs. >16 µg/mL; abstract presentation) [31]. Oritavancin had good in vitro activity against S. aureus, E. faecalis and E. faecium (MIC90 values 0.06, 0.12 and 0.12 µg/mL, respectively; n = 25, 13, 32) strains with elevated linezolid MICs [32].
Oritavancin had a MIC90 value of 0.06 µg/mL against MRSA isolates (n = 14) harbouring a novel gene, mecC, which was within a doubling dilution of its MIC90 value against isolates carrying the typical mecA gene, which confers methicillin resistance [33]. Oritavancin also showed good in vitro activity (MIC90 0.25 µg/mL) against community-acquired MRSA isolates (n = 58), regardless of their genetic markers (presence or absence of Panton-Valentine leukocidin gene or presence of staphylococcal chromosome cassette mec type II or IV) [34].
2.2.2 Bactericidal Activity
In time-kill analyses, oritavancin at physiologically relevant concentrations demonstrated rapid, concentration-dependent bactericidal activity against Gram-positive pathogens associated with ABSSSI [33, 35–42]. For example, oritavancin at 16 µg/mL was bactericidal [i.e. ≥3 log10 reductions in colony-forming unit (CFU)/mL from baseline] against MSSA, MRSA and VRSA (within 2 h), VISA (within 24 h), vancomycin-susceptible E. faecalis (within 6 h), and VRE (within 6 and 24 h against VanB and VanA strains); in comparison, vancomycin was bactericidal against MSSA and MRSA at 24 h [36]. Oritavancin showed bactericidal activity against a daptomycin-nonsusceptible MRSA strain (within 8 h) [39], exponentially-growing as well as stationary-phase and biofilm-producing S. aureus [36, 38], intracellular S. aureus [43], and S. pyogenes, including erythromycin-resistant strains (within 0.25–3 h) [35]. A simulated single oritavancin dose of 1200 mg produced significantly (p < 0.05) lower area under the bacterial-kill curve at 24 h than vancomycin 1000 mg twice daily against three MRSA strains [40]. Preliminary results (abstract presentations) suggest that oritavancin may be more potent than dalbavancin against MRSA (including non-dividing) isolates, with respect to the rate and/or extent of killing [41, 42, 44].
In time-kill assays, oritavancin showed synergistic activity with gentamicin or linezolid against MRSA (comprising VISA and hVISA strains) [37], VISA [45] and VRSA [45], with rifampin against 7 of 9 MRSA strains [37] and VRSA [45], and with gentamicin, moxifloxacin or rifampin against MSSA [45]. In vitro, there was no antagonism between oritavancin and gentamicin, moxifloxacin, linezolid or rifampin [8, 37].
2.3 Resistance Issues
There was no evidence for the emergence of bacterial resistance to oritavancin in surveillance (Table 1) or clinical studies [8]. In surveillance studies, including a longitudinal analysis [46], MIC90 values and susceptibility rates against specified bacteria have generally remained constant for oritavancin over the past several years. In vitro, emergence of S. aureus and E. faecalis strains resistant to oritavancin has been seen in serial passage studies [8]. The mechanism of resistance to oritavancin is not fully understood [10].
2.4 Pharmacokinetic/Pharmacodynamic Considerations
The oritavancin area under the plasma concentration–time curve (AUC) from time zero to 72 h (AUC72h) to MIC90 ratio correlated well with its efficacy in phase 3 trials (abstract presentation) [47]. The AUC72h:MIC90 ratio threshold for achieving post-therapy clinical cure (defined in Table 2) was determined to be 11,982 in patients with S. aureus infections, with 82.6 and 96.2 % of patients with AUC72h:MIC90 ratios below and above this target, respectively, achieving clinical success (p = 0.03 for between-group comparison). The mean overall model-predicted probability of achieving clinical success was 95.4 % across oritavancin MIC values of 0.06–0.5 µg/mL against S. aureus [47].
2.5 In Vivo Activity
Consistent with its in vitro activity, oritavancin exhibited potent bactericidal effects in animal models of infection with S. aureus (including MRSA and MSSA), E. faecalis [vancomycin-susceptible and -resistant (both VanA- and VanB-type) strains] or E. faecium (VanA-type) [48, 49]. For example, a single human-equivalent dose of 1200 mg reduced MSSA or MRSA counts from baseline by ≥2.7 log10 CFU/thigh in a neutropenic murine thigh infection model [49]. The single-dose regimen was significantly (p < 0.05) more effective than daily regimens in this model [48, 49]. Subsequently, single-dose regimens were evaluated in a phase 2 study (Sect. 4).
3 Pharmacokinetic Properties of Oritavancin
The pharmacokinetics of oritavancin are linear at doses of up to 1200 mg [8] and are best described using a three-compartment model (α and β distributional phases followed by a terminal elimination phase), with zero-order intravenous infusion and first-order linear elimination [50, 51].
In a population pharmacokinetic analysis [51] of the phase 3 trials (which used the approved oritavancin dose of a single intravenous infusion of 1200 mg in patients with ABSSSI; Sect. 4), the model-derived mean maximum plasma concentration was 138 µg/mL and AUC72h was 1530 µg · h/mL. AUC72h is the main AUC parameter of interest for oritavancin (Sect. 2.4) and was consistent with the timing of the assessment of early clinical outcomes (i.e. 48–72 h after the start of therapy) in the phase 3 trials.
Oritavancin is highly (≈85 %) bound to human plasma proteins [8] and is extensively distributed into tissues (estimated total and mean steady-state volume of distribution 87.6 and 97.8 L) [8, 51]. Oritavancin showed modest penetration into cantharide-induced skin blister fluid in healthy volunteers; after a single intravenous infusion of 800 mg, the ratio of mean area under the blister fluid concentration–time curve at 24 h to that of plasma was 0.185; however, mean oritavancin concentrations in blister fluid exceeded its MIC90 value against S. aureus [52].
