
Abstract
Intravenous dalbavancin (Dalvance™; Xydalba™) is approved for use in adult patients with acute bacterial skin and skin structure infections (ABSSSI), with the recommended regimen being a 1000 mg dose followed 1 week later by a 500 mg dose. In the multinational DISCOVER 1 and 2 trials in adult patients with ABSSSI, dalbavancin treatment was noninferior to vancomycin (for ≥3 days with an option to switch to oral linezolid to complete a 10- to 14-day course) in terms of early clinical success rates (assessed 48–72 h after initiation of treatment; primary endpoint required by the FDA to assess noninferiority in registration trials of ABSSSI). Clinical response rates were also similar in both treatment groups at the end of treatment (day 14–15), irrespective of geographic region or baseline characteristics, including by infection type, diabetes mellitus status, systemic inflammatory response syndrome status, causative pathogen and renal function. Dalbavancin was generally well tolerated, with adverse events generally being of mild to moderate intensity and transient. With its broad spectrum of activity against clinically relevant Gram-positive pathogens and its favourable pharmacokinetic profile that permits a convenient two-dose, once-weekly regimen with no requirement for therapeutic drug monitoring, dalbavancin is a promising emerging option for the treatment of ABSSSI in adult patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Excellent in vitro activity against a broad spectrum of Gram-positive bacteria, including methicillin-resistant and methicillin-susceptible Staphylococcus aureus, and streptococci bacteria |
No evidence for the emergence of dalbavancin-resistant isolates |
Intravenous (IV) 30-min infusion; convenient two-dose, once-weekly regimen, with good tissue penetration (e.g. into skin blister fluid, bone and articular cartilage) |
Noninferior to IV vancomycin (for ≥3 days with an option to switch to oral linezolid) for clinical success rate 48–72 h after initiation of treatment |
At the end of therapy (day 14–15), the clinical success rate is similar to that with vancomycin-linezolid |
Generally well tolerated |
1 Introduction
Acute bacterial skin and skin structure infections (ABSSSI) involve the skin and subcutaneous tissue, fascia and or muscle layers, with disease severity ranging from mild, superficial infections to severe, potentially life-threatening infections [1]. Key pathogens associated with ABSSSI include Gram-positive organisms such as Staphylococcus aureus and Streptococcus pyogenes strains and, particularly in polymicrobial infections, Gram-negative and anaerobic bacteria [2]. The increasing global prevalence of drug-resistant pathogens associated with community-acquired and nosocomial Gram-positive infections, especially staphylococci and enterococci strains, has added to the challenge of treating these infections [2–6].
Vancomycin (glycopeptide antibacterial) and teicoplanin (first generation lipoglycopeptide antibacterial, a subclass of the glycopeptides; not approved in USA) have been the mainstay of parenteral antibacterial therapy for serious drug-resistant Gram-positive infections, including methicillin-resistant S. aureus (MRSA) infections [1, 5–7]. However, in addition to an increased risk of nephrotoxicity with high doses of vancomycin and the need for therapeutic drug monitoring, its use is potentially limited by the emergence of vancomycin-intermediate and -resistant S. aureus and enterococci strains (albeit these isolates remain relatively uncommon), with vancomycin-resistant isolates associated with treatment failures and poor clinical outcomes [5, 6, 8]. Antibacterial drugs developed to overcome glycopeptide resistance, such as daptomycin and linezolid, have also been associated with the emergence of resistance [5, 8].
The increasing prevalence of multidrug-resistant pathogens, including vancomycin-resistant pathogens, led to the development of the second generation lipoglycopeptides (e.g. dalbavancin, oritavancin and telavancin; all administered intravenously) [3, 4]. The presence of lipophilic/hydrophobic side chains mean that, relative to vancomycin, lipoglycopeptide antibacterials exhibit increased in vitro potency against Gram-positive bacteria and a low potential for the emergence of resistance. All lipoglycopeptides are bactericidal, whereas vancomycin exhibits mainly bacteriostatic activity [3, 4]. Intravenous dalbavancin (Dalvance™; Xydalba™) has recently been approved in the USA [9] and EU [10] for the treatment of adult patients with ABSSSI. This article provides a narrative review of its clinical use in adult patients with ABSSSI and overviews the pharmacological properties and in vitro antibacterial activity of the drug.
2 Pharmacodynamic Properties of Dalbavancin
Dalbavancin, a semisynthetic lipoglycopeptide, binds to the C terminal d-alanyl-d-alanine subgroup of the stem pentapeptide in nascent cell wall peptidoglycan and thereby inhibits cell wall synthesis by preventing transglycosylation and transpeptidation of the peptidoglycan chains [3, 4]. As a consequence, the integrity of the cell membrane is disrupted, leading to rapid cell death [3]. The addition of a lipophilic side chain prolongs the half-life of dalbavancin (Sect. 3), anchors the drug to the cell membrane, improves its affinity for the d-alanyl-d-alanine subgroup and enhances its antibacterial activity (Sect. 2.1.1).
2.1 Antibacterial Activity
2.1.1 In Vitro Activity
This section focuses on the antibacterial activity of dalbavancin against Gram-positive microorganisms associated with ABSSSI specified in the US manufacturer’s prescribing information [i.e. S. aureus, including MRSA and methicillin-susceptible S. aureus (MSSA), S. pyogenes, Streptococcus agalactiae and Streptococcus anginosus group (including S. anginosus, S. intermedius and S. constellatus)] [9]. Susceptibility is based on the US FDA-approved interpretative breakpoint (≤0.12 μg/mL) against these indicated species/groups [9, 11]; the current absence of data on resistant isolates precludes defining any other category [9]. The European Committee on Antimicrobial Susceptibility Testing interpretative breakpoint for dalbavancin for Staphylococcus spp., β-haemolytic streptococci of Groups A, B, C and G, and S. anginosus group for susceptibility is ≤0.125 μg/mL, with a resistant breakpoint of >0.125 μg/mL [10]. Dalbavancin shows no antibacterial activity against Gram-negative organisms [10]. Only studies that utilized microbroth dilution techniques are discussed, since disk diffusion is currently not considered to be a reliable method for determining the in vitro activity of dalbavancin [9].