Nonclinical studies show that oritavancin is not metabolized [8]. In humans, unchanged oritavancin is slowly excreted in urine and faeces (<5 and <1 %, respectively, over 14 days) [8]. In patients with ABSSSI, oritavancin had a prolonged terminal elimination half-life (245 h), with a clearance of 0.445 L/h; despite the long terminal half-life, AUC72h was 55 % of the overall exposure (i.e. AUC∞) [51].
In healthy volunteers receiving a single dose of 1200 mg, oritavancin nonspecifically and weakly inhibited cytochrome P450 (CYP) enzymes CYP2C9 and CYP2C19, and weakly induced CYP3A4 and CYP2D6 [8]. Therefore, oritavancin may alter exposure to drugs with a narrow therapeutic window that are mainly metabolized by these enzymes; for example, the mean AUC for warfarin (a CYP2C9 substrate) increased by 31 % with concomitant oritavancin. In vitro, oritavancin is not a substrate or an inhibitor of P-glycoprotein [8].
The pharmacokinetics of oritavancin were not affected to a clinically relevant extent by mild to moderate renal impairment (creatinine clearance >29 mL/min), moderate hepatic impairment (Child-Pugh class B), presence of diabetes, age, height, weight, gender or race; hence, dosage adjustment is not required [8, 51]. The effects of severe renal or hepatic impairment on the pharmacokinetics of oritavancin have not been evaluated [8]. In vitro, oritavancin was not removed from blood by haemodialysis [8], suggesting that dosage adjustment may not be required in patients undergoing haemodialysis.
4 Therapeutic Efficacy of Oritavancin
As stated by Corey et al. [6], two initial phase 3 trials evaluating a daily regimen of intravenous oritavancin (200 or 300 mg daily for 3–7 days) did not provide sufficient evidence for the efficacy and safety of oritavancin in patients with ABSSSI. However in a subsequent randomized, double-blind, multicentre phase 2 study (n = 302) in patients with ABSSSI, oritavancin administered as a single 1200 mg dose or 800 mg on day 1 with an optional 400 mg on day 5 was noninferior to a daily regimen (200 mg daily for 3–7 days) in terms of clinical response rates at the test-of-cure visit in the clinically evaluable population (81.5 and 77.5 vs. 72.4 %, respectively) [53]. Based on these results, the efficacy of a single dose of oritavancin 1200 mg was compared with that of twice-daily intravenous vancomycin in two identically designed double-blind, noninferiority, multinational, phase 3 trials (SOLO I [6] and SOLO II [54]) in adults with ABSSSI. Combined analyses of these trials are available as abstracts [55–62] and in the US prescribing information [33]. Discussion in this section focuses on the SOLO trials.
Eligible patients in the SOLO trials were aged ≥18 years and had a diagnosis of ABSSSI (proven or suspected to be caused by a Gram-positive pathogen) that was expected to require ≥7 days of intravenous therapy [6, 54]. The diagnosis of ABSSSI required the presence of a wound infection, cellulitis/erysipelas or major cutaneous abscess, each lesion surrounded by erythema, oedema and/or induration of ≥75 cm2. At least two signs of ABSSSI (purulent drainage or discharge, erythema, fluctuance, heat or localized warmth, oedema/induration, pain or tenderness to palpation) and one or more signs of systemic inflammation (proximal lymph node swelling and tenderness, body temperature ≥38.0 or <36.0 °C, white blood cell count >10,000 cells/mm3, bandemia >10 %, or C-reactive protein level above the upper limit of normal) had to be present. Patients without signs of systemic inflammation could be enrolled if they were aged >70 years, had diabetes requiring treatment, or had received immunosuppressive or chemotherapeutic agents in the previous 3 months. Patients who received antibacterials that had activity against Gram-positive pathogens within 14 days prior to randomization were among those excluded [6, 54].
Randomized treatment regimens are shown in Table 2. The primary endpoint was a composite outcome assessed at the US FDA recommended timepoint of 48–72 h in the modified intent-to-treat (mITT) population (Table 2) [6, 54]. Secondary endpoints included early reduction in lesion size and post-therapy clinical cure rate (Table 2), which are recommended by the FDA and the European Medicines Agency, respectively, in ABSSSI trials.
Patient demographic and baseline clinical characteristics did not differ markedly between the treatment groups within each trial [6, 54]. In SOLO I, 49.9 % of patients had cellulitis/erysipelas, 29.5 % had major cutaneous abscess and 20.6 % had wound infection [6]. The corresponding proportions in SOLO II were 30.9, 32.5 and 36.5 %, respectively [54]. At baseline, 19.7 and 9.1 % of patients had diabetes in SOLO I [6] and II [54], respectively. The median lesion size in the oritavancin and vancomycin groups was 248.0 and 225.6 cm2 in SOLO I [6], and 287.8 and 308.8 cm2 in SOLO II [6]. A baseline pathogen was isolated in 61.1 and 60.5 % of patients in the oritavancin and vancomycin groups, respectively, in SOLO I, and 69.8 and 70.1 % in SOLO II, with S. aureus being the most common [6, 54].
4.1 Individual Trials
Oritavancin was noninferior to vancomycin in terms of the composite outcome at the early clinical evaluation in the mITT population in both SOLO I and II trials (Table 2; primary endpoint) [6, 54]. Similar results were seen in the clinically evaluable population (n = 791 [6] and 835 [54] in SOLO I and II). In subgroup analyses of the mITT population, the proportion of patients with major cutaneous abscess achieving the primary endpoint was lower in the oritavancin than in the vancomycin group in SOLO II (81.0 vs. 89.9 %; difference −9.0; 95 % CI −16.5 to −1.4) [54]. Apart from this, in individual trials, there were no significant between-group differences by lesion type, geographic region (North America, Eastern or Western Europe, Asia), risk factors [diabetes or systemic inflammatory response syndrome (SIRS)], age (<65 or ≥65 years), sex, body mass index, race or baseline pathogen (≥1 pathogen, MRSA, MSSA, S. anginosus group) [6, 54]. The overall incidence of treatment failure for the primary endpoint in the oritavancin and vancomycin groups was 14.3 and 16.3 %, respectively, in SOLO I [6] and 17.3 and 14.1 % in SOLO II [54]. The pattern of reasons for the failure was generally similar between the groups, with fever between 48 and 72 h being the most common reason in both groups [6, 54].