Dalbavancin exhibits bactericidal activity [3, 4, 12–14] and demonstrates excellent in vitro activity against a broad spectrum of Gram-positive clinical isolates collected since 2002 as part of global [15, 16], US [11, 17, 18] (one [11] of which was a pooled analysis of isolates collected in the other two studies [17, 18]) and Canadian [19] surveillance studies (Table 1). In these surveillance studies, based on the minimum inhibitory concentration (MIC) required to inhibit the growth of 90 % of isolates (MIC90), dalbavancin exhibited more potent in vitro activity than vancomycin against these targeted strains; however, more than 99 % of clinical isolates were susceptible to both drugs [15, 16, 19]. In the largest study, MIC90 values for against both MSSA (n = 27,052) and MRSA (n = 19,721) isolates were 0.06 μg/mL for dalbavancin and 1 μg/mL for vancomycin [15].
In vitro activity data for clinical isolates collected in the phase 3 DISCOVER trials (Sect. 4.1) were consistent with those in the surveillance studies [20]. MIC90 values for dalbavancin were 0.06 μg/mL against S. aureus (n = 511), MSSA (n = 361), MRSA (n = 135) and S. pyogenes (n = 77), 0.03 μg/mL against S. agalactiae (n = 20) and 0.015 μg/mL against S. constellatus (n = 25) [20].
Dalbavancin also showed very good in vitro activity against clinical isolates of vancomycin-susceptible Enterococcus faecium and vancomycin-susceptible Enterococcus faecalis isolates [11, 15, 18]. At least 90 % of E. faecium and E. faecalis vancomycin-susceptible isolates exhibited an in vitro MIC less than or equal to the dalbavancin susceptibility breakpoint of ≤0.12 μg/mL [9]. The clinical significance of these in vitro data remains to be determined, since there are currently no adequate well-controlled clinical studies of the efficacy and safety of dalbavancin in clinical infections caused by these species [9]. Dalbavancin does not exhibit significant in vitro activity against vancomycin-resistant enterococci containing vanA gene mutations [3, 5]. The reduced in vitro susceptibility of dalbavancin and most other lipoglycopeptides (except oritavancin) to VanA-type but not VanB-type resistant enterococci, reflects that unlike vancomycin which activates both the vanA and vanB genes, lipoglycopeptides only activate the vanA gene [3, 5].
Currently, there is no evidence for the emergence of bacterial isolates resistant to dalbavancin, based on in vitro, animal and clinical studies [9, 13, 20]. More than 99 % of clinical isolates collected over the past decade were susceptible to dalbavancin (Table 1). In the DISCOVER trials, relative to baseline, no Gram-positive isolates identified after treatment with dalbavancin or vancomycin exhibited a greater than twofold increase in in vitro susceptibility [20].
In broth microdilution checkerboard assays, no synergistic or antagonist interactions were observed between dalbavancin and aztreonam, clindamycin, gentamicin, levofloxacin, linezolid, quinupristin/dalfopristin, rifampin or vancomycin [21], or with azithromycin [22]. Dalbavancin was synergistic or partially synergistic with oxacillin for staphylococci, including against methicillin-resistant strains and vancomycin-intermediate S. aureus isolates [21].
2.1.2 Activity in Animal Models
The in vitro activity of dalbavancin is supported by evidence from animal models of S. aureus infections [23–25], including models of MRSA infection [24] and of infection with S. aureus with or without reduced susceptibility to vancomycin and teicoplanin [23]. For example, in a rat granuloma pouch infection model, within 24 h of administration of a single intravenous dalbavancin 10 mg/kg dose, viable MRSA counts were reduced by >2 log colony forming units (CFU)/mL and MSSA counts to below the limits of detection [24]. No bacterial regrowth of MRSA was observed for up to 120 h, with no regrowth of MSSA for at least 96 h [24].
2.1.3 Pharmacodynamic/Pharmacokinetic Considerations
The inter-relationships between pharmacodynamic and pharmacokinetic parameters are an important consideration in predicting antibacterial activity [26]. Glycopeptides typically exhibit concentration-dependent bactericidal activity, with the maximum plasma concentration (Cmax):MIC ratio and the area under the concentration-time curve (AUC) from time zero to 24 h:MIC ratio shown to correlate with efficacy in in vitro studies and animal models [25–28].
There was a significant (p < 0.05) correlation between a successful clinical response at the test-of-cure (TOC; 14 ± 2 days post treatment) visit and the average AUC (AUCavg):MIC ratio, using data from a phase 3 study in patients with ABSSSI for the MIC values and a population pharmacokinetic model [29]. At the TOC visit, 89.3 and 98.4 % of patients achieved clinical success at AUCavg:MIC ratios of <21,267 and ≥21,267, respectively, in the overall population (n = 192 evaluable). Similarly, in a subgroup of patients with S. aureus infections at baseline (n = 177), 89.1 and 100 % of patients achieved clinical success at these respective AUCavg:MIC ratios. The AUCavg was derived by dividing the AUC from time zero to 120 h by 5 [29].
2.2 Effects on Cardiac Electrophysiology
In a thorough QT study in healthy volunteers, a 30-min intravenous infusion of dalbavancin 1000 or 1500 mg had no clinically relevant effects on heart rate, the PR or QRS intervals or the corrected QT (QTc) interval [30].
3 Pharmacokinetic Properties of Dalbavancin
The pharmacokinetics of dalbavancin are best described using a three-compartment model (α and β distribution phases followed by a terminal elimination phase) [10]. The distributional half-life, which constitutes most of the clinically-relevant concentration-time profile, ranged from 5 to 7 days and is consistent with once-weekly dosing [10].
In healthy adult volunteers, dalbavancin exhibits linear, dose-proportional pharmacokinetics following single intravenous doses of 140–1120 mg (30-min infusion) [14]. In two phase 1 studies, mean Cmax values following a single dalbavancin 1000 mg dose were 248.8 and 287.3 mg/L [31]. There was no apparent accumulation of dalbavancin following multiple doses of dalbavancin (1000 mg on day 1 and then 7 weeks of once-weekly 500 mg doses) [32]. Minimum plasma concentrations of dalbavancin on day 22 were similar to those observed on day 8 (i.e. at end of the dosing interval prior to the weekly dose), indicating that steady-state concentrations of the drug were attained by day 8 [32].