Oritavancin was also noninferior to vancomycin with respect to the percentage of patients achieving a ≥20 % reduction in lesion size at 48–72 h and clinical cure rate 7–14 days after the end of therapy in individual trials (Table 2) [6, 54]. In SOLO I, there were no significant between-group differences in lesion size reduction by lesion type and risk factors such as diabetes, SIRS, age ≥65 years or renal insufficiency (abstract presentation [63]), or by baseline MRSA or MSSA [6]. In SOLO II, there were no significant between-group differences in clinical cure rate by lesion type, geographic region, presence of SIRS, age, sex, race, and baseline MRSA or MSSA; however, in patients with diabetes, the clinical cure rate was significantly lower with oritavancin than with vancomycin (69.6 vs. 88.9 %; treatment difference −19.3; 95 % CI −35.5 to −3.2; n = 46 and 45) [54].
4.2 Combined Analyses
Combined analyses of SOLO trials also confirmed the efficacy of oritavancin in the overall population and in prespecified subgroups by treatment setting, infection type and baseline pathogen (Table 2). Of note, among patients with baseline MRSA, significantly more oritavancin than vancomycin recipients achieved a ≥20 % reduction in their lesion size at 48–72 h [59].
Oritavancin was effective in the inpatient as well as the outpatient setting, irrespective of ABSSSI severity [56, 62]. In SOLO trials, ≈40 % of patients (all from the USA [62]) were treated entirely in the outpatient setting, and the early and post-therapy clinical outcomes in this population were generally similar to those of the overall population (Table 2) [56]. Clinical outcomes did not differ markedly between treatment groups based on disease severity (Eron class I, II or III) or treatment setting (inpatient or outpatient); the majority (≈71 %) of outpatients were deemed to belong to Eron class II or III, reflecting a moderate level of disease severity [62].
Among inpatients, post-therapy clinical cure rates did not differ markedly between oritavancin and vancomycin recipients in the USA (81 and 78 %; n = 1165) and Eastern European countries (86 and 84 %; n = 202) [55]. The average length of hospital stay was shorter in the USA (6.0 and 6.4 days, respectively) than in the Eastern European countries (14.9 and 14.7 days) [55].
There was high concordance between early and post-therapy clinical outcomes in SOLO trials, with 87 % of patients who achieved the composite outcome and 85 % of those who achieved a ≥20 % reduction in the baseline lesion size achieving clinical cure at the post-therapy evaluation [60].
Oritavancin produced high microbiological response at the post-therapy evaluation [58]. In the microbiological intent-to-treat population, eradication or presumed eradication rates in the oritavancin and vancomycin groups by baseline pathogen were: MRSA 91.4 and 93.9 %, respectively (n = 186 and 181); MSSA 93.2 and 93.9 % (n = 235 and 244); S. pyogenes 92.6 and 85.2 % (n = 27 and 27); S. anginosus group 88.5 and 90.5 % (n = 26 and 42); and, E. faecalis 66.7 and 88.9 % (n = 12 and 9) [58].
5 Tolerability of Oritavancin
Oritavancin was generally well tolerated in patients with ABSSSI in SOLO I and II trials, with most treatment-emergent adverse events being mild in severity [6, 8, 54]. Of note, an extended (60 days) safety follow-up for oritavancin did not identify any prolonged or delayed adverse events, suggesting that the extended half-life of oritavancin does not markedly affect its safety profile [6, 54]. In SOLO trials, relatively few patients in the oritavancin and vancomycin groups discontinued treatment because of an adverse event (3.8 vs. 5.8 % [6]; 3.6 vs. 2.6 % [54]). In a pooled analysis of these trials, cellulitis (0.4 %) and osteomyelitis (0.3 %) were the most common adverse reactions leading to discontinuation of oritavancin [8].
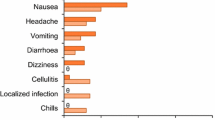
In the pooled analysis, 55.3 and 56.9 % of patients in the oritavancin (n = 976) and vancomycin (n = 983) groups experienced one or more adverse reactions [8]. The most common adverse reactions (incidence ≥1.5 % in the oritavancin group) that occurred in the oritavancin and vancomycin groups were nausea (9.9 vs. 10.5 %), headache (7.1 vs. 6.7 %), vomiting (4.6 vs. 4.7 %), limb or subcutaneous abscess (3.8 vs. 2.3 %), diarrhoea (3.7 vs. 3.4 %), elevated alanine transaminase (2.8 vs. 1.5 %) or aspartate aminotransferase (1.8 vs. 1.5 %), dizziness (2.7 vs. 2.6 %), infusion-site phlebitis (2.5 vs. 1.5 %), tachycardia (2.5 vs. 1.1 %) and infusion-site reaction (1.9 vs. 3.5 %) [8]. Where reported [6], Red-Man syndrome did not occur in oritavancin recipients. In SOLO I and II, osteomyelitis was reported as an adverse event in six oritavancin recipients and one vancomycin recipient in total [6, 54]. Treatment-related adverse events occurred in less than one-third of patients in the oritavancin and vancomycin groups (22.8 vs. 31.4 % [6]; 21.7 vs. 25.5 % [54]).
Serious adverse reactions occurred in ≈6 % of patients in both treatment groups, with cellulitis being the most common (≈1 % in both groups) [8]. There were no reports of serious elevations in liver enzymes or discontinuation of study drugs because of these adverse events [6, 54]. Two patients in the oritavancin group and three patients in the vancomycin group died during SOLO trials; no deaths were considered to be related to study drug by the investigator [6, 54].
In a pooled analysis of SOLO trials, the distribution of time to onset and duration of adverse events were generally similar between oritavancin and vancomycin groups [64]. The median time to onset and the duration was 2.0 and 2.0 days for oritavancin-related adverse events, and 1.0 and 6.0 days for oritavancin-related serious adverse events [64].
The incidence of laboratory abnormalities, vital signs and electrocardiographic findings did not differ markedly between oritavancin and vancomycin recipients [6, 54]. In a thorough QT study in healthy volunteers (n = 135), a single 1600 mg dose of oritavancin did not prolong the corrected QT interval to a clinically relevant extent [8].
In the two initial phase 3 trials (Sect. 4), once-daily oritavancin was not associated with potential glycopeptide-related adverse events, such as nephrotoxicity, ototoxicity, vestibular toxicity or haematologic toxicity [65].
6 Dosage and Administration of Oritavancin
Intravenous oritavancin is approved in the USA [8] and EU [9] for the treatment of ABSSSI in adults. In the USA, it is indicated for patients with ABSSSI caused or suspected to be caused by susceptible isolates of specific Gram-positive bacteria (Table 1). In order to reduce the risk of antibacterial resistance and maintain the effectiveness of oritavancin and other antibacterial drugs, oritavancin should only be used to treat infections that are proven or strongly suspected to be caused by susceptible bacteria [8, 9].
The recommended dosage of oritavancin is a single 1200 mg administered as a 3-h infusion [8, 9]. Infusion related reactions, such as pruritus, urticaria or flushing, can occur with oritavancin; if they occur, slowing or interrupting the infusion should be considered. Oritavancin should be used in patients taking chronic warfarin only when the benefits outweigh the risks of bleeding. The drug may artificially prolong activated partial thromboplastin time for up to 48 h, and prothrombin time and International Normalized Ratio for up to 24 h. Clostridium difficile-associated diarrhoea (CDAD) can occur with systemic antibacterial drugs, including oritavancin, and therefore, patients should be evaluated for CDAD if diarrhoea occurs during treatment. Patients should be monitored for signs and symptoms of osteomyelitis, and if osteomyelitis is diagnosed or suspected, an appropriate alternate antibacterial therapy should be initiated. Use of intravenous unfractionated heparin sodium is contraindicated for 48 h after oritavancin administration and oritavancin is contraindicated in patients with a hypersensitivity to oritavancin [8, 9]. Local prescribing information should be consulted for detailed information, including contraindications, precautions, drug interactions and use in special patient populations.
7 Place of Oritavancin in Acute Bacterial Skin and Skin Structure Infections
Current Infectious Diseases Society of America practice guidelines for the management of severe skin and soft tissue infections recommend a number of parenteral empirical treatment options, including vancomycin, daptomycin, linezolid, telavancin, ceftaroline and clindamycin [4]. As oritavancin was approved only recently, it was not included in these guidelines, nor were dalbavancin and tedizolid. There are no specific treatment guidelines for ABSSSI in Europe. However, a retrospective assessment of clinical practice patterns in Europe during 2010–2011 found that the majority of hospitalized adult patients with ABSSSI who required intravenous therapy were initially treated empirically (≈82 %; n = 1995), most commonly with penicillins with or without a β-lactamase inhibitor (≈60 %) [2]. Vancomycin, daptomycin and linezolid were the most commonly used anti-MRSA agents. Of note, ≈40 % of patients required their initial therapy to be subsequently modified and the most common reason for this was insufficient clinical response or treatment failure (17 %) [2].
Oritavancin has multiple mechanisms of action resulting in rapid, concentration-dependent bactericidal activity in vitro (Sect. 2.1). It shows potent in vitro activity against susceptible Gram-positive pathogens associated with ABSSSI (Table 1) and has a low potential for the emergence of bacterial resistance (Sect. 2.3). Oritavancin is also active against VISA, VRSA and VRE; unlike other glycopeptides, it retains its activity against VanA-type VRE (Sect. 2.2.1).
The prolonged plasma terminal half-life (245 h) of oritavancin allows for convenient single-dose therapy for ABSSSI (Sect. 3). Dalbavancin also has a prolonged terminal half-life (mean 348–372 h), although it was evaluated only as a two-dose regimen in key clinical trials [66]. In comparison, telavancin is administered once daily [67] and vancomycin every 6 or 12 h [68] for up to 14 days.
Oritavancin dosage adjustment is not required for moderate renal or hepatic impairment, age, weight, gender or race, while the other glycopeptides require dosage adjustment for renal impairment [66–68]. Although, penetration of oritavancin into skin blister fluid is modest relative to dalbavancin [66] or telavancin [67], oritavancin concentration in blister fluid exceeds its MIC90 value of S. aureus. There is a potential risk of bleeding with concomitant use of oritavancin and warfarin. The safety of oritavancin in ABSSSI patients receiving chronic warfarin treatment is currently being evaluated in a phase 4 clinical trial (NCT02452918). Oritavancin is also known to interfere with some coagulation tests. While potential drug–drug or drug-laboratory test interactions are also reported for vancomycin [68] and telavancin [67], dalbavancin has a low potential for such interactions [69].
In two large phase 3 registration (SOLO I and II) trials in adult patients with ABSSSI, oritavancin was noninferior to vancomycin in terms of the primary composite outcome at an FDA-recommended early timepoint of 48–72 h after initiating therapy, with ≥79 % of patients in both treatment groups achieving this endpoint (Sect. 4). Similar results were reported for various subgroup analyses in either one or both trials. Oritavancin was also noninferior to vancomycin in terms of a ≥20 % reduction from baseline in lesion size at the early timepoint and clinical cure rate 7–14 days after the end of therapy. There was good concordance between early and post-therapy clinical outcomes. Oritavancin was also effective in the outpatient setting, including in patients with severe ABSSSI. In the absence of head-to-head comparative studies, a Bayesian network meta-analysis of 52 randomized controlled trials suggests that most antibacterials used for the treatment of ABSSSI, including oritavancin, may have similar efficacy [70].