Dalbavancin is extensively (≈93 %) and reversibly bound to human plasma proteins, primarily to albumin [9]. The extent of protein binding is not altered by the concentration of dalbavancin, or renal or hepatic impairment [9]. Based on a population pharmacokinetic analysis using data from three clinical trials in patients with ABSSSI, the estimated central, peripheral and steady-state volumes of distribution of dalbavancin were 4.03, 11.8 and 15.9 L, respectively [33].
After intravenous infusion, dalbavancin shows good tissue penetration, including into skin blister fluid [34], bone [32] and articular cartilage [32]. The mean penetration of dalbavancin into cantharidin-induced skin blisters was 59.6 % after a single 1000 mg dose in healthy adult volunteers [34], with mean blister fluid concentrations (>30.3 mg/L at 7 days post dose) exceeding the MIC90 values for target bacteria associated with ABSSSI (including for drug-resistant staphylococci and streptococci strains) (Table 1) [34]. Plasma dalbavancin concentrations were also maintained above the relevant MIC90 values for at least 12 days in patients with ABSSSI treated with dalbavancin 1000 mg on day 1 and 500 mg on day 8 [35]. In adults scheduled for elective orthopaedic surgery, mean concentrations of dalbavancin in the bone, skin and articular cartilage are predicted to exceed the MIC90 of S. aureus strains [32]. After a single 1000 mg dose, mean dalbavancin concentrations at 12 h in the synovium, synovial fluid, bone and skin were 25.0 μg/g, 22.9 μg/mL, 6.3 μg/g and 19.4 μg/g, respectively, with corresponding concentrations at day 14 of 15.9 μg/g, 6.2 μg/mL, 4.1 μg/g and 13.8 μg/g [32].
Metabolites of dalbavancin have not been observed in significant amounts in human plasma [9, 10]. The metabolites hydroxy-dalbavancin and mannosyl aglycone have been detected in the urine (<25 % of administered dose); the metabolic pathways for these metabolites have not been identified. After a single 1000 mg dose, ≈20 % of the drug was eliminated in the faeces over the 70-day post dose period. An average of 19–33 % of the dose was excreted in the urine as unchanged drug and 8–12 % as hydroxy-dalbavancin over the 42-day post dose period. Dalbavancin has a prolonged terminal elimination half-life (mean 14.4–15.5 days) [9, 10].
Dalbavancin has a low potential for drug-drug interactions, based on preclinical studies [9]. In a population pharmacokinetic analysis, dalbavancin pharmacokinetics were not affected by coadministration of known CYP substrates, inducers or inhibitor or by individual drugs such as acetaminophen, aztreonam, fentanyl, metronidazole, furosemide, proton pump inhibitors, midazolam or simvastatin [9].
3.1 In Specific Populations
In adults, there are no clinically relevant effects of gender or age on the pharmacokinetics of dalbavancin [9, 10]. The pharmacokinetics of dalbavancin in children aged <12 years have not been established [9, 10]. In children aged 12–17 years, dalbavancin-exposure profiles were slightly lower (≈30 % lower) than those observed in adults with ABSSSI (historical data) [36].
Compared with adults with normal renal function, mean plasma clearance of dalbavancin was reduced by 35 and 47 % in adults with moderate [creatinine clearance (CRCL) 30–49 mL/min] and severe renal impairment (CRCL <30 mL/min) [9, 10, 31]. No dosage adjustments of dalbavancin are required in patients with a CRCL of >30 mL/min and in those on haemodialysis [9, 10]. The dosage of dalbavancin should be reduced in patients with severe renal impairment (CRCL <30 mL/min) who are not receiving regularly scheduled haemodialysis (Sect. 6) [9, 10].
The pharmacokinetic profile of dalbavancin was not altered to a clinically relevant extent in patients with varying degrees of hepatic impairment compared with adults with normal hepatic function [9, 10, 31]. Clinical experience in patients with moderate or severe hepatic impairment is limited; thus, caution is advised when dalbavancin is prescribed in these patient populations [9, 10].
4 Therapeutic Efficacy of Dalbavancin
The efficacy of intravenous dalbavancin versus intravenous vancomycin (for ≥3 days with an option to switch to oral linezolid thereafter; i.e. the vancomycin-linezolid group) [37] or oral linezolid [38] in the treatment of adult patients with an ABSSSI [37] or complicated skin and skin structure infections (CSSSI) [38] was investigated in three double-blind, double-dummy, multinational, phase 3, noninferiority trials. Two of these trials (DISCOVER 1 and 2) are reported in the same publication, along with pooled and prespecified subgroup analyses of these two identically designed trials [37]. Other pooled prespecified exploratory and post hoc subgroup analyses of DISCOVER 1 and 2 [39–43] are also briefly discussed (available as abstract presentations).
Discussion focuses on the DISCOVER 1 and 2 trials (Sect. 4.1), which used the recent US FDA recommended timepoint (i.e. 48–72 h after initiation of treatment; early success) to assess clinical response for determination of noninferiority in registration ABSSSI trials [37]. Historical timepoints for assessing clinical response rates are at the end of treatment (EOT) or thereafter, at the test-of-cure (TOC) visit (typically on days 14–28) [37]. The initial phase 3 trial (VER001-09) [38] of dalbavancin treatment in patients with CSSSI evaluated clinical response rates at the EOT and TOC visits (reviewed previously in Drugs [44]), with the clinical response rate at the TOC visit in the dalbavancin group noninferior to that in the linezolid group (primary endpoint) [88.9 vs. 91.2 %; the lower limit of 95 % CI was within the prespecified noninferiority criterion]. All phase 3 trials assessed clinical success rates at the EOT and TOC/short-term follow-up (SFU) visits [37, 38].
4.1 DISCOVER 1 and 2 Trials
In the DISCOVER trials, key eligibility criteria included a diagnosis of ABSSSI as determined by the presence of cellulitis, a major abscess or a wound infection, each of which were associated with ≥75 cm2 of erythema [37]. Eligible patients were adults who were thought to require 3 days of intravenous therapy and had one or more systemic signs of infection within 24 h prior to randomization, including a body temperature of >38 °C, a white cell count of >12,000 cells/mm3 or more than 10 % band forms on the white-cell differential count. At least two of the following local signs had to be present in addition to erythema: purulent drainage or discharge, fluctuance, heat or localized warmth, tenderness on palpation, and swelling or induration. Patients were excluded if they had received antibiotic treatment within 14 days prior to randomization. Patients were randomized to two doses of dalbavancin (1000 mg then 500 mg) given one week apart or twice-daily vancomycin for ≥3 days, with vancomycin recipients having an option to switch to oral linezolid twice daily thereafter; treatment continued for 10–14 days [37].