Oritavancin was generally well tolerated in adults with ABSSSI in SOLO trials, with most treatment-emergent adverse events being of mild severity (Sect. 5). The most common adverse reactions occurring in oritavancin recipients included nausea, headache, vomiting, limb and subcutaneous abscesses, and diarrhoea. Treatment-related adverse events occurred in less than one-quarter of oritavancin recipients, with <4 % of patients discontinuing treatment because of an adverse reaction. Overall, the safety profile of oritavancin was similar to that of vancomycin. The extended plasma half-life of oritavancin was not associated with any prolonged or delayed adverse events in SOLO trials. However, the safety of oritavancin in real-world clinical use remains to be seen.
CDAD, hypersensitivity and infusion related reactions have been known to occur with most glycopeptide antibacterials, including oritavancin (Sect. 5). In SOLO trials, more cases of osteomyelitis were reported with oritavancin than with vancomycin (6 vs. 1); however, in SOLO II, all five cases in the oritavancin group occurred within 1–9 days after treatment, suggesting that osteomyelitis may have been present at time of screening [54]. Specific adverse events with other glycopeptide antibacterials include nephrotoxicity with vancomycin and telavancin [67], and elevated alanine transaminase levels with dalbavancin [69]. Telavancin also has a boxed warning for foetal risk, based on animal studies [67].
Budget impact model analyses (abstract presentations) conducted from a USA [71] and UK [72] hospital perspective suggest that using oritavancin in some patients (≈26 and ≈4 %, respectively) with moderate to severe ABSSSI or skin and soft tissue infections would reduce the total annual cost of medical care relative to the current clinical practice, driven by reduced healthcare resource utilization. However, the cost effectiveness of oritavancin relative to the standard-of-care agents in ABSSSI is yet to be established.
In conclusion, intravenous oritavancin was effective and generally well tolerated in adult patients with ABSSSI in two, large well-controlled clinical trials. The convenient single-dose therapy with oritavancin may provide some advantages: shorter, or elimination of, hospital stays, which may also reduce the risk of nosocomial infections; a reduction in healthcare resource utilization and cost; potential for use in outpatient parenteral antimicrobial therapy; no need for peripherally inserted central catheters; and, elimination of patient compliance concerns, complex therapeutic drug monitoring and dosage adjustments. However, head-to-head clinical studies and robust pharmacoeconomic analyses are required to definitively position this drug relative to other antibacterials used for ABSSSI.
Data selection sources:
Relevant medical literature (including published and unpublished data) on oritavancin was identified by searching databases including MEDLINE (from 1946), PubMed (from 1946) and EMBASE (from 1996) [searches last updated 7 September 2015], bibliographies from published literature, clinical trial registries/databases and websites. Additional information was also requested from the company developing the drug.
Search terms: Oritavancin, Nuvocid, ORBACTIV, LY 333328, skin.
Study selection: Studies in patients with acute bacterial skin and skin structure infections who received oritavancin. When available, large, well designed, comparative trials with appropriate statistical methodology were preferred. Relevant pharmacodynamic and pharmacokinetic data are also included.
References
US FDA. Guidance for industry. Acute bacterial skin and skin structure infections: developing drugs for treatment. 2013. http://www.fda.gov/. Accessed 25 Jun 2015.
Garau J, Ostermann H, Medina J, et al. Current management of patients hospitalized with complicated skin and soft tissue infections across Europe (2010–2011): assessment of clinical practice patterns and real-life effectiveness of antibiotics from the REACH study. Clin Microbiol Infect. 2013;19(9):E377–85.
Pollack CV Jr, Amin A, Ford WT Jr, et al. Acute bacterial skin and skin structure infections (ABSSSI): practice guidelines for management and care transitions in the emergency department and hospital. J Emerg Med. 2015;48(4):508–19.
Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society of America. Clin Infect Dis. 2014;59(2):147–59.
Khachatryan A, Patel D, Stephens J, et al. Rising US hospital admissions for Gram+ acute bacterial skin and skin structure infections (ABSSSI) [abstract no. 58]. J Hosp Med. 2014;9(Suppl 2).
Corey GR, Kabler H, Mehra P, et al. Single-dose oritavancin in the treatment of acute bacterial skin infections. N Engl J Med. 2014;370(23):2180–90.
Butler MS, Hansford KA, Blaskovich MA, et al. Glycopeptide antibiotics: back to the future. J Antibiot. 2014;67(9):631–44.
The Medicines Company. Orbactiv (oritavancin) for injection, for intravenous use: US prescribing information. 2014. http://www.orbactiv.com. Accessed 26 Jun 2015.
European Medicines Agency. Orbactiv 400 mg powder for concentrate for solution for infusion: summary of product characteristics. 2015. http://www.ema.europa.eu. Accessed 26 Jun 2015.
Zhanel GG, Schweizer F, Karlowsky JA. Oritavancin: mechanism of action. Clin Infect Dis. 2012;54(Suppl 3):S214–9.
Belley A, McKay GA, Arhin FF, et al. Oritavancin disrupts membrane integrity of Staphylococcus aureus and vancomycin-resistant enterococci to effect rapid bacterial killing. Antimicrob Agents Chemother. 2010;54(12):5369–71.
Belley A, Harris R, Beveridge T, et al. Ultrastructural effects of oritavancin on methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus. Antimicrob Agents Chemother. 2009;53(2):800–4.