Within each trial, there were no statistically significant between-group differences in patient characteristics or disease status at baseline, with the exception of the proportion of patients with diabetes mellitus enrolled in DISCOVER 2 (9.4 % in the dalbavancin group vs. 16.8 % in the vancomycin-linezolid group; p = 0.003) [37]. At baseline, across both trials, the mean age of patients was ≈49.6 years; 13 % of patients had diabetes; ≈25.6 % had major abscess infections, ≈53.6 % had cellulitis and ≈20.8 % had wound or surgical site infections; ≈51.2 % had systemic inflammatory response syndrome (SIRS). The median size of the infected area was 351 cm2 in DISCOVER 1 and 336 cm2 in DISCOVER 2. The primary endpoint was the success rate 48–72 h after initiating therapy (i.e. early clinical response) in the intent-to-treat (ITT) population (Table 2) [37].
Early clinical response rates in the dalbavancin group were noninferior to the vancomycin-linezolid group in both the individual studies and in pooled analyses (primary endpoint), with ≥77 % of patients in both treatment groups achieving early success indicative of a clinical response (Table 2) [37]. In prespecified sensitivity analyses of the primary endpoint in the ITT population, the proportion of patients with a ≥20 % reduction in lesion size at 48–72 h was similar in the dalbavancin and vancomycin-linezolid groups in DISCOVER 1 (89.9 vs. 90.9 %; absolute difference −1.0; 95 % CI −5.7 to 4.0), DISCOVER 2 (87.6 vs. 85.9 %; absolute difference 1.7; 95 % CI −3.2 to 6.7) and for the pooled data (88.6 vs. 88.1 %; absolute difference 0.6; 95 % CI −2.9 to 4.1) [37]. In prespecified pooled subgroup analyses for the primary outcome, there was also no between-group differences in terms of early clinical response rates by geographic region [43] or baseline infection type [37] (Table 2). The most common reason for patients being classified as treatment failures 48–72 h after initiating treatment was missing data (41 % of 134 treatment failures in the dalbavancin group vs. 39 % of 132 treatment failures in the vancomycin-linezolid group). The definition of missing data included no fever, but temperature protocol-specified criteria not met (i.e. temperature not recorded or taken outside the protocol-specified time window). Both groups had a similar pattern of reasons for treatment failure [37].
There were also no significant differences between the dalbavancin and vancomycin-linezolid group for the secondary outcome of clinical status at the EOT (day 14–15) in individual trials and/or across trials, including in the pooled clinical per-protocol (CPP) population (Table 2), by infection type (Table 2; CPP population), by geographic region (Table 2) [43], by diabetes status at baseline (CPP population), by SIRS status at baseline (CPP population) or by baseline pathogen (assessed in a subset of patients with monomicrobial infections who were in the microbiological per-protocol population; the latter included patients who had ≥1 Gram-positive pathogen isolated at baseline) [37]. For example in pooled analyses, clinical response rates in the dalbavancin and vancomycin-linezolid groups were similar in those with S. aureus (97.9 vs. 96.6 %; n = 191 and 177), MRSA (97.3 vs. 98.0 %; n = 74 and 50) or S. pyogenes (100 vs. 92.3 %; n = 19 and 13) infections at baseline, in patients with diabetes (84.5 vs. 88.2 %; n = 71 and 76) or without diabetes (91.6 vs. 92.7 %; n = 499 and 469) at baseline and in patients with SIRS (86.8 vs. 90.7 %; n = 296 and 290) or without SIRS (94.9 vs. 93.7 %; n = 274 and 255) at baseline [37].
Based on a pooled analysis of DISCOVER 1 and 2 that evaluated the concordance of an early clinical response with clinical success at the EOT, 90.3 % of patients (945 of 1046 patients) who achieved an early clinical response and 70.9 % (129 of 182 patients) who failed to achieve an early clinical response were cured at the EOT [42]. Positive predictive values for an early clinical response predicting clinical success at the EOT exceeded 90 %, irrespective of the early response parameters utilized (i.e. cessation of spread ± absence of fever; cessation of spread + worsening pain; absence of fever; ≥20 % reduction in lesion size). Conversely, the cessation of lesion spread with an assessment of pain (negative predictive value of 80 %) could potentially predict which patients will ultimately fail therapy [42].
With dalbavancin treatment, early clinical response rates did not differ significantly based on baseline renal function, including between patients with normal renal function and those with varying degrees of renal impairment (80 vs. 80–82 % of patients achieved an early clinical response), in post hoc pooled analyses of the DISCOVER trials [41]. In patients with a CRCL of <30, 30–59, 60–89 or ≥90 mL/min (n = 35, 249, 392 and 627, respectively), there were also no significant differences in early clinical response rates between the dalbavancin and vancomycin-linezolid group. Respective between-group differences based on renal function were 6.7 % (95 % CI −21.4 to 36.3), 5.4 % (95 % CI −5.1 to 15.9), −2.9 % (95 % CI −10.4 to 4.6) and 0.8 % (95 % CI −5.5 to 7.1) [41].
Clinical response rates at the EOT were also similar in the dalbavancin and vancomycin-linezolid groups in patients who were treated in the outpatient setting for all doses (90.6 vs 90.5 %; n = 138 and 126) [40]. In this post hoc analysis of the DISCOVER trials, outpatient treatment occurred more frequently in North America than in Europe, Asia and South Africa (99.6 vs. 0.4 % of outpatients; no p value reported) [40].
At the EOT, clinical response rates were similar between the dalbavancin and vancomycin group in patients who had received intravenous therapy (i.e. ≥10 days’ dalbavancin or vancomycin + oral placebo) without switching to oral treatment in a post hoc analysis of the DISCOVER trials (68.9 vs. 66.7 %; n = 61 and 54 ITT) [39].
In a pooled analysis of three phase 3 trials in patients with ABSSSI and a phase 2 study in patients with catheter-related infections, dalbavancin treatment resulted in clearance of bacteremia and clinical response rates that were similar to those in the comparator arms at the EOT in evaluable patients who had Gram-positive bacteremia at baseline (abstract presentation) [45]. For example, in patients with S. aureus bacteremia at baseline, 100 and 95 % of patients in the dalbavancin (n = 24) and comparator (n = 20) groups had documented clearance of S. aureus bacteremia at the EOT, with clinical success rates of 86.4 and 78.3 % [45].
At the SFU (days 26–30), there was no significant between-group difference in clinical response rates (i.e. clinical status; as defined for EOT in Table 2) [9]. In ITT analyses, clinical response rates at the SFU in the dalbavancin (n = 288) and vancomycin-linezolid (n = 285) group were 83.7 and 88.1 % in DISCOVER 1, with respective rates of 88.1 and 84.5 % (n = 371 and 368) in DISCOVER 2 [9].
5 Tolerability of Dalbavancin
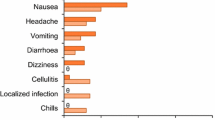
Dalbavancin was generally well tolerated in patients with ABSSSI participating in phase 2 and 3 clinical trials, with most adverse events being of mild to moderate severity and transient (median duration 4 vs. 3 days in the vancomycin-linezolid group [37]) [37, 38]. In a pooled analysis of seven phase 2 and 3 trials, the most common selected adverse reactions (incidence >2 %) occurring in the dalbavancin (n = 1778) and comparator (n = 1224) groups were nausea (5.5 vs. 6.4 %), headache (4.7 vs. 4.8 %), diarrhoea (4.4 vs. 5.9 %), vomiting (2.8 vs. 3.0 %), rash (2.7 vs. 2.4 %) and pruritus (2.1 vs. 3.3 %) [9]. The median duration of adverse reactions was 4 days in both the dalbavancin and comparator groups, with 3.0 and 2.8 % of patients discontinuing treatment because of an adverse reaction; the comparator drugs included linezolid, cefazolin, cephalexin and vancomycin. Serious adverse events occurred in 6.1 % of patients receiving dalbavancin and 6.5 % of patients in the comparator arms [9].
There was a numerically lower incidence of nephrotoxicity during intravenous dalbavancin therapy than during intravenous vancomycin therapy (3.3 vs. 9.3 %; p = 0.06; n = 637 and 54 evaluable), based on a pooled subgroup ITT analysis of patients who received ≥10 days of intravenous vancomycin without switching to oral linezolid in DISCOVER 1 and 2 (abstract presentation) [39]. Nephrotoxicity was defined as a 50 % increase from baseline in serum creatinine levels or an absolute increase in serum creatinine level of 0.5 mg/dL.
Overall, abnormalities in liver tests [alanine aminotransferase (ALT), aspartate aminotransferase (AST), bilirubin] occurred with a similar frequency in the dalbavancin and comparator groups in a pooled analysis of phase 2 and 3 trials [9]. In patients with normal ALT levels at baseline, elevations in ALT levels of >3× the upper limit of normal (ULN) occurred in 12 dalbavancin recipients (0.8 %) and two comparator recipients (0.2 %); amongst these patients, eight dalbavancin recipients and one comparator recipient had underlying conditions that could affect liver enzyme levels. Elevations in ALT levels of >10× ULN occurred in three patients in the dalbavancin group and no patients in the comparator group in these trials, with one dalbavancin recipient participating in a phase 1 trial having post baseline ALT levels of >20× ULN. All elevations in ALT levels were reversible [9]. Increased γ-glutamyl transferase levels were reported in 1.1 % of patients; the degree of increase was not stated [10].
6 Dosage and Administration of Dalbavancin
In the USA [9] and EU [10], intravenous dalbavancin is indicated for the treatment of adult patients with ABSSSI, with the US prescribing information [9] specifying infections caused by susceptible isolates of the following Gram-positive micro-organisms: S. aureus (including methicillin-susceptible and -resistant strains), S. pyogenes, S. agalactiae and S. anginosus group (including S. anginosus, S. intermedius, S. constellatus). To reduce the risk of drug-resistant bacteria and maintain the effectiveness of dalbavancin and other antibacterial drugs, dalbavancin should only be used to treat infections that are proven or strongly suspected to be caused by susceptible pathogens [9, 10].
The recommended initial dose of dalbavancin is 1000 mg followed 1 week later by a 500 mg dose, administered as a 30-min infusion [9, 10]. In patients with a CLCR of <30 mL/min and not receiving regularly scheduled haemodialysis, the dosage of dalbavancin should be reduced to 750 mg followed 1 week later by a dose of 375 mg. Dalbavancin is contraindicated in patients with a hypersensitivity to the drug and caution should be exercised when administering the drug to patients with a known hypersensitivity to glycopeptides. Rapid intravenous infusions of glycopeptide antibacterial drugs may cause a reaction that resembles Red-Man Syndrome; stopping or slowing the infusion may result in cessation of these reactions. If diarrhoea occurs during treatment, evaluate patients for potential Clostridium difficile infection, as cases of C. difficile-associated diarrhoea (CDAD) have been reported with almost all systemic antibacterial agents, including dalbavancin [9, 10].
7 Place of Dalbavancin in the Management of Acute Bacterial Skin and Skin Structure Infections
Current Infectious Diseases Society of America treatment guidelines for the management of severe ABSSSI recommend several first-line empirical treatment options, including vancomycin, linezolid, daptomycin, telavancin and clindamycin [1, 6]. The approval of dalbavancin for the treatment of adults with ABSSSI is too recent for the drug to have been considered for inclusion in these guidelines [9, 10]. Oritavancin, which is active against Gram-positive bacteria, was also recently approved in the USA [46] and the EU [47] for use in adult patients with ABSSSI. Antibacterials from other classes that have recently been approved for use in the treatment of ABSSSI, include drugs with activity against Gram-positive bacteria (daptomycin and tedizolid) and with activity against both Gram-positive and -negative bacteria (ceftaroline and tigecycline) [7].