Arhin FF, Moeck G, Sahm DF. Comparison of oritavancin (ORI) in vitro susceptibility of Staphylococcus aureus (SA) isolates from phase 3 studies for acute bacterial skin and skin-structure infections and from a surveillance study in the US and Europe [abstract no. F-961]. In: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy. 2014.
Jones RN, Turnidge JD, Moeck G, et al. Use of in vitro vancomycin testing results to predict susceptibility to oritavancin, a new long-acting lipoglycopeptide. Antimicrob Agents Chemother. 2015;59(4):2405–9.
Mendes RE, Farrell DJ, Sader HS, et al. Activity of oritavancin against Gram-positive clinical isolates responsible for documented skin and soft-tissue infections in European and US hospitals (2010–13). J Antimicrob Chemother. 2015;70(2):498–504.
Morrissey I, Seifert H, Canton R, et al. Activity of oritavancin against methicillin-resistant staphylococci, vancomycin-resistant enterococci and beta-haemolytic streptococci collected from western European countries in 2011. J Antimicrob Chemother. 2013;68(1):164–7.
Mendes RE, Farrell DJ, Sader HS, et al. Oritavancin microbiologic features and activity results from the surveillance program in the United States. Clin Infect Dis. 2012;54(Suppl 3):S203–13.
Mendes RE, Sader HS, Jones RN, et al. Longitudinal (2001–2013) analysis of vancomycin-resistant enterococci causing invasive infections in European and USA hospitals, including a contemporary (2010–2013) analysis of oritavancin in vitro potency [abstract no. O100]. In: 25th European Congress of Clinical Microbiology and Infectious Diseases. 2015.
Biedenbach D, Lynch T, Arhin F, et al. Oritavancin and comparator activities against S. aureus and Streptococcus spp. isolated from skin and skin structure infections in Europe and North America 2011–2014 [abstract no. P0709]. In: 25th European Congress of Clinical Microbiology and Infectious Diseases. 2015.
Mendes RE, Sader HS, Flamm RK, et al. Oritavancin activity against Staphylococcus aureus causing invasive infections in US and European hospitals: a 5-year international surveillance program. Antimicrob Agents Chemother. 2014;58(5):2921–4.
Mendes RE, Farrell DJ, Streit JM, et al. Current analysis of oritavancin potency when tested against vancomycin-resistant and -susceptible enterococcal clinical isolates recovered from European medical centres (2009–2013) [abstract no. eP171]. In: 24th European Congress of Clinical Microbiology and Infectious Disease. 2014.
Mendes RE, Farrell DJ, Flamm RK, et al. Activity of oritavancin and comparator agents against multidrug-resistant staphylococcal and streptococcal isolates responsible for documented infections in European hospitals (2011–2013) [abstract no. eP172]. In: 24th European Congress of Clinical Microbiology and Infectious Disease. 2014.
Mendes RE, Flamm RK, Sader HS, et al. Activity of oritavancin tested against Gram-positive clinical isolates responsible for documented skin and soft tissue infections in European hospitals (2011–2013) [abstract no. P1580]. In: 24th European Congress of Clinical Microbiology and Infectious Disease. 2014.
Mendes RE, Sader HS, Flamm RK, et al. Oritavancin activity against gram-positive clinical isolates responsible for documented skin and skin structure infections in USA and European hospitals (2012–2013) [abstract no. 470]. In: Infectious Diseases Week Meeting. 2014.
Mendes RE, Flamm RK, Farrell DJ, et al. Oritavancin in vitro activity against the most prevalent antibiogram resistance patterns among methicillin-resistant Staphylococcus aureus, including multidrug-resistant strains from patients in European hospitals (2010–2013) [abstract no. P0708]. In: 25th European Congress of Clinical Microbiology and Infectious Diseases. 2015.
Arhin FF, Draghi DC, Pillar CM, et al. Comparative in vitro activity profile of oritavancin against recent gram-positive clinical isolates. Antimicrob Agents Chemother. 2009;53(11):4762–71.
Karlowsky J, Walkty A, Baxter M, et al. In vitro activity of oritavancin against Gram-positive pathogens isolated in Canadian hospital laboratories from 2011 to 2013. Diagn Microbiol Infect Dis. 2014;80(4):311–5.
Mendes RE, Sader HS, Flamm RK, et al. Activity of oritavancin tested against uncommonly isolated Gram-positive pathogens responsible for documented infections in hospitals worldwide. J Antimicrob Chemother. 2014;69(6):1579–81.
Arhin FF, Sarmiento I, Parr TR Jr, et al. Comparative in vitro activity of oritavancin against Staphylococcus aureus strains that are resistant, intermediate or heteroresistant to vancomycin. J Antimicrob Chemother. 2009;64(4):868–70.
Mendes RE, Farrell DJ, Sader HS, et al. In vitro activity of oritavancin against streptococcal clinical isolates, including macrolide- and/or lincosamide (constitutive)-resistant and multidrug-resistant isolates from European hospitals (2010–2013) [abstract no. P0707]. In: 25th European Congress of Clinical Microbiology and Infectious Diseases. 2015.
Sweeney BD, Shinabarger DL, Pillar CM, et al. Comparative in vitro activity of new long-acting lipoglycopeptides and other agents against enterococci, including vancomycin-resistant isolates [abstract no. C-567]. In: 55th Interscience Conference on Antimicrobial Agents and Chemotherapy. 2015.
Mendes RE, Farrell DJ, Sader HS, et al. Oritavancin in vitro activity against a collection of molecularly-characterized staphylococci and enterococci displaying elevated linezolid MICs [abstract no. C-560]. In: 55th Interscience Conference on Antimicrobial Agents and Chemotherapy. 2015.
Arhin FF, Sarmiento I, Moeck G. In vitro activities of oritavancin and comparators against meticillin-resistant Staphylococcus aureus (MRSA) isolates harbouring the novel mecC gene. Int J Antimicrob Agents. 2014;44(1):65–8.