Dalbavancin shows excellent in vitro activity against a broad spectrum of Gram-positive bacteria, including MRSA isolates, with good tissue penetration and a low potential for the emergence of bacterial resistance (Sect. 2.1.1). The prolonged distribution phase means that plasma concentrations of dalbavancin remain above the MIC of common ABSSSI pathogens for at least 7 days (Sect. 3) and this, along with the prolonged elimination half-life (Sect. 3), permits a convenient two-dose, once-weekly administration schedule (Sect. 6), with no requirement for therapeutic drug monitoring. The potential for using a single-dose dalbavancin regimen to treat ABSSSI is currently being investigated in a double-blind, multinational, phase 3, noninferiority trial (DUR001-303; n = 698), as per FDA guidelines for developing treatments for ABSSSI [48]. Preliminary results indicated that a single 1500 mg dose of dalbavancin was noninferior to the currently approved two-dose regimen (Sect. 6), with a similar proportion of patients achieving a ≥20 % decrease in lesion size 48–72 h after initiation of therapy (81.4 vs. 84.2 %; between group difference −2.9; 95 % CI −8.5 to 2.8) (primary endpoint) [48]. Oritavancin also has a convenient dosage regimen involving a single intravenous 3-h infusion [46], whereas vancomycin is administered twice daily [49] and telavancin once daily [50].
The potential for drug-drug interactions is an important factor when considering treatment options. Dalbavancin has a low potential for drug-drug interactions (Sect. 3) and does not alter the QTc interval (Sect. 2.2). Concomitant administration of vancomycin and local anaesthetics has been associated with anaphylactoid reactions, erythema and histamine-like flushing, and concurrent and/or sequential use of potentially nephrotoxic and/or neurotoxic drugs requires careful monitoring [49]. Telavancin treatment should be avoided in patients with QTc prolongation and should be used with caution in patients taking concomitant drugs known to prolong the QTc interval [50]. Like oritavancin (for up to 48 h post administration) [46], telavancin interferes with some laboratory coagulation tests, including prothrombin time, International Normalized Ratio and activated partial thromboplastin time [50]. Oritavancin should only be used in patients taking chronic warfarin if the benefits outweigh the risks of bleeding; the use of intravenous unfractionated heparin is contraindicated for 48 h after oritavancin administration [46].
In the large, multinational, DISCOVER 1 and 2 trials in adult patients with ABSSSI, dalbavancin was noninferior to vancomycin (for ≥3 days, with an option to switch to oral linezolid) in terms of early clinical response rates, with ≥77 % of patients in both treatment groups achieving early success indicative of a clinical response (Sect. 4.1). There was also no between-group difference in clinical response rates at the EOT, irrespective of geographic region or baseline characteristics, including by infection type, diabetes status, SIRS status, causative pathogen and renal function (Sect. 4.1). Early clinical success, the recently defined FDA endpoint for determining noninferiority in ABSSSI trials, showed concordance with cure at the EOT for positive predictive values (Sect. 4.1). Limited data also indicate that dalbavancin treatment may be effectively administered in the outpatient setting, with clinical response rates at the EOT in both groups of ≥90.5 % (Sect. 4.1).
Dalbavancin was generally well tolerated in adult patients with ABSSSI (Sect. 5). Most adverse events occurring during dalbavancin treatment were of mild to moderate intensity and transient, with 3 % of patients discontinuing treatment because of an adverse reaction. The most common adverse reactions (incidence ≥4 %) were nausea, headache and diarrhoea. Overall, liver test abnormalities were uncommon and occurred with a similar frequency in the dalbavancin and comparator arms in clinical trials. For instance, in patients with normal ALT levels at baseline, 0.8 % of dalbavancin recipients had an increase in ALT level of >3× ULN, all of which were reversible (Sect. 5).
Another important determinant in the choice of drug is its tolerability and safety, with specific safety issues differing between individual drugs. As with all glycopeptide antibacterials, cases of CDAD, hypersensitivity and rapid infusion reactions have been known to occur with dalbavancin treatment (Sect. 5). Vancomycin is also associated with an increased risk of ototoxicity and nephrotoxicity (in patients with renal dysfunction or taking concomitant aminoglycosides) [49]. Of interest, intravenous dalbavancin was associated with a numerically lower incidence of nephrotoxicity than intravenous vancomycin (≥10 days’ treatment) in a pooled subgroup analysis of DISCOVER 1 and 2 (Sect. 5). Telavancin has also been associated with an increased risk of nephrotoxicity and of new or worsening renal impairment and, based on data from animal studies, should not be used during pregnancy [50]. Oritavancin may be associated with an increased risk of osteomyelitis, with more cases of osteomyelitis reported in oritavancin than vancomycin recipients in phase 3 clinical trials in patients with ABSSSI [46]. Although dalbavancin has a prolonged half-life (Sect. 3), as is also the case with oritavancin (≈10 days) [46], the median duration of adverse reactions (4 days) during dalbavancin treatment was similar to that of comparators in clinical trials (Sect. 5).
In contemporary healthcare systems, pharmacoeconomic considerations play an important role in determining the choice of pharmacotherapy. The increasing prevalence of multidrug-resistant bacterial infections, including ABSSSI, has significant impacts on morbidity and mortality, and poses a considerable cost from a healthcare perspective [5]. Given its recent approval, robust pharmacoeconomic studies evaluating dalbavancin treatment in ABSSSI are currently lacking, albeit the convenient two-dose, once-weekly regimen and the potential for use of dalbavancin in the outpatient setting may reduce hospital costs and be more acceptable to patients.
In conclusion, in the multinational DISCOVER 1 and 2 registration trials, intravenous dalbavancin was an effective and generally well tolerated treatment in adult patients with ABSSSI. In these trials, dalbavancin was noninferior to vancomycin (3 days’ therapy with an option to switch to oral linezolid to complete a 10–14 day course) in terms of early clinical success rates. Clinical response rates were also similar in both treatment groups at the EOT, irrespective of geographic region or baseline characteristics, including by infection type, diabetes status, SIRS status, causative pathogen and renal function. Adverse events occurring during dalbavancin therapy were generally of mild to moderate intensity and transient, with the most common adverse reactions being nausea, headache and diarrhoea. With its broad spectrum of activity against clinically relevant Gram-positive pathogens and favourable pharmacokinetic profile that permits a convenient two-dose, once-weekly regimen with no requirement for therapeutic drug monitoring, dalbavancin is a promising emerging option for the treatment of ABSSSI in adult patients.
Data selection sources:
Relevant medical literature (including published and unpublished data) on dalbavancin was identified by searching databases including MEDLINE (from 1946), PubMed (from 1946) and EMBASE (from 1996) [searches last updated 9 Jun 2015], bibliographies from published literature, clinical trial registries/databases and websites. Additional information was also requested from the company developing the drug.
Search terms: Dalbavancin, Dalvance, Xydalba, skin.
Study selection: Studies in patients with skin and skin structure infections who received dalbavancin. When available, large, well designed, comparative trials with appropriate statistical methodology were preferred. Relevant pharmacodynamic and pharmacokinetic data are also included.
References
Stevens DL, Bisno AL, Chambers HF, et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update of the Infectious Diseases Society of America. Clin Infect Dis. 2014;59(2):147–59.
Moran GJ, Abrahamian FM, LoVecchio F, et al. Acute bacterial skin infections: developments since the 2005 Infectious Diseases Society of America (IDSA) guidelines. J Emerg Med. 2013;44(6):e-397–412.
Zhanel GG, Calic D, Schweizer F, et al. New lipoglycopeptides: a comparative review of dalbavancin, oritavancin and telavancin. Drugs. 2010;70(7):859–86.
Guskey MT, Tsuji BT. A comparative review of the lipoglycopeptides: oritavancin, dalbavancin, and telavancin. Pharmacotherapy. 2010;30(1):80–94.
Bailey J, Summers KM. Dalbavancin: a new lipoglycopeptide antibiotic. Am J Health Syst Pharm. 2008;65(7):599–610.
Liu C, Bayer A, Cosgrove SE, et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. 2011;52(3):e18–55.
Dryden MS. Alternative clinical indications for novel antibiotics licensed for skin and soft tissue infection. Curr Opin Infect Dis. 2015;28:117–24.
Rodvold KA, McConeghy KW. Methicillin-resistant Staphylococcus aureus therapy: past, present, and future. Clin Drug Investig. 2014;58(Suppl 1):S20–7.
Durata Therapeutics US Ltd. Dalvance (dalbavancin) for injection, for intravenous use: US prescribing information. 2014. http://www.dalvance.com/. Accessed 3 Mar 2015.
European Medicines Agency. Xydalba 500 mg powder for concentrate for solution for infusion: summary of product chracteristics. 2015 http://www.ema.europa.eu. Accessed 18 Mar 2015.
Jones RN, Farrell DJ, Flamm RK, et al. Surrogate analysis of vancomycin to predict susceptible categorization of dalbavancin. Diagn Microbiol Infect Dis. 2015;82(1):73–7.
Lin G, Credito K, Ednie LM, et al. Antistaphylococcal activity of dalbavancin, an experimental glycopeptide. Antimicrob Agents Chemother. 2005;49(2):770–2.
Lopez S, Hackbarth C, Romano G, et al. In vitro antistaphylococcal activity of dalbavancin, a novel glycopeptide. J Antimicrob Chemother. 2005;55(Suppl 2):ii21–4.
Leighton A, Gottlieb AB, Dorr MB, et al. Tolerability, pharmacokinetics, and serum bactericidal activity of intravenous dalbavancin in healthy volunteers. Antimicrob Agents Chemother. 2004;48(3):940–5.
Biedenbach DJ, Bell JM, Sader HS, et al. Activities of dalbavancin against a worldwide collection of 81,673 Gram-positive bacterial isolates. Antimicrob Agents Chemother. 2009;53(3):1260–3.
Streit JM, Fritsche TR, Sader HS, et al. Worldwide assessment of dalbavancin activity and spectrum against over 6,000 clinical isolates. Diagn Microbiol Infect Dis. 2004;48(2):137–43.
Jones RN, Sader HS, Flamm RK. Update of dalbavancin spectrum and potency in the USA: report from the SENTRY Antimicrobial Surveillance Program (2011). Diagn Microbiol Infect Dis. 2013;75(3):304–7.
Jones RN, Flamm RK, Sader HS. Surveillance of dalbavancin potency and spectrum in the United States (2012). Diagn Microbiol Infect Dis. 2013;76(1):122–3.
Karlowsky JA, Adam HJ, Poutanen SM, et al. In vitro activity of dalbavancin and telavancin against staphylococci and streptococci isolated from patients in Canadian hospitals: results of the CANWARD 2007-2009 study. Diagn Microbiol Infect Dis. 2011;69(3):342–7.
Dunne M, Boucher H, Wilcox MH, et al. Microbiologic analysis of target pathogens identified in the dalbavancin DISCOVER program [abstract no. 1338 plus poster]. In: Infectious Diseases Week Meeting. 2013.
Johnson DM, Fritsche TR, Sader HS, et al. Evaluation of dalbavancin in combination with nine antimicrobial agents to detect enhanced or antagonistic interactions. Int J Antimicrob Agents. 2006;27(6):557–60.
Koeth LM, DiFranco-Fisher JM, Dunne M, et al. Dalbavancin and azithromycin synergy/antagonism study by checkerboard MIC [abstract no. D-877]. In: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy. 2014.
Lefort A, Pavie J, Garry L, et al. Activities of dalbavancin in vitro and in a rabbit model of experimental endocarditis due to Staphylococcus aureus with or without reduced susceptibility to vancomycin and teicoplanin. Antimicrob Agents Chemother. 2004;48(3):1061–4.
Jabés D, Candiani G, Romano G, et al. Efficacy of dalbavancin against methicillin-resistant Staphylococccus aureus in the rat granuloma pouch infection model. Antimicrob Agents Chemother. 2004;48(4):1118–23.
Andes D, Craig WA. In vivo pharmacodynamic activity of the glycopeptide dalbavancin. Antimicrob Agents Chemother. 2007;51(5):1633–42.
Dorr MB, Jabes D, Cavaleri M, et al. Human pharmacokinetics and rationale for once-weekly dosing of dalbavancin, a semisynthetic glycopeptide. J Antimicrob Chemother. 2005;55(Suppl 2):ii25–30.
Dowell JA, Goldstein BP, Buckwalter M. Pharmacokinetic-pharmacodynamic modeling of dalbavancin, a novel glycopeptide antibiotic. J Clin Pharmacol. 2008;48(9):1063–8.