Arhin FF, Kurepina N, Sarmiento I, et al. Comparative in vitro activity of oritavancin against recent, genetically diverse, community-associated meticillin-resistant Staphylococcus aureus (MRSA) isolates. Int J Antimicrob Agents. 2010;35(1):93–4.
Arhin FF, McKay GA, Beaulieu S, et al. Time-kill kinetics of oritavancin and comparator agents against Streptococcus pyogenes. Int J Antimicrob Agents. 2009;34(6):550–4.
McKay GA, Beaulieu S, Arhin FF, et al. Time-kill kinetics of oritavancin and comparator agents against Staphylococcus aureus, Enterococcus faecalis and Enterococcus faecium. J Antimicrob Chemother. 2009;63(6):1191–9.
Lin G, Pankuch G, Appelbaum PC, et al. Antistaphylococcal activity of oritavancin and its synergistic effect in combination with other antimicrobial agents. Antimicrob Agents Chemother. 2014;58(10):6251–4.
Belley A, Neesham-Grenon E, McKay G, et al. Oritavancin kills stationary-phase and biofilm Staphylococcus aureus cells in vitro. Antimicrob Agents Chemother. 2009;53(3):918–25.
Vidaillac C, Parra-Ruiz J, Rybak MJ. In vitro time-kill analysis of oritavancin against clinical isolates of methicillin-resistant Staphylococcus aureus with reduced susceptibility to daptomycin. Diagn Microbiol Infect Dis. 2011;71(4):470–3.
Belley A, Arhin FF, Sarmiento I, et al. Pharmacodynamics of a simulated single 1200-milligram dose of oritavancin in an in vitro pharmacokinetic/pharmacodynamic model of methicillin-resistant Staphylococcus aureus infection. Antimicrob Agents Chemother. 2013;57(1):205–11.
Belley A, Lalonde-Seguin D, Arhin D, et al. Comparison of the pharmacodynamics (PD) of the long-acting lipoglycopeptides oritavancin (ORI) and dalbavancin (DAL) against methicillin-resistant Staphylococcus aureus (MRSA) in an in vitro pharmacokinetic (PK)/PD model (IVPM) [abstract no. A-491]. In: 55th Interscience Conference on Antimicrobial Agents and Chemotherapy. 2015.
Belley A, Lalonde-Seguin D, Arhin F, et al. In vitro bactericidal activity of oritavancin, dalbavancin and vancomycin against non-dividing methicillin-resistant Staphylococcus aureus (MRSA) [abstract no. A-493]. In: 55th Interscience Conference on Antimicrobial Agents and Chemotherapy. 2015.
Van Bambeke F, Carryn S, Seral C, et al. Cellular pharmacokinetics and pharmacodynamics of the glycopeptide antibiotic oritavancin (LY333328) in a model of J774 mouse macrophages. Antimicrob Agents Chemother. 2004;48(8):2853–60.
Sweeney BD, Shinabarger DL, Pillar CM, et al. Comparison of the in vitro bactericidal activity of seven agents against MRSA [abstract no. D-1173]. In: 55th Interscience Conference on Antimicrobial Agents and Chemotherapy. 2015.
Belley A, Neesham-Grenon E, Arhin FF, et al. Assessment by time-kill methodology of the synergistic effects of oritavancin in combination with other antimicrobial agents against Staphylococcus aureus. Antimicrob Agents Chemother. 2008;52(10):3820–2.
Arhin FF, Moeck G, Draghi DC, et al. Longitudinal analysis of the in vitro activity profile of oritavancin and comparator glycopeptides against Gram-positive organisms from Europe: 2005–2008. Int J Antimicrob Agents. 2010;36(5):474–6.
Bhavnani SM, Hammel JP, Rubino CM, et al. Oritavancin (ORI) pharmacokinetic-pharmacodynamic (PK-PD) analyses for efficacy based on data from patients with acute bacterial skin and skin structure infections (ABSSSI) enrolled in SOLO I and II [abstract no. A-1309]. In: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy. 2014.
Ambrose PG, Drusano GL, Craig WA. In vivo activity of oritavancin in animal infection models and rationale for a new dosing regimen in humans. Clin Infect Dis. 2012;54(Suppl 3):S220–8.
Lehoux D, Laquerre K, Ostiguy V, et al. A single 1200 mg human equivalent dose of oritavancin is highly efficacious in the neutropenic mouse thigh infection model [abstract no. P1568]. In: 20th European Congress of Clinical Microbiology and Infectious Diseases. 2010.
Rubino CM, Van Wart SA, Bhavnani SM, et al. Oritavancin population pharmacokinetics in healthy subjects and patients with complicated skin and skin structure infections or bacteremia. Antimicrob Agents Chemother. 2009;53(10):4422–8.
Rubino CM, Bhavnani SM, Moeck G, et al. Population pharmacokinetic analysis for a single 1200-milligram dose of oritavancin using data from two pivotal phase 3 clinical trials. Antimicrob Agents Chemother. 2015;59(6):3365–72.
Fetterly GJ, Ong CM, Bhavnani SM, et al. Pharmacokientics of oritavancin in plasma and skin blister fluid following administration of a 200-milligram dose for 3 days or a single 800-milligram dose. Antimicrob Agents Chemother. 2005;49:148–52.
Dunbar LM, Milata J, McClure T, et al. Comparison of the efficacy and safety of oritavancin front-loaded dosing regimens to daily dosing: an analysis of the SIMPLIFI trial. Antimicrob Agents Chemother. 2011;55(7):3476–84.
Corey GR, Good S, Jiang H, et al. Single-dose oritavancin versus 7–10 days of vancomycin in the treatment of Gram-positive acute bacterial skin and skin structure infections: the SOLO II noninferiority study. Clin Infect Dis. 2015;60(2):254–62.
LaPensee K, Fan W, Fiset C, et al. Efficacy and hospitalization length of stay of single dose oritavancin compared to 7–10 days of vancomycin in patients with acute bacterial skin and skin structure infections in the US and Eastern Europe [abstract no. PIN4]. Value Health. 2014;17(7):A664.
LaPensee K, Fan W, Lodise T, et al. Efficacy and safety of single dose oritavancin compared to 7–10 days of vancomycin in patients with acute bacterial skin and skin structure infections treated in the outpatient setting [abstract no. L-1729]. In: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy. 2014.
Corey R, Wikler M, Moeck M, et al. The SOLO studies: a single-dose of oritavancin (ORI) compared to 7–10 days of vancomycin (VAN) in the treatment of acute bacterial skin and skin structure infections (ABSSSI) [abstract no. 1331]. In: Infectious Diseases Week Meeting. 2014.
Moeck G, Arhin FF, Jiang H, et al. Microbiological responses by pathogen in phase 3 studies of single dose oritavancin for acute bacterial skin and skin structure infection (ABSSSI) [abstract no. L-1725]. In: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy. 2014.
Holland TL, Wikler M, Moeck G, et al. A single dose of oritavancin (ORI) compared to 7–10 days of vancomycin (VAN) in the treatment of acute bacterial skin and skin structure infections (ABSSSI): an analysis of the MRSA subpopulation in the SOLO non-inferiority studies [abstract no. 170]. J Hosp Med. 2014;9(Suppl 2).
Corey GR, Good S, Jiang H, et al. Concordance between early (48–72 hours) and late (7–14 days post therapy) clinical response: an analysis from the SOLO non-inferiority clinical studies [abstract no. eP418]. In: 24th European Congress of Clinical Microbiology and Infectious Diseases. 2014.
Pollack CV Jr, Corey GR, Good S, et al. Efficacy outcomes by lesion type in studies of a single dose of oritavancin compared to 7 to 10 days of vancomycin [abstract no. 252]. Ann Emerg Med. 2014;64(Suppl 4):S90.
Holland TL, Fan W, Wikler ME, et al. Treatment of acute bacterial skin and skin structure infections in the hospital and outpatient settings: analysis of oritavancin efficacy using Eron severity classification [abstract no. 323 plus poster]. J Hosp Med. 2015;10(Suppl 2).
LaPensee K, Lodise TP, Fan W, et al. Comparison of early achievement of >20% reduction in lesion size of acute bacterial skin and skin structure infections (ABSSSI) between patients who received oritavancin versus vancomycin in SOLO I [abstract no. 1329 plus poster]. In: Infectious Diseases Week Meeting. 2013.
Corey R, O’ riordan W, Huang N, et al. Time to onset and duration of adverse events in patients with acute bacterial skin and skin structure infections treated with oritavancin—the SOLO studies [abstract no. 685]. In: IDWeek. 2014.
Targanta Therapeutics Corporation. Nuvocid® (oritavancin diphosphate for injection) for treatment of complicated skin and skin structure infections. 2008. http://www.fda.gov/. Accessed 2 Sep 2015.
Scott LJ. Dalbavancin: a review in acute bacterial skin and skin structure infections. Drugs. 2015;75(11):1281–91.
Theravance Biopharma US Inc. Vibativ® (telavancin) for injection, for intravenous use: US prescribing information. 2009. http://www.vibativ.com. Accessed 26 Jun 2015.
US FDA. Vancomycin hydrochloride injection, solution: US prescribing information. 2008. http://www.fda.gov. Accessed 29 Jun 2015.
Durata Therapeutics US Ltd. Dalvance™ (dalbavancin) for injection, for intravenous use: US prescribing information. 2014. http://www.dalvance.com. Accessed 26 Jun 2015.
Thom H, Thompson JC, Scott DA, et al. Comparative efficacy of antibiotics for the treatment of acute bacterial skin and skin structure infections (ABSSSI): a systematic review and network meta-analysis. Curr Med Res Opin. 2015;31(8):1539–51.
Wu C, Fortier KJ, LaPensee K, et al. A US Hospital economic impact model for oritavancin in ABSSSI patients with risk of MRSA infections [abstract no. PSS10]. Value Health. 2014;17(7):A605.
Wu C, Jensen IS, Cyr PL, et al. Use of oritavancin for the treatment of skin and soft tissue infections: a UK hospital budget impact analysis [abstract no. PIN32]. Value Health. 2015;18(3):A233.
European Committee on Antimicrobial Susceptibility Testing. Addendum: clinical breakpoints and QC recommendations for new agents dalbavancin, oritavancin and tedizolid. 2015. http://www.eucast.org. Accessed 1 Sep 2015.
Acknowledgments
During the peer review process, the manufacturer of oritavancin was also offered an opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Conflict of interest
Yahiya Syed and Lesley Scott are salaried employees of Adis/Springer, are responsible for the article content and declare no relevant conflicts of interest.
Additional information
The manuscript was reviewed by: N. E. Holmes, Department of Infectious Diseases, Austin Health, Heidelberg, Australia; R. N. Jones, JMI Laboratories, North Liberty, IA, USA; B. M. Lomaestro, Department of Pharmacy, Albany Medical Center Hospital, Albany, NY, USA; C. V. Pollack, Department of Emergency Medicine, Thomas Jefferson University, Philadelphia, PA, USA; K. A. Rodvold, Colleges of Pharmacy and Medicine, University of Illinois at Chicago, Chicago, IL, USA; F. Van Bambeke, Pharmacologie cellulaire et moléculaire, Louvain Drug Research Institute, Université catholique de Louvain, Brussels, Belgium.
Rights and permissions
About this article

Cite this article
Syed, Y.Y., Scott, L.J. Oritavancin: A Review in Acute Bacterial Skin and Skin Structure Infections. Drugs 75, 1891–1902 (2015). https://doi.org/10.1007/s40265-015-0478-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-015-0478-7