Bowker KE, Noel AR, MacGowan AP. Pharmacodynamics of dalbavancin studied in an in vitro pharmacokinetic system. J Antimicob Chemother. 2006;58(4):802–5.
Bhavnani SM, Hammel JP, Rubino CM, et al. Pharmacokinetic-pharmacodynamic (PK-PD) analyses for the efficacy of dalbavancin (DAL) using phase 3 data from patients with acute bacterial skin and skin structure infections (ABSSSI) [abstract no. A-1186]. In: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy. 2014.
Dunne MW, Zhou M, Darpo B. A thorough QT study with dalbavancin: a novel lipoglycopeptide antibiotic for the treatment of acute bacterial skin and skin-structure infections. Int J Antimicrob Agents. 2015;45(4):393–8.
Marbury T, Dowell JA, Seltzer E, et al. Pharmacokinetics of dalbavancin in patients with renal or hepatic impairment. J Clin Pharmacol. 2009;49:465–76.
Dunne MW, Puttagunta S, Sprenger CR, et al. Extended-duration dosing and distribution of dalbavancin into bone and articular tissue Antimicrob Agents Chemother. 2015;59(4):1849–55.
Buckwalter MS, Dowell JA. Population pharmacokinetic analysis of dalbavancin, a novel lipoglycopeptide. J Clin Pharmacol. 2005;45(11):1279–87.
Nicolau DP, Sun HK, Seltzer E, et al. Pharmacokinetics of dalbavancin in plamsa and skin blister fluid. J Antimicob Chemother. 2007;60(3):681–4.
Seltzer E, Dorr MB, Goldstein BP, et al. Once-weekly dlabavancin versus standard-of-care antimicrobial regimens for treatment of skin and soft-tissue infections. Clin Infect Dis. 2003;37(10):1298–303.
Bradley JSMD, Puttagunta SMD, Rubino CMP, et al. Pharmacokinetics, safety and tolerability of single dose dalbavancin in children 12 through 17 years of age. Pediatr Infect Dis J. 2014. doi:10.1097/INF.0000000000000646.
Boucher HW, Wilcox M, Talbot GH, et al. Once-weekly dalbavancin versus daily conventional therapy for skin infection. N Engl J Med. 2014;370(23):2169–79.
Jauregui LE, Babazadeh S, Seltzer E, et al. Randomized, double-blind comparison of once-weekly dalbavancin versus twice-daily linezolid therapy for the treatment of complicated skin and skin structure infections. Clin Infect Dis. 2005;41(10):1407–15.
Puttagunta S, Boucher H, Talbot G, et al. Dalbavancin vs vancomycin for the treatment of acute bacterial skin and skin structure infections (ABSSSI): a subanalysis from the DISCOVER studies [abstract no. 266]. Open Forum Infect Dis. 2014;1(Suppl 1):S113.
Dunne MW, Puttagunta S, Wilcox M, et al. Treatment of acute bacterial skin and skin structure infection (ABSSSI) with dalbavancin in an outpatient setting [abstract no. eP408]. In: 24th European Congress of Clinical Microbiology and Infectious Disease. 2014.
Puttagunta S, Talbot GH, Boucher H, et al. Safety and efficacy of dalbavancin in patients with renal impairment treated for skin infections [abstract no. L-1731]. In: 54th Interscience Conference on Antimicrobial Agents and Chemotherapy. 2014.
Dunne M, Puttagunta S, Wilcox M. Concordance of clinical response at 48-72 hours after initiation of therapy and end of treatment (EOT) in patients with acute bacterial skin and skin structure infection (abSSSI) in the DISCOVER studies [abstract no. 1340 plus poster]. In: Infectious Diseases Week Meeting. 2013.
Puttagunta S, Talbot G, Wilcox M, et al. Geographic differences in the presentation and outcomes of acute bacterial skin and skin structure infections (ABSSSI) in the DISCOVER program [abstract no. P1818]. In: 24th European Congress of Clinical Microbiology and Infectious Disease. 2014.
Anderson VR, Keating GM. Dalbavancin. Drugs. 2008;68(5):639–48.
Dunne M, Puttagunta S. Clearance of Staphylococcus aureus bacteremia in patients treated with dalbavancin [abstract no. 1343 plus poster]. In: Infectious Diseases Week Meeting. 2013.
The Medicines Company. Orbactiv (oritavancin) for injection, for intravenous use: US prescribing information. 2014. http://www.themedicinescompany.com. Accessed 18 Mar 2015.
European Medicines Agency. Orbactiv 400 mg powder for concentrate solution for infusion: summary of product characteristics. 2015. http://www.ema.europa.eu. Accessed 11 May 2015.
PRNewswire. Actavis announces topline phase 3 clinical trial results for single-dose Dalvance® (dalbavancin) in the treatment of ABSSSI. 2015. http://www.prnewswire.com/news-releases/. Accessed 12 May 2015.
US FDA. Vancomycin hydrochloride injection, solution: US prescribing information. 2008. http://www.accessdata.fda.gov. Accessed 19 Mar 2015.
Theravance Biopharma US Inc. Vibativ® (telavancin) for injection, for intravenous use: US prescribing information. 2014. http://www.vibativ.com/. Accessed 19 Mar 2015.
Disclosure
The preparation of this review was not supported by any external funding. During the peer review process, the manufacturer of the agent under review was offered an opportunity to comment on this article. Changes resulting from comments received were made by the author on the basis of scientific and editorial merit. Lesley Scott is a salaried employee of Adis/Springer.
Author information
Authors and Affiliations
Corresponding author
Additional information
The manuscript was reviewed by: H. W. Boucher, Division of Geographic Medicine and Infectious Diseases, Tufts Medical Center, Boston, MA, USA; B. M. Lomaestro, Albany Medical Center Hospital, Albany, NY, USA; M. Zeitlinger, Medical University of Vienna, Clinical Pharmacology, Vienna, Austria.
Rights and permissions
About this article

Cite this article
Scott, L.J. Dalbavancin: A Review in Acute Bacterial Skin and Skin Structure Infections. Drugs 75, 1281–1291 (2015). https://doi.org/10.1007/s40265-015-0430-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-015-0430-